Co-reporter:Hao Cai, Xiaojing Huang, Shengtao Xu, Hao Shen, Pengfei Zhang, Yue Huang, Jieyun Jiang, Yijun Sun, Bo Jiang, Xiaoming Wu, Hequan Yao, Jingyi Xu
European Journal of Medicinal Chemistry 2016 Volume 108() pp:89-103
Publication Date(Web):27 January 2016
DOI:10.1016/j.ejmech.2015.11.013
•Novel COX-2/5-LOX dual inhibitors were designed via pharmacophore hybrid approach.•Most compounds showed COX-2/5-LOX inhibitory and anti-proliferative activities.•The compound 15c was found to induce apoptosis and G2/M phase cell cycle arrest.•The most potent compound 22b significantly inhibited tumor growth in vivo.•The docking studies revealed possible binding mode of these compounds.Inflammation plays a key role in cancer initiation and propagation. Cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX), two important enzymes in inflammatory responses are up-regulated in various tumor types. Dual inhibition of COX-2 and 5-LOX constitutes a rational concept for the design of more efficacious anti-tumor agents with an improved safety profile. We have previously reported a series of diaryl-1,2,4-triazole derivatives as selective COX-2 inhibitors. Herein, we hybridized the diaryl-1,2,4-triazoles with caffeic acid (CA) which was reported to display 5-LOX inhibitory and anti-tumor activities, affording a novel class of COX-2/5-LOX dual inhibitors as anti-tumor drug candidates. Most of these compounds exhibited potent COX-2/5-LOX inhibitory and antiproliferative activities in vitro. And the most potent compound 22b could significantly inhibit tumor growth in vivo. Furthermore, mechanistic investigation showed that the representative compound 15c blocked cell cycle in G2 phase and induced apoptosis in human non-small cell lung cancer A549 cells in a dose-dependent manner. Our preliminary investigation results would provide new clues for the cancer theatment with COX-2/5-LOX dual inhibitors.
Co-reporter:Haipin Zhou, Kuo Gai, Aijun Lin, Jinyi Xu, Xiaoming Wu and Hequan Yao
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 4) pp:1243-1248
Publication Date(Web):14 Nov 2014
DOI:10.1039/C4OB01844C
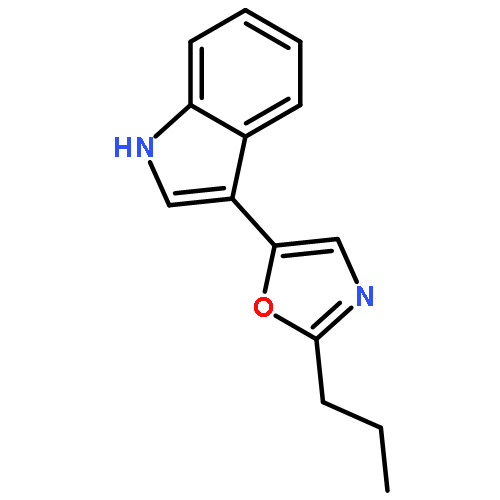
An efficient and straightforward method for the production of 5-(3-indolyl)azoles incorporating the privileged structures indoles and azoles via palladium-catalyzed double C–H bond cleavage under mild conditions was disclosed. As expected, this protocol provided an easy method for the synthesis of indole alkaloids pimprinine and WS-30581 A in moderate yields.
Co-reporter:Yanchun Zhang;Jinyi Xu;Yunman Li;Hequan Yao;Xiaoming Wu
Chemical Biology & Drug Design 2015 Volume 85( Issue 5) pp:541-548
Publication Date(Web):
DOI:10.1111/cbdd.12442
Two series of novel NO-releasing benzimidazole derivatives (8a–e, 9a–g) were designed and synthesized by coupling nitro ester and furoxan NO-donor moieties with benzimidazole biphenyl skeleton. The NO-releasing assay indicated that all the target compounds had different level of NO-releasing ability. Furthermore, the isolated organ assay (rat aortic strips) was used to evaluate the antagonism of Ang II-induced vasoconstriction ability. It was observed that the pA2 values of compounds 8e and 9e were better than that of lead compound 6. Moreover, the pharmacological investigation showed that the antagonism of Ang II-induced pressure response by oral administration of compound 8e was obviously superior to that of lead compound 6, and comparable to that of the positive control losartan. These results suggested that NO-releasing hybrids may provide a promising approach for the discovery of novel antihypertensive agents.
Co-reporter:Long Liu, Cheng-Qian Wang, Dan Liu, Wei-Gang He, Jin-Yi Xu, Ai-Jun Lin, He-Quan Yao, Genzoh Tanabe, Osamu Muraoka, Wei-Jia Xie, and Xiao-Ming Wu
Organic Letters 2014 Volume 16(Issue 19) pp:5004-5007
Publication Date(Web):September 15, 2014
DOI:10.1021/ol5022838
A novel synthetic approach to construct various 3,6-anhydrohexosides via an intramolecular cyclization of corresponding triflates is described. The nucleophilic attack from C3 p-methoxybenzylated hydroxyl to C6 trifluoromethanesulfonate on triflate structures triggered the cyclization reaction to provide 3,6-anhydrohexosides in excellent yields, making the strategy more efficient with respect to the reported protocols. By applying this methodology, a concise first total synthesis of natural product isolated from leaves of Sauropus rostratus was accomplished.
Co-reporter:Bo Jiang, Xiaojing Huang, Hequan Yao, Jieyun Jiang, Xiaoming Wu, Siyi Jiang, Qiujuan Wang, Tao Lu and Jinyi Xu
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 13) pp:2114-2127
Publication Date(Web):14 Jan 2014
DOI:10.1039/C3OB41936C
A series of hybrids from diaryl-1,2,4-triazole and hydroxamic acid or N-hydroxyurea were synthesized and evaluated as novel anti-inflammatory agents. The biological data showed that (i) all the compounds showed dual COX-2/5-LOX inhibitory activities in vitro, and 15e showed optimal inhibitory activities (COX-2: IC50 = 0.15 μM, 5-LOX: IC50 = 0.85 μM), (ii) 15e selectively inhibited COX-2 relative to COX-1 with selectivity index (SI = 0.012) comparable to celecoxib (SI = 0.015), (iii) 15e exhibited potent anti-inflammatory activity (inhibition: 54.1%) which was comparable to the reference drug celecoxib (inhibition: 46.7%) in a xylene-induced ear edema assay, and (iv) 15e displayed promising analgesic activity in acetic acid-induced writhing response and hot-plate assay. Finally, a molecular modeling study revealed the binding interactions of 15e with COX-2 and 5-LOX. Our findings suggest that 15e may be a promising anti-inflammatory agent for further evaluation.
Co-reporter:Dahong Li, Hao Cai, Bowen Jiang, Guyue Liu, Yuetong Wang, Lei Wang, Hequan Yao, Xiaoming Wu, Yijun Sun, Jinyi Xu
European Journal of Medicinal Chemistry 2013 Volume 59() pp:322-328
Publication Date(Web):January 2013
DOI:10.1016/j.ejmech.2012.11.002
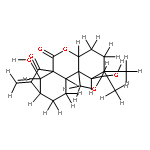
A series of novel spirolactone-type diterpenoid derivatives of oridonin (12a–j) were designed and synthesized. All the target compounds showed improved anti-proliferative activity against a panel of human cancer cell lines and the most effective compound 12j was more potent than positive control Taxol in K562 and Bel-7402 cells with IC50 values of 0.39 μM and 1.39 μM, respectively. The cellular mechanisms showed that compound 12j induced apoptosis at low micromolar concentrations in human hepatoma Bel-7402 cells. These results demonstrate that the spirolactone-type diterpenoid derivatives of oridonin have optimized growth inhibitory activity against cancer cells and interesting apoptosis-inducing ability.Graphical abstractThe most effective synthetic spirolactone-type diterpenoid analog 12j exhibited similar anti-proliferative activity as the positive control Taxol and induced apoptosis at low micromolar concentrations in human hepatoma Bel-7402 cells.Highlights► Spirolactone-type diterpenoids could be got from commercial available oridonin. ► A series of derivatives with improved anti-proliferative activities were synthesized. ► Compound 12j showed similar anti-proliferative activity as Taxol in vitro. ► Induction of apoptosis and influence of cell cycle by 12j were investigated. ► The structure–activity relationships of the derivatives were concluded.
