Co-reporter:Yu-Shan Cheng and James C. Sacchettini
Biochemistry 2016 Volume 55(Issue 7) pp:1107-1119
Publication Date(Web):February 5, 2016
DOI:10.1021/acs.biochem.5b00993
Mycobacterium tuberculosis (Mtb) Rv2671 is annotated as a 5-amino-6-ribitylamino-2,4(1H,3H)-pyrimidinedione 5′-phosphate (AROPP) reductase (RibD) in the riboflavin biosynthetic pathway. Recently, a strain of Mtb with a mutation in the 5′ untranslated region of Rv2671, which resulted in its overexpression, was found to be resistant to dihydrofolate reductase (DHFR) inhibitors including the anti-Mtb drug para-aminosalicylic acid (PAS). In this study, a biochemical analysis of Rv2671 showed that it was able to catalyze the reduction of dihydrofolate (DHF) to tetrahydrofolate (THF), which explained why the overexpression of Rv2671 was sufficient to confer PAS resistance. We solved the structure of Rv2671 in complex with the NADP+ and tetrahydrofolate (THF), which revealed the structural basis for the DHFR activity. The structures of Rv2671 complexed with two DHFR inhibitors, trimethoprim and trimetrexate, provided additional details of the substrate binding pocket and elucidated the differences between their inhibitory activities. Finally, Rv2671 was unable to catalyze the reduction of AROPP, which indicated that Rv2671 and its closely related orthologues are not involved in riboflavin biosynthesis.
Co-reporter:Inna V. Krieger, Joel S. Freundlich, Vijay B. Gawandi, Justin P. Roberts, Vidyadhar B. Gawandi, Qingan Sun, Joshua L. Owen, Maria T. Fraile, Sofia I. Huss, Jose-Luis Lavandera, Thomas R. Ioerger, James C. Sacchettini
Chemistry & Biology 2012 Volume 19(Issue 12) pp:1556-1567
Publication Date(Web):21 December 2012
DOI:10.1016/j.chembiol.2012.09.018
The glyoxylate shunt plays an important role in fatty acid metabolism and has been shown to be critical to survival of several pathogens involved in chronic infections. For Mycobacterium tuberculosis (Mtb), a strain with a defective glyoxylate shunt was previously shown to be unable to establish infection in a mouse model. We report the development of phenyl-diketo acid (PDKA) inhibitors of malate synthase (GlcB), one of two glyoxylate shunt enzymes, using structure-based methods. PDKA inhibitors were active against Mtb grown on acetate, and overexpression of GlcB ameliorated this inhibition. Crystal structures of complexes of GlcB with PDKA inhibitors guided optimization of potency. A selected PDKA compound demonstrated efficacy in a mouse model of tuberculosis. The discovery of these PDKA derivatives provides chemical validation of GlcB as an attractive target for tuberculosis therapeutics.Highlights► Discovery and stabilization of phenyl diketo-acid (PDKA) inhibitors for GlcB ► Exploring binding interactions and improving potency through structure-based design ► PDKAs are bactericidal to Mtb grown on fatty acids and carbohydrates ► Targeting GlcB with PDKA reduces bacterial load in a mouse model of tuberculosis
Co-reporter:Sanghamitra Dey, James M. Lane, Richard E. Lee, Eric J. Rubin and James C. Sacchettini
Biochemistry 2010 Volume 49(Issue 31) pp:
Publication Date(Web):June 21, 2010
DOI:10.1021/bi902097j
Mycobacterium tuberculosis (Mtb) depends on biotin synthesis for survival during infection. In the absence of biotin, disruption of the biotin biosynthesis pathway results in cell death rather than growth arrest, an unusual phenotype for an Mtb auxotroph. Humans lack the enzymes for biotin production, making the proteins of this essential Mtb pathway promising drug targets. To this end, we have determined the crystal structures of the second and third enzymes of the Mtb biotin biosynthetic pathway, 7,8-diaminopelargonic acid synthase (DAPAS) and dethiobiotin synthetase (DTBS), at respective resolutions of 2.2 and 1.85 Å. Superimposition of the DAPAS structures bound either to the SAM analogue sinefungin or to 7-keto-8-aminopelargonic acid (KAPA) allowed us to map the putative binding site for the substrates and to propose a mechanism by which the enzyme accommodates