Co-reporter:Minmin Huang;Zhoujie Luo;Tong Zhu;Jian Chen;John Zenghui Zhang
RSC Advances (2011-Present) 2017 vol. 7(Issue 81) pp:51521-51527
Publication Date(Web):2017/11/02
DOI:10.1039/C7RA09485J
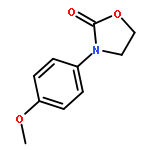
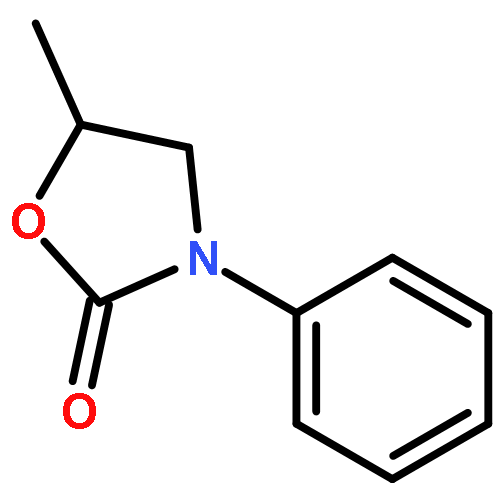
The reaction mechanisms of one-pot conversion of carbon dioxide, ethylene oxide and amines to 3-substituted-2-oxazolidinones catalyzed by the binary ionic liquids of BmimBr and BmimOAc were investigated using DFT methods. In this work, we focus on exploring how the different substituents in amines affect the yields of 3-substituted-2-oxazolidinones. The comparison of calculated free energy profiles and pathways reveals that the electronic structures of the substitutional groups in amines have a substantial influence on the nucleophilic properties of nitrogen atoms of key intermediates, which leads to a discrepancy in the activation barriers. The comparison of the calculated activation barriers of key steps and experimental yields indicates that an anticorrelation relationship exists between them. The current theoretical study inspires us to design new substrates for CO2 conversion by modulating the substituents in substrates.
Co-reporter:Shunying Liu;Jun Jiang;Jianghui Chen;Qinghua Wei;Wenfeng Yao;Wenhao Hu
Chemical Science (2010-Present) 2017 vol. 8(Issue 6) pp:4312-4317
Publication Date(Web):2017/05/30
DOI:10.1039/C7SC00257B
Metal-associated carbenes from diazo compounds promote many useful chemistry transformations in modern organic chemistry. However, compared to α-aryldiazoacetate-derived carbenes (ArDCs), the synthetic application of α-alkyldiazoacetate-derived carbenes (AlDCs) is greatly limited due to intramolecular α-H transfer (elimination) that results in alkenes as the main by-products. An intriguing α-alkyldiazoacetate-involved three-component reaction has been developed following DFT calculation inspiration to provide β-hydroxyl α-alkyl-α-amino acid derivatives in good yields. The intramolecular α-H shift of an α-alkyldiazoacetate-derived carbene was successfully suppressed by the association of a Rh(I) complex to form the corresponding active ammonium ylide, which was trapped before the fast 1,2-H transfer process. A Rh(I)-chiral diene complex was identified as an effective catalyst to give an asymmetric version of the reaction with good enantioselectivity. This reaction provides insight into extending the efficient transformation of α-alkyldiazoacetate-derived carbenes and their synthetic application.