Co-reporter:Dan Liu, Weijia Xie, Long Liu, Hequan Yao, Jinyi Xu, Genzoh Tanabe, Osamu Muraoka, Xiaoming Wu
Tetrahedron Letters 2013 Volume 54(Issue 47) pp:6333-6336
Publication Date(Web):20 November 2013
DOI:10.1016/j.tetlet.2013.09.044
Coupling reaction between thiosugar and triflate as the key protocol to synthesize neoponkoranol, a naturally occurring potent α-glucosidase inhibitor, and its related sulfonium salts was optimized by applying different esters as protecting group, with the yields of desired products being greatly improved. Our proposed mechanism of the coupling reaction indicated that the nucleophilicity of C3-hydroxyl moiety on monosaccharide structure is closely related to the reaction mode.
Co-reporter:Lei Zhao, Ziyuan Li, Lin Chang, Jinyi Xu, Hequan Yao, and Xiaoming Wu
Organic Letters 2012 Volume 14(Issue 8) pp:2066-2069
Publication Date(Web):March 30, 2012
DOI:10.1021/ol300584m
An efficient construction of fused indolines with a 2-quaternary center through a palladium-catalyzed intramolecular Heck reaction of N-(2(2-halobenzoxyl)-2,3-disubstituted indoles is disclosed. This protocol provided a straightforward access to diverse fused indolines with good functional group tolerance.
Co-reporter:Renren Bai, Zhen Wei, Jie Liu, Weijia Xie, Hequan Yao, Xiaoming Wu, Jieyun Jiang, Qiujuan Wang, Jinyi Xu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 15) pp:4661-4667
Publication Date(Web):1 August 2012
DOI:10.1016/j.bmc.2012.06.011
A series of 4′-[(benzimidazole-1-yl)methyl]biphenyl-2-sulfonamide derivatives (Ia–Il) were synthesized and biologically evaluated. It was found that Ig, the most active compound, antagonized both Ang II AT1 and endothelin ETA receptors (AT1 IC50 = 8.5, ETA IC50 = 8.9 nM), and was more potent than losartan in RHRs with no significant effect on heart rate. The preliminary structure–activity relationships were also discussed in the present paper.A series of 4′-[(benzimidazole-1-yl)methyl]biphenyl-2-sulfonamide derivatives (Ia–Il) were synthesized and biologically evaluated. The most prospective compound Ig was found to have the most effective antagonism for Ang II AT1 and endothelin ETA receptors, exhibiting more potent activity (AT1 IC50 = 8.5 nM, ETA IC50 = 8.9 nM) than losartan and equivalent activity to bosentan, and was more potent than losartan in RHRs with no significant effect on heart rate.
Co-reporter:Yong Wu, Chen Shi, Xiaowei Sun, Xiaoming Wu, Hongbin Sun
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 14) pp:4238-4249
Publication Date(Web):15 July 2011
DOI:10.1016/j.bmc.2011.05.059
Eighteen octane-carboxamide based renin inhibitors with extended segments for mimicking P3′ unit of angiotensinogen have been synthesized. The biological evaluation identified novel renin inhibitors with more potent activity than aliskiren. Molecular docking studies showed that the extended amide-tails matched the P3′ position of angiotensinogen and exerted interactions with the S3′ site of renin. An unexpected π–π stacking interaction was observed during docking study for compound 9r, which could be a reasonable explanation for the outstanding potency of this compound. Further study is in progress to reveal a feasibility for developing novel renin inhibitors based on the possible non-classical interactions between the ligands and the new subsite of renin.
Co-reporter:Haifeng Gan, Yunyu Lu, Yue Huang, Lijun Ni, Jinyi Xu, Hequan Yao, Xiaoming Wu
Tetrahedron Letters 2011 Volume 52(Issue 12) pp:1320-1324
Publication Date(Web):23 March 2011
DOI:10.1016/j.tetlet.2011.01.058
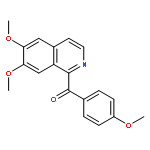
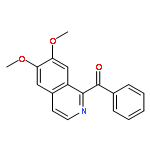
An environmental-benign methodology to synthesize 1-benzoylisoquinolines from 1-benzyl-3, 4-dihydroisoquinolines or 1-benzyl-1,2,3,4-tetrahydroisoquinolines using dioxygen as an oxidant was developed. This methodology in combination with Bischler-Napieralski reaction leads to a facile synthesis of 1-benzoylisoquinolines from phenylacetic acids and phenylethanamines.
Co-reporter:Changhua Tang, Ziyuan Li, Yiyun Wang, Jinyi Xu, Lingyi Kong, Hequan Yao, Xiaoming Wu
Tetrahedron Letters 2011 Volume 52(Issue 26) pp:3275-3278
Publication Date(Web):29 June 2011
DOI:10.1016/j.tetlet.2011.04.069
The first total synthesis of a sesquiterpenoid, tenuifolin, was achieved in seven linear steps. Phenyliodine(III) bis(trifluoacetate) (PIFA) mediated oxidative biaryl coupling was employed as a key step to construct the central seven-membered ring with a double bond. The double bond formation was also exploited.
Co-reporter:Ziyuan Li;Yiyun Wang;Changhua Tang;Jinyi Xu;Xiaoming Wu;Hequan Yao
Chinese Journal of Chemistry 2010 Volume 28( Issue 7) pp:1301-1305
Publication Date(Web):
DOI:10.1002/cjoc.201090225
Abstract
Dehydrogenation by IBX/p-TsOH is applied to the conversion of 3-benzoyl propionates/propionamides to 3-benzoyl acrylates/acrylamides in moderate to excellent yields. The reaction time for the dehydrogenation of 3-benzoyl propionamides was remarkably shorter than that for the dehydrogenation of esters.
Co-reporter:Zhang Yu Yao, Hao Zhang, Huan Ming Sheng, Xiao Ming Wu, Hong Bin Sun
Chinese Chemical Letters 2010 Volume 21(Issue 11) pp:1334-1337
Publication Date(Web):November 2010
DOI:10.1016/j.cclet.2010.06.022
Dihydrotetrabenazine (DTBZ) is the major pharmacologically active form of tetrabenazine (TBZ), which was approved by FDA for the treatment of chorea associated with Huntington's disease (HD). An unexpected Hoffmann elimination was observed during the treatment of DTBZ with sodium hydrogen and alkyl halides, leading to the formation of both eliminated products (major) and hydroxyl-alkylated products (minor).
Co-reporter:Yan Chun Zhang, Jin Pei Zhou, Xiao Ming Wu, Wei Hong Pan
Chinese Chemical Letters 2009 Volume 20(Issue 3) pp:302-305
Publication Date(Web):March 2009
DOI:10.1016/j.cclet.2008.11.012
A series of novel nitric oxide-donating derivatives (7a–e, 8a–e) were synthesized by coupling furoxan and nitric oxide with irbesartan analogue and their cytotoxicity against BEL7402 cells in vitro were evaluated by MTT method. It was found that 8c exhibits the most cytotoxic activities with IC50 value of 12.5 μmol/L. The hybrids of AT1 antagonist and nitric oxide donor appear to have beneficial effects on antitumor.
Co-reporter:Jie Liu, Hao Ren, Jinyi Xu, Renren Bai, Qi Yan, Wenlong Huang, Xiaoming Wu, Jihua Fu, Qiujuan Wang, Qian Wu, Rong Fu
