Co-reporter:Cathleen Jendrny ;Dr. Annette G. Beck-Sickinger
ChemBioChem 2016 Volume 17( Issue 8) pp:719-726
Publication Date(Web):
DOI:10.1002/cbic.201500539
Abstract
Serpin proteins irreversibly inhibit serine proteases, but only a small part of the serpin reactive-center loop (RCL) is responsible for the initial protein–protein interaction (PPI). To develop peptidic protease inhibitors, kallikrein-related peptidases 7 (KLK7) and 5 (KLK5) were chosen. Firstly, we demonstrated that short peptides derived from RCL sequences can be cleaved by KLK7 in a substrate-like manner. Next, these substrates were grafted onto the protease-binding loop of sunflower trypsin inhibitor-1 (SFTI-1). Peptides based on kallistatin, α1-antichymotrypsin, and protein C inhibitor (PCI) inhibited KLK7 with Ki=0.4, 0.5, and 0.7 μm, respectively. In contrast, the trypsin-like KLK5 was only blocked by the peptide derived from PCI (Ki=0.6 μm). Thus, serpin function can be mimicked by introducing its PPI site into the rigid structure of the SFTI-1 scaffold. This approach might be applicable not only to KLKs but also to other serine protease members, thus opening up new therapeutic fields.
Co-reporter:Cathleen Jendrny ;Dr. Annette G. Beck-Sickinger
ChemBioChem 2016 Volume 17( Issue 8) pp:
Publication Date(Web):
DOI:10.1002/cbic.201600146
Co-reporter:Verena M. Ahrens, Katja B. Kostelnik, Robert Rennert, David Böhme, Stefan Kalkhof, David Kosel, Lutz Weber, Martin von Bergen, Annette G. Beck-Sickinger
Journal of Controlled Release 2015 Volume 209() pp:170-178
Publication Date(Web):10 July 2015
DOI:10.1016/j.jconrel.2015.04.037
Myxobacterial tubulysins are promising chemotherapeutics inhibiting microtubule polymerization, however, high unspecific toxicity so far prevents their application in therapy. For selective cancer cell targeting, here the coupling of a synthetic cytolysin to the hY1-receptor preferring peptide [F7,P34]-neuropeptide Y (NPY) using a labile disulfide linker is described. Since hY1-receptors are overexpressed in breast tumors and internalize rapidly, this system has high potential as peptide–drug shuttle system. Molecular characterization of the cytolysin–[F7,P34]-NPY bioconjugate revealed potent receptor activation and receptor-selective internalization, while viability studies verified toxicity. Triple SILAC studies comparing free cytolysin with the bioconjugate demonstrated an intracellular mechanism of action regardless of the delivery pathway. Treatments resulted in a regulation of proteins implemented in cell cycle arrest confirming the tubulysin-like effect of the cytolysin. Thus, the cytolysin–peptide bioconjugate fused by a cleavable linker enables a receptor-specific delivery as well as a potent intracellular drug-release with high cytotoxic activity.
Co-reporter:Ulrike Reinhardt, Jonathan Lotze, Karin Mörl, Annette G. Beck-Sickinger, and Oliver Seitz
Bioconjugate Chemistry 2015 Volume 26(Issue 10) pp:2106
Publication Date(Web):September 14, 2015
DOI:10.1021/acs.bioconjchem.5b00387
Fluorescently labeled proteins enable the microscopic imaging of protein localization and function in live cells. In labeling reactions targeted against specific tag sequences, the size of the fluorophore-tag is of major concern. The tag should be small to prevent interference with protein function. Furthermore, rapid and covalent labeling methods are desired to enable the analysis of fast biological processes. Herein, we describe the development of a method in which the formation of a parallel coiled coil triggers the transfer of a fluorescence dye from a thioester-linked coil peptide conjugate onto a cysteine-modified coil peptide. This labeling method requires only small tag sequences (max 23 aa) and occurs with high tag specificity. We show that size matching of the coil peptides and a suitable thioester reactivity allow the acyl transfer reaction to proceed within minutes (rather than hours). We demonstrate the versatility of this method by applying it to the labeling of different G-protein coupled membrane receptors including the human neuropeptide Y receptors 1, 2, 4, 5, the neuropeptide FF receptors 1 and 2, and the dopamine receptor 1. The labeled receptors are fully functional and able to bind the respective ligand with high affinity. Activity is not impaired as demonstrated by activation, internalization, and recycling experiments.
Co-reporter:Katja B. Kostelnik, Sylvia Els-Heindl, Nora Klöting, Sven Baumann, Martin von Bergen, Annette G. Beck-Sickinger
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 14) pp:3925-3932
Publication Date(Web):15 July 2015
DOI:10.1016/j.bmc.2014.12.008
The constitutive activity of the ghrelin receptor is of high physiological and pathophysiological relevance. In-depth structure–activity relationship studies revealed a palmitoylated ghrelin receptor ligand that displays an in vitro binding affinity in the low nanomolar range. Activity studies revealed inverse agonistic as well as antagonistic properties and in vitro metabolic analysis indicated a high stability in blood serum and liver homogenate. For metabolic testing in vivo, a combined approach of stable isotopic labeling and mass spectrometry-based analysis was established. Therefore, a heavy isotopic version of the peptide containing a 13C-labeled palmitic acid was synthesized and a 1:1 ratio of a 12C/13C-peptide mixture was injected into rats. Biological samples were analyzed by multiple reaction monitoring allowing simultaneous peptide detection and quantification. Measurements revealed a suitable bioavailability over 24 h in rat serum and subsequent high-resolution mass spectrometry investigations showed only negligible degradation and slow body clearance. Hence, this method combination allowed the identification and evaluation of a highly potent and metabolically stable ghrelin receptor ligand in vivo.
Co-reporter:Dr. Verena M. Ahrens;Dr. René Frank;Solveig Boehnke;Dr. Christian L. Schütz;Dr. Gabriele Hampel;Dorothée S. Iffl;Dr. Nicolas H. Bings;Dr. Evamarie Hey-Hawkins;Dr. Annette G. Beck-Sickinger
ChemMedChem 2015 Volume 10( Issue 1) pp:164-172
Publication Date(Web):
DOI:10.1002/cmdc.201402368
Abstract
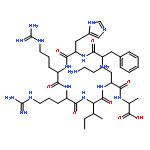
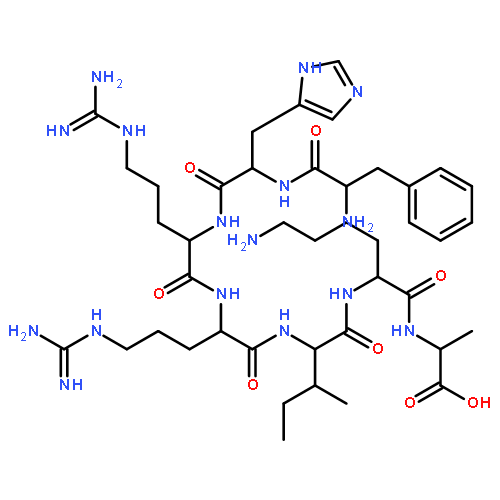
Peptidic ligands selectively targeting distinct G protein-coupled receptors that are highly expressed in tumor tissue represent a promising approach in drug delivery. Receptor-preferring analogues of neuropeptide Y (NPY) bind and activate the human Y1 receptor subtype (hY1 receptor), which is found in 90 % of breast cancer tissue and in all breast-cancer-derived metastases. Herein, novel highly boron-loaded Y1-receptor-preferring peptide analogues are described as smart shuttle systems for carbaboranes as 10B-containing moieties. Various positions in the peptide were screened for their susceptibility to carbaborane modification, and the most promising positions were chosen to create a multi-carbaborane peptide containing 30 boron atoms per peptide with excellent activation and internalization patterns at the hY1 receptor. Boron uptake studies by inductively coupled plasma mass spectrometry revealed successful uptake of the multi-carbaborane peptide into hY1-receptor-expressing cells, exceeding the required amount of 109 boron atoms per cell. This result demonstrates that the NPY/hY receptor system can act as an effective transport system for boron-containing moieties.
Co-reporter:David Böhme ;Dr. Annette G. Beck-Sickinger
ChemMedChem 2015 Volume 10( Issue 5) pp:804-814
Publication Date(Web):
DOI:10.1002/cmdc.201402514
Abstract
The side effects of chemotherapy can be overcome by linking toxic agents to tumor-targeting peptides with cleavable linkers. Herein, this concept is demonstrated by addressing the human Y1 receptor (hY1R), overexpressed in breast tumors, with analogues of the hY1R-preferring [F7,P34]NPY. First, carboxytetramethylrhodamine was connected to [F7,P34]NPY by an amide, ester, disulfide, or enzymatic linkage. Live imaging revealed hY1R-mediated delivery and allowed visualization of time-dependent intracellular release. Next, the fluorophore was replaced by the toxic agent methotrexate (MTX). In addition to linkage through the amide, ester, disulfide bond, or enzymatic cleavage site, a novel disulfide/ester linker was designed and coupled to [F7,P34]NPY by solid-phase peptide synthesis. Internalization studies showed hY1R subtype selective uptake, and cell viability experiments demonstrated hY1R-mediated toxicity that was clearly dependent on the linkage type. Fast release profiles for fluorophore-[F7,P34]NPY analogues correlated with high toxicities of MTX conjugates carrying the same linker types and emphasize the relevance of new structures connecting the toxophore and the carrier.
