Co-reporter:Jun Li, Ana Carolina Dantas Machado, Ming Guo, Jared M. Sagendorf, Zhan Zhou, Longying Jiang, Xiaojuan Chen, Daichao Wu, Lingzhi Qu, Zhuchu Chen, Lin Chen, Remo Rohs, and Yongheng Chen
Biochemistry July 25, 2017 Volume 56(Issue 29) pp:3745-3745
Publication Date(Web):June 23, 2017
DOI:10.1021/acs.biochem.7b00211
FOXA2, a member of the forkhead family of transcription factors, plays essential roles in liver development and bile acid homeostasis. In this study, we report a 2.8 Å co-crystal structure of the FOXA2 DNA-binding domain (FOXA2-DBD) bound to a DNA duplex containing a forkhead consensus binding site (GTAAACA). The FOXA2-DBD adopts the canonical winged-helix fold, with helix H3 and wing 1 regions mainly mediating the DNA recognition. Although the wing 2 region was not defined in the structure, isothermal titration calorimetry assays suggested that this region was required for optimal DNA binding. Structure comparison with the FOXA3-DBD bound to DNA revealed more major groove contacts and fewer minor groove contacts in the FOXA2 structure than in the FOXA3 structure. Structure comparison with the FOXO1-DBD bound to DNA showed that different forkhead proteins could induce different DNA conformations upon binding to identical DNA sequences. Our findings provide the structural basis for FOXA2 protein binding to a consensus forkhead site and elucidate how members of the forkhead protein family bind different DNA sites.
Co-reporter:Liang Guo;Aidong Han;Jue Cao;Darren L. Bates
PNAS 2007 Volume 104 (Issue 11 ) pp:4297-4302
Publication Date(Web):2007-03-13
DOI:10.1073/pnas.0608041104
Glutamine-rich sequences exist in a wide range of proteins across multiple species. A subset of glutamine-rich sequences has
been shown to form amyloid fibers implicated in human diseases. The physiological functions of these sequence motifs are not
well understood, partly because of the lack of structural information. Here we have determined a high-resolution structure
of a glutamine-rich domain from human histone deacetylase 4 (HDAC4) by x-ray crystallography. The glutamine-rich domain of
HDAC4 (19 glutamines of 68 residues) folds into a straight α-helix that assembles as a tetramer. In contrast to most coiled
coil proteins, the HDAC4 tetramer lacks regularly arranged apolar residues and an extended hydrophobic core. Instead, the
protein interfaces consist of multiple hydrophobic patches interspersed with polar interaction networks, wherein clusters
of glutamines engage in extensive intra- and interhelical interactions. In solution, the HDAC4 tetramer undergoes rapid equilibrium
with monomer and intermediate species. Structure-guided mutations that expand or disrupt hydrophobic patches drive the equilibrium
toward the tetramer or monomer, respectively. We propose that a general role of glutamine-rich motifs be to mediate protein–protein
interactions characteristic of a large component of polar interaction networks that may facilitate reversible assembly and
disassembly of protein complexes.
Co-reporter:Daichao Wu, Lingzhi Qu, Yang Fu, Jun Li, Longying Jiang, Xiaojuan Chen, Ming Guo, Zhuchu Chen, Lin Chen, Yongheng Chen
Protein Expression and Purification (December 2016) Volume 128() pp:67-72
Publication Date(Web):1 December 2016
DOI:10.1016/j.pep.2016.08.010
•The kinase domain of PINK1 was successfully expressed in Pichia pastoris.•The induction conditions of recombinant PINK1 transformant were optimized.•Recombinant PINK1 was obtained after two steps purification.•The purity of purified recombinant PINK1 was higher than 95%.•Recombinant PINK1 was able to phosphorylate ubiquitin.PTEN-induced putative kinase 1 (PINK1) is a Ser/Thr kinase that specifically localizes on the mitochondrial membrane. It cooperates with Parkin to regulate mitochondrial quality control. Mutations in PINK1 protein which account for 8–15% of Parkinson's disease (PD), are the second most common cause of early-onset Autosomal Recessive Parkinson's disease (AR-PD). The lack of methods for PINK1 heterologous expression and purification has slowed progress in the AR-PD research field. To pave the way for direct structural study of this important protein, in this study, we developed an efficient expression system of recombinant PINK1 kinase domain (rPINK1) using Pichia pastoris (P. pastoris). Our results showed that rPINK1 is best expressed in P. pastoris at 25 °C induction. Additionally, we determined that the optimal induction time was 72 h and the optimal induction methanol concentration was 1% for the expression of rPINK1 in P. pastoris. Subsequent purification by Ni affinity chromatography (Ni-NTA) and cation-exchange chromatography (Mono S) produced the protein with purity higher than 95%. The pure rPINK1 was active to phosphorylate ubiquitin in a substrate phosphorylation assay. Overall, these studies provide the first effective method for heterologous expression and purification of the rPINK1 with a high purity. These findings can help contribute to further researches on the interactions study and biochemical characterization of PINK1.
Co-reporter:Darren L. Bates, Kristen K.B. Barthel, Yongqing Wu, Reza Kalhor, ... Lin Chen
Structure (7 May 2008) Volume 16(Issue 5) pp:684-694
Publication Date(Web):7 May 2008
DOI:10.1016/j.str.2008.01.020
The host factor, nuclear factor of activated T-cells (NFAT), regulates the transcription and replication of HIV-1. Here, we have determined the crystal structure of the DNA binding domain of NFAT bound to the HIV-1 long terminal repeat (LTR) tandem κB enhancer element at 3.05 Å resolution. NFAT binds as a dimer to the upstream κB site (Core II), but as a monomer to the 3′ end of the downstream κB site (Core I). The DNA shows a significant bend near the 5′ end of Core I, where a lysine residue from NFAT bound to the 3′ end of Core II inserts into the minor groove and seems to cause DNA bases to flip out. Consistent with this structural feature, the 5′ end of Core I become hypersensitive to dimethylsulfate in the in vivo footprinting upon transcriptional activation of the HIV-1 LTR. Our studies provide a basis for further investigating the functional mechanisms of NFAT in HIV-1 transcription and replication.
Co-reporter:Yongqing Wu, Raja Dey, Aidong Han, Nimanthi Jayathilaka, ... Lin Chen
Journal of Molecular Biology (26 March 2010) Volume 397(Issue 2) pp:520-533
Publication Date(Web):26 March 2010
DOI:10.1016/j.jmb.2010.01.067