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 6) pp:1822-1824
Publication Date(Web):15 March 2009
DOI:10.1016/j.bmcl.2008.12.102
This letter describes the total synthesis, preliminary biological evaluation and mechanism studies of a novel and structurally unique isochromanone, (±)7,8-dihydroxy-3-methyl-isochromanone-4 (1), a nature product contained in banana (Musa sapientum L.) peel. The bioassay showed that compound 1 displays potent antihypertensive activity in renal hypertensive rats and further mechanism studies revealed that it is an ACE inhibitor.The total synthesis of (±)7,8-dihydroxy-3-methyl-isochromanone-4 (1) is described and this compound displays potent antihypertensive activity and moderate ACE inhibitory activity.
Co-reporter:Peiqing Zhu, Yi Bi, Jinyi Xu, Zan Li, Jun Liu, Luyong Zhang, Wencai Ye, Xiaoming Wu
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 24) pp:6966-6969
Publication Date(Web):15 December 2009
DOI:10.1016/j.bmcl.2009.10.055
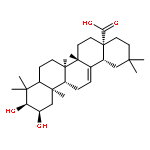
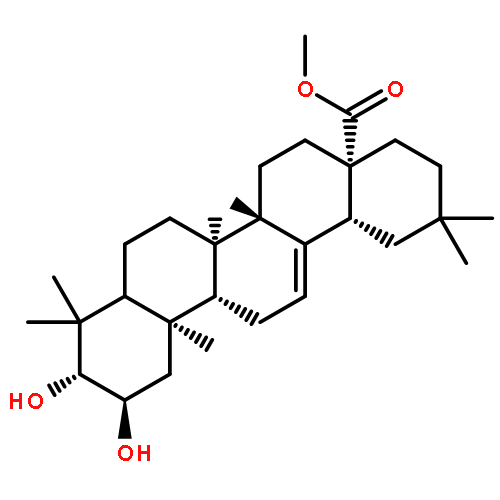
A series of 23-hydroxybetulinic acid derivatives were prepared and tested in vitro as a new class of inhibitors of glycogen phosphorylase (GP). Within this series of compounds, 12b (IC50 = 3.5 μM) is the most potent GPa inhibitor. The preliminary SAR results of the 23-hydroxybetulinic acid derivatives are discussed.A series of 23-hydroxybetulinic acid derivatives were prepared and evaluated as a new class of inhibitors of glycogen phosphorylase (GP), among which 12b was the most potent GPa inhibitor (IC50 = 3.5 μM).
Co-reporter:Jinyi Xu, JingYi Yang, Qian Ran, Lei Wang, Jie Liu, Zhixuan Wang, Xiaoming Wu, Weiyi Hua, Shengtao Yuan, Luyong Zhang, Mingqin Shen, Yongfang Ding
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 16) pp:4741-4744
Publication Date(Web):15 August 2008
DOI:10.1016/j.bmcl.2008.06.097
Novel 1-O- and 14-O-derivatives of oridonin were synthesized and biologically evaluated. All of the derivatives exhibited stronger cytotoxicity against six cancer cell lines (BGC-7901, SW-480, HL-60, BEL-7402, A549, and B16) than oridonin in vitro, and some of them were more potent than oridonin and cyclophosphamide in vivo. Compounds Ib and IIg were the most potent with the IC50 values of 0.84 μM for Ib in HL-60 cell and 1.00 μM for IIg in BEL-7402 cell.1-O- and 14-O-derivatives of oridonin exhibited stronger cytotoxicity against six cancer cell lines than oridonin in vitro, compounds Ib and IIg were more potent than oridonin and cyclophosphamide in vivo.
Co-reporter:Jin Yi Xu, Yi Zeng, Qian Ran, Zhen Wei, Yi Bi, Qian Hui He, Qiu Juan Wang, Song Hu, Jing Zhang, Ming Yue Tang, Wei Yi Hua, Xiao Ming Wu
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 10) pp:2921-2926
Publication Date(Web):15 May 2007
DOI:10.1016/j.bmcl.2007.02.042
A series of 2-alkylbenzimidazoles bearing a N-phenylpyrrole moiety were synthesized and evaluated as a novel class of AT1 receptor antagonists. Among them, compounds 10a and 10g inhibited [125I] AngII-binding affinity to AT1 receptor at nanomolar level and potently inhibited the Ang II-induced pressor response by oral administration. Moreover, evaluation in spontaneously hypertensive rats showed that 10a is an orally active AT1 receptor antagonist.2-Alkylbenzimidazoles bearing a N-phenylpyrrole moiety 10a and 10g inhibited [125I] AngII-binding affinity to AT1 receptor at nanomolar level and evaluation in spontaneously hypertensive rats showed that 10a is an orally active AT1 receptor antagonist.
Co-reporter:Jin Yi Xu, Qian Ran, Wei Yi Hua, Xiao Ming Wu, Qiu Juan Wang, Jing Zhang
Chinese Chemical Letters 2007 Volume 18(Issue 3) pp:251-254
Publication Date(Web):March 2007
DOI:10.1016/j.cclet.2006.12.029
A series of 2-alkylbenzimidazole derivatives 9a–n have been designed and synthesized as a novel class of non-peptide angiotensin II AT1 receptor antagonists. The synthesized compounds were evaluated for their antagonism of angiotensin II, induced contraction in the rabbit thoracic aortic ring and the results showed that compounds 9a, 9g and 9j exhibited potent antagonistic activity of AT1 receptor.
Co-reporter:Yi Bi, Jinyi Xu, Xiaoming Wu, Wencai Ye, Shengtao Yuan, Luyong Zhang
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 5) pp:1475-1478
Publication Date(Web):1 March 2007
DOI:10.1016/j.bmcl.2006.09.096
New 17-carboxylic acid modified 23-hydroxy betulinic acid ester derivatives were prepared and tested for cytotoxic activity on five cancer cell lines in vitro: all tested compounds showed stronger cytotoxic activity than 23-hydroxy betulinic acid and betulinic acid. In addition, compound 5a was tested for anti-tumor activity in vivo: it had much better anti-tumor activity than 23-OH betulinic acid and had similar anti-tumor activity with cyclophosphamide and 5-fluorouracil.The derivatives 5a–f of 23-hydroxy betulinic acid have better cytotoxicity than 23-hydroxy betulinic acid and betulinic acid in vitro. And they are also more potent than 23-hydroxy betulinic acid in vivo. The research results have been applied China Patent: CN 10040277.2, 2006.
Co-reporter:Bo Jiang, Xiaojing Huang, Hequan Yao, Jieyun Jiang, Xiaoming Wu, Siyi Jiang, Qiujuan Wang, Tao Lu and Jinyi Xu
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 13) pp:NaN2127-2127
Publication Date(Web):2014/01/14
DOI:10.1039/C3OB41936C
A series of hybrids from diaryl-1,2,4-triazole and hydroxamic acid or N-hydroxyurea were synthesized and evaluated as novel anti-inflammatory agents. The biological data showed that (i) all the compounds showed dual COX-2/5-LOX inhibitory activities in vitro, and 15e showed optimal inhibitory activities (COX-2: IC50 = 0.15 μM, 5-LOX: IC50 = 0.85 μM), (ii) 15e selectively inhibited COX-2 relative to COX-1 with selectivity index (SI = 0.012) comparable to celecoxib (SI = 0.015), (iii) 15e exhibited potent anti-inflammatory activity (inhibition: 54.1%) which was comparable to the reference drug celecoxib (inhibition: 46.7%) in a xylene-induced ear edema assay, and (iv) 15e displayed promising analgesic activity in acetic acid-induced writhing response and hot-plate assay. Finally, a molecular modeling study revealed the binding interactions of 15e with COX-2 and 5-LOX. Our findings suggest that 15e may be a promising anti-inflammatory agent for further evaluation.
Co-reporter:Haipin Zhou, Kuo Gai, Aijun Lin, Jinyi Xu, Xiaoming Wu and Hequan Yao
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 4) pp:NaN1248-1248
Publication Date(Web):2014/11/14
DOI:10.1039/C4OB01844C
An efficient and straightforward method for the production of 5-(3-indolyl)azoles incorporating the privileged structures indoles and azoles via palladium-catalyzed double C–H bond cleavage under mild conditions was disclosed. As expected, this protocol provided an easy method for the synthesis of indole alkaloids pimprinine and WS-30581 A in moderate yields.