Co-reporter:Max Steinhagen, Peter-Georg Hoffmeister, Karoline Nordsieck, Rudi Hötzel, Lars Baumann, Michael C. Hacker, Michaela Schulz-Siegmund, and Annette G. Beck-Sickinger
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 8) pp:5891
Publication Date(Web):March 19, 2014
DOI:10.1021/am500794q
Preparation of smart materials by coatings of established surfaces with biomolecules will lead to the next generation of functionalized biomaterials. Rejection of implants is still a major problem in medical applications but masking the implant material with protein coatings is a promising approach. These layers not only disguise the material but also equip it with a certain biological function. The anti-inflammatory chemokine stromal cell-derived factor 1α (SDF-1α) is well suited to take over this function, because it efficiently attracts stem cells and promotes their differentiation and proliferation. At least the initial stem cell homing requires the formation of a concentration gradient. Thus, a reliable and robust release mechanism of SDF-1α from the material is essential. Several proteases, most notably matrix metalloproteinases, are upregulated during inflammation, which, in principle, can be exploited for a tightly controlled release of SDF-1α. Herein, we present the covalent immobilization of M-[S4V]-SDF-1α on novel biodegradable polymer films, which consist of heterobifunctional poly(ethylene glycol) and oligolactide-based functionalized macromers. A peptidic linker with a trimeric matrix metalloproteinase 9 (MMP-9) cleavage site (MCS) was used as connection and the linkage between the three components was achieved by combination of expressed protein ligation and Cu(I) catalyzed azide/alkyne cycloaddition. The MCS was used for MMP-9 mediated release of M-[S4V]-SDF-1α from the biomaterial and the released SDF-1α derivative was biologically active and induced strong cell migration, which demonstrates the great potential of this system.Keywords: cross-linked macromer; immobilization; matrix metalloproteinase 9; release; Stromal cell-derived factor 1α; tissue regeneration;
Co-reporter:Veronika Mäde;Dr. Kathrin Bellmann-Sickert;Anette Kaiser;Dr. Jens Meiler;Dr. Annette G. Beck-Sickinger
ChemMedChem 2014 Volume 9( Issue 11) pp:2463-2474
Publication Date(Web):
DOI:10.1002/cmdc.201402235
Abstract
Pancreatic polypeptide (PP) is a satiety-inducing gut hormone targeting predominantly the Y4 receptor within the neuropeptide Y multiligand/multireceptor family. Palmitoylated PP-based ligands have already been reported to exert prolonged satiety-inducing effects in animal models. Here, we suggest that other lipidation sites and different fatty acid chain lengths may affect receptor selectivity and metabolic stability. Activity tests revealed significantly enhanced potency of long fatty acid conjugates on all four Y receptors with a preference of position 22 over 30 at Y1, Y2 and Y5 receptors. Improved Y receptor selectivity was observed for two short fatty acid analogues. Moreover, [K30(E-Prop)]hPP2−36 (15) displayed enhanced stability in blood plasma and liver homogenates. Thus, short chain lipidation of hPP at key residue 30 is a promising approach for anti-obesity therapy because of maintained selectivity and a sixfold increased plasma half-life.
Co-reporter:Veronika Mäde;Dr. Kathrin Bellmann-Sickert;Anette Kaiser;Dr. Jens Meiler;Dr. Annette G. Beck-Sickinger
ChemMedChem 2014 Volume 9( Issue 11) pp:
Publication Date(Web):
DOI:10.1002/cmdc.201490043
Co-reporter:Veronika Mäde;Stefanie Babilon;Dr. Navjeet Jolly;Lizzy Wanka;Dr. Kathrin Bellmann-Sickert;Dr. Luis E. DiazGimenez;Dr. Karin Mörl;Dr. Helen M. Cox;Dr. Vsevolod V. Gurevich;Dr. Annette G. Beck-Sickinger
Angewandte Chemie International Edition 2014 Volume 53( Issue 38) pp:10067-10071
Publication Date(Web):
DOI:10.1002/anie.201403750
Abstract
Although G protein-coupled receptors (GPCRs) are targeted by more clinically used drugs than any other type of protein, their ligand development is particularly challenging. Humans have four neuropeptide Y receptors: hY1R and hY5R are orexigenic, while hY2R and hY4R are anorexigenic, and represent important anti-obesity drug targets. We show for the first time that PEGylation and lipidation, chemical modifications that prolong the plasma half-lives of peptides, confer additional benefits. Both modifications enhance pancreatic polypeptide preference for hY2R/hY4R over hY1R/hY5R. Lipidation biases the ligand towards arrestin recruitment and internalization, whereas PEGylation confers the opposite bias. These effects were independent of the cell system and modified residue. We thus provide novel insights into the mode of action of peptide modifications and open innovative venues for generating peptide agonists with extended therapeutic potential.
Co-reporter:Veronika Mäde;Stefanie Babilon;Dr. Navjeet Jolly;Lizzy Wanka;Dr. Kathrin Bellmann-Sickert;Dr. Luis E. DiazGimenez;Dr. Karin Mörl;Dr. Helen M. Cox;Dr. Vsevolod V. Gurevich;Dr. Annette G. Beck-Sickinger
Angewandte Chemie 2014 Volume 126( Issue 38) pp:10231-10235
Publication Date(Web):
DOI:10.1002/ange.201403750
Abstract
Although G protein-coupled receptors (GPCRs) are targeted by more clinically used drugs than any other type of protein, their ligand development is particularly challenging. Humans have four neuropeptide Y receptors: hY1R and hY5R are orexigenic, while hY2R and hY4R are anorexigenic, and represent important anti-obesity drug targets. We show for the first time that PEGylation and lipidation, chemical modifications that prolong the plasma half-lives of peptides, confer additional benefits. Both modifications enhance pancreatic polypeptide preference for hY2R/hY4R over hY1R/hY5R. Lipidation biases the ligand towards arrestin recruitment and internalization, whereas PEGylation confers the opposite bias. These effects were independent of the cell system and modified residue. We thus provide novel insights into the mode of action of peptide modifications and open innovative venues for generating peptide agonists with extended therapeutic potential.
Co-reporter:Max Steinhagen, Katja Zunker, Karoline Nordsieck, Annette G. Beck-Sickinger
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 12) pp:3504-3510
Publication Date(Web):15 June 2013
DOI:10.1016/j.bmc.2013.03.039
Recently, sortase A (SrtA) from Staphyloccus aureus moved into the focus of bioscience because of its ability to incorporate site specific modifications into proteins. The enzyme was mostly used to modify target proteins in an analytical scale, to study biomolecules in their cellular context. In this study, we show the applicability of SrtA mediated ligation for site specific modification of proteins in a large scale. Therefore, the reaction was first optimized using peptides and subsequently new reaction conditions were applied for the large scale biotinylation of interleukin-8. Furthermore, we established C-terminal immobilization of the SrtA on a PEG based resin and could demonstrate maintaining enzymatic activity. Immobilized SrtA significantly facilitates previous ligation protocols as the enzyme can be easily recycled. Also, the removal of excess reaction solution and the whole washing process is significantly accelerated, as centrifugation or filtration techniques can be applied instead of time-consuming chromatography steps.
Co-reporter:Daniel Rathmann, Xavier Pedragosa-Badia, Annette G. Beck-Sickinger
Analytical Biochemistry 2013 Volume 439(Issue 2) pp:173-183
Publication Date(Web):15 August 2013
DOI:10.1016/j.ab.2013.04.015
Mutagenic investigations of expressed membrane proteins are routine, but the variety of modifications is limited by the twenty canonical amino acids. We describe an easy and effective cysteine substitution mutagenesis method to modify and investigate distinct amino acids in vitro. The approach combines the substituted cysteine accessibility method (SCAM) with a functional signal transduction readout system using different thiol-specific reagents. We applied this approach to the prolactin-releasing peptide receptor (PrRPR) to facilitate biochemical structure–activity relationship studies of eight crucial positions. Especially for D6.59C, the treatment with the positively charged methanethiosulfonate (MTS) ethylammonium led to an induced basal activity, whereas the coupling of the negatively charged MTS ethylsulfonate nearly reconstituted full activity, obviously by mimicking the wild-type charged side chain. At E5.26C, W5.28C, Y5.38C, and Q7.35C, accessibility was observed but hindered transfer into the active receptor conformation. Accordingly, the combination of SCAM and signaling assay is feasible and can be adapted to other G-protein-coupled receptors (GPCRs). This method circumvents the laborious way of inserting non-proteinogenic amino acids to investigate activity and ligand binding, with rising numbers of MTS reagents allowing selective side chain modification. This method pinpoints to residues being accessible but also presents potential molecular positions to investigate the global conformation.
Co-reporter:Kerstin Burkert;Stefanie Babilon;Verena Ahrens;Annette Beck-Sickinger
BIOspektrum 2013 Volume 19( Issue 5) pp:499-501
Publication Date(Web):2013 September
DOI:10.1007/s12268-013-0345-6
G-protein coupled receptors (GPCRs) are involved in many physiological and pathophysiological roles. Many important drugs act on GPCRs, either by blocking them (antagonists) or by mimicking the natural ligand (agonists). As GPCRs have been shown to internalize a new concept is currently investigated: application of agonistic ligands fused to drugs to allow selective uptake into specific cells. This has been proven for selective uptake into tumour cells but may be expanded to other systems that selectively express GPCRs.