Myocyte enhancer factor 2 (MEF2) regulates specific gene expression in diverse developmental programs and adaptive responses. MEF2 recognizes DNA and interacts with transcription cofactors through a highly conserved N-terminal domain referred to as the MADS-box/MEF2 domain. Here we present the crystal structure of the MADS-box/MEF2 domain of MEF2A bound to DNA. In contrast to previous structural studies showing that the MEF2 domain of MEF2A is partially unstructured, the present study reveals that the MEF2 domain participates with the MADS-box in both dimerization and DNA binding as a single domain. The sequence divergence at and immediately following the C-terminal end of the MEF2 domain may allow different MEF2 dimers to recognize different DNA sequences in the flanking regions. The current structure also suggests that the ligand-binding pocket previously observed in the Cabin1–MEF2B–DNA complex and the HDAC9 (histone deacetylase 9)–MEF2B–DNA complex is not induced by cofactor binding but rather preformed by intrinsic folding. However, the structure of the ligand-binding pocket does undergo subtle but significant conformational changes upon cofactor binding. On the basis of these observations, we generated a homology model of MEF2 bound to a myocardin family protein, MASTR, that acts as a potent coactivator of MEF2-dependent gene expression. The model shows excellent shape and chemical complementarity at the binding interface and is consistent with existing mutagenesis data. The apo structure presented here can also serve as a target for virtual screening and soaking studies of small molecules that can modulate the function of MEF2 as research tools and therapeutic leads.
Co-reporter:Hozefa S. Bandukwala, Yongqing Wu, Markus Feuerer, Yongheng Chen, ... Lin Chen
Immunity (22 April 2011) Volume 34(Issue 4) pp:
Publication Date(Web):22 April 2011
DOI:10.1016/j.immuni.2011.04.011
Co-reporter:Hozefa S. Bandukwala, Yongqing Wu, Markus Feuerer, Yongheng Chen, ... Lin Chen
Immunity (22 April 2011) Volume 34(Issue 4) pp:479-491
Publication Date(Web):22 April 2011
DOI:10.1016/j.immuni.2011.02.017
The transcription factor FOXP3 is essential for the suppressive function of regulatory T cells that are required for maintaining self-tolerance. We have solved the crystal structure of the FOXP3 forkhead domain as a ternary complex with the DNA-binding domain of the transcription factor NFAT1 and a DNA oligonucleotide from the interleukin-2 promoter. A striking feature of this structure is that FOXP3 forms a domain-swapped dimer that bridges two molecules of DNA. Structure-guided or autoimmune disease (IPEX)-associated mutations in the domain-swap interface diminished dimer formation by the FOXP3 forkhead domain without compromising FOXP3 DNA binding. These mutations also eliminated T cell-suppressive activity conferred by FOXP3, both in vitro and in a murine model of autoimmune diabetes in vivo. We conclude that FOXP3-mediated suppressor function requires dimerization through the forkhead domain and that mutations in the dimer interface can lead to the systemic autoimmunity observed in IPEX patients.Highlights► The forkhead domain of FOXP3 exists predominantly as a domain-swapped dimer ► The domain-swapped dimer of FOXP3 simultaneously binds two distinct pieces of DNA ► Disruption of domain swapping inhibits dimerization without affecting DNA binding ► Disruption of domain swapping abrogates FOXP3-mediated suppressor functions
Co-reporter:James C. Stroud, Amy Oltman, Aidong Han, Darren L. Bates, Lin Chen
Journal of Molecular Biology (16 October 2009) Volume 393(Issue 1) pp:98-112
Publication Date(Web):16 October 2009
DOI:10.1016/j.jmb.2009.08.023
The activation and latency of human immunodeficiency virus type 1 (HIV-1) are tightly controlled by the transcriptional activity of its long terminal repeat (LTR) region. The LTR is regulated by viral proteins as well as host factors, including the nuclear factor kappaB (NF-κB) that becomes activated in virus-infected cells. The two tandem NF-κB sites of the LTR are among the most highly conserved sequence elements of the HIV-1 genome. Puzzlingly, these sites are arranged in a manner that seems to preclude simultaneous binding of both sites by NF-κB, although previous biochemical work suggests otherwise. Here, we have determined the crystal structure of p50:RelA bound to the tandem κB element of the HIV-1 LTR as a dimeric dimer, providing direct structural evidence that NF-κB can occupy both sites simultaneously. The two p50:RelA dimers bind the adjacent κB sites and interact through a protein contact that is accommodated by DNA bending. The two dimers clamp DNA from opposite faces of the double helix and form a topological trap of the bound DNA. Consistent with these structural features, our biochemical analyses indicate that p50:RelA binds the HIV-1 LTR tandem κB sites with an apparent anti-cooperativity but enhanced kinetic stability. The slow on and off rates we observe may be relevant to viral latency because viral activation requires sustained NF-κB activation. Furthermore, our work demonstrates that the specific arrangement of the two κB sites on the HIV-1 LTR can modulate the assembly kinetics of the higher-order NF-κB complex on the viral promoter. This phenomenon is unlikely restricted to the HIV-1 LTR but probably represents a general mechanism for the function of composite DNA elements in transcription.
Co-reporter:Yongheng Chen, Raja Dey, Lin Chen
Structure (10 February 2010) Volume 18(Issue 2) pp:246-256
Publication Date(Web):10 February 2010
DOI:10.1016/j.str.2009.11.011
Recent studies suggest that p53 binds predominantly to consensus sites composed of two decameric half-sites with zero spacing in vivo. Here we report the crystal structure of the p53 core domain bound to a full consensus site as a tetramer at 2.13Å resolution. Comparison with previously reported structures of p53 dimer:DNA complexes and a chemically trapped p53 tetramer:DNA complex reveals that DNA binding by the p53 core domain is a cooperative self-assembling process accompanied by structural changes of the p53 dimer and DNA. Each p53 monomer interacts with its two neighboring subunits through two different protein-protein interfaces. The DNA is largely B-form and shows no discernible bend, but the central base-pairs between the two half-sites display a significant slide. The extensive protein-protein and protein-DNA interactions explain the high cooperativity and kinetic stability of p53 binding to contiguous decameric sites and the conservation of such binding-site configuration in vivo.

