![1H-Benzimidazole, 1-[(4-bromophenyl)methyl]-](http://img.cochemist.com/ccimg/73800/73798-61-5.png)
![1H-Benzimidazole, 1-[(4-bromophenyl)methyl]-](http://img.cochemist.com/ccimg/73800/73798-61-5_b.png)


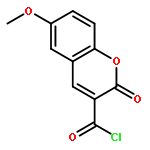





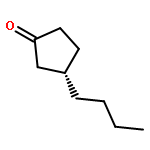
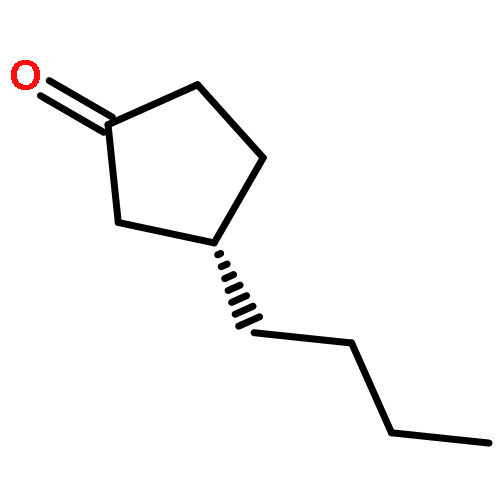
![Isoquinoline,3,4-dihydro-6,7-dimethoxy-1-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/72600/72527-29-8.png)
![Isoquinoline,3,4-dihydro-6,7-dimethoxy-1-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/72600/72527-29-8_b.png)