Co-reporter:Yuwei Zhang, Zexing Cao, John Zenghui Zhang, and Fei Xia
Journal of Chemical Information and Modeling 2017 Volume 57(Issue 2) pp:
Publication Date(Web):January 27, 2017
DOI:10.1021/acs.jcim.6b00683
Construction of coarse-grained (CG) models for large biomolecules used for multiscale simulations demands a rigorous definition of CG sites for them. Several coarse-graining methods such as the simulated annealing and steepest descent (SASD) based on the essential dynamics coarse-graining (ED-CG) or the stepwise local iterative optimization (SLIO) based on the fluctuation maximization coarse-graining (FM-CG), were developed to do it. However, the practical applications of these methods such as SASD based on ED-CG are subject to limitations because they are too expensive. In this work, we extend the applicability of ED-CG by combining it with the SLIO algorithm. A comprehensive comparison of optimized results and accuracy of various algorithms based on ED-CG show that SLIO is the fastest as well as the most accurate algorithm among them. ED-CG combined with SLIO could give converged results as the number of CG sites increases, which demonstrates that it is another efficient method for coarse-graining large biomolecules. The construction of CG sites for Ras protein by using MD fluctuations demonstrates that the CG sites derived from FM-CG can reflect the fluctuation properties of secondary structures in Ras accurately.
Co-reporter:Jun Zhou, Siying Lv, Dan Zhang, Fei XiaWenhao Hu
The Journal of Organic Chemistry 2017 Volume 82(Issue 5) pp:
Publication Date(Web):February 7, 2017
DOI:10.1021/acs.joc.6b03017
It has been long recognized that, in chemical glycosylation, the anomeric reactivity of glycosyl donor can be influenced greatly by protecting groups. As opposed to the effects of protecting groups, we report herein a finding on how O-glycosyl substituent can affect the reactivity of oligosaccharyl donor, which in turn should have impact on convergent assembly of oligosaccharide. During our synthetic efforts toward Pichia holstii oligomannoside, a type of α-1,3-linked dimannosyl thioglycosides was found to exhibit unexpected low reactivity toward the activation of NIS/TMSOTf. This observation prompted us to perform a series of comparative reactivity studies, which attributed the donor deactivation to the presence of 3-O-glycosyl substituent, by comparison with O-acetyl group and O-glycosidic linkages at C-4/C-6 positions. To rationalize the unusual phenomenon, we hypothesize that O-glycosyl moiety at C-3 could destabilize the oxocarbenium ion intermediate by additionally increasing the O2–C2–C3–O3 torsional strain, which was further supported by DFT calculation of the hypothetical 4H3-like oxocarbeniums. The observed deactivating influence provides a basis for estimation of donor reactivity and logical selection of synthetic strategy in oligosaccharide synthesis. Following this finding, we opted to use an iterative strategy for the synthesis of targeted pentamannoside 1 by using monomeric thiomannosides that ensured sufficient reactivity.
Co-reporter:Yuwei Zhang, Zexing Cao, Fei Xia
Chemical Physics Letters 2017 Volume 681(Volume 681) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.cplett.2017.05.039
•The resolution of UCG model is lower than a residue-based CG model.•The UCG model can reproduce the fluctuation property of all-atom model accurately.•The UCG model can be used for the simulation of large biomaterials.Ultra-Coarse-Grained (UCG) model whose resolution is less than residue-based coarse-grained model, is being developed in recent years. In this work, we attempt to construct a UCG model with a classical Gō-like potential. The heterogeneous parameters of local and nonlocal interactions in the energy potential function are derived by using fluctuation matching method. The simulated results with UCG models indicate that it could reproduce the root mean square fluctuations as accurate as that of long-time all-atom simulations. Therefore, the UCG model has great potential in the simulation of huge biological systems such as biomaterials.TOC: An ultra-coarse-grained model of Ras protein.Download high-res image (179KB)Download full-size image
Co-reporter:Yuan Liu, Zhunzhun Yu, John Zenghui Zhang, Lu Liu, Fei Xia and Junliang Zhang
Chemical Science 2016 vol. 7(Issue 3) pp:1988-1995
Publication Date(Web):27 Nov 2015
DOI:10.1039/C5SC04319K
In past decade, gold revealed more and more unique properties in carbene chemistry. It was disclosed in our recent communication (J. Am. Chem. Soc. 2014, 136, 6904) that gold carbenes have unprecedented chemo- and site-selectivity and ligand effect toward the functionalization of C–H bonds in phenols. In this full article, we report a comprehensively combined theoretical and experimental study on the mechanism of the insertion of gold carbenes into C–H and O–H bonds in phenol. It significantly revealed that the ligands have an important effect on C–H insertion and the reaction proceeds through a pathway involving the formation of an enolate-like intermediate. Moreover, two water molecules serving as a proton shuttle are believed to be the key issue for achieving chemoselective C–H functionalization, which is strongly supported by the DFT calculations and control experiments. It is the first time that a clear explanation is given about the prominent catalysis of gold carbenes toward C–H functionalization based on a theoretical and experimental study.