their disparate structures. Comparison of the DTBS structures bound to the substrate 7,8-diaminopelargonic acid (DAPA) or to ADP and the product dethiobiotin (DTB) permitted derivation of an enzyme mechanism. There are significant differences between the Mtb enzymes and those of other organisms; the Bacillus subtilis DAPAS, presented here at a high resolution of 2.2 Å, has active site variations and the Escherichia coli and Helicobacter pylori DTBS have alterations in their overall folds. We have begun to exploit the unique characteristics of the Mtb structures to design specific inhibitors against the biotin biosynthesis pathway in Mtb.
Co-reporter:JoelS. Freundlich Dr.;Feng Wang;Catherine Vilchèze Dr.;Gulcin Gulten;Robert Langley;GuyA. Schiehser Dr.;DavidP. Jacobus Dr.;WilliamR. Jacobs Jr. Dr.;JamesC. Sacchettini Dr.
ChemMedChem 2009 Volume 4( Issue 2) pp:241-248
Publication Date(Web):
DOI:10.1002/cmdc.200800261
Co-reporter:James C. Sacchettini,
Eric J. Rubin
&
Joel S. Freundlich
Nature Reviews Microbiology 2008 6(1) pp:41
Publication Date(Web):2008-01-01
DOI:10.1038/nrmicro1816
Tuberculosis (TB) claims a life every 10 seconds and global mortality rates are increasing despite the use of chemotherapy. But why have we not progressed towards the eradication of the disease? There is no simple answer, although apathy, politics, poverty and our inability to fight the chronic infection have all contributed. Drug resistance and HIV-1 are also greatly influencing the current TB battle plans, as our understanding of their complicity grows. In this Review, recent efforts to fight TB will be described, specifically focusing on how drug discovery could combat the resistance and persistence that make TB worthy of the moniker 'The Great White Plague'.
Co-reporter:Sanghamitra Dey, Rodney L. Burton, Gregory A. Grant and James C. Sacchettini
Biochemistry 2008 Volume 47(Issue 32) pp:
Publication Date(Web):July 16, 2008
DOI:10.1021/bi800212b
The crystal structure of Mycobacterium tuberculosis d-3-phosphoglycerate dehydrogenase has been solved with bound effector, l-serine, and substrate, hydroxypyruvic acid phosphate, at resolutions of 2.7 and 2.4 Å, respectively. The subunits display the same extreme asymmetry as seen in the apo-structure and provide insight into the mode of serine binding and closure of the active site. Mutagenesis studies confirm the identity of the main residues involved in serine binding and suggest that the poly glycine stretch in the loop that contains the locus for the 160° rotation that leads to subunit asymmetry may have a larger role in folding than in catalysis. The lack of electron density for the cofactor, NADH, in any of the crystals examined led us to study binding by stopped flow kinetic analysis. The kinetic data suggest that productive NADH binding, that would support catalytic turnover, is dependent on the presence of substrate. This observation, along with the binding of substrate in the active site, but in an unproductive conformation, suggests a possible mechanism where initial binding of substrate leads to enhanced interaction with cofactor accompanied by a rearrangement of catalytically critical residue side chains. Furthermore, comparison to the structure of a truncated form of human d-3-phosphoglycerate dehydrogenase with cofactor and a substrate analog, provides insight into the conformational changes that occur during catalysis.
Co-reporter:Nilofar N. MohamedMohaideen, Satheesh K. Palaninathan, Paul M. Morin, Brad J. Williams, Miriam Braunstein, Shane E. Tichy, Joseph Locker, David H. Russell, William R. Jacobs Jr. and James C. Sacchettini
Biochemistry 2008 Volume 47(Issue 23) pp:
Publication Date(Web):May 15, 2008
DOI:10.1021/bi701929m