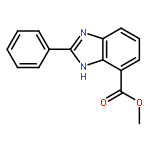









![1H-Imidazo[4,5-b]pyridine, 2-butyl-](http://img.cochemist.com/ccimg/68200/68175-10-0.png)
![1H-Imidazo[4,5-b]pyridine, 2-butyl-](http://img.cochemist.com/ccimg/68200/68175-10-0_b.png)
![1H-Imidazo[4,5-b]pyridine, 2-propyl-](http://img.cochemist.com/ccimg/68200/68175-09-7.png)
![1H-Imidazo[4,5-b]pyridine, 2-propyl-](http://img.cochemist.com/ccimg/68200/68175-09-7_b.png)
![2-ethyl-3H-Imidazo[4,5-b]pyridine](http://img.cochemist.com/ccimg/68200/68175-08-6.png)
![2-ethyl-3H-Imidazo[4,5-b]pyridine](http://img.cochemist.com/ccimg/68200/68175-08-6_b.png)













![ISOQUINOLINE, 1-[(4-CHLOROPHENYL)METHYL]-3,4-DIHYDRO-6,7-DIMETHOXY-](http://img.cochemist.com/ccimg/47300/47216-54-6.png)
![ISOQUINOLINE, 1-[(4-CHLOROPHENYL)METHYL]-3,4-DIHYDRO-6,7-DIMETHOXY-](http://img.cochemist.com/ccimg/47300/47216-54-6_b.png)


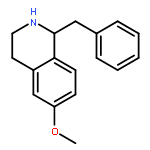
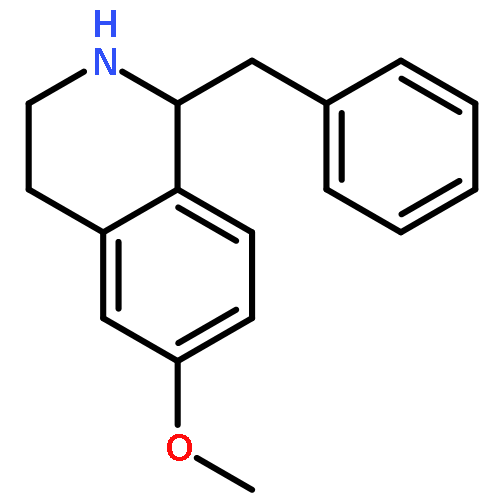



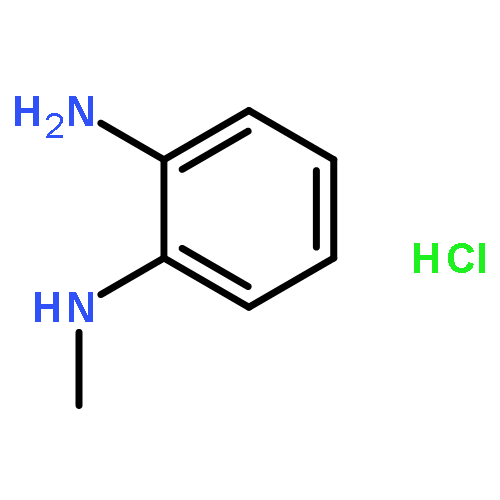
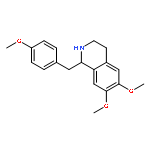

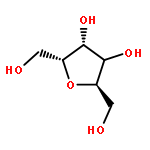





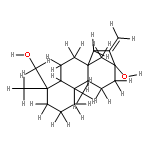





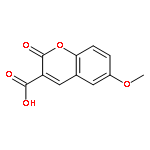





















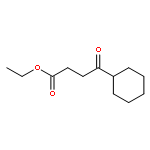
![Oxirane, [[2-(phenylmethoxy)phenoxy]methyl]-](http://img.cochemist.com/ccimg/22600/22530-53-6.png)
![Oxirane, [[2-(phenylmethoxy)phenoxy]methyl]-](http://img.cochemist.com/ccimg/22600/22530-53-6_b.png)





![Butanoic acid,2,3-dihydroxy-4-[(2-nitrophenyl)amino]-4-oxo-,[R-(R*,R*)]- (9CI)](http://img.cochemist.com/ccimg/19600/19523-81-0.png)
![Butanoic acid,2,3-dihydroxy-4-[(2-nitrophenyl)amino]-4-oxo-,[R-(R*,R*)]- (9CI)](http://img.cochemist.com/ccimg/19600/19523-81-0_b.png)











![Isoquinoline,3,4-dihydro-6,7-dimethoxy-1-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/10300/10268-39-0.png)
![Isoquinoline,3,4-dihydro-6,7-dimethoxy-1-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/10300/10268-39-0_b.png)
![Isoquinoline, 1,2,3,4-tetrahydro-6,7-dimethoxy-1-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/10300/10268-26-5.png)
![Isoquinoline, 1,2,3,4-tetrahydro-6,7-dimethoxy-1-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/10300/10268-26-5_b.png)




















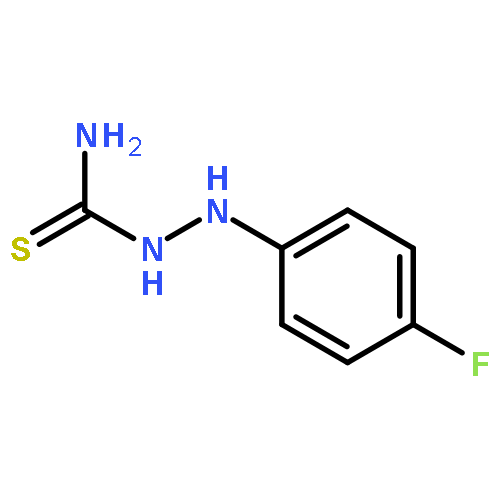
![Hydrazinecarbothioamide, 2-[(2-nitrophenyl)methylene]-](http://img.cochemist.com/ccimg/6100/6072-63-5.png)
![Hydrazinecarbothioamide, 2-[(2-nitrophenyl)methylene]-](http://img.cochemist.com/ccimg/6100/6072-63-5_b.png)


















![3-[1-(1H-INDOL-3-YL)-3-METHYL-2-BUTEN-1-YL]-7-(3-METHYL-2-BUTEN-1<WBR />-YL)-1H-INDOLE](http://img.cochemist.com/ccimg/3800/3757-06-0.png)
![3-[1-(1H-INDOL-3-YL)-3-METHYL-2-BUTEN-1-YL]-7-(3-METHYL-2-BUTEN-1<WBR />-YL)-1H-INDOLE](http://img.cochemist.com/ccimg/3800/3757-06-0_b.png)
![2H-Benzo[a]quinolizin-2-ol,1,3,4,6,7,11b-hexahydro-9,10-dimethoxy-3-(2-methylpropyl)-](http://img.cochemist.com/ccimg/3500/3466-75-9.png)
![2H-Benzo[a]quinolizin-2-ol,1,3,4,6,7,11b-hexahydro-9,10-dimethoxy-3-(2-methylpropyl)-](http://img.cochemist.com/ccimg/3500/3466-75-9_b.png)






















