Co-reporter:Ping Wu, Hao Xu, Zhi Li, Yang Zhou, Yingxia Li, Wei Zhang
Tetrahedron Letters 2017 Volume 58, Issue 33(Issue 33) pp:
Publication Date(Web):16 August 2017
DOI:10.1016/j.tetlet.2017.07.046
•Two oxazoline analogues of apratoxin E have been prepared with late-stage formation of oxazoline ring.•Oxazoline bioisosteres were about 6-fold less potent than their thiazoline parent compounds.•The positive impact of 30R on activity was observed, with about 2-fold enhancement than corresponding 30S epimer.30R- and 30S-oxazoline analogues of apratoxin E have been prepared with late-stage formation of an oxazoline ring. These two compounds have a potent inhibitory effect on HCT-116 cell proliferation with IC50 values of 345 and 638 nM, respectively. These results suggest that apratoxin E oxazoline bioisosteres are approximately 6-fold less potent than their thiazoline parent compounds. A positive impact of the 30R conformation on antiproliferative activity was also observed, with approximately 2-fold enhancement when compared to the 30S epimer.Download high-res image (96KB)Download full-size image
Co-reporter:Ping Wu, Hao Xu, Zhi Li, Yang Zhou, Yingxia Li, Wei Zhang
Tetrahedron Letters 2017 Volume 58, Issue 44(Issue 44) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.tetlet.2017.09.059
Co-reporter:Yuhua Ma, Haisheng He, Fei Xia, Yingxia Li, Yan Lu, Daofeng Chen, Jianping Qi, Yi Lu, Wei Zhang, Wei Wu
Nanomedicine: Nanotechnology, Biology and Medicine 2017 Volume 13, Issue 8(Issue 8) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.nano.2017.07.014
•LDCs-SLNs improve the oral bioavailability of silybin by 5–7 times in comparison with a fast-release formulation.•LDCs might be broken down in the GI tract and within enterocytes to release silybin, which is then transported to the circulation.•The contribution of absorption of integral SLNs via the M cell pathway is negligible to the bulk bioavailability•Longer lipid chains create more steric hindrance and slow degradation and biodistribution.Lipid-drug conjugates (LDCs) of a poorly soluble and poorly permeable drug silybin (SB) and lipids with different chain lengths (6C, 12C, 18C) are synthesized and formulated into solid lipid nanoparticles (SLNs). The in vivo fate of LDCs as well as SLNs is investigated by tracking either SB or LDCs or SLNs. LDCs are prone to be hydrolyzed by lipases either in simulated gastrointestinal media or in Caco-2 cell lines in a lipid chain length-dependent mode. The oral bioavailability of SB is enhanced by 5–7-fold in comparison with a fast-release formulation. No integral LDCs are detected in plasma confirms the readily degradable nature of LDCs. The absorption of LDCs by enteric epithelia and subsequent transportation into circulation might play a leading role in absorption enhancement, whereas the contribution of then M-cell pathway is not as remarkable. A shorter lipid chain favors earlier lipolysis and faster absorption along the intestine-to-circulation path.LDCs-containing SLNs can be degraded to release LDCs, which are then taken up by enterocytes, further broken down to SB, and finally transported to systemic circulation. This enterocyte-to-portal vein route accounts for a major part of enhanced oral bioavailability, whereas the contribution of the M cell/lymphatics route is not as remarkable.Download high-res image (206KB)Download full-size image
Co-reporter:Ping Wu, Weijing Cai, Qi-Yin Chen, Senhan Xu, Ruwen Yin, Yingxia Li, Wei Zhang, and Hendrik Luesch
Organic Letters 2016 Volume 18(Issue 20) pp:5400-5403
Publication Date(Web):October 10, 2016
DOI:10.1021/acs.orglett.6b02780