Co-reporter:Min Li, John Zenghui Zhang, and Fei Xia
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 4) pp:2091-2100
Publication Date(Web):March 1, 2016
DOI:10.1021/acs.jctc.6b00016
Coarse-grained (CG) models are valuable tools for the study of functions of large biomolecules on large length and time scales. The definition of CG representations for huge biomolecules is always a formidable challenge. In this work, we propose a new method called fluctuation maximization coarse-graining (FM-CG) to construct the CG sites of biomolecules. The defined residual in FM-CG converges to a maximal value as the number of CG sites increases, allowing an optimal CG model to be rigorously defined on the basis of the maximum. More importantly, we developed a robust algorithm called stepwise local iterative optimization (SLIO) to accelerate the process of coarse-graining large biomolecules. By means of the efficient SLIO algorithm, the computational cost of coarse-graining large biomolecules is reduced to within the time scale of seconds, which is far lower than that of conventional simulated annealing. The coarse-graining of two huge systems, chaperonin GroEL and lengsin, indicates that our new methods can coarse-grain huge biomolecular systems with up to 10 000 residues within the time scale of minutes. The further parametrization of CG sites derived from FM-CG allows us to construct the corresponding CG models for studies of the functions of huge biomolecular systems.
Co-reporter:Yuan Liu, Zhunzhun Yu, Zhoujie Luo, John Zenghui Zhang, Lu Liu, and Fei Xia
The Journal of Physical Chemistry A 2016 Volume 120(Issue 11) pp:1925-1932
Publication Date(Web):March 1, 2016
DOI:10.1021/acs.jpca.6b00636
It was recently reported that the gold-carbenes have an unprecedented catalysis toward the functionalization of C(sp2)–H bonds of aromatic compounds. However, the associated mechanisms of C(sp2)–H bonds inserted by gold-carbenes have not been comprehensively understood. We carried out a detailed mechanistic investigation of gold-carbene insertion into the C(sp2)–H bond of anisole by means of theoretical calculations and control experiments. It significantly reveals that the aromatic C(sp2)–H bond activation starts with the electrophilic addition of aromatic carbon toward the carbene carbon and subsequently followed the [1,3]-proton shift to form an enol intermediate. The rearrangement of enol proceeds through the mechanisms of proton transfer assisted by water molecules or enol intermediates, which are supported by our control experiments. It was also found that the C(sp3)–H insertions of alkanes by gold-carbenes proceed through a concerted process via a three-centered transition state. The further comparison of different mechanisms provides a clear theoretical scheme to account for the difference in aromatic C(sp2)–H and alkyl C(sp3)–H bond activation, which is instructive for the further experimental functionalization of C–H bonds by gold-carbenes.