![Phosphonic acid, ethenyl-, bis[2-(1H-benzimidazol-1-yl)ethyl] ester](http://img.cochemist.com/ccimg/70800/70745-16-3.png)
![Phosphonic acid, ethenyl-, bis[2-(1H-benzimidazol-1-yl)ethyl] ester](http://img.cochemist.com/ccimg/70800/70745-16-3_b.png)
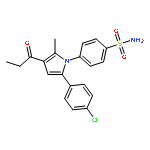
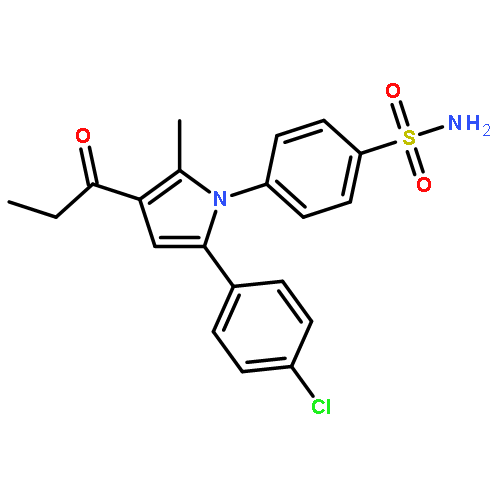
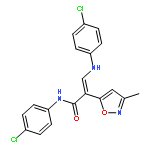
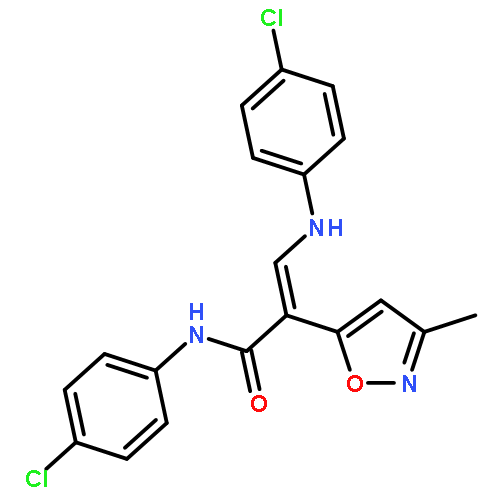
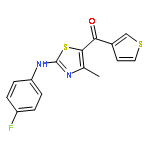
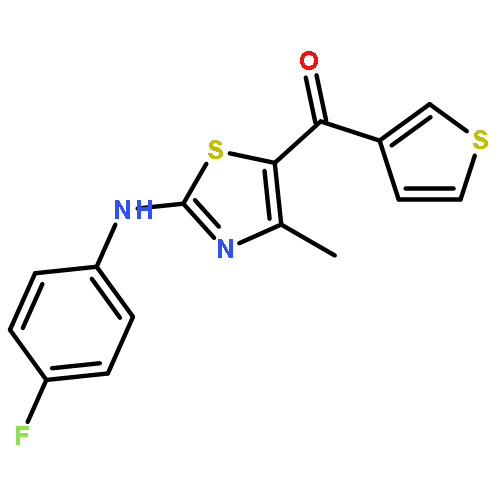