Apratoxin E provided the inspiration for the design of apratoxin A/E hybrids under preclinical development. Through total synthesis using two different strategies, it was determined that the originally proposed configuration of the thiazoline at C30 is opposite from that in apratoxin A, in contrast to previous assumptions on biosynthetic grounds. The epimer and true natural apratoxin E were synthesized, and the biological activities were evaluated.
Co-reporter:Yadong Zhu, Pengfei Qian, Jiyang Yang, Shaohua Chen, Yanwei Hu, Ping Wu, Wei Wang, Wei Zhang and Shilei Zhang
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 16) pp:4769-4775
Publication Date(Web):06 Mar 2015
DOI:10.1039/C5OB00202H
An efficient aminocatalytic enantioselective Michael addition of readily available cyclic hemiacetals to nitroolefins has been developed. The strategy serves as a powerful approach to synthetically valuable chiral 3-substituted tetrahydrofurans (THFs) and tetrahydropyrans (THPs). The synthetic utilities of the versatile Michael adducts also have been demonstrated in the synthesis of 2,3-disubstituted cyclic ethers, α-substituted lactones and venlafaxine analogues.
Co-reporter:Jian Liu, Wuyan Chen, Yechun Xu, Sumei Ren, Wei Zhang, Yingxia Li
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 9) pp:1963-1974
Publication Date(Web):1 May 2015
DOI:10.1016/j.bmc.2015.03.034
Nineteen new derivatives based on the structure of marine natural product tasiamide B were designed, synthesized, and evaluated for their inhibitory activity against BACE1, a potential therapeutic target for Alzheimer’s disease. The hydrophobic substituents Val at P3 position, Leu at P1′ position, Ala at P2′ position, and Phe at P3′ position were found to significantly affect the inhibition. Free carboxylic acid at C-terminus was also found to be important to the activity. In addition, the structure–activity relationships (SARs) were supported by molecular docking simulation.
Co-reporter:Ping Wu, Senhan Xu, Hao Xu, Haiyan Hu, Wei Zhang
Tetrahedron Letters (15 February 2017) Volume 58(Issue 7) pp:618-621
Publication Date(Web):15 February 2017
DOI:10.1016/j.tetlet.2016.12.088
Co-reporter:Senhan Xu, Ping Wu and Wei Zhang
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 48) pp:NaN11395-11395
Publication Date(Web):2016/11/14
DOI:10.1039/C6OB02200F
α-Bromo ketones are versatile intermediates of high practical utility. Traditional approaches to these compounds are restricted to a relatively hazardous/complex reagent combination, a long reaction time, the use of non-environmentally friendly solvents, or a limited substrate scope. Herein, we describe the development of a new methodology for the preparation of α-bromo ketones from alkenes using 1,3-dibromo-5,5-dimethylhydantoin (DBH) as a bromine source and an oxidant simultaneously. This easy to carry out two-step one-pot protocol proceeds in water and provides high yield of a great variety of α-bromo ketones. Addition of an amine to the intermediate α-bromo ketone further enables the preparation of α-amino ketones in a one-pot sequence.
Co-reporter:Yadong Zhu, Pengfei Qian, Jiyang Yang, Shaohua Chen, Yanwei Hu, Ping Wu, Wei Wang, Wei Zhang and Shilei Zhang
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 16) pp:NaN4775-4775
Publication Date(Web):2015/03/06
DOI:10.1039/C5OB00202H
An efficient aminocatalytic enantioselective Michael addition of readily available cyclic hemiacetals to nitroolefins has been developed. The strategy serves as a powerful approach to synthetically valuable chiral 3-substituted tetrahydrofurans (THFs) and tetrahydropyrans (THPs). The synthetic utilities of the versatile Michael adducts also have been demonstrated in the synthesis of 2,3-disubstituted cyclic ethers, α-substituted lactones and venlafaxine analogues.