Co-reporter:Yuan Liu, Zhoujie Luo, John Zenghui Zhang, and Fei Xia
The Journal of Physical Chemistry A 2016 Volume 120(Issue 32) pp:6485-6492
Publication Date(Web):July 29, 2016
DOI:10.1021/acs.jpca.6b05735
The reaction of diazo compounds with transition-metal carbenes is an efficient way to achieve the functionalization of chemical bonds in organic molecules, especially for the C–H and O–H bonds. However, the selective mechanisms of C–H and O–H bond insertions by various metal carbenes such as Rh and Cu complexes are not quite clear. In this work, we performed a comprehensively theoretical investigation of the phenol C–H and O–H bonds inserted by Rh and Cu carbenes by using DFT calculations. The calculated results reveal that the nucleophilic additions of phenols to the Rh and Cu carbenes in the C–H bond insertions are the rate-determining steps of whole reactions, which are higher than the barriers in the O–H insertions. In the process of intramolecular [1,3]-H transfer, the Rh and Cu ligands in their carbenes tend to dissociate into solution rather than the intramolecular migration due to their weak metal–carbon bonds. A deeply theoretical analysis of the electronic structures of Rh, Cu, and Au carbenes as well as their complexes elucidated their differences in the chemoselectivity of C–H and O–H insertion products, which agrees with the experimental observations well.
Co-reporter:Binshen Wang;Zhoujie Luo;Elnazeer H. M. Elageed;Shi Wu;Yongya Zhang;Xiaopei Wu;Dr. Fei Xia;Guirong Zhang ;Dr. Guohua Gao
ChemCatChem 2016 Volume 8( Issue 4) pp:830-838
Publication Date(Web):
DOI:10.1002/cctc.201500928
Abstract
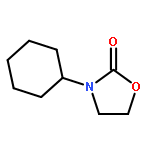
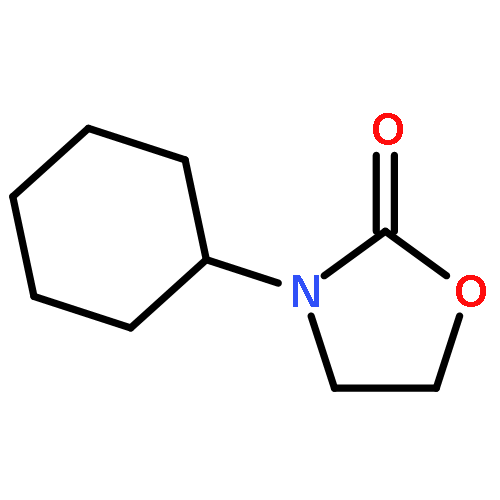
An intermolecular synergistic catalytic combination of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and a DBU-derived bromide ionic liquid has been developed for the conversion of CO2, epoxides, and amines under metal- and solvent-free conditions. Various 3-aryl-2-oxazolidinones are produced in moderate to excellent yields within a short reaction time. NMR spectroscopy and DFT calculations demonstrate that DBU as a hydrogen bond acceptor and the ionic liquid as a hydrogen bond donor activate the substrates cooperatively by inducing hydrogen bonds to promote the reaction effectively. Based on these results, a possible reaction mechanism on the synergistic catalysis of DBU and the ionic liquid is proposed. In addition, the reaction of CS2, ethylene oxide, and aniline catalyzed by the combination of DBU and the DBU-derived ionic liquid also proceeds smoothly, which opens a hitherto unreported route to [1,3]dithiolan-2-ylidenephenylamine in a straightforward way.
Co-reporter:Yidong Wang, Peichao Zhang, Yuan Liu, Fei Xia and Junliang Zhang
Chemical Science 2015 vol. 6(Issue 10) pp:5564-5570
Publication Date(Web):23 Jun 2015
DOI:10.1039/C5SC01827G
A highly enantioselective [2+2] versus a [4+2]-cycloaddition of 3-styrylindoles to N-allenamides catalyzed by identical gold(I)/chiral phosphoramidite complexes is presented, which provides facile access to synthetically valuable, optically active substituted cyclobutanes and tetrahydrocarbazoles. The cycloaddition mode unexpectedly depends on the electronic nature of the N-substituent 3-styrylindoles, the origin of which could be well rationalized using DFT calculations and experimental results. To the best of our knowledge, the present work represents the first example of such an impressive substituent effect in tuning the reaction mode with high chemo-, regio- and enantioselectivity in asymmetric gold catalysis.