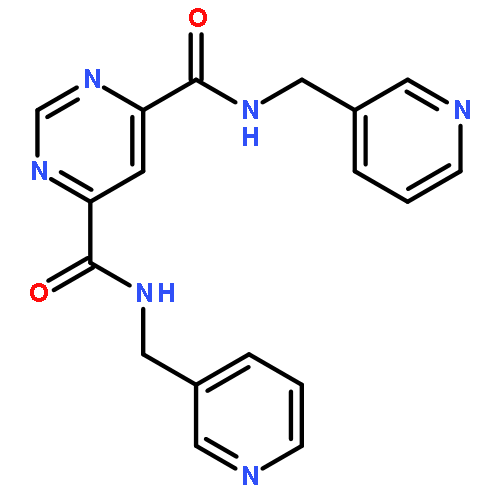
![Acetic acid, [[4-(aminomethyl)phenyl]amino]oxo-](http://img.cochemist.com/ccimg/840500/840454-25-3.png)
![Acetic acid, [[4-(aminomethyl)phenyl]amino]oxo-](http://img.cochemist.com/ccimg/840500/840454-25-3_b.png)


![2-Naphthalenecarboxamide,6-(aminoiminomethyl)-N-[3,4-dihydro-1-(1-methylethyl)-7-isoquinolinyl]-](http://img.cochemist.com/ccimg/653700/653604-64-9.png)
![2-Naphthalenecarboxamide,6-(aminoiminomethyl)-N-[3,4-dihydro-1-(1-methylethyl)-7-isoquinolinyl]-](http://img.cochemist.com/ccimg/653700/653604-64-9_b.png)
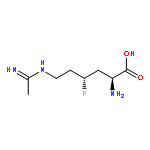
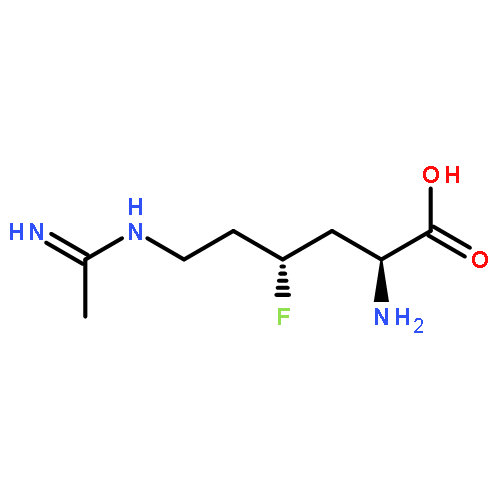


![Sulfamic acid, 2-bromo-4-[[(4-cyanophenyl)-4H-1,2,4-triazol-4-ylamino]methyl]phenyl ester](/data/chemimg/3769700/537674-89-8.png)
![Sulfamic acid, 2-bromo-4-[[(4-cyanophenyl)-4H-1,2,4-triazol-4-ylamino]methyl]phenyl ester](/data/chemimg/3769700/537674-89-8_b.png)
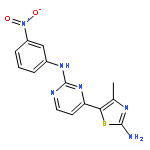
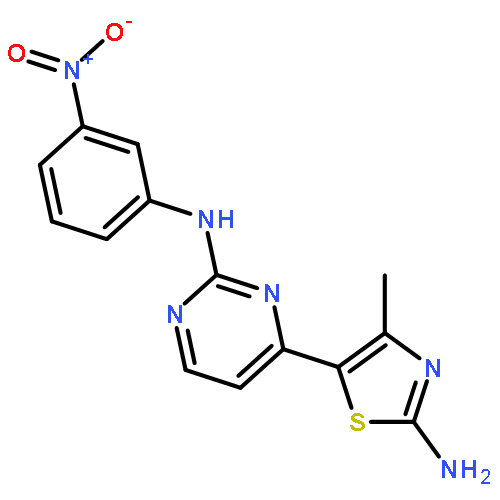


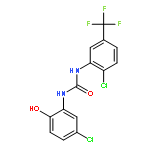
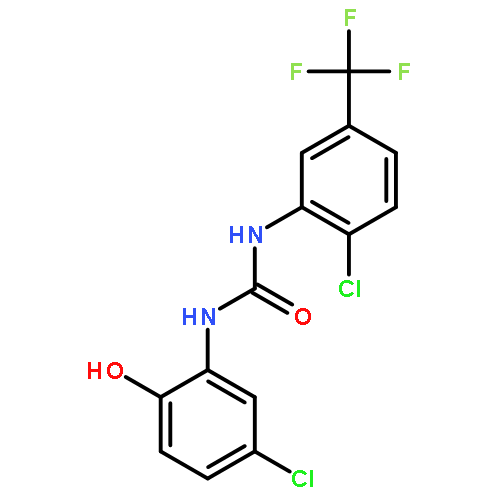
![[1,1'-BIPHENYL]-2,3-DIOL, 2',6'-DICHLORO-](http://img.cochemist.com/ccimg/500800/500798-65-2.png)
![[1,1'-BIPHENYL]-2,3-DIOL, 2',6'-DICHLORO-](http://img.cochemist.com/ccimg/500800/500798-65-2_b.png)
![Boronic acid, [[[(2-ethoxy-1-naphthalenyl)carbonyl]amino]methyl]-](http://img.cochemist.com/ccimg/438600/438567-77-2.png)
![Boronic acid, [[[(2-ethoxy-1-naphthalenyl)carbonyl]amino]methyl]-](http://img.cochemist.com/ccimg/438600/438567-77-2_b.png)
![3-[3-(2,3-DIHYDROXYPROPYLAMINO)PHENYL]-4-(5-FLUORO-1-METHYLINDOL-3-YL)PYRROLE-2,5-DIONE](http://img.cochemist.com/ccimg/396100/396091-16-0.png)
![3-[3-(2,3-DIHYDROXYPROPYLAMINO)PHENYL]-4-(5-FLUORO-1-METHYLINDOL-3-YL)PYRROLE-2,5-DIONE](http://img.cochemist.com/ccimg/396100/396091-16-0_b.png)
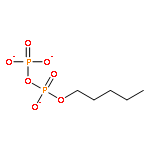
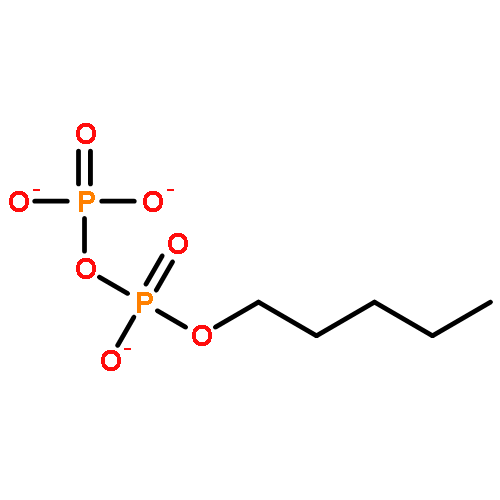
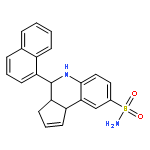
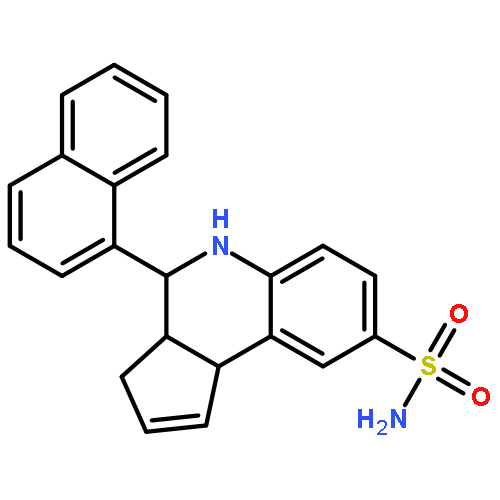