The high-temperature requirement A (HtrA) family of serine proteases has been shown to play an important role in the environmental and cellular stress damage control system in Escherichia coli. Mycobacterium tuberculosis (Mtb) has three putative HtrA-like proteases, HtrA1, HtrA2, and HtrA3. The deletion of htrA2 gives attenuated virulence in a mouse model of TB. Biochemical analysis reveals that HtrA2 can function both as a protease and as a chaperone. The three-dimensional structure of HtrA2 determined at 2.0 Å resolution shows that the protease domains form the central core of the trimer and the PDZ domains extend to the periphery. Unlike E. coli DegS and DegP, the protease is naturally active due to the formation of the serine protease-like catalytic triad and its uniquely designed oxyanion hole. Both protease and PDZ binding pockets of each HtrA2 molecule are occupied by autoproteolytic peptide products and reveal clues for a novel autoregulatory mechanism that might have significant importance in HtrA-associated virulence of Mtb.
Co-reporter:Feng Wang, Robert Langley, Gulcin Gulten, Lei Wang, James C. Sacchettini
Chemistry & Biology 2007 Volume 14(Issue 5) pp:543-551
Publication Date(Web):29 May 2007
DOI:10.1016/j.chembiol.2007.04.005
Rv0098 is part of an operon, Rv0096–Rv0101, from Mycobacterium tuberculosis (Mtb) that is essential for Mtb's survival in mouse macrophages. This operon also contains an acyl carrier protein and one of the only two nonribosomal peptide synthases in Mtb. Rv0098 is annotated in the genome as a hypothetical protein and was proposed to be an acyl-coenzyme A (CoA) dehydratase. The structure of Rv0098, together with subsequent biochemical analysis, indicated that Rv0098 is a long-chain fatty acyl-CoA thioesterase (FcoT). However, FcoT lacks a general base or a nucleophile that is always found in the catalytic site of type II and type I thioesterases, respectively. The active site of Mtb FcoT reveals the structural basis for its substrate specificity for long-chain acyl-CoA and allows us to propose a catalytic mechanism for the enzyme. The characterization of Mtb FcoT provides a putative function of this operon that is crucial for Mtb pathogenicity.
Co-reporter:Min Xu;Arockiasamy Arulandu;Douglas K. Struck;Stephanie Swanson;Ry Young
Science 2005 Vol 307(5706) pp:113-117
Publication Date(Web):07 Jan 2005
DOI:10.1126/science.1105143
Abstract
The P1 lysozyme Lyz is secreted to the periplasm of Escherichia coli and accumulates in an inactive membrane-tethered form. Genetic and biochemical experiments show that, when released from the bilayer, Lyz is activated by an intramolecular thiol-disulfide isomerization, which requires a cysteine in its N-terminal SAR (signal-arrest-release) domain. Crystal structures confirm the alternative disulfide linkages in the two forms of Lyz and reveal dramatic conformational differences in the catalytic domain. Thus, the exported P1 endolysin is kept inactive by three levels of control—topological, conformational, and covalent—until its release from the membrane is triggered by the P1 holin.
Co-reporter:
Nature Structural and Molecular Biology 2000 7(2) pp:141 - 146
Publication Date(Web):
DOI:10.1038/72413
Co-reporter:
Nature Structural and Molecular Biology 2000 7(4) pp:312 - 321
Publication Date(Web):
DOI:10.1038/74082
Co-reporter:
Nature Structural and Molecular Biology 2000 7(8) pp:663-668
Publication Date(Web):
DOI:10.1038/77964
Isocitrate lyase (ICL) plays a pivotal role in the persistence of Mycobacterium tuberculosis in mice by sustaining intracellular infection in inflammatory macrophages. The enzyme allows net carbon gain by diverting acetyl-CoA from -oxidation of fatty acids into the glyoxylate shunt pathway. Given its potential as a drug target against persistent infections, we solved its structure without ligand and in complex with two inhibitors. Covalent modification of an active site residue, Cys 191, by the inhibitor 3-bromopyruvate traps the enzyme in a catalytic conformation with the active site completely inaccessible to solvent. The structure of a C191S mutant of the enzyme with the inhibitor 3-nitropropionate provides further insight into the reaction mechanism.
Co-reporter:Xiaojun Li, Qingan Sun, Cai Jiang, Kailu Yang, ... James C. Sacchettini
Structure (6 October 2015) Volume 23(Issue 10) pp:1858-1865
Publication Date(Web):6 October 2015