![Ethyl 2-[4-(trifluoromethyl)phenyl]thiazole-4-carboxylate](http://img.cochemist.com/ccimg/175300/175204-88-3.png)
![Ethyl 2-[4-(trifluoromethyl)phenyl]thiazole-4-carboxylate](http://img.cochemist.com/ccimg/175300/175204-88-3_b.png)



















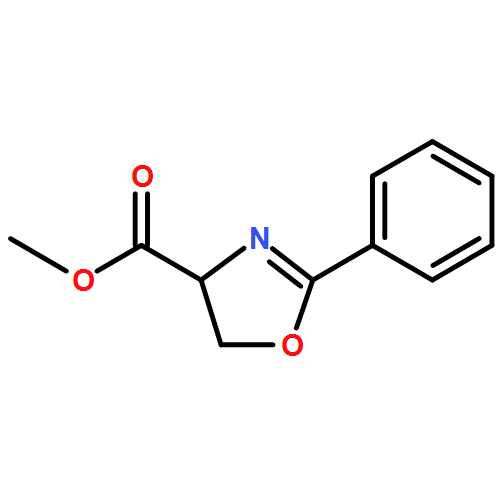


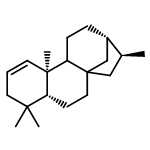
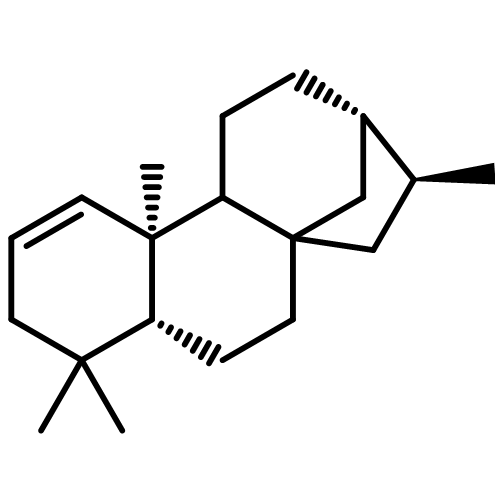
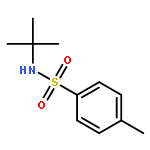






![ACETATE, 2,2',2'',2'''-[(1R,2R)-1,2-CYCLOHEXANEDIYLDINITRILO]TETR<WBR />AKIS-, COPPER(2+) SALT (1:1)](http://img.cochemist.com/ccimg/300900/300800-07-1.png)
![ACETATE, 2,2',2'',2'''-[(1R,2R)-1,2-CYCLOHEXANEDIYLDINITRILO]TETR<WBR />AKIS-, COPPER(2+) SALT (1:1)](http://img.cochemist.com/ccimg/300900/300800-07-1_b.png)
![METHYL 1-[4-(BROMOMETHYL)PHENYL]PYRROLE-2-CARBOXYLATE](http://img.cochemist.com/ccimg/144100/144062-63-5.png)
![METHYL 1-[4-(BROMOMETHYL)PHENYL]PYRROLE-2-CARBOXYLATE](http://img.cochemist.com/ccimg/144100/144062-63-5_b.png)







![1-[4-(BROMOMETHYL)PHENYL]PYRROLE-2-CARBONITRILE](http://img.cochemist.com/ccimg/142100/142044-78-8.png)
![1-[4-(BROMOMETHYL)PHENYL]PYRROLE-2-CARBONITRILE](http://img.cochemist.com/ccimg/142100/142044-78-8_b.png)







![4-Thiazolecarbonylchloride, 2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/857300/857284-28-7.png)
![4-Thiazolecarbonylchloride, 2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/857300/857284-28-7_b.png)




![9H-CARBAZOLE, 9-[(2-BROMOPHENYL)METHYL]-](http://img.cochemist.com/ccimg/757300/757233-20-8.png)
![9H-CARBAZOLE, 9-[(2-BROMOPHENYL)METHYL]-](http://img.cochemist.com/ccimg/757300/757233-20-8_b.png)
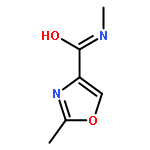
![4-OXAZOLECARBOXYLIC ACID, 2-[4-(TRIFLUOROMETHYL)PHENYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/753500/753479-58-2.png)
![4-OXAZOLECARBOXYLIC ACID, 2-[4-(TRIFLUOROMETHYL)PHENYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/753500/753479-58-2_b.png)










![1-{2-[4-(Trifluoromethyl)phenyl]-1,3-thiazol-4-yl}ethan-1-one](http://img.cochemist.com/ccimg/263600/263564-37-0.png)
![1-{2-[4-(Trifluoromethyl)phenyl]-1,3-thiazol-4-yl}ethan-1-one](http://img.cochemist.com/ccimg/263600/263564-37-0_b.png)

![Ethanone, 1-[2-[4-(trifluoromethyl)phenyl]-4-thiazolyl]-, oxime](http://img.cochemist.com/ccimg/206700/206653-23-8.png)
![Ethanone, 1-[2-[4-(trifluoromethyl)phenyl]-4-thiazolyl]-, oxime](http://img.cochemist.com/ccimg/206700/206653-23-8_b.png)






![5H-Indolo[3,2-c]quinoline,5-methyl-](http://img.cochemist.com/ccimg/165500/165467-65-2.png)
![5H-Indolo[3,2-c]quinoline,5-methyl-](http://img.cochemist.com/ccimg/165500/165467-65-2_b.png)

![Hydrazinecarbothioamide, 2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/147200/147162-44-5.png)
![Hydrazinecarbothioamide, 2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/147200/147162-44-5_b.png)


![1H-Indole, 5-methoxy-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/139800/139717-71-8.png)
![1H-Indole, 5-methoxy-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/139800/139717-71-8_b.png)