![2-Naphthalenecarboxylic acid, 3-[(carboxycarbonyl)amino]-](http://img.cochemist.com/ccimg/244000/243989-49-3.png)
![2-Naphthalenecarboxylic acid, 3-[(carboxycarbonyl)amino]-](http://img.cochemist.com/ccimg/244000/243989-49-3_b.png)
![1H-Indole-5-carboxylic acid, 6-[(carboxycarbonyl)amino]-](http://img.cochemist.com/ccimg/244000/243967-44-4.png)
![1H-Indole-5-carboxylic acid, 6-[(carboxycarbonyl)amino]-](http://img.cochemist.com/ccimg/244000/243967-44-4_b.png)
![Benzamide, 4-(aminosulfonyl)-N-[(2,3,4-trifluorophenyl)methyl]-](http://img.cochemist.com/ccimg/235100/235088-56-9.png)
![Benzamide, 4-(aminosulfonyl)-N-[(2,3,4-trifluorophenyl)methyl]-](http://img.cochemist.com/ccimg/235100/235088-56-9_b.png)
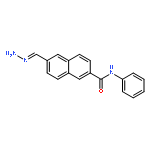
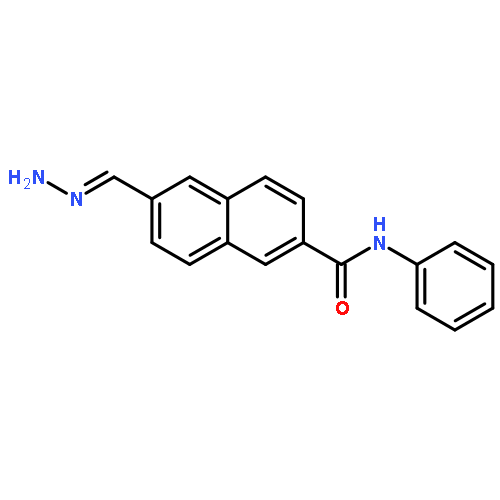
![Benzenesulfonamide,4-[5-(4-bromophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]-](http://img.cochemist.com/ccimg/170600/170569-93-4.png)
![Benzenesulfonamide,4-[5-(4-bromophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]-](http://img.cochemist.com/ccimg/170600/170569-93-4_b.png)
![Benzoic acid, 4-(acetylamino)-3-[(aminoiminomethyl)amino]-](http://img.cochemist.com/ccimg/170500/170447-93-5.png)
![Benzoic acid, 4-(acetylamino)-3-[(aminoiminomethyl)amino]-](http://img.cochemist.com/ccimg/170500/170447-93-5_b.png)
![Benzenecarboximidamide,4-[[[(4-phenoxyphenyl)amino]carbonyl]amino]-](http://img.cochemist.com/ccimg/162100/162021-02-5.png)
![Benzenecarboximidamide,4-[[[(4-phenoxyphenyl)amino]carbonyl]amino]-](http://img.cochemist.com/ccimg/162100/162021-02-5_b.png)


![6-Heptenoic acid,7-[3-(4-fluorophenyl)-1-(1-methylethyl)-1H-indol-2-yl]-3,5-dihydroxy-,(3S,5R,6E)-](http://img.cochemist.com/ccimg/155300/155229-76-8.png)
![6-Heptenoic acid,7-[3-(4-fluorophenyl)-1-(1-methylethyl)-1H-indol-2-yl]-3,5-dihydroxy-,(3S,5R,6E)-](http://img.cochemist.com/ccimg/155300/155229-76-8_b.png)
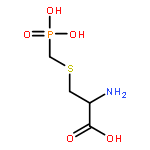