DOI:10.1016/j.str.2015.07.014
•Crystal structure of a ribosomal silencing factor, RsfS, from Mtb•Cryo-EM structures of the Mtb ribosome's large subunit with or without RsfS bound•RsfS dimer dissociates into monomers in order to bind to L14 of the 50S subunit•RsfS inhibits the association of 30S subunit and blocks protein synthesisThe ribosomal silencing factor RsfS slows cell growth by inhibiting protein synthesis during periods of diminished nutrient availability. The crystal structure of Mycobacterium tuberculosis (Mtb) RsfS, together with the cryo-electron microscopy (EM) structure of the large subunit 50S of Mtb ribosome, reveals how inhibition of protein synthesis by RsfS occurs. RsfS binds to the 50S at L14, which, when occupied, blocks the association of the small subunit 30S. Although Mtb RsfS is a dimer in solution, only a single subunit binds to 50S. The overlap between the dimer interface and the L14 binding interface confirms that the RsfS dimer must first dissociate to a monomer in order to bind to L14. RsfS interacts primarily through electrostatic and hydrogen bonding to L14. The EM structure shows extended rRNA density that it is not found in the Escherichia coli ribosome, the most striking of these being the extended RNA helix of H54a.Download high-res image (218KB)Download full-size image
Co-reporter:Wayne Harshbarger, Chase Miller, Chandler Diedrich, James Sacchettini
Structure (3 February 2015) Volume 23(Issue 2) pp:418-424
Publication Date(Web):3 February 2015
DOI:10.1016/j.str.2014.11.017
•Unbound and ligand bound structures of human constitutive 20S proteasome•Explanation for chymotrypsin-like selectivity of carfilzomib•Facilitate rational drug design against either constitutive or immunoproteasomesProteasome inhibition is highly effective as a treatment for multiple myeloma, and recently carfilzomib was granted US FDA approval for the treatment of relapsed and refractory multiple myeloma. Here, we report the X-ray crystal structure of the human constitutive 20S proteasome with and without carfilzomib bound at 2.9 and 2.6 Å, respectively. Our data indicate that the S3 and S4 binding pockets play a pivotal role in carfilzomib’s selectivity for chymotrypsin-like sites. Structural comparison with the mouse immunoproteasome crystal structure reveals amino acid substitutions that explain carfilzomib’s slight preference for chymotrypsin-like subunits of constitutive proteasomes. In addition, comparison of the human proteasome:carfilzomib complex with the mouse proteasome:PR-957 complex reveals new details that explain why PR-957 is selective for immunoproteasomes. Together, the data presented here support the design of inhibitors for either constitutive or immunoproteasomes, with implications for the treatment of cancers as well as autoimmune and neurodegenerative diseases.
Co-reporter:Satheesh K. Palaninathan, Nilofar N. Mohamedmohaideen, William C. Snee, Jeffery W. Kelly, James C. Sacchettini
Journal of Molecular Biology (24 October 2008) Volume 382(Issue 5) pp:1157-1167
Publication Date(Web):24 October 2008
DOI:10.1016/j.jmb.2008.07.029
Acidification of the transthyretin (TTR) tetramer facilitates dissociation and conformational changes in the protein, allowing alternatively folded monomers to self-assemble into insoluble amyloid fibers by a downhill polymerization mechanism in vitro. To investigate the influence of acidification on the quaternary and tertiary structures of TTR, crystal structures of wild-type human TTR at pH 4.0 and pH 3.5 have been determined to 1.7 Å resolution. The acidic pH crystals are isomorphous to most of the previously reported TTR structures, containing two subunits in the asymmetric unit (the so-called A and B subunits) but forming a tetramer through crystallographic symmetry. The pH 4.0 crystal structure reveals that the native fold of the tetramer remains mostly undisturbed. In particular, subunit A of the TTR pH 4.0 structure is very similar to the wild-type TTR pH 7.4 structure with an r.m.s.d. of 0.38 Å. In contrast, subunit B of