Co-reporter:John T. Heiker;Nora Klöting;Peter Kovacs
Cellular and Molecular Life Sciences 2013 Volume 70( Issue 14) pp:2569-2583
Publication Date(Web):2013 July
DOI:10.1007/s00018-013-1258-8
The molecular target of the adipokine vaspin (visceral adipose tissue-derived serpin; serpinA12) and its mode of action are unknown. Here, we provide the vaspin crystal structure and identify human kallikrein 7 (hK7) as a first protease target of vaspin inhibited by classical serpin mechanism with high specificity in vitro. We detect vaspin–hK7 complexes in human plasma and find co-expression of both proteins in murine pancreatic β-cells. We further demonstrate that hK7 cleaves human insulin in the A- and B-chain. Vaspin treatment of isolated pancreatic islets leads to increased insulin concentration in the media upon glucose stimulation without influencing insulin secretion. By application of vaspin and generated inactive mutants, we find the significantly improved glucose tolerance in C57BL/6NTac and db/db mice treated with recombinant vaspin fully dependent on the vaspin serpin activity and not related to vaspin-mediated changes in insulin sensitivity as determined by euglycemic-hyperinsulinemic clamp studies. Improved glucose metabolism could be mediated by increased insulin plasma concentrations 150 min after a glucose challenge in db/db mice, supporting the hypothesis that vaspin may inhibit insulin degradation by hK7 in the circulation. In conclusion, we demonstrate the inhibitory serpin nature and the first protease target of the adipose tissue-derived serpin vaspin, and our findings suggest hK7 inhibition by vaspin as an underlying physiological mechanism for its compensatory actions on obesity-induced insulin resistance.
Co-reporter:Dr. Lars Baumann ;Dr. Annette G. Beck-Sickinger
Angewandte Chemie 2013 Volume 125( Issue 36) pp:9729-9732
Publication Date(Web):
DOI:10.1002/ange.201302242
Co-reporter:Stephanie H. DeLuca;Daniel Rathmann;Jens Meiler
Biopolymers 2013 Volume 99( Issue 5) pp:314-325
Publication Date(Web):
DOI:10.1002/bip.22162
Abstract
The prolactin releasing peptide (PrRP) is involved in regulating food intake and body weight homeostasis, but molecular details on the activation of the PrRP receptor remain unclear. C-terminal segments of PrRP with 20 (PrRP20) and 13 (PrRP8-20) amino acids, respectively, have been suggested to be fully active. The data presented herein indicate this is true for the wildtype receptor only; a 5-10-fold loss of activity was found for PrRP8-20 compared to PrRP20 at two extracellular loop mutants of the receptor. To gain insight into the secondary structure of PrRP, we used CD spectroscopy performed in TFE and SDS. Additionally, previously reported NMR data, combined with ROSETTANMR, were employed to determine the structure of amidated PrRP20. The structural ensemble agrees with the spectroscopic data for the full-length peptide, which exists in an equilibrium between α- and 310-helix. We demonstrate that PrRP8-20's reduced propensity to form an α-helix correlates with its reduced biological activity on mutant receptors. Further, distinct amino acid replacements in PrRP significantly decrease affinity and activity but have no influence on the secondary structure of the peptide. We conclude that formation of a primarily α-helical C-terminal region of PrRP is critical for receptor activation. © 2012 Wiley Periodicals, Inc. Biopolymers 99: 273–281, 2013.
Co-reporter:Dr. Lars Baumann ;Dr. Annette G. Beck-Sickinger
Angewandte Chemie International Edition 2013 Volume 52( Issue 36) pp:9550-9553
Publication Date(Web):
DOI:10.1002/anie.201302242
Co-reporter:Stephan Schultz ;Dr. Annette G. Beck-Sickinger
ChemMedChem 2013 Volume 8( Issue 4) pp:549-559
Publication Date(Web):
DOI:10.1002/cmdc.201200448
Abstract
Obesity is one of the main human epidemics today. The increase in fat accumulation, which is associated with obesity, may significantly change the expression of several bioactive molecules known as adipokines. These adipokines interact not only with adipose tissue, but also with metabolically relevant organs such as liver and muscle. Understanding the molecular basics of potential novel targets might help to improve the therapeutic treatment of people who suffer from obesity. Herein we summarize the state of the art of two novel adipokines and their impaired or protective action in obesity: chemerin and vaspin. Their expression patterns, signal transduction activity, and resulting functions within the human body are introduced. We also discuss various possibilities to target these adipokines, which may represent promising new targets for the treatment of obesity by small and synthetic compounds.
Co-reporter:Lars Baumann, Silvana Prokoph, Christian Gabriel, Uwe Freudenberg, Carsten Werner, Annette G. Beck-Sickinger
Journal of Controlled Release 2012 Volume 162(Issue 1) pp:68-75
Publication Date(Web):20 August 2012
DOI:10.1016/j.jconrel.2012.04.049
The CXC chemokine stromal cell-derived factor-1α (SDF-1α, CXCL12) has been proven to recruit CXCR4 positive stem and progenitor cells of different sources to defected heart sites, with significant clinical benefits. However, the rapid proteolytic inactivation by inflammation-related proteases, inaccurate drug delivery or inappropriate local concentrations belong to the largest disadvantages for feasible application. Herein, we present a switchable, biased-like SDF-1α variant, AAV-[S4V]-SDF-1α, whose distinct activity is coupled to the inflammation-associated presence of dipeptidylpeptidase-4 (DPP-4), which cleaves an alanine-alanine dipeptide from the precursor. We decorated starPEG-heparin hydrogels with our novel SDF-1α variant and tested them for immobilization efficiency, time-dependent protein release as well as mobilization of early endothelial progenitor cells (eEPCs) in vitro. We found higher migration rates compared to conventional SDF-1α. In summary, we provide a conceptual work on cooperative effects of enzymatically activatable SDF-1α and starPEG-heparin hydrogels.
Co-reporter:Maria Findeisen ; Cäcilia Würker ; Daniel Rathmann ; René Meier ; Jens Meiler ; Roger Olsson
Journal of Medicinal Chemistry 2012 Volume 55(Issue 13) pp:6124-6136
Publication Date(Web):June 18, 2012
DOI:10.1021/jm300535s
The binding pocket of both NPFF receptors was investigated, focusing on subtype-selective behavior. By use of four nonpeptidic compounds and the peptide mimetics RF9 and BIBP3226, agonistic and antagonistic properties were characterized. A set of Ala receptor mutants was generated. The binding pocket was narrowed down to the upper part of transmembrane helices V, VI, VII and the extracellular loop 2. Positions 5.27 and 6.59 have been shown to have a strong impact on receptor activation and were suggested to form an acidic, negatively charged binding pocket in both NPFF receptor subtypes. Additionally, position 7.35 was identified to play an important role in functional selectivity. According to docking experiments, the aryl group of AC-216 interacts with position 7.35 in the NPFF1 but not in the NPFF2 receptor. These results provide distinct insights into the receptor specific binding pockets, which is necessary for the development of drugs to address the NPFF system.
Co-reporter:Sylvia Els ; Enrico Schild ; Pia Steen Petersen ; Tom-Marten Kilian ; Jacek Mokrosinski ; Thomas M. Frimurer ; Constance Chollet ; Thue W. Schwartz ; Birgitte Holst
Journal of Medicinal Chemistry 2012 Volume 55(Issue 17) pp:7437-7449
Publication Date(Web):August 27, 2012
DOI:10.1021/jm300414b
The ghrelin receptor displays a high constitutive activity suggested to be involved in the regulation of appetite and food intake. Here, we have created peptides with small changes in the core binding motif -wFw- of the hexapeptide KwFwLL-NH2 that can swap the peptide behavior from inverse agonism to agonism, indicating the importance of this sequence. Introduction of β-(3-benzothienyl)-d-alanine (d-Bth), 3,3-diphenyl-d-alanine (d-Dip) and 1-naphthyl-d-alanine (d-1-Nal) at position 2 resulted in highly potent and efficient inverse agonists, whereas the substitution of d-tryptophane at position 4 with 1-naphthyl-d-alanine (d-1-Nal) and 2-naphthyl-d-alanine (d-2-Nal) induces agonism in functional assays. Competitive binding studies showed a high affinity of the inverse agonist K-(d-1-Nal)-FwLL-NH2 at the ghrelin receptor. Moreover, mutagenesis studies of the receptor revealed key positions for the switch between inverse agonist and agonist response. Hence, only minor changes in the peptide sequence can decide between agonism and inverse agonism and have a major impact on the biological activity.
Co-reporter:Rayk Hassert, Mareen Pagel, Zhou Ming, Tilmann Häupl, Bernd Abel, Klaus Braun, Manfred Wiessler, and Annette G. Beck-Sickinger
Bioconjugate Chemistry 2012 Volume 23(Issue 10) pp:2129
Publication Date(Web):September 18, 2012
DOI:10.1021/bc3003875
Multifunctionality is gaining more and more importance in the field of improved biomaterials. Especially peptides feature a broad chemical variability and are versatile mediators between inorganic surfaces and living cells. Here, we synthesized a unique peptide that binds to SiO2 with nM affinity. We equipped the peptide with the bioactive integrin binding c[RGDfK]-ligand and a fluorescent probe by stepwise Diels–Alder reaction with inverse electron demand and copper(I) catalyzed azide–alkyne cycloaddition. For the first time, we report the generation of a multifunctional peptide by combining these innovative coupling reactions. The resulting peptide displayed an outstanding binding to silicon oxide and induced a significant increase in cell spreading and cell viability of osteoblasts on the oxidized silicon surface.