![N-[3-[5-CHLORO-2-[2-METHOXY-4-(4-METHYLPIPERAZIN-1-YL)ANILINO]PYRIMIDIN-4-YL]OXYPHENYL]PROP-2-ENAMIDE](http://img.cochemist.com/ccimg/1213300/1213269-23-8.png)
![N-[3-[5-CHLORO-2-[2-METHOXY-4-(4-METHYLPIPERAZIN-1-YL)ANILINO]PYRIMIDIN-4-YL]OXYPHENYL]PROP-2-ENAMIDE](http://img.cochemist.com/ccimg/1213300/1213269-23-8_b.png)
![3-Pyridinecarboxamide,N-[4-[(6,7-dimethoxy-4-quinolinyl)oxy]-3-fluorophenyl]-1-(4-fluorophenyl)-1,2-dihydro-2-oxo-](http://img.cochemist.com/ccimg/929300/929252-41-5.png)
![3-Pyridinecarboxamide,N-[4-[(6,7-dimethoxy-4-quinolinyl)oxy]-3-fluorophenyl]-1-(4-fluorophenyl)-1,2-dihydro-2-oxo-](http://img.cochemist.com/ccimg/929300/929252-41-5_b.png)







![2-HEPTEN-1-OL, 5-[(4-METHOXYPHENYL)METHOXY]-3,6,6-TRIMETHYL-, (2E,5S)-](http://img.cochemist.com/ccimg/878700/878626-77-8.png)
![2-HEPTEN-1-OL, 5-[(4-METHOXYPHENYL)METHOXY]-3,6,6-TRIMETHYL-, (2E,5S)-](http://img.cochemist.com/ccimg/878700/878626-77-8_b.png)
![1H-PYRROLO[2,3-D]PYRIMIDINE, 4-(2-FLUORO-4-NITROPHENOXY)-](http://img.cochemist.com/ccimg/875800/875764-99-1.png)
![1H-PYRROLO[2,3-D]PYRIMIDINE, 4-(2-FLUORO-4-NITROPHENOXY)-](http://img.cochemist.com/ccimg/875800/875764-99-1_b.png)





![Heptanal, 3,6,6-trimethyl-5-[(triethylsilyl)oxy]-, (3R,5S)-](http://img.cochemist.com/ccimg/780800/780770-95-8.png)
![Heptanal, 3,6,6-trimethyl-5-[(triethylsilyl)oxy]-, (3R,5S)-](http://img.cochemist.com/ccimg/780800/780770-95-8_b.png)






![Heptanal, 5-[(4-methoxyphenyl)methoxy]-3,6,6-trimethyl-, (3R,5S)-](http://img.cochemist.com/ccimg/676400/676324-77-9.png)
![Heptanal, 5-[(4-methoxyphenyl)methoxy]-3,6,6-trimethyl-, (3R,5S)-](http://img.cochemist.com/ccimg/676400/676324-77-9_b.png)
![1-HEPTANOL, 5-[(4-METHOXYPHENYL)METHOXY]-3,6,6-TRIMETHYL-, (3S,5S)-](http://img.cochemist.com/ccimg/676400/676324-76-8.png)
![1-HEPTANOL, 5-[(4-METHOXYPHENYL)METHOXY]-3,6,6-TRIMETHYL-, (3S,5S)-](http://img.cochemist.com/ccimg/676400/676324-76-8_b.png)
![Pentanal, 3-[(4-methoxyphenyl)methoxy]-4,4-dimethyl-, (3S)-](http://img.cochemist.com/ccimg/676400/676324-73-5.png)
![Pentanal, 3-[(4-methoxyphenyl)methoxy]-4,4-dimethyl-, (3S)-](http://img.cochemist.com/ccimg/676400/676324-73-5_b.png)


![1-[3-[(DIMETHYLAMINO)METHYL]PHENYL]ETHANONE](http://img.cochemist.com/ccimg/628400/628305-89-5.png)
![1-[3-[(DIMETHYLAMINO)METHYL]PHENYL]ETHANONE](http://img.cochemist.com/ccimg/628400/628305-89-5_b.png)
![2-HEXANONE, 4-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-5,5-DIMETHYL-, (4S)-](http://img.cochemist.com/ccimg/612500/612492-55-4.png)
![2-HEXANONE, 4-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-5,5-DIMETHYL-, (4S)-](http://img.cochemist.com/ccimg/612500/612492-55-4_b.png)


















![Ruthenium, bis(acetato-kO,kO')[1,1'-(1S)-[1,1'-binaphthalene]-2,2'-diylbis[1,1-diphenylphosphine-kP]]-, (OC-6-22)-](http://img.cochemist.com/ccimg/262000/261948-85-0.png)
![Ruthenium, bis(acetato-kO,kO')[1,1'-(1S)-[1,1'-binaphthalene]-2,2'-diylbis[1,1-diphenylphosphine-kP]]-, (OC-6-22)-](http://img.cochemist.com/ccimg/262000/261948-85-0_b.png)
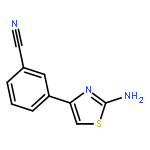







































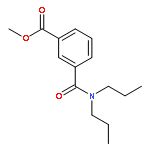





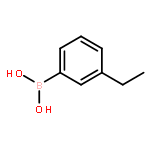





![4-Amino-1-[(5S)-5-(hydroxymethyl)tetrahydro-2-furanyl]-2(1H)-pyri midinone](http://img.cochemist.com/ccimg/83900/83869-56-1.png)
![4-Amino-1-[(5S)-5-(hydroxymethyl)tetrahydro-2-furanyl]-2(1H)-pyri midinone](http://img.cochemist.com/ccimg/83900/83869-56-1_b.png)
![ACETIC ACID, [[(4-METHYLPHENYL)AMINO]SULFONYL]-, ETHYL ESTER](http://img.cochemist.com/ccimg/78400/78374-64-8.png)
![ACETIC ACID, [[(4-METHYLPHENYL)AMINO]SULFONYL]-, ETHYL ESTER](http://img.cochemist.com/ccimg/78400/78374-64-8_b.png)
![ACETIC ACID, [[(4-METHYLPHENYL)AMINO]SULFONYL]-](http://img.cochemist.com/ccimg/78400/78374-09-1.png)
![ACETIC ACID, [[(4-METHYLPHENYL)AMINO]SULFONYL]-](http://img.cochemist.com/ccimg/78400/78374-09-1_b.png)