the TTR pH 4.0 structure exhibits several significant changes. The EF-helix (residues 75–81) and the adjacent EF-loop (residues 82–90) show an r.m.s.d. greater than 2.0 Å. The acidic residues within this region (Glu72, Asp74, Glu89, and Glu92) undergo significant conformational changes that instigate movement of the EF helix–loop region and make residues Lys70, Lys76, His88, and His90 orient their side chains toward these acidic residues. In particular, Glu89 undergoes a maximum deviation of 5.6 Å, occupying Phe87's initial position in the wild-type TTR pH 7.4 structure, and points its side chain into a hydrophobic pocket of the neighboring subunit. In the pH 3.5 structure, the EF helix–loop region is completely disordered. These results demonstrate that acidic conditions increase the susceptibility of the EF helix–loop region of the TTR B subunit to undergo conformational changes and unfold, likely destabilizing the tetramer and identifying at least the initial conformational changes likely occurring within the tetramer that leads to the amyloidogenic monomer.
Co-reporter:Sudharsan Sridharan, Lei Wang, Alistair K. Brown, Lynn G. Dover, ... James C. Sacchettini
Journal of Molecular Biology (16 February 2007) Volume 366(Issue 2) pp:469-480
Publication Date(Web):16 February 2007
DOI:10.1016/j.jmb.2006.11.006
Mycolic acids are long chain α-alkyl branched, β-hydroxy fatty acids that represent a characteristic component of the Mycobacterium tuberculosis cell wall. Through their covalent attachment to peptidoglycan via an arabinogalactan polysaccharide, they provide the basis for an essential outer envelope membrane. Mycobacteria possess two fatty acid synthases (FAS); FAS-I carries out de novo synthesis of fatty acids while FAS-II is considered to elongate medium chain length fatty acyl primers to provide long chain (C56) precursors of mycolic acids. Here we report the crystal structure of Mycobacterium tuberculosis β-ketoacyl acyl carrier protein synthase (ACP) II mtKasB, a mycobacterial elongation condensing enzyme involved in FAS-II. This enzyme, along with the M. tuberculosis β-ketoacyl ACP synthase I mtKasA, catalyzes the Claisen-type condensation reaction responsible for fatty acyl elongation in FAS-II and are potential targets for development of novel anti-tubercular drugs. The crystal structure refined to 2.4 Å resolution revealed that, like other KAS-II enzymes, mtKasB adopts a thiolase fold but contains unique structural features in the capping region that may be crucial to its preference for longer fatty acyl chains than its counterparts from other bacteria. Modeling of mtKasA using the mtKasB structure as a template predicts the overall structures to be almost identical, but a larger entrance to the active site tunnel is envisaged that might contribute to the greater sensitivity of mtKasA to the inhibitor thiolactomycin (TLM). Modeling of TLM binding in mtKasB shows that the drug fits the active site poorly and results of enzyme inhibition assays using TLM analogues are wholly consistent with our structural observations. Consequently, the structure described here further highlights the potential of TLM as an anti-tubercular lead compound and will aid further exploration of the TLM scaffold towards the design of novel compounds, which inhibit mycobacterial KAS enzymes more effectively.
Co-reporter:Stephanie Swanson, Kuppan Gokulan, James C. Sacchettini
Structure (15 July 2009) Volume 17(Issue 7) pp:914-915
Publication Date(Web):15 July 2009
DOI:10.1016/j.str.2009.06.006
Mycobacterium tuberculosis, the causative agent of tuberculosis, a disease that has been plaguing humanity for centuries, has a unique cell wall composition believed to be critical for pathogenicity. Luckner et al. (2009) now describe the structure of KasA, an enzyme involved in cell wall biosynthesis.
Co-reporter:Xiaojun Li, Qingan Sun, Cai Jiang, Kailu Yang, ... James C. Sacchettini
Structure (1 December 2015) Volume 23(Issue 12) pp:2387
Publication Date(Web):1 December 2015
DOI:10.1016/j.str.2015.11.002
.jpg)