Co-reporter:Rayk Hassert;Peter-Georg Hoffmeister;Mareen Pagel;Michael Hacker;Michaela Schulz-Siegmund
Chemistry & Biodiversity 2012 Volume 9( Issue 11) pp:2648-2658
Publication Date(Web):
DOI:10.1002/cbdv.201200290
Abstract
Cyclic Arg-Gly-Asp (RGD) peptides show remarkable affinity and specificity to integrin receptors and mediate important physiological effects in tumor angiogenesis. Additionally, they are one of the keyplayers in improving the biocompatibility of biomaterials. The fully biodegradable polymer poly(lactic-co-glycolic acid) (PLGA) is frequently used for biomedical implants and can be applied as nanoparticles for drug delivery. The aim of this work was the generation of a lipidated c[RGDfK] peptide including a second functionality for coating of hydrophobic PLGA. Therefore, we established a general and straightforward strategy for the introduction of two different modifications into the same c[RGDfK] peptide. This allowed the generation of a palmitoylated integrin-binding lipopeptide that shows high affinity to PLGA. Additionally, we coupled 5(6)-carboxyfluorescein to the second site for modification to enable sensitive quantification of the immobilized lipopeptide on PLGA. In conclusion, we present a synthesis protocol that enables the preparation of c[RGDfK] lipopeptides with a strong affinity to PLGA and an additional site for modifications. This will provide the opportunity to introduce a variety of effector molecules site-specifically to the c[RGDfK] lipopeptide, which will enable the introduction of multifunctionality into c[RGDfK]-coated PLGA devices or nanoparticles.
Co-reporter:Karoline Nordsieck;Annelie Pichert;Dr. Sergey A. Samsonov;Dr. Lars Thomas;Dr. Christian Berger;Dr. M. Teresa Pisabarro; Dr. Daniel Huster; Dr. Annette G. Beck-Sickinger
ChemBioChem 2012 Volume 13( Issue 17) pp:2558-2566
Publication Date(Web):
DOI:10.1002/cbic.201200467
Abstract
The interactions between regulatory proteins such as interleukin-8 (IL-8) and glycosaminoglycans are of great interest both for the general understanding of regulatory processes in biology and for the development of implant coatings and innovative materials that suppress undesired immune responses and improve wound healing. In previous work, a number of residues of IL-8 that interact strongly with several glycosaminoglycans (GAGs) have been identified. In particular, the negatively charged Glu75 was reported to be involved in interactions with charged GAGs. To improve understanding of the role of this residue, we generated a selectively 15N-labeled E75K variant of IL-8(1–77) by expressed protein ligation. NMR and fluorescence spectroscopy in combination with molecular modeling were applied to evaluate the particular role of residue 75 in interactions with GAGs. Remarkably, more residues in the variant responded to GAG binding than in the wild-type. For the first time, we identified amino acids 34 to 36 as additional residues in the loop region of IL-8(1–77) that participate in the interactions with GAGs. These findings indicate that the N terminus of the E75K variant is more important as a second binding site for GAGs than that of the wild-type IL-8(1–77).
Co-reporter:Dr. Annette G. Beck-Sickinger;Dr. Nediljko Budisa
Angewandte Chemie 2012 Volume 124( Issue 2) pp:317-319
Publication Date(Web):
DOI:10.1002/ange.201107211
Co-reporter:Dr. Annette G. Beck-Sickinger;Dr. Nediljko Budisa
Angewandte Chemie International Edition 2012 Volume 51( Issue 2) pp:310-312
Publication Date(Web):
DOI:10.1002/anie.201107211
Co-reporter:Verena M. Ahrens ; René Frank ; Sven Stadlbauer ; Annette G. Beck-Sickinger ;Evamarie Hey-Hawkins
Journal of Medicinal Chemistry 2011 Volume 54(Issue 7) pp:2368-2377
Publication Date(Web):March 11, 2011
DOI:10.1021/jm101514m
Nontoxic ortho-carbaborane is one of the most promising structure for boron neutron capture therapy (BNCT). For directed uptake of ortho-carbaborane by tumor cells, receptor-subtype selective neuropeptide Y (NPY) and its derivatives were modified with ortho-carbaborane. The derivative [F7, P34]-NPY has been shown to be a breast cancer selective ligand that binds to the Y1-receptor subtype, whereas [Ahx(5−24)]-NPY selectively addresses Y2-receptor subtypes that are found in neuroblastoma cells. ortho-Carbaboranyl propionic acid was synthesized and linked to the ε-amino group of Nα-Fmoc protected l-lysine. The characterization of the compounds was performed by NMR, IR, and MS studies. The carbaborane-modified amino acid was incorporated into NPY, [F7, P34]-NPY, and [Ahx(5−24)]-NPY by an optimized solid phase peptide synthesis using Fmoc protection. Binding studies and IP accumulation assays confirmed nanomolar affinity and activity of the modified analogues despite of the large carbaborane cluster. Internalization studies revealed excellent and receptor subtype specific uptake of the conjugates into respective cells.
Co-reporter:Kathrin Bellmann-Sickert ; Christian E. Elling ; Andreas N. Madsen ; Paul B. Little ; Karsten Lundgren ; Lars-Ole Gerlach ; Ralf Bergmann ; Birgitte Holst ; Thue W. Schwartz
Journal of Medicinal Chemistry 2011 Volume 54(Issue 8) pp:2658-2667
Publication Date(Web):March 17, 2011
DOI:10.1021/jm101357e
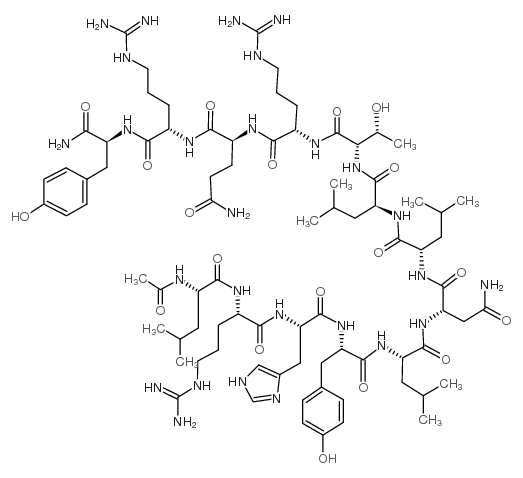
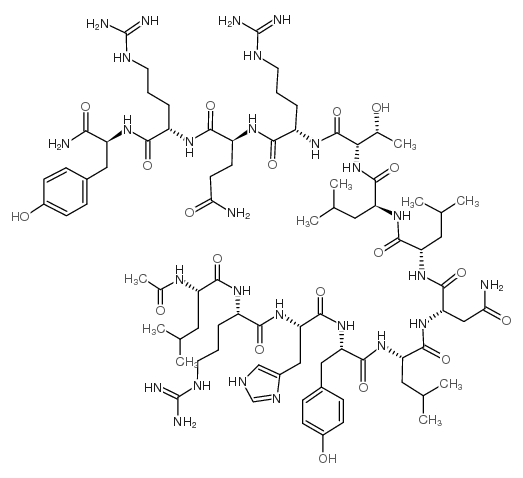
The main disadvantages of peptide pharmaceuticals are their rapid degradation and excretion, their low hydrophilicity, and low shelf lifes. These bottlenecks can be circumvented by acylation with fatty acids (lipidation) or polyethylene glycol (PEGylation). Here, we describe the modification of a human pancreatic polypeptide analogue specific for the human (h)Y2 and hY4 receptor with PEGs of different size and palmitic acid. Receptor specificity was demonstrated by competitive binding studies. Modifications had only a small influence on binding affinities and no influence on secondary structure. Both modifications improved pharmacokinetic properties of the hPP analogue in vivo and in vitro, however, lipidation showed a greater resistance to degradation and excretion than PEGylation. Furthermore, the lipidated peptide is taken up and degraded solely by the liver but not the kidneys. Lipidation resulted in prolonged action of the hPP analogue in respect of reducing food intake in mice after subcutaneous administration. Therefore, the lipidated hPP analogue could constitute a potential new therapeutic agent against obesity.
Co-reporter:Max Steinhagen;Dr. Kai Holl-Nell; Dr. Morten Meldal; Dr. Annette G. Beck-Sickinger
ChemBioChem 2011 Volume 12( Issue 16) pp:2426-2430
Publication Date(Web):
DOI:10.1002/cbic.201100434
Co-reporter:Kathrin Bellmann-Sickert;Dr. Annette G. Beck-Sickinger
ChemMedChem 2011 Volume 6( Issue 1) pp:193-200
Publication Date(Web):
DOI:10.1002/cmdc.201000403
Abstract
The chemokine stromal cell-derived factor-1 α (SDF1 α) is strongly involved in organogenesis, as well as inflammation and tissue repair, and acts by attracting different kinds of stem and progenitor cells. Therefore, it constitutes an interesting compound for drug development in regenerative medicine. However, it is prone to inactivation by proteolytic cleavage in human serum. Accordingly, it has to be stabilized against enzymatic degradation for any therapeutic application. We synthesized a palmitoylated SDF1 α analogue by native chemical ligation. Both the N-terminal thioester and the C-terminal palmitoylated fragment were prepared by solid-phase peptide synthesis. The activity of the refolded and pure compound was determined by an inositol phosphate turnover assay and revealed no loss in receptor activation. Additionally, resistance to proteolytic degradation was investigated in porcine liver homogenates and showed a near sevenfold increased half time. This study is a proof of principle approach for the lipidation of SDF1 α and provides a basis for further engineering of the chemokine in order to increase its therapeutic value.