![Spiro[pyrrolidine-3,4'(1'H)-pyrrolo[1,2-a]pyrazine]-1',2,3',5(2'H)-tetrone,2'-[(4-bromo-2-fluorophenyl)methyl]-, (3R)-](http://img.cochemist.com/ccimg/147300/147254-64-6.png)
![Spiro[pyrrolidine-3,4'(1'H)-pyrrolo[1,2-a]pyrazine]-1',2,3',5(2'H)-tetrone,2'-[(4-bromo-2-fluorophenyl)methyl]-, (3R)-](http://img.cochemist.com/ccimg/147300/147254-64-6_b.png)
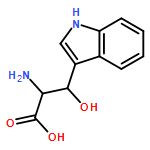
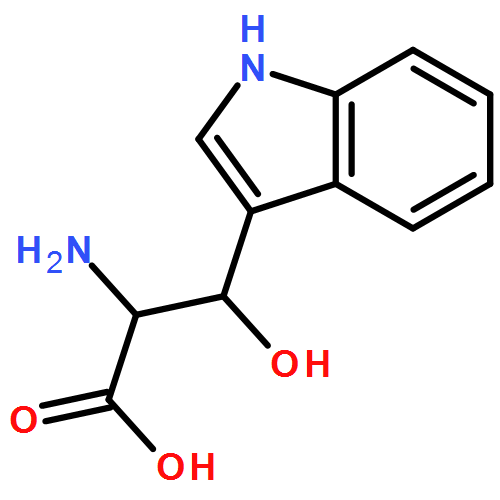
![1H-Indole-1-aceticacid, 2-[[[4-(2-chlorophenyl)-2-thiazolyl]amino]carbonyl]-](http://img.cochemist.com/ccimg/136400/136381-85-6.png)
![1H-Indole-1-aceticacid, 2-[[[4-(2-chlorophenyl)-2-thiazolyl]amino]carbonyl]-](http://img.cochemist.com/ccimg/136400/136381-85-6_b.png)
![(2S,4S)-6-Fluoro-2',5'-dioxospiro[chroman-4,4'-imidazolidine]-2-carboxamide](http://img.cochemist.com/ccimg/136100/136087-85-9.png)
![(2S,4S)-6-Fluoro-2',5'-dioxospiro[chroman-4,4'-imidazolidine]-2-carboxamide](http://img.cochemist.com/ccimg/136100/136087-85-9_b.png)

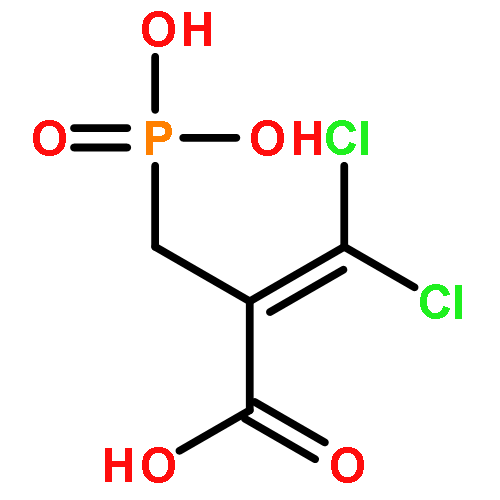
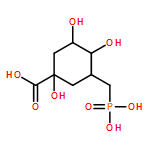
![Glycine, N-[[(4-cyanophenyl)amino][[(1S)-1-phenylethyl]amino]methylene]-](/data/chemimg/1677700/116869-20-6.png)
![Glycine, N-[[(4-cyanophenyl)amino][[(1S)-1-phenylethyl]amino]methylene]-](/data/chemimg/1677700/116869-20-6_b.png)





![1(2H)-Quinazolineaceticacid, 3-[(4-bromo-2-fluorophenyl)methyl]-7-chloro-3,4-dihydro-2,4-dioxo-](http://img.cochemist.com/ccimg/112800/112733-06-9.png)
![1(2H)-Quinazolineaceticacid, 3-[(4-bromo-2-fluorophenyl)methyl]-7-chloro-3,4-dihydro-2,4-dioxo-](http://img.cochemist.com/ccimg/112800/112733-06-9_b.png)
![Adenosine,5'-[[(3S)-3-amino-3-carboxypropyl]methylamino]-5'-deoxy- (9CI)](http://img.cochemist.com/ccimg/111800/111770-79-7.png)
![Adenosine,5'-[[(3S)-3-amino-3-carboxypropyl]methylamino]-5'-deoxy- (9CI)](http://img.cochemist.com/ccimg/111800/111770-79-7_b.png)


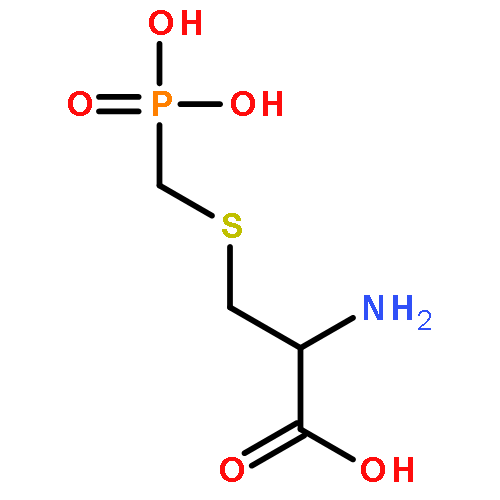
![Nδ-[(Sulfoamino)(amino)phosphinyl]-L-ornithine](http://img.cochemist.com/ccimg/93300/93289-64-6.png)
![Nδ-[(Sulfoamino)(amino)phosphinyl]-L-ornithine](http://img.cochemist.com/ccimg/93300/93289-64-6_b.png)