![Quinazoline, 6-chloro-4-[4-(4-chloro-2-nitrophenyl)-1H-pyrazol-1-yl]-](http://img.cochemist.com/ccimg/586400/586368-64-1.png)
![Quinazoline, 6-chloro-4-[4-(4-chloro-2-nitrophenyl)-1H-pyrazol-1-yl]-](http://img.cochemist.com/ccimg/586400/586368-64-1_b.png)




![PHENOL, 3-[4-(4-CHLORO-2-NITROPHENYL)-1H-PYRAZOL-1-YL]-](http://img.cochemist.com/ccimg/586400/586368-60-7.png)
![PHENOL, 3-[4-(4-CHLORO-2-NITROPHENYL)-1H-PYRAZOL-1-YL]-](http://img.cochemist.com/ccimg/586400/586368-60-7_b.png)
![Benzoic acid, 2-[4-(4-chloro-2-nitrophenyl)-1H-pyrazol-1-yl]-, ethyl ester](http://img.cochemist.com/ccimg/586400/586368-55-0.png)
![Benzoic acid, 2-[4-(4-chloro-2-nitrophenyl)-1H-pyrazol-1-yl]-, ethyl ester](http://img.cochemist.com/ccimg/586400/586368-55-0_b.png)








![Propanoic acid, 3-[(9H-fluoren-9-ylideneamino)oxy]-2-methyl-, (2S)-](http://img.cochemist.com/ccimg/430500/430424-93-4.png)
![Propanoic acid, 3-[(9H-fluoren-9-ylideneamino)oxy]-2-methyl-, (2S)-](http://img.cochemist.com/ccimg/430500/430424-93-4_b.png)
![PROPANOIC ACID, 3-[(9H-FLUOREN-9-YLIDENEAMINO)OXY]-2-METHYL-, (2R)-](http://img.cochemist.com/ccimg/430500/430424-92-3.png)
![PROPANOIC ACID, 3-[(9H-FLUOREN-9-YLIDENEAMINO)OXY]-2-METHYL-, (2R)-](http://img.cochemist.com/ccimg/430500/430424-92-3_b.png)