Co-reporter:Le Xu, Da-Ding Huang, Chen-Geng Li, Xinyi Ji, Shaoqing Jin, Zhaochi Feng, Fei Xia, Xiaohong Li, Fengtao Fan, Can Li and Peng Wu
Chemical Communications 2015 vol. 51(Issue 43) pp:9010-9013
Publication Date(Web):17 Apr 2015
DOI:10.1039/C5CC02321A
A novel organic–inorganic layered titanosilicate consisting of Ti-containing MWW-type nanosheets and piperidine ligands was constructed. It exhibited an unprecedented high catalytic activity and recyclability in alkene epoxidation.
Co-reporter:Min Li, John Z.H. Zhang, Fei Xia
Chemical Physics Letters 2015 Volume 618() pp:102-107
Publication Date(Web):2 January 2015
DOI:10.1016/j.cplett.2014.11.006
Co-reporter:Le Xu, Da-Ding Huang, Chen-Geng Li, Xinyi Ji, Shaoqing Jin, Zhaochi Feng, Fei Xia, Xiaohong Li, Fengtao Fan, Can Li and Peng Wu
Chemical Communications 2015 - vol. 51(Issue 43) pp:NaN9013-9013
Publication Date(Web):2015/04/17
DOI:10.1039/C5CC02321A
A novel organic–inorganic layered titanosilicate consisting of Ti-containing MWW-type nanosheets and piperidine ligands was constructed. It exhibited an unprecedented high catalytic activity and recyclability in alkene epoxidation.
Co-reporter:Yidong Wang, Peichao Zhang, Yuan Liu, Fei Xia and Junliang Zhang
Chemical Science (2010-Present) 2015 - vol. 6(Issue 10) pp:NaN5570-5570
Publication Date(Web):2015/06/23
DOI:10.1039/C5SC01827G
A highly enantioselective [2+2] versus a [4+2]-cycloaddition of 3-styrylindoles to N-allenamides catalyzed by identical gold(I)/chiral phosphoramidite complexes is presented, which provides facile access to synthetically valuable, optically active substituted cyclobutanes and tetrahydrocarbazoles. The cycloaddition mode unexpectedly depends on the electronic nature of the N-substituent 3-styrylindoles, the origin of which could be well rationalized using DFT calculations and experimental results. To the best of our knowledge, the present work represents the first example of such an impressive substituent effect in tuning the reaction mode with high chemo-, regio- and enantioselectivity in asymmetric gold catalysis.
Co-reporter:Shunying Liu, Jun Jiang, Jianghui Chen, Qinghua Wei, Wenfeng Yao, Fei Xia and Wenhao Hu
Chemical Science (2010-Present) 2017 - vol. 8(Issue 6) pp:NaN4317-4317
Publication Date(Web):2017/03/22
DOI:10.1039/C7SC00257B
Metal-associated carbenes from diazo compounds promote many useful chemistry transformations in modern organic chemistry. However, compared to α-aryldiazoacetate-derived carbenes (ArDCs), the synthetic application of α-alkyldiazoacetate-derived carbenes (AlDCs) is greatly limited due to intramolecular α-H transfer (elimination) that results in alkenes as the main by-products. An intriguing α-alkyldiazoacetate-involved three-component reaction has been developed following DFT calculation inspiration to provide β-hydroxyl α-alkyl-α-amino acid derivatives in good yields. The intramolecular α-H shift of an α-alkyldiazoacetate-derived carbene was successfully suppressed by the association of a Rh(I) complex to form the corresponding active ammonium ylide, which was trapped before the fast 1,2-H transfer process. A Rh(I)-chiral diene complex was identified as an effective catalyst to give an asymmetric version of the reaction with good enantioselectivity. This reaction provides insight into extending the efficient transformation of α-alkyldiazoacetate-derived carbenes and their synthetic application.