![BORONIC ACID, [3-[(IODOACETYL)AMINO]PHENYL]-](http://img.cochemist.com/ccimg/87200/87199-19-7.png)
![BORONIC ACID, [3-[(IODOACETYL)AMINO]PHENYL]-](http://img.cochemist.com/ccimg/87200/87199-19-7_b.png)





![[[HYDROXY(PHOSPHONOOXY)PHOSPHORYL]AMINO]PHOSPHONIC ACID](http://img.cochemist.com/ccimg/79900/79828-60-7.png)
![[[HYDROXY(PHOSPHONOOXY)PHOSPHORYL]AMINO]PHOSPHONIC ACID](http://img.cochemist.com/ccimg/79900/79828-60-7_b.png)





![L-LYSINE, N6-[(1R)-1-CARBOXYETHYL]-](http://img.cochemist.com/ccimg/68900/68852-49-3.png)
![L-LYSINE, N6-[(1R)-1-CARBOXYETHYL]-](http://img.cochemist.com/ccimg/68900/68852-49-3_b.png)
![2,3-dihydroxypropyl 2-[(7-chloroquinolin-4-yl)amino]benzoate,hydrochloride](http://img.cochemist.com/ccimg/65600/65513-72-6.png)
![2,3-dihydroxypropyl 2-[(7-chloroquinolin-4-yl)amino]benzoate,hydrochloride](http://img.cochemist.com/ccimg/65600/65513-72-6_b.png)

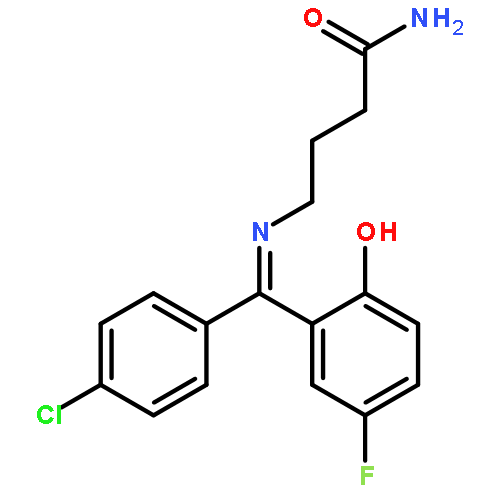
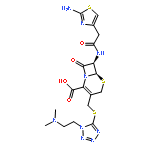
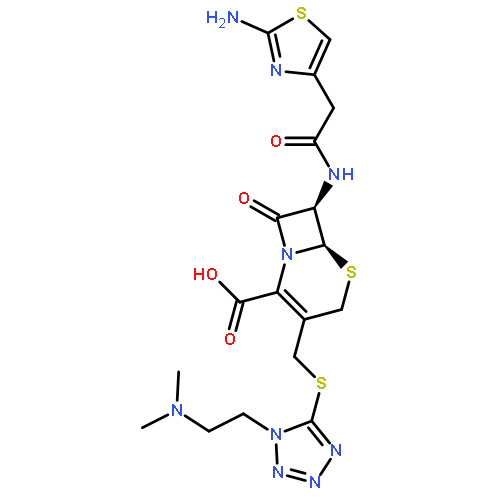
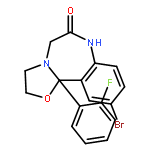













![Pyridinium,1-[(4-amino-2-methyl-5-pyrimidinyl)methyl]-2-methyl-3-(4,6,6-trihydroxy-4,6-dioxido-3,5-dioxa-4,6-diphosphahex-1-yl)-](http://img.cochemist.com/ccimg/36500/36467-78-4.png)
![Pyridinium,1-[(4-amino-2-methyl-5-pyrimidinyl)methyl]-2-methyl-3-(4,6,6-trihydroxy-4,6-dioxido-3,5-dioxa-4,6-diphosphahex-1-yl)-](http://img.cochemist.com/ccimg/36500/36467-78-4_b.png)




![4H-[1,3]Oxazino[3,2-d][1,4]benzodiazepine-4,7(6H)-dione,11-chloro-8,12b-dihydro-2,8-dimethyl-12b-phenyl-](http://img.cochemist.com/ccimg/27300/27223-35-4.png)
![4H-[1,3]Oxazino[3,2-d][1,4]benzodiazepine-4,7(6H)-dione,11-chloro-8,12b-dihydro-2,8-dimethyl-12b-phenyl-](http://img.cochemist.com/ccimg/27300/27223-35-4_b.png)




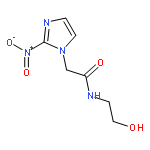
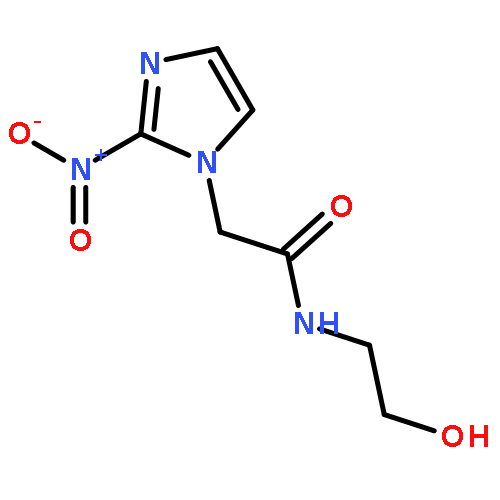
![1,3-Cyclohexadiene-1-carboxylicacid, 5-[(1-carboxyethenyl)oxy]-6-hydroxy-, (5S,6S)-](http://img.cochemist.com/ccimg/22700/22642-82-6.png)
![1,3-Cyclohexadiene-1-carboxylicacid, 5-[(1-carboxyethenyl)oxy]-6-hydroxy-, (5S,6S)-](http://img.cochemist.com/ccimg/22700/22642-82-6_b.png)