Co-reporter:Maria Findeisen;Daniel Rathmann;Dr. Annette G. Beck-Sickinger
ChemMedChem 2011 Volume 6( Issue 6) pp:1081-1093
Publication Date(Web):
DOI:10.1002/cmdc.201100089
Abstract
Selectivity is a major issue in closely related multiligand/multireceptor systems. In this study we investigated the RFamide systems of hNPFF1R and hNPFF2R that bind the endogenous peptide hormones NPFF, NPAF, NPVF, and NPSF. By use of a systematic approach, we characterized the role of the C-terminal dipeptide with respect to agonistic properties using synthesized [Xaa 7]NPFF and [Xaa 8]NPFF analogues. We were able to identify only slight differences in potency upon changing the position of Arg 7, as all modifications resulted in identical behavior at the NPFF1R and NPFF2R. However, the C-terminal Phe 8 was able to be replaced by Trp or His with only a minor loss in potency at the NPFF2R relative to the NPFF1R. Analogues with shorter side chains, such as α-amino-4-guanidino butyric acid ([Agb 7]NPFF) or phenylglycine ([Phg 8]NPFF), decreased efficacy for the NPFF1R to 25–31 % of the maximal response, suggesting that these agonist–receptor complexes are more susceptible to structural modifications. In contrast, mutations to the conserved Asp 6.59 residue in the third extracellular loop of both receptors revealed a higher sensitivity toward the hNPFF2R receptor than toward hNPFF1R. These data provide new insight into the subtype-specific agonistic activation of the NPFF1 and NPFF2 receptors that are necessary for the development of selective agonists.
Co-reporter:Cathleen Juhl;Karin Mörl
Molecular and Cellular Biochemistry 2011 Volume 356( Issue 1-2) pp:
Publication Date(Web):2011 October
DOI:10.1007/s11010-011-0941-z
Adiponectin is an adipose tissue-derived hormone that is involved in the inhibition of metabolic syndrome, protection of hypertension, and suppression of atherosclerosis. Since these effects are not understood in detail, adiponectin signaling has to be clarified for therapeutic applications. Adiponectin activities are mediated by its two receptors adiponectin receptor 1 and adiponectin receptor 2, which consist of seven transmembrane helices. Previous studies revealed the beta subunit of protein kinase CK2 as an interaction partner of the adiponectin receptor 1 N-terminus using a yeast-two-hybrid screen, co-immunoprecipitation, ELISA experiments, and co-localization studies. Inhibition of CK2 activity by 2-dimethylamino-4,5,6,7-tetrabromo-1H-benz-imidazole led to a decrease of ACC phosphorylation and indicates an important role of CK2 in adiponectin signaling. CK2 is characterized as a heterotetramer that consists of two regulatory beta and two catalytic alpha subunits, but a holoenzyme-independent role for both subunits is described as well. Therefore, we analyzed the role of the catalytic subunit in this interaction by co-immunoprecipitation and bimolecular fluorescence complementation studies and found CK2 alpha as an interaction partner of the receptor. Treatment with full-length adiponectin resulted in no dissociation of the catalytic alpha subunit. Consequently, our data suggest an interaction of the adiponectin receptor 1 with the tetrameric complex and identified protein kinase CK2 as a key player in adiponectin signaling.
Co-reporter:IrfanU. Khan Dr.;Denise Zwanziger;Ilka Böhme Dr.;Muhammad Javed;Hamid Naseer;SyedW. Hyder;AnnetteG. Beck-Sickinger Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 6) pp:1155-1158
Publication Date(Web):
DOI:10.1002/anie.200905008
Co-reporter:IrfanU. Khan Dr.;Denise Zwanziger;Ilka Böhme Dr.;Muhammad Javed;Hamid Naseer;SyedW. Hyder;AnnetteG. Beck-Sickinger Dr.
Angewandte Chemie 2010 Volume 122( Issue 6) pp:1174-1177
Publication Date(Web):
DOI:10.1002/ange.200905008
Co-reporter:Michael Haack
Chemical Biology & Drug Design 2009 Volume 73( Issue 6) pp:573-583
Publication Date(Web):
DOI:10.1111/j.1747-0285.2009.00823.x
Despite a considerable sequence identity of the three mammalian hormones of the neuropeptide Y family, namely neuropeptide Y, peptide YY and pancreatic polypeptide, their structure in solution is described to be different. A so-called pancreatic polypeptide-fold has been identified for pancreatic polypeptide, whereas the structure of the N-terminal segment of neuropeptide Y is unknown. This element is important for the binding of neuropeptide Y to two of its relevant receptors, Y1 and Y5, but not to the Y2 receptor subtype. In this study now, three doubly fluorescent-labeled analogs of neuropeptide Y have been synthesized that still bind to the Y5 receptor with high affinity to investigate the conformation in solution and, for the first time, to probe the conformational changes upon binding of the ligand to its receptor in cell membrane preparations. The results obtained from the fluorescence resonance energy transfer investigations clearly show considerable differences in transfer efficiency that depend both on the solvent as well as on the peptide concentration. However, the studies do not support a pancreatic polypeptide-like folding of neuropeptide Y in the presence of membranes that express the human Y5 receptor subtype.
Co-reporter:Ilka Böhme;Annette G Beck-Sickinger
Cell Communication and Signaling 2009 Volume 7( Issue 1) pp:
Publication Date(Web):2009 December
DOI:10.1186/1478-811X-7-16
The investigation of biological systems highly depends on the possibilities that allow scientists to visualize and quantify biomolecules and their related activities in real-time and non-invasively. G-protein coupled receptors represent a family of very dynamic and highly regulated transmembrane proteins that are involved in various important physiological processes. Since their localization is not confined to the cell surface they have been a very attractive "moving target" and the understanding of their intracellular pathways as well as the identified protein-protein-interactions has had implications for therapeutic interventions. Recent and ongoing advances in both the establishment of a variety of labeling methods and the improvement of measuring and analyzing instrumentation, have made fluorescence techniques to an indispensable tool for GPCR imaging. The illumination of their complex life cycle, which includes receptor biosynthesis, membrane targeting, ligand binding, signaling, internalization, recycling and degradation, will provide new insights into the relationship between spatial receptor distribution and function. This review covers the existing technologies to track GPCRs in living cells. Fluorescent ligands, antibodies, auto-fluorescent proteins as well as the evolving technologies for chemical labeling with peptide- and protein-tags are described and their major applications concerning the GPCR life cycle are presented.
Co-reporter:Denise Zwanziger, Irfan Ullah Khan, Ines Neundorf, Stephanie Sieger, Lutz Lehmann, Matthias Friebe, Ludger Dinkelborg and Annette G. Beck-Sickinger
Bioconjugate Chemistry 2008 Volume 19(Issue 7) pp:1430
Publication Date(Web):June 24, 2008
DOI:10.1021/bc7004297
The successful use of peptides as potential radiopharmaceuticals essentially requires the modification of the bioactive peptide hormones to introduce chelators for radiolabeling. In this study, four Y1/Y2 receptor-selective NPY analogues with different receptor subtype specificities have been investigated. For in vitro studies, the cold metal surrogate was used. Gallium and indium complexes were introduced by using 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid as bifunctional chelator. The peptides were synthesized by solid-phase peptide synthesis (SPPS), the chelator was coupled either at the N-terminus or at the Nε side chain of Lys4 of the resin-bound peptide, and the labeling was performed in solution after cleavage. Competitive binding assays showed high binding affinity of the receptor-selective analogues at NPY receptor expressing cells. To test internalization of the novel peptide analogues and the metabolic stability in human blood plasma, the corresponding 5(6)-carboxyfluorescein (CF) analogues were prepared and investigated. One of the most promising analogues, the Y1-receptor selective [Lys(DOTA)4, Phe7, Pro34]NPY was labeled with 111In and injected into nude mice that bear MCF-7 breast cancer xenografts, and biodistribution studies were performed. In vitro and in vivo studies suggest that receptor-selective analogues of NPY have promising characteristics for future applications in nuclear medicine for breast tumor diagnosis and therapy.