![Pyrimidine, 2-[4-(2,4-dinitrophenyl)-1H-pyrazol-1-yl]-4-(trifluoromethyl)-](http://img.cochemist.com/ccimg/353600/353522-07-3.png)
![Pyrimidine, 2-[4-(2,4-dinitrophenyl)-1H-pyrazol-1-yl]-4-(trifluoromethyl)-](http://img.cochemist.com/ccimg/353600/353522-07-3_b.png)










![Glycine, N-[[(4-cyanophenyl)amino][[(1S)-1-phenylethyl]amino]methylene]-](/data/chemimg/1677700/116869-20-6.png)
![Glycine, N-[[(4-cyanophenyl)amino][[(1S)-1-phenylethyl]amino]methylene]-](/data/chemimg/1677700/116869-20-6_b.png)
![Propanoic acid, 3-[(9H-fluoren-9-ylideneamino)oxy]-](http://img.cochemist.com/ccimg/103600/103586-60-3.png)
![Propanoic acid, 3-[(9H-fluoren-9-ylideneamino)oxy]-](http://img.cochemist.com/ccimg/103600/103586-60-3_b.png)








![(2r,3z,5r)-3-(2-hydroxyethylidene)-7-oxo-4-oxa-1-azabicyclo[3.2.0]heptane-2-carboxylic Acid](http://img.cochemist.com/ccimg/58100/58001-44-8.png)
![(2r,3z,5r)-3-(2-hydroxyethylidene)-7-oxo-4-oxa-1-azabicyclo[3.2.0]heptane-2-carboxylic Acid](http://img.cochemist.com/ccimg/58100/58001-44-8_b.png)


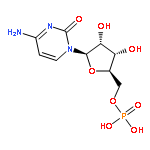
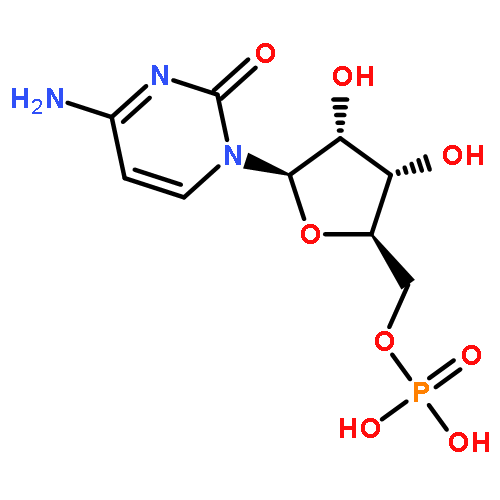
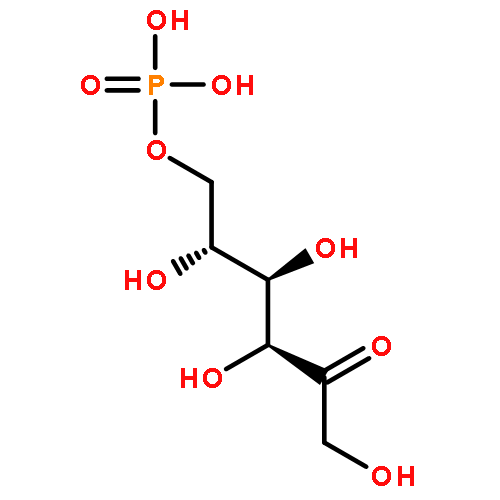
![Glutamic acid,N-[4-[[(2-amino-3,4,5,6,7,8-hexahydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-](http://img.cochemist.com/ccimg/29400/29347-89-5.png)
![Glutamic acid,N-[4-[[(2-amino-3,4,5,6,7,8-hexahydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-](http://img.cochemist.com/ccimg/29400/29347-89-5_b.png)







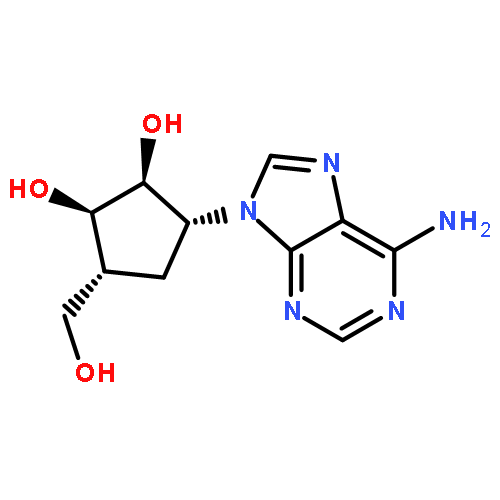









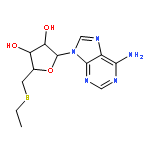
![1H-Imidazo[4,5-c]pyridin-4-amine,1-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/6800/6736-58-9.png)
![1H-Imidazo[4,5-c]pyridin-4-amine,1-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/6800/6736-58-9_b.png)
![Diphosphoric acid,P-[(2E,6E,10E)-3,7,11,15-tetramethyl-2,6,10,14-hexadecatetraen-1-yl] ester](http://img.cochemist.com/ccimg/6700/6699-20-3.png)
![Diphosphoric acid,P-[(2E,6E,10E)-3,7,11,15-tetramethyl-2,6,10,14-hexadecatetraen-1-yl] ester](http://img.cochemist.com/ccimg/6700/6699-20-3_b.png)