![D-Glucose, O-b-D-galactopyranosyl-(1®4)-O-2-(acetylamino)-2-deoxy-b-D-glucopyranosyl-(1®2)-O-a-D-mannopyranosyl-(1®3)-O-[O-b-D-galactopyranosyl-(1®4)-O-2-(acetylamino)-2-deoxy-b-D-glucopyranosyl-(1®2)-a-D-mannopyranosyl-(1®6)]-O-b-D-mannopyranosyl-(1®4)-O-2-(acetylamino)-2-deoxy-b-D-glucopyranosyl-(1®4)-2-(acetylamino)-2-deoxy-](http://img.cochemist.com/ccimg/71500/71496-53-2.png)
![D-Glucose, O-b-D-galactopyranosyl-(1®4)-O-2-(acetylamino)-2-deoxy-b-D-glucopyranosyl-(1®2)-O-a-D-mannopyranosyl-(1®3)-O-[O-b-D-galactopyranosyl-(1®4)-O-2-(acetylamino)-2-deoxy-b-D-glucopyranosyl-(1®2)-a-D-mannopyranosyl-(1®6)]-O-b-D-mannopyranosyl-(1®4)-O-2-(acetylamino)-2-deoxy-b-D-glucopyranosyl-(1®4)-2-(acetylamino)-2-deoxy-](http://img.cochemist.com/ccimg/71500/71496-53-2_b.png)
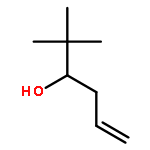
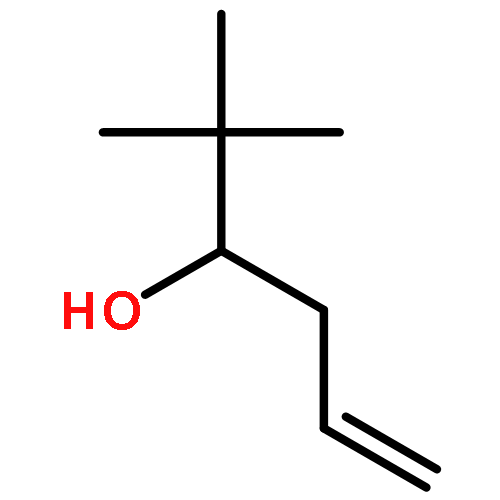
![Propanedioic acid,2-[(2-nitrophenyl)methylene]-, 1,3-dimethyl ester](http://img.cochemist.com/ccimg/66000/65974-52-9.png)
![Propanedioic acid,2-[(2-nitrophenyl)methylene]-, 1,3-dimethyl ester](http://img.cochemist.com/ccimg/66000/65974-52-9_b.png)


![Acetic acid, [(phenylamino)sulfonyl]-, ethyl ester](http://img.cochemist.com/ccimg/56200/56146-84-0.png)
![Acetic acid, [(phenylamino)sulfonyl]-, ethyl ester](http://img.cochemist.com/ccimg/56200/56146-84-0_b.png)




![ALANINE, N-[(1,1-DIMETHYLETHOXY)CARBONYL]LEUCYL-, METHYL ESTER](http://img.cochemist.com/ccimg/43200/43189-51-1.png)
![ALANINE, N-[(1,1-DIMETHYLETHOXY)CARBONYL]LEUCYL-, METHYL ESTER](http://img.cochemist.com/ccimg/43200/43189-51-1_b.png)
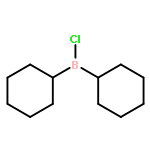
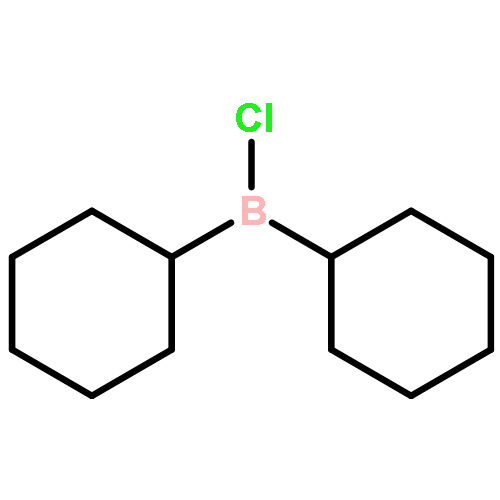
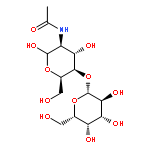