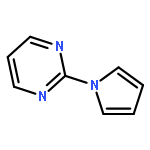
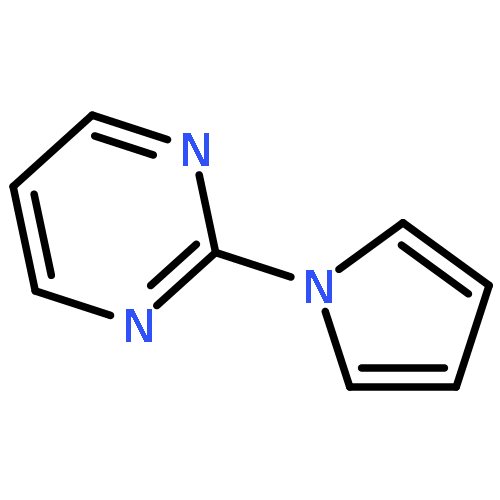
![1H-Indole, 4-methyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/113000/112970-65-7.png)
![1H-Indole, 4-methyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/113000/112970-65-7_b.png)


![1H-Indole, 5-methyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/107800/107734-07-6.png)
![1H-Indole, 5-methyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/107800/107734-07-6_b.png)


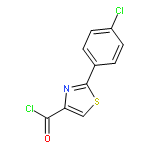

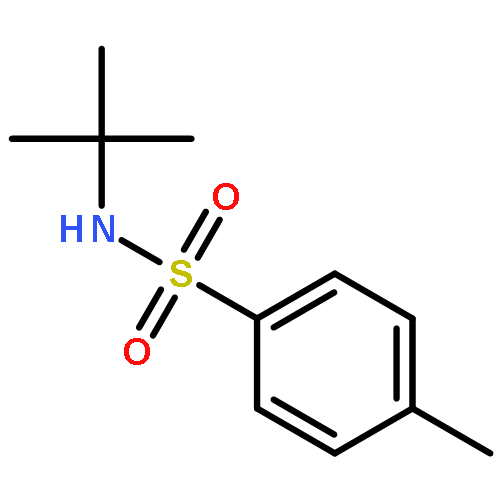
![Benzoic acid,4-[[(1,1-dimethylethyl)amino]sulfonyl]-](http://img.cochemist.com/ccimg/100000/99987-05-0.png)
![Benzoic acid,4-[[(1,1-dimethylethyl)amino]sulfonyl]-](http://img.cochemist.com/ccimg/100000/99987-05-0_b.png)
![2-Propenoyl chloride, 3-[3,4-bis(acetyloxy)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/98700/98631-72-2.png)
![2-Propenoyl chloride, 3-[3,4-bis(acetyloxy)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/98700/98631-72-2_b.png)
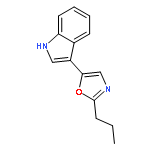





![2-Propenoic acid, 3-[3,4-bis(acetyloxy)phenyl]-, (E)-](http://img.cochemist.com/ccimg/88700/88623-81-8.png)
![2-Propenoic acid, 3-[3,4-bis(acetyloxy)phenyl]-, (E)-](http://img.cochemist.com/ccimg/88700/88623-81-8_b.png)
![methyl 4-[bis(2-chloroethyl)amino]-L-phenylalaninate](http://img.cochemist.com/ccimg/88500/88457-23-2.png)
![methyl 4-[bis(2-chloroethyl)amino]-L-phenylalaninate](http://img.cochemist.com/ccimg/88500/88457-23-2_b.png)
![6H-INDOLO[3,2-C]QUINOLIN-6-ONE, 5,11-DIHYDRO-5-METHYL-](http://img.cochemist.com/ccimg/85200/85149-47-9.png)
![6H-INDOLO[3,2-C]QUINOLIN-6-ONE, 5,11-DIHYDRO-5-METHYL-](http://img.cochemist.com/ccimg/85200/85149-47-9_b.png)


![2,5-Cyclohexadien-1-one, 2,6-bis(1,1-dimethylethyl)-4-[(4-methoxyphenyl)methylene]-](/data/chemimg/663300/71711-98-3.png)
![2,5-Cyclohexadien-1-one, 2,6-bis(1,1-dimethylethyl)-4-[(4-methoxyphenyl)methylene]-](/data/chemimg/663300/71711-98-3_b.png)

![Ethanone, 1-[2-(4-methoxyphenyl)-4-thiazolyl]-](http://img.cochemist.com/ccimg/65900/65823-91-8.png)
![Ethanone, 1-[2-(4-methoxyphenyl)-4-thiazolyl]-](http://img.cochemist.com/ccimg/65900/65823-91-8_b.png)












![Benzaldehyde, 2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/52100/52095-44-0.png)
![Benzaldehyde, 2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/52100/52095-44-0_b.png)









![2-Propenoic acid, 3-[4-(1,1-dimethylethyl)phenyl]-, methyl ester, (2E)-](http://img.cochemist.com/ccimg/36300/36215-20-0.png)
![2-Propenoic acid, 3-[4-(1,1-dimethylethyl)phenyl]-, methyl ester, (2E)-](http://img.cochemist.com/ccimg/36300/36215-20-0_b.png)


![L-ALANINE, 3-(P-[BIS(2-CHLOROETHYL)AMINO]PHENYL)-N-FORMYL-](http://img.cochemist.com/ccimg/35900/35849-41-3.png)
![L-ALANINE, 3-(P-[BIS(2-CHLOROETHYL)AMINO]PHENYL)-N-FORMYL-](http://img.cochemist.com/ccimg/35900/35849-41-3_b.png)

![2-Propenoic acid, 3-[4-(acetyloxy)-3-methoxyphenyl]-, (2E)-](/data/chemimg/1442400/34749-55-8.png)
![2-Propenoic acid, 3-[4-(acetyloxy)-3-methoxyphenyl]-, (2E)-](/data/chemimg/1442400/34749-55-8_b.png)







![Oxayohimban-21-one, 19,20-didehydro-16-ethenyl-17-[(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)oxy]-, (15β,16α,17β)-](/data/chemimg/1799800/23141-26-6.png)
![Oxayohimban-21-one, 19,20-didehydro-16-ethenyl-17-[(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)oxy]-, (15β,16α,17β)-](/data/chemimg/1799800/23141-26-6_b.png)
![5H-Indolo[2,3-a]pyrano[3,4-g]quinolizin-5-one,1-ethenyl-2-(b-D-glucopyranosyloxy)-1,2,7,8,13,13b,14,14a-octahydro-,(1R,2S,13bS,14aS)-](http://img.cochemist.com/ccimg/23200/23141-25-5.png)
![5H-Indolo[2,3-a]pyrano[3,4-g]quinolizin-5-one,1-ethenyl-2-(b-D-glucopyranosyloxy)-1,2,7,8,13,13b,14,14a-octahydro-,(1R,2S,13bS,14aS)-](http://img.cochemist.com/ccimg/23200/23141-25-5_b.png)