Co-reporter:Yuan Liu, Zhunzhun Yu, John Zenghui Zhang, Lu Liu, Fei Xia and Junliang Zhang
Chemical Science (2010-Present) 2016 - vol. 7(Issue 3) pp:NaN1995-1995
Publication Date(Web):2015/11/27
DOI:10.1039/C5SC04319K
In past decade, gold revealed more and more unique properties in carbene chemistry. It was disclosed in our recent communication (J. Am. Chem. Soc. 2014, 136, 6904) that gold carbenes have unprecedented chemo- and site-selectivity and ligand effect toward the functionalization of C–H bonds in phenols. In this full article, we report a comprehensively combined theoretical and experimental study on the mechanism of the insertion of gold carbenes into C–H and O–H bonds in phenol. It significantly revealed that the ligands have an important effect on C–H insertion and the reaction proceeds through a pathway involving the formation of an enolate-like intermediate. Moreover, two water molecules serving as a proton shuttle are believed to be the key issue for achieving chemoselective C–H functionalization, which is strongly supported by the DFT calculations and control experiments. It is the first time that a clear explanation is given about the prominent catalysis of gold carbenes toward C–H functionalization based on a theoretical and experimental study.












![Morpholine, 4-[1-(4-methylphenyl)ethenyl]-](http://img.cochemist.com/ccimg/56000/55949-65-0.png)
![Morpholine, 4-[1-(4-methylphenyl)ethenyl]-](http://img.cochemist.com/ccimg/56000/55949-65-0_b.png)

![Benzenamine, N-[(4-nitrophenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/1700/1614-00-2.png)
![Benzenamine, N-[(4-nitrophenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/1700/1614-00-2_b.png)





![[(2r,3r,4s,5s,6r)-5-acetoxy-3,4-dibenzyloxy-6-(2,2,2-trichloroeth Animidoyl)oxy-tetrahydropyran-2-yl]methyl Acetate](http://img.cochemist.com/ccimg/197700/197649-28-8.png)
![[(2r,3r,4s,5s,6r)-5-acetoxy-3,4-dibenzyloxy-6-(2,2,2-trichloroeth Animidoyl)oxy-tetrahydropyran-2-yl]methyl Acetate](http://img.cochemist.com/ccimg/197700/197649-28-8_b.png)
![Benzenamine, N-[(4-methylphenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/1700/1613-98-5.png)
![Benzenamine, N-[(4-methylphenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/1700/1613-98-5_b.png)
![Benzenamine, N-[(4-methoxyphenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/1700/1613-96-3.png)
![Benzenamine, N-[(4-methoxyphenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/1700/1613-96-3_b.png)
![Bicyclo[2.2.2]octa-2,5-diene, 2,5-diphenyl-, (1R,4R)-](http://img.cochemist.com/ccimg/797000/796966-15-9.png)
![Bicyclo[2.2.2]octa-2,5-diene, 2,5-diphenyl-, (1R,4R)-](http://img.cochemist.com/ccimg/797000/796966-15-9_b.png)


![Benzenamine, 4-methyl-N-[(4-nitrophenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/33500/33442-37-4.png)
![Benzenamine, 4-methyl-N-[(4-nitrophenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/33500/33442-37-4_b.png)
















![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,2,6-bis[3,5-bis(trifluoromethyl)phenyl]-4-hydroxy-, 4-oxide, (11bR)-](/data/chemimg/114800/791616-62-1.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,2,6-bis[3,5-bis(trifluoromethyl)phenyl]-4-hydroxy-, 4-oxide, (11bR)-](/data/chemimg/114800/791616-62-1_b.png)





![N-[(E)-(3-nitrophenyl)methylidene]aniline](http://img.cochemist.com/ccimg/5700/5676-82-4.png)
![N-[(E)-(3-nitrophenyl)methylidene]aniline](http://img.cochemist.com/ccimg/5700/5676-82-4_b.png)