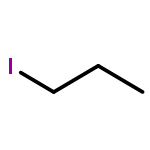

![L-Isoleucine, N-[(1,1-dimethylethoxy)carbonyl]-N-methyl-, methyl ester](http://img.cochemist.com/ccimg/24200/24164-05-4.png)
![L-Isoleucine, N-[(1,1-dimethylethoxy)carbonyl]-N-methyl-, methyl ester](http://img.cochemist.com/ccimg/24200/24164-05-4_b.png)




















![ACETIC ACID, [(PHENYLAMINO)SULFONYL]-](http://img.cochemist.com/ccimg/7200/7117-21-7.png)
![ACETIC ACID, [(PHENYLAMINO)SULFONYL]-](http://img.cochemist.com/ccimg/7200/7117-21-7_b.png)






![Benzothiazolium,3-ethyl-2-[7-(3-ethyl-2(3H)-benzothiazolylidene)-1,3,5-heptatrien-1-yl]-,iodide (1:1)](/data/chemimg/28100/3071-70-3.png)
![Benzothiazolium,3-ethyl-2-[7-(3-ethyl-2(3H)-benzothiazolylidene)-1,3,5-heptatrien-1-yl]-,iodide (1:1)](/data/chemimg/28100/3071-70-3_b.png)









![3-Pyridinecarboxamide, N-[4-[(2-amino-3-chloro-4-pyridinyl)oxy]-3-fluorophenyl]-4-ethoxy-1-(4-fluorophenyl)-1,2-dihydro-2-oxo-](/data/chemimg/315000/1025720-94-8.png)
![3-Pyridinecarboxamide, N-[4-[(2-amino-3-chloro-4-pyridinyl)oxy]-3-fluorophenyl]-4-ethoxy-1-(4-fluorophenyl)-1,2-dihydro-2-oxo-](/data/chemimg/315000/1025720-94-8_b.png)
![Benzenamine, 3-fluoro-4-(1H-pyrrolo[2,3-d]pyrimidin-4-yloxy)-](http://img.cochemist.com/ccimg/875800/875765-00-7.png)
![Benzenamine, 3-fluoro-4-(1H-pyrrolo[2,3-d]pyrimidin-4-yloxy)-](http://img.cochemist.com/ccimg/875800/875765-00-7_b.png)
![Benzenamine, 4-[(6,7-dimethoxy-4-quinolinyl)oxy]-3-fluoro-](http://img.cochemist.com/ccimg/347200/347161-74-4.png)
![Benzenamine, 4-[(6,7-dimethoxy-4-quinolinyl)oxy]-3-fluoro-](http://img.cochemist.com/ccimg/347200/347161-74-4_b.png)


![Hexanamide,N-[2-hydroxy-1-(hydroxymethyl)-3-heptadecen-1-yl]-6-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-](/data/chemimg/985200/86701-10-2.png)
![Hexanamide,N-[2-hydroxy-1-(hydroxymethyl)-3-heptadecen-1-yl]-6-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-](/data/chemimg/985200/86701-10-2_b.png)






![Pentacyclo[9.5.1.13,9.15,15.17,13]octasiloxane, 1,3,5,7,9,11,13,15-octakis(3-iodopropyl)-](/data/chemimg/3783300/161678-43-9.png)
![Pentacyclo[9.5.1.13,9.15,15.17,13]octasiloxane, 1,3,5,7,9,11,13,15-octakis(3-iodopropyl)-](/data/chemimg/3783300/161678-43-9_b.png)
![Pentacyclo[9.5.1.13,9.15,15.17,13]octasiloxane, 1,3,5,7,9,11,13,15-octakis(3-chloropropyl)-](/data/chemimg/130100/161678-38-2.png)
![Pentacyclo[9.5.1.13,9.15,15.17,13]octasiloxane, 1,3,5,7,9,11,13,15-octakis(3-chloropropyl)-](/data/chemimg/130100/161678-38-2_b.png)