Co-reporter:Diana Lindner, Jan van Dieck, Nicole Merten, Karin Mörl, Robert Günther, Hans-Jörg Hofmann and Annette G. Beck-Sickinger
Biochemistry 2008 Volume 47(Issue 22) pp:
Publication Date(Web):May 6, 2008
DOI:10.1021/bi800181k
Many G protein-coupled receptors belong to families of different receptor subtypes, which are recognized by a variety of distinct ligands. To study such a multireceptor/multiligand system, we investigated the Y-receptor family. This family consists of four G protein-coupled Y receptors in humans (hY1R, hY2R, hY4R, and hY5R) and is activated by the so-called NPY hormone family, which itself consists of three native peptide ligands named neuropeptide Y (NPY), pancreatic polypeptide (PP), and peptide YY (PYY). The hY5R shows high affinity for all ligands, although for PP binding, the affinity is slightly decreased. As a rational explanation, we suggest that Tyr27 is lost as a contact point between PP and the hY5R in contrast to NPY or PYY. Furthermore, several important residues for ligand binding were identified by the first extensive mutagenesis study of the hY5R. Using a complementary mutagenesis approach, we were able to discover a novel interaction point between hY5R and NPY. The interaction between NPY(Arg25) and hY5R(Asp2.68) as well as between NPY(Arg33) and hY5R(Asp6.59) is maintained in the binding of PYY and PP to hY5R but different to the PP-hY4R and NPY-hY1R contact points. Therefore, we provide evidence that the receptor subtype and not the pre-orientated conformation of the ligand at the membrane decides the binding mode. Furthermore, the first hY5R model was set up on the basis of the crystal structure of bovine rhodopsin. We can show that most of the residues identified to be critical for ligand binding are located within the now postulated binding pocket.
Co-reporter:Robert Rennert;Ines Neundorf Dr.;Heinz-Georg Jahnke;Philipp Suchowerskyj;Pascal Dournaud Dr.;Andrea Robitzki Dr.;AnnetteG. Beck-Sickinger Dr.
ChemMedChem 2008 Volume 3( Issue 2) pp:241-253
Publication Date(Web):
DOI:10.1002/cmdc.200700216
Abstract
Now that the human genome has been decoded, the demand for novel therapeutic concepts, such as gene and stem cell therapy, is higher than ever before. Although new and better pharmaceutical agents are available, their efficient delivery to the intracellular site of action is still a serious challenge. A possible solution to this problem is the use of cell-penetrating peptides as delivery vectors, including derivatives of human calcitonin (hCT). The aim of this study was to synthesise novel branched hCT-derived peptides for the noncovalent delivery of nucleic acids. The uptake of the resulting oligocationic peptides into various cell lines as well as primary cells was monitored by fluorescence microscopy. To determine the appropriate peptide–plasmid charge ratios for efficient cell transfection, electromobility shift assays were carried out. Finally, flow cytometric and fluorescence microscopic studies of gene expression highlighted two novel hCT-derived peptides as highly effective in the delivery of noncovalently complexed plasmid DNA. Thus, the absence of cytotoxicity paired with highly efficient cell internalisation and transfection rates, in primary cells as well, make both peptides powerful candidates as drug delivery vectors, especially for plasmid DNA, for both in vivo and ex vivo therapeutic applications.
Co-reporter:Franka Pluder;Andrea Sinz
Chemical Biology & Drug Design 2007 Volume 69(Issue 1) pp:
Publication Date(Web):20 FEB 2007
DOI:10.1111/j.1747-0285.2007.00466.x
Calcitonin gene-related peptide is a 37 amino acid neuropeptide. Although calcitonin gene-related peptide activates a G-protein-coupled receptor, recent evidence suggests that calcitonin gene-related peptide induces more complex signaling cascades including the activation of MAP kinases [Eur J Pharmacol; 389:125–130 (2000), Neuropeptides; 34:229–233 (2000)]. However, the molecular mechanisms of this activation still remain to be elucidated. For the first time we applied a proteomics approach in order to identify molecular targets of calcitonin gene-related peptide downstream signaling in the neuroblastoma cell line SK-N-MC and identified proteins that changed either their expression, location, or their post-translational modifications in a time-dependent manner after calcitonin gene-related peptide stimulation.
Co-reporter:Franka Pluder, Annette G. Beck-Sickinger
Analytical Biochemistry 2007 Volume 361(Issue 2) pp:299-301
Publication Date(Web):15 February 2007
DOI:10.1016/j.ab.2006.11.003
Co-reporter:Ralf David
European Biophysics Journal 2007 Volume 36( Issue 4-5) pp:385-391
Publication Date(Web):2007 April
DOI:10.1007/s00249-006-0100-8
The three-dimensional structure of human interleukin-8 (hIL-8) was determined by the use of NMR and X-ray methodology. At high concentrations interleukin-8 and many other chemokines form a non-covalent homodimer. Several studies have been performed to investigate the relevance of the dimer on receptor activation and led to contradictory results. In order to obtain a better understanding of the dimerisation process, covalently linked homo- and heterodimers were produced by photo-induced dimerisation of hIL-8 analogues that contain the photo-activatable amino acid p-benzoyl-phenylalanine (Bpa) at different positions. Whereas the N-terminal fragment (1–54) was expressed as recombinant thioester, the C-terminal fragments (55–77) that contain Bpa either at position 65 or 74 were obtained by solid-phase peptide synthesis. The segments were combined by expressed protein ligation and led to full length IL-8 variants containing the non-proteinogenic amino acid Bpa at single positions. IP3 activity tests showed high biological activity for the CXCR1–GFP receptor for both variants comparable to that of the native ligand. The refolded and purified ligation-products were used for dimer formation by UV-irradiation. The analysis of the reaction mixture was performed by gel-electrophoresis and mass spectrometry and showed that dimer formation of IL-8 occurred in a position dependent manner. [Bpa74]hIL-8 has a high tendency to form covalent dimers whereas no dimer formation was observed for the variant with Bpa at position 65. Accordingly one residue of the dimerisation interface could be identified.
Co-reporter:Kai Holl-Nell and Dr.
ChemBioChem 2007 Volume 8(Issue 9) pp:
Publication Date(Web):16 MAY 2007
DOI:10.1002/cbic.200700056
The difference between site-specific and random immobilisation of the aldo/keto reductase AKR1A1 was explored. AKR1A1 was recombinantly expressed as a thioester by the intein strategy. The thioester was selectively modified with a biotin label by the expressed protein ligation method, and subsequent immobilisation on streptavidin templates was performed. Adsorption of wild-type AKR1A1 to streptavidin templates and of biotinylated AKR1A1 to uncoated templates was used to study randomly immobilised enzymes. Investigation of the kinetic parameters revealed remarkably improved activity for the site-specifically immobilised enzyme, which was comparable to that of the wild-type enzyme in solution and 60–300-fold greater than that of the randomly immobilized enzymes. Furthermore, the enzyme was surprisingly stable. No loss of activity was observed for over a week, and even after 50 days more than 35 % of activity was maintained.
Co-reporter:Felix Kratz;Ulrike Krauss
Journal of Molecular Recognition 2003 Volume 16(Issue 5) pp:280-287
Publication Date(Web):22 SEP 2003
DOI:10.1002/jmr.638
Severe and often therapy-limiting side effects are a major obstacle in cancer chemotherapy. New delivery concepts reducing systemic side effects are needed in order to optimize anticancer therapies. Several approaches have been followed, most of them concentrating on macromolecular carriers like liposomes, monoclonal antibodies, serum proteins or polyethylene glycol. We present here a novel type of anthracycline conjugate, using a small carrier peptide derived from the peptide hormone human calcitonin (hCT). The carrier peptide hCT(9–32) has so far been shown to be capable of transporting fluorophores or proteins across cellular membranes. Two different carrier peptide–daunorubicin conjugates were prepared, one with an acid-stable amide bond, the second with an acid-labile hydrazone bond. In vitro studies with daunorubicin linked to the carrier peptide via an acid-labile hydrazone bond demonstrated comparable cytotoxicity to daunorubicin in various daunorubicin sensitive cell lines (neuroblastoma cell lines SK-N-MC and SMS-KAN; HEK 293 T cells). In addition, fluorescence microscopy provided further insight into the mechanism of uptake of the carrier peptide hCT(9–32), indicating that endosomal compartments with reduced pH are involved in the intracellular release of daunorubicin. Copyright © 2003 John Wiley & Sons, Ltd.
Co-reporter:Zuzana Machova Dr.;Regula von Eggelkraut-Gottanka Pharm.;Nicole Wehofsky Dr.;Frank Bordusa Dr. Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 40) pp:
Publication Date(Web):30 SEP 2003
DOI:10.1002/anie.200351774
Enzymatic synthesis and expressed protein ligation were combined in a method for protein synthesis in which protein thioesters with specific leaving groups could be used by protease V8 from Staphylococcus aureus to catalyze the formation of peptide bonds. This method was applied to the synthesis of fluorescence-labeled (Fl) analogues of proneuropeptide Y (proNPY; see scheme). CBD=chitin binding domain.
Co-reporter:Regula von Eggelkraut-Gottanka Dipl.-Pharm.;Zuzana Machova Dipl.-Ing.;Eric Grouzmann PD Dr. Dr.
ChemBioChem 2003 Volume 4(Issue 5) pp:
Publication Date(Web):5 MAY 2003
DOI:10.1002/cbic.200390063
The cover picture shows how a combination of recombinant synthesis and chemical synthesis has been used to obtain chemically modified proteins. N-terminal protein segments of pro-neuropeptide Y (proNPY) were produced as intein-fusion proteins in Escherischia coli in order to obtain thioesters. C-terminal segments were synthesized by parallel automated peptide synthesis and derivatized to obtain carboxyfluorescein- (CF) and biotin-labeled peptides. Native chemical ligation yielded chemically modified full-length analogues of proNPY that can be used to monitor the biosynthesis of neuropeptide Y. Futher information can be found in the article by Beck-Sickinger and co-workers on p. 425 ff.
Co-reporter:Regula von Eggelkraut-Gottanka Dipl.-Pharm.;Zuzana Machova Dipl.-Ing.;Eric Grouzmann PD Dr. Dr.