![Benzoic acid,4-[bis(2-hydroxyethyl)amino]-, ethyl ester](http://img.cochemist.com/ccimg/15800/15716-30-0.png)
![Benzoic acid,4-[bis(2-hydroxyethyl)amino]-, ethyl ester](http://img.cochemist.com/ccimg/15800/15716-30-0_b.png)




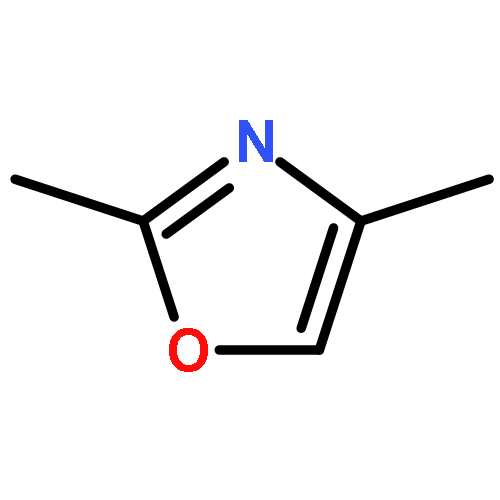
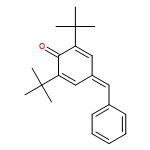
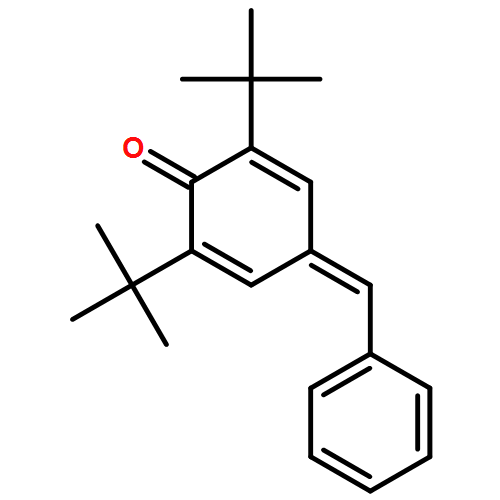


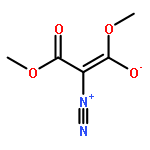




![2-Propanone, 1-diazo-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/2800/2725-60-2.png)
![2-Propanone, 1-diazo-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/2800/2725-60-2_b.png)
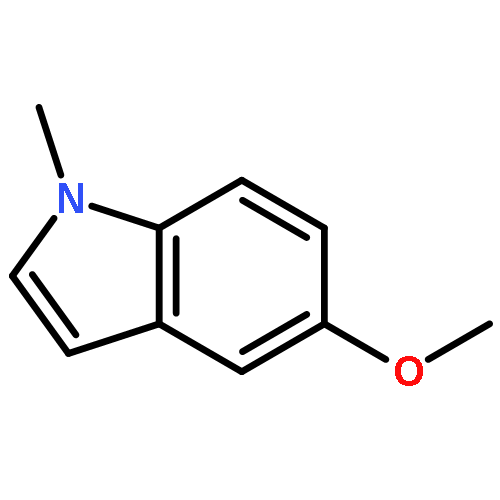




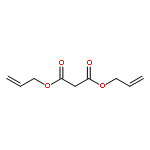
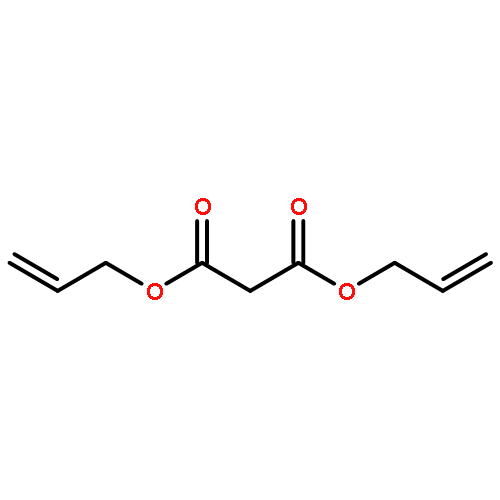


![Benzoic acid,4-[bis(2-chloroethyl)amino]-](http://img.cochemist.com/ccimg/1200/1141-37-3.png)
![Benzoic acid,4-[bis(2-chloroethyl)amino]-](http://img.cochemist.com/ccimg/1200/1141-37-3_b.png)
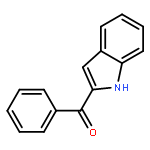


![METHANONE, [4-HYDROXY-3,5-BIS(1-METHYLETHYL)PHENYL]PHENYL-](http://img.cochemist.com/ccimg/800/738-15-8.png)
![METHANONE, [4-HYDROXY-3,5-BIS(1-METHYLETHYL)PHENYL]PHENYL-](http://img.cochemist.com/ccimg/800/738-15-8_b.png)














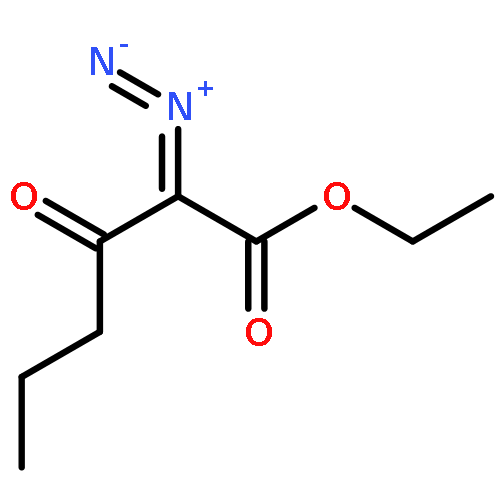
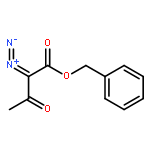
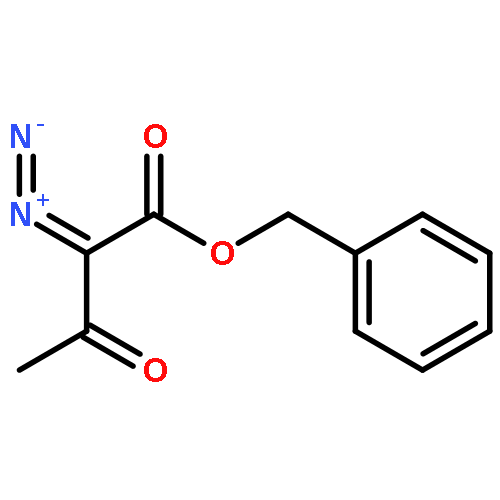




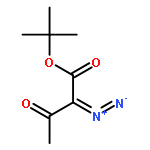
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)