![Benzoic acid,4-[[(2-amino-3,4,7,8-tetrahydro-4-oxo-6-pteridinyl)methyl]amino]-](http://img.cochemist.com/ccimg/2200/2134-76-1.png)
![Benzoic acid,4-[[(2-amino-3,4,7,8-tetrahydro-4-oxo-6-pteridinyl)methyl]amino]-](http://img.cochemist.com/ccimg/2200/2134-76-1_b.png)





![(1AR,4R,4AR,7S,7AS,7BR)-1,1,4,7-TETRAMETHYLDECAHYDRO-1H-CYCLOPROP<WBR />A[E]AZULENE-4,7-DIOL](http://img.cochemist.com/ccimg/800/763-10-0.png)
![(1AR,4R,4AR,7S,7AS,7BR)-1,1,4,7-TETRAMETHYLDECAHYDRO-1H-CYCLOPROP<WBR />A[E]AZULENE-4,7-DIOL](http://img.cochemist.com/ccimg/800/763-10-0_b.png)
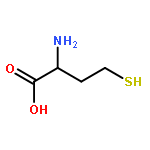
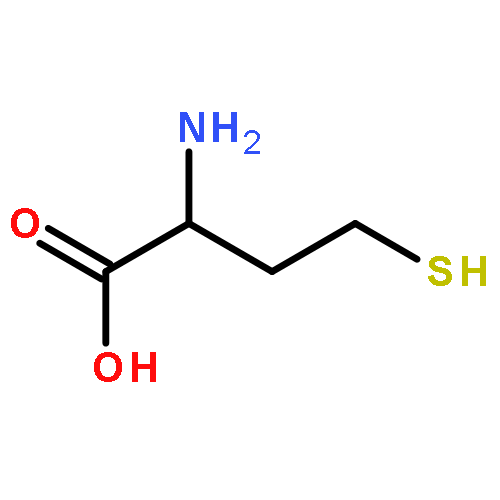








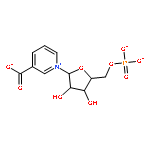






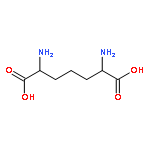
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9_b.png)


![10H-Phenoxazine-4,6-dicarboxylic acid, 10-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/264000/263908-32-3.png)
![10H-Phenoxazine-4,6-dicarboxylic acid, 10-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/264000/263908-32-3_b.png)
![Glycine,N-[[(4-cyanophenyl)amino][(diphenylmethyl)amino]methylene]-](http://img.cochemist.com/ccimg/138500/138460-25-0.png)
![Glycine,N-[[(4-cyanophenyl)amino][(diphenylmethyl)amino]methylene]-](http://img.cochemist.com/ccimg/138500/138460-25-0_b.png)


![(2R)-5-hydroxy-2,4-dimethyl-2-[(1E)-2-methylbuta-1,3-dienyl]thiophen-3-one](http://img.cochemist.com/ccimg/82100/82079-32-1.png)
![(2R)-5-hydroxy-2,4-dimethyl-2-[(1E)-2-methylbuta-1,3-dienyl]thiophen-3-one](http://img.cochemist.com/ccimg/82100/82079-32-1_b.png)


![2,4-Quinazolinediamine,5-methyl-6-[[(3,4,5-trimethoxyphenyl)amino]methyl]-](http://img.cochemist.com/ccimg/52200/52128-35-5.png)
![2,4-Quinazolinediamine,5-methyl-6-[[(3,4,5-trimethoxyphenyl)amino]methyl]-](http://img.cochemist.com/ccimg/52200/52128-35-5_b.png)
![2-[2-(TRIFLUOROMETHYL)ANILINO]BENZOIC ACID](http://img.cochemist.com/ccimg/32700/32621-47-9.png)
![2-[2-(TRIFLUOROMETHYL)ANILINO]BENZOIC ACID](http://img.cochemist.com/ccimg/32700/32621-47-9_b.png)