ChemBioChem 2003 Volume 4(Issue 5) pp:
Publication Date(Web):5 MAY 2003
DOI:10.1002/cbic.200200546
Enzymatic cleavage of prohormone neuropeptide Y (proNPY) leads to mature neuropeptide Y (NPY), a widely distributed neuropeptide with multiple functions both peripherally and centrally. A single dibasic pair of amino acids, Lys38-Arg39, represents the recognition motif for a class of hormone-processing enzymes known as prohormone convertases (PCs). Two members of this PC family, PC1/3 and PC2, are involved in proNPY cleavage. The aim of this work was to establish an effective method for the generation of full-length 69-amino acid proNPY analogues for further studies of prohormone convertase interaction. We have chosen two ligation sites in order to perform the semisynthesis of proNPY analogues by expressed protein ligation (EPL). By using the intein-mediated purification system (IMPACT) with improved conditions for intein splicing, we were able to isolate proNPY 1–40 and proNPY 1–54 fragments as Cterminal thioesters. Peptides bearing Nterminal cysteine instead of the naturally occurring Ser41and Thr55residues, respectively, were generated by solid-phase peptide synthesis. Moreover, labels (carboxyfluorescein and biotin) were inserted into the peptide sequences. The synthesis of the [C41]proNPY 41–69 fragment, which proved to be a difficult peptide sequence, could be achieved by the incorporation of two pseudo-proline derivatives. Western blot analysis revealed that all five proNPY analogues are recognized by monoclonal antibodies directed against NPY as well as against the Cflanking peptide of NPY (CPON).
Co-reporter:Zuzana Machova Dr.;Regula von Eggelkraut-Gottanka Pharm.;Nicole Wehofsky Dr.;Frank Bordusa Dr. Dr.
Angewandte Chemie 2003 Volume 115(Issue 40) pp:
Publication Date(Web):30 SEP 2003
DOI:10.1002/ange.200351774
Enzymatische Synthese und exprimierte Proteinligation werden hier zur Proteinsynthese kombiniert, wobei Proteinthioester mit spezifischen Abgangsgruppen von der Protease V8 aus Staphylococcus aureus zur Bildung von Peptidbindungen genutzt werden. Auf diesem Weg wurden Fluoreszenz-markierte Analoga des Pro-Neuropeptids Y synthetisiert.
Co-reporter:Norman Koglin Dipl.-Biochem.;Chiara Zorn Dr.;Raphael Beumer Dr.;Chiara Cabrele Dr.;Christian Bubert Dr.;Norbert Sewald Dr.;Oliver Reiser Dr. Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 2) pp:
Publication Date(Web):16 JAN 2003
DOI:10.1002/anie.200390078
Unique, conformationally restricted β-amino acids are a constituent of truncated linear peptides such as 1, which have been obtained for the first time; these species show high and selective affinity toward the Y1 receptor. The high affinity of these ligands is dependent on the position and the configuration of this β-aminocyclopropane carboxylic acid.
Co-reporter:Norman Koglin Dipl.-Biochem.;Chiara Zorn Dr.;Raphael Beumer Dr.;Chiara Cabrele Dr.;Christian Bubert Dr.;Norbert Sewald Dr.;Oliver Reiser Dr. Dr.
Angewandte Chemie 2003 Volume 115(Issue 2) pp:
Publication Date(Web):16 JAN 2003
DOI:10.1002/ange.200390046
Als einzigartige konformativ eingeschränkte β-Aminosäuren erwiesen sich β-Aminocyclopropancarbonsäuren. Erstmals wurden lineare Peptide wie 1 hergestellt, die diese Aminosäuren enthalten und hohe sowie selektive Affinität gegenüber dem Y1-Rezeptor aufweisen. Die Affinität dieser Liganden ist abhängig von der Position und der Konfiguration der β-Aminocyclopropancarbonsäuren.
Co-reporter:Zuzana Machova Dipl.-Biochem.;Christiane Mühle Dipl.Biochem.;Ulrike Krauss Dipl.-Pharm.;Rachel Tréhin Dipl.-Pharm.;Annette Koch Dipl.-Pharm.;Hans P. Merkle Dr. Dr.
ChemBioChem 2002 Volume 3(Issue 7) pp:
Publication Date(Web):24 JUN 2002
DOI:10.1002/1439-7633(20020703)3:7<672::AID-CBIC672>3.0.CO;2-D
Carrier peptides offer new opportunities to overcome problems in cellular drug delivery. Their objectives are improved cellular uptake or permeation of biological membranes, which are important pharmacokinetic features for the cellular distribution of therapeutics. Previously, human calcitonin (hCT) and selected C-terminal hCT fragments have been shown to be internalized and to permeate the epithelium of the nasal mucosa. To assess the potential of hCT-derived carrier peptides for cellular internalization of a model protein we fused enhanced green fluorescent protein (EGFP) and the [C8]hCT8–32 fragment by using expressed protein ligation (EPL). EGFP thioester was obtained by intein-mediated purification with an affinity chitin-binding tag (the IMPACT system, based on protein splicing). Internalization of EGFP-[C8]hCT8–32 by excised bovine nasal mucosa was monitored by confocal laser scanning microscopy. This novel conjugate displayed internalization into some sectors of the mucosa, whereas EGFP itself was not capable of translocation. Thus, we demonstrate successful internalization of a model protein through ligation to an hCT-derived carrier peptide, which has potential for the delivery of therapeutics. At this point the respective mechanism of translocation is unknown.
Co-reporter:Cathleen Juhl, David Kosel, Annette G. Beck-Sickinger
Cellular Signalling (September 2012) Volume 24(Issue 9) pp:1762-1769
Publication Date(Web):1 September 2012
DOI:10.1016/j.cellsig.2012.05.002
The anterograde trafficking of GPCR has been described as a tightly controlled process involving specific amino acid sequences that mediate the receptor transport. In this study, we investigated whether the cell surface delivery of the adiponectin receptor 1, a newly identified class of heptahelix receptors different from G protein-coupled receptors, is regulated. Sequential N-terminal deletion revealed that the export of the AdipoR1 from the endoplasmic reticulum (ER) is controlled by distinct parts of the receptor N-terminus. Strong evidence is provided that the ER exit is mediated by two specific sequences, a F(X)3F(X)3F and a D(X)3LL motif. Disruption of these motifs led to a substantial accumulation of the AdipoR1 in the ER. Mutation of similar motifs in the AdipoR1 C-terminus did not result in aberrant receptor localization, suggesting that these motifs are sequence and position specific to the AdipoR1 N-terminus. Further analysis of the regulation mechanism identified an interaction with the chaperone BiP and additionally, strong evidence is provided that both motifs exert different biological function in the AdipoR1 ER export. In conclusion, our data demonstrate that the receptor transport shares similar ER exit motifs although AdipoR are structurally different from GPCR. However, since even two specific sequences are identified, the anterograde trafficking of the AdipoR1 seems to be regulated in a more complex manner.Highlights► The AdipoR1 N-terminus controls the anterograde transport. ► The ER exit is mediated by two sequences, a F(X)3F(X)3F and a D(X)3LL motif. ► Both motifs are sequence and position specific to the AdipoR1 N-terminus. ► AdipoR and GPCR share similar regulation motifs but distinct mechanisms.
Co-reporter:Kathrin Bellmann-Sickert, Annette G. Beck-Sickinger
Trends in Pharmacological Sciences (September 2010) Volume 31(Issue 9) pp:434-441
Publication Date(Web):1 September 2010
DOI:10.1016/j.tips.2010.06.003
Major indications for use of peptide-based therapeutics include endocrine functions (especially diabetes mellitus and obesity), infectious diseases, and cancer. Whereas some peptide pharmaceuticals are drugs, acting as agonists or antagonists to directly treat cancer, others (including peptide diagnostics and tumour-targeting pharmaceuticals) use peptides to ‘shuttle’ a chemotherapeutic agent or a tracer to the tumour and allow sensitive imaging or targeted therapy. Significant progress has been made in the last few years to overcome disadvantages in peptide design such as short half-life, fast proteolytic cleavage, and low oral bioavailability. These advances include peptide PEGylation, lipidisation or multimerisation; the introduction of peptidomimetic elements into the sequences; and innovative uptake strategies such as liposomal, capsule or subcutaneous formulations. This review focuses on peptides targeting G protein-coupled receptors that are promising drug candidates or that have recently entered the pharmaceutical market.
Co-reporter:John T. Heiker, Cornelia M. Wottawah, Cathleen Juhl, David Kosel, Karin Mörl, Annette G. Beck-Sickinger
Cellular Signalling (June 2009) Volume 21(Issue 6) pp:936-942
Publication Date(Web):1 June 2009
DOI:10.1016/j.cellsig.2009.02.003
Adiponectin is an adipokine with anti-atherogenic, anti-diabetic and insulin sensitizing properties. Its effects on energy homeostasis, glucose and lipid metabolism are mediated by two ubiquitously expressed seven-transmembrane receptors, AdipoR1 and -R2. With the exception of APPL1 and RACK1, no intracellular binding partners of adiponectin receptors are reported and thus signaling pathways downstream of these receptors remain largely unknown. To incorporate adiponectins protective potential in drug development it is essential to understand adiponectin signaling cascades in detail. A yeast two-hybrid approach employing AdipoR1s cytoplasmatic N-terminus led to the identification of the regulatory subunit of protein kinase CK2. We confirmed the interaction in co-immunoprecipitation, ELISA experiments and co-localization analysis in mammalian cells. Furthermore we could localize the interaction site in an N-terminal basic region close to the transmembrane domain. In adiponectin stimulation experiments of C2C12 mouse myotubes and MCF7 cells incorporating CK2 inhibitor 2-dimethylamino-4,5,6,7-tetrabromo-1H-benz-imidazole (DMAT) we found a modulator role of CK2 in adiponectin signaling. Accordingly we identified the regulatory subunit of protein kinase CK2 as a novel intracellular partner of AdipoR1 and have strong evidence of CK2 as an effector molecule in adiponectin signaling. Since CK2 is involved in signaling cascades of other adipokines and hormones, e.g. leptin and insulin, our findings suggest a possible key function in crosstalk between adiponectin and insulin signaling pathways and could provide further insight into the anti-diabetic effects of adiponectin.
Co-reporter:Diana Lindner, Cornelia Walther, Anja Tennemann, Annette G. Beck-Sickinger
Cellular Signalling (January 2009) Volume 21(Issue 1) pp:61-68
Publication Date(Web):1 January 2009
DOI:10.1016/j.cellsig.2008.09.007
The N terminus is the most variable element in G protein-coupled receptors (GPCRs), ranging from seven residues up to ~ 5900 residues. For family B and C GPCRs it is described that at least part of the ligand binding site is located within the N terminus. Here we investigated the role of the N terminus in the neuropeptide Y receptor family, which belongs to the class A of GPCRs. We cloned differentially truncated Y receptor mutants, in which the N terminus was partially or completely deleted. We found, that eight amino acids are sufficient for full ligand binding and signal transduction activity. Interestingly, we could show that no specific amino acids but rather the extension of the first transmembrane helix by any residues is sufficient for receptor activity but also for membrane integration in case of the hY1 and the hY4 receptors. In contrast, the complete deletion of the N terminus in the hY2 receptors resulted in a mutant that is fully integrated in the membrane but does not bind the ligand very well and internalizes much slower compared to the wild type receptor. Interestingly, also these effects could be reverted by any N-terminal extension. Accordingly, the most important function of the N termini seems to be the stabilization of the first transmembrane helix to ensure the correct receptor structure, which obviously is essential for ligand binding, integration into the cell membrane and receptor internalization.
Co-reporter:Robert Rennert, Ines Neundorf, Annette G. Beck-Sickinger
Advanced Drug Delivery Reviews (1 March 2008) Volume 60(Issues 4–5) pp:485-498
Publication Date(Web):1 March 2008
DOI:10.1016/j.addr.2007.09.008
Among the family of the so-called cell-penetrating peptides (CPP) sequences derived from the native peptide hormone human calcitonin (hCT) have recently proven to translocate different bioactive molecules across cellular membranes. Herein, we give an extensive summary of the development of hCT-derived carrier peptides, beginning with the therapeutic nasal administration of full-length hCT. Hence, N-terminally truncated hCT fragments were investigated and subsequently optimised to extend their field of application. The latest generation of hCT-derived carrier peptides are highly effective, branched peptides. The current state of the art is reviewed concerning the structural requirements, mechanistic assumptions and metabolic features of these peptides as well as experiments proving their excellent carrier potential.

![Europium(3+),[N,N'-bis(2-aminoethyl)-1,14,39,40,41,42,43,44-octaazaoctacyclo[12.12.12.13,7.18,12.116,20.121,25.128,32.133,37]tetratetraconta-3,5,7(44),8,10,12(43),16,18,20(42),21,23,25(41),28,30,32(40),33,35,37(39)-octadecaene-5,10-dicarboxamide-kN1,kN14,kN39,kN40,kN41,kN42,kN43,kN44]-](/data/chemimg/427300/125433-96-7.png)
![Europium(3+),[N,N'-bis(2-aminoethyl)-1,14,39,40,41,42,43,44-octaazaoctacyclo[12.12.12.13,7.18,12.116,20.121,25.128,32.133,37]tetratetraconta-3,5,7(44),8,10,12(43),16,18,20(42),21,23,25(41),28,30,32(40),33,35,37(39)-octadecaene-5,10-dicarboxamide-kN1,kN14,kN39,kN40,kN41,kN42,kN43,kN44]-](/data/chemimg/427300/125433-96-7_b.png)
![N-[9-(2-carboxyphenyl)-6-(dimethylamino)-3H-xanthen-3-ylidene]-N-methylmethanaminium perchlorate](http://img.cochemist.com/ccimg/62700/62669-72-1.png)
![N-[9-(2-carboxyphenyl)-6-(dimethylamino)-3H-xanthen-3-ylidene]-N-methylmethanaminium perchlorate](http://img.cochemist.com/ccimg/62700/62669-72-1_b.png)
![(S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(2,2-dimethylbenzo[d][1,3]dioxol-5-yl)propanoic acid](http://img.cochemist.com/ccimg/852300/852288-18-7.png)
![(S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(2,2-dimethylbenzo[d][1,3]dioxol-5-yl)propanoic acid](http://img.cochemist.com/ccimg/852300/852288-18-7_b.png)






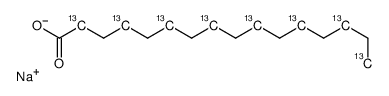
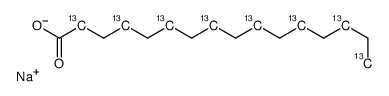
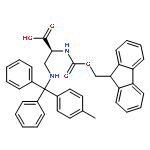
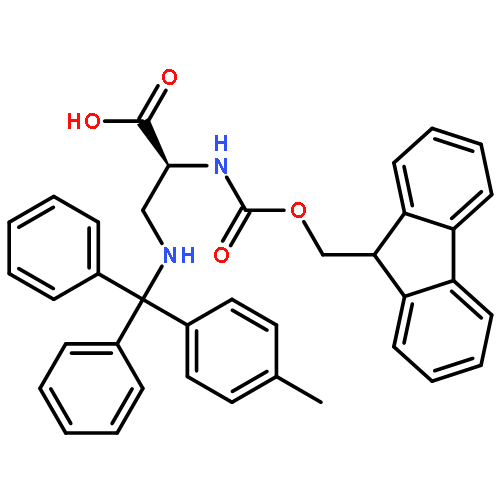


![L-Norleucine, 6-azido-N-[(9H-fluoren-9-ylmethoxy)carbonyl]-](/data/chemimg/116900/159610-89-6.png)
![L-Norleucine, 6-azido-N-[(9H-fluoren-9-ylmethoxy)carbonyl]-](/data/chemimg/116900/159610-89-6_b.png)












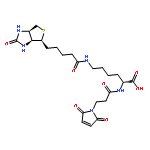
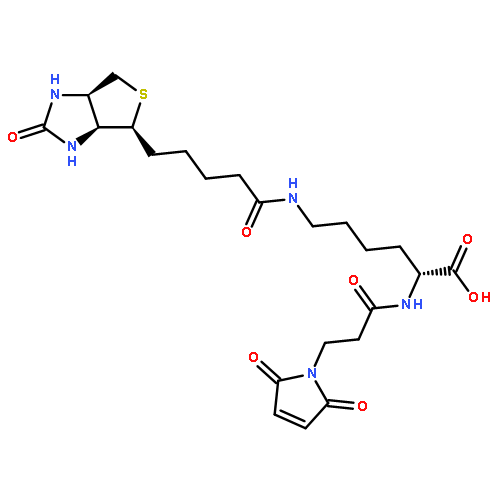
![b-Alanine, N,N-dimethyl-,(3R,4aR,5S,6S,6aS,10S,10aR,10bS)-5-(acetyloxy)-3-ethenyldodecahydro-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-1H-naphtho[2,1-b]pyran-6-ylester, hydrochloride (1:1)](http://img.cochemist.com/ccimg/138700/138605-00-2.png)
![b-Alanine, N,N-dimethyl-,(3R,4aR,5S,6S,6aS,10S,10aR,10bS)-5-(acetyloxy)-3-ethenyldodecahydro-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-1H-naphtho[2,1-b]pyran-6-ylester, hydrochloride (1:1)](http://img.cochemist.com/ccimg/138700/138605-00-2_b.png)
![N-[(9h-fluoren-9-ylmethoxy)carbonyl]-3-hydroxy-l-tyrosine](http://img.cochemist.com/ccimg/137100/137018-93-0.png)
![N-[(9h-fluoren-9-ylmethoxy)carbonyl]-3-hydroxy-l-tyrosine](http://img.cochemist.com/ccimg/137100/137018-93-0_b.png)
![Ethanamine, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/114300/134179-38-7.png)
![Ethanamine, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/114300/134179-38-7_b.png)














![3-[3-(4-Biphenylyl)-1,2,3,4-tetrahydro-1-naphthalenyl]-2-hydroxy- 4H-chromen-4-one](http://img.cochemist.com/ccimg/59800/59763-91-6.png)
![3-[3-(4-Biphenylyl)-1,2,3,4-tetrahydro-1-naphthalenyl]-2-hydroxy- 4H-chromen-4-one](http://img.cochemist.com/ccimg/59800/59763-91-6_b.png)