![{[({3-hydroxy-2-methyl-5-[(phosphonooxy)methyl]pyridin-4-yl}methyl)amino]oxy}acetic acid](http://img.cochemist.com/ccimg/20000/19988-33-1.png)
![{[({3-hydroxy-2-methyl-5-[(phosphonooxy)methyl]pyridin-4-yl}methyl)amino]oxy}acetic acid](http://img.cochemist.com/ccimg/20000/19988-33-1_b.png)

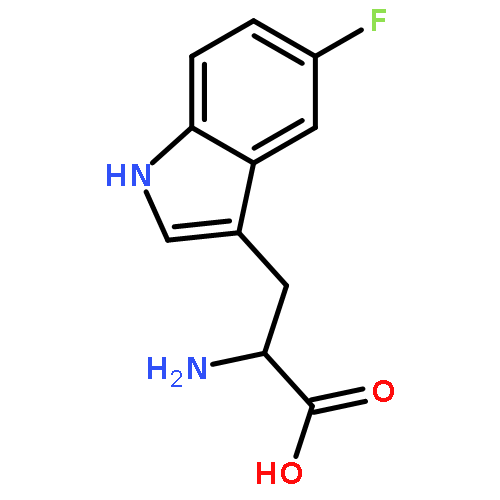





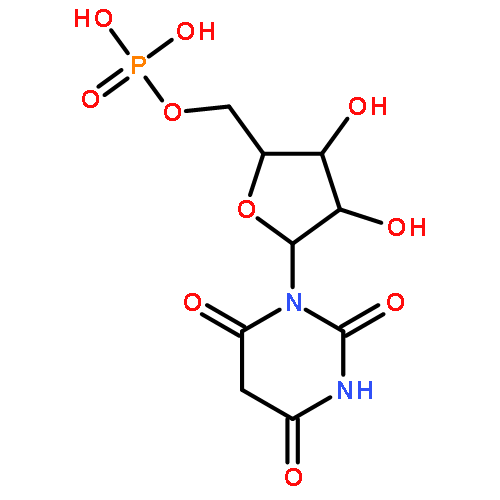
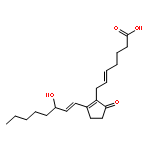


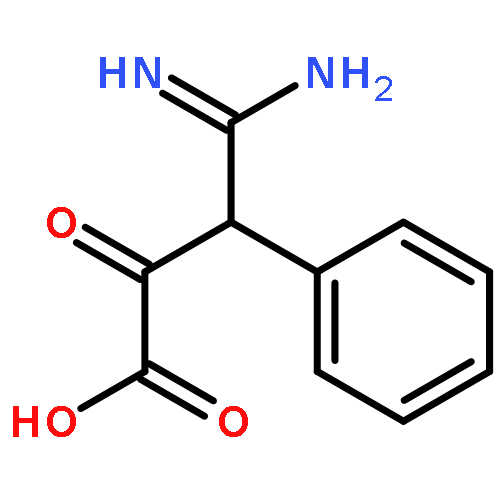








![Diphosphoric acid,P-[(2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl] ester](http://img.cochemist.com/ccimg/6900/6863-06-5.png)
![Diphosphoric acid,P-[(2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl] ester](http://img.cochemist.com/ccimg/6900/6863-06-5_b.png)

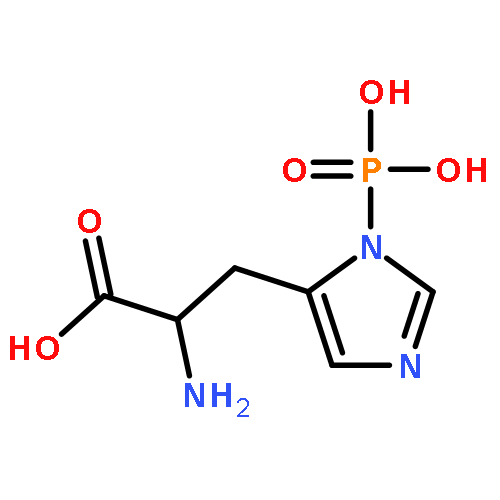


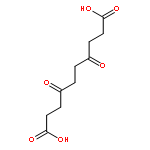



![2-AMINO-5-[[1-(CARBOXYMETHYLAMINO)-1-OXO-3-SULFOPROPAN-2-YL]AMINO]-5-OXOPENTANOIC ACID](http://img.cochemist.com/ccimg/3800/3773-07-7.png)
![2-AMINO-5-[[1-(CARBOXYMETHYLAMINO)-1-OXO-3-SULFOPROPAN-2-YL]AMINO]-5-OXOPENTANOIC ACID](http://img.cochemist.com/ccimg/3800/3773-07-7_b.png)
![9H-Thioxanthen-9-one,1-[[2-(diethylamino)ethyl]amino]-4-(hydroxymethyl)-](http://img.cochemist.com/ccimg/3200/3105-97-3.png)
![9H-Thioxanthen-9-one,1-[[2-(diethylamino)ethyl]amino]-4-(hydroxymethyl)-](http://img.cochemist.com/ccimg/3200/3105-97-3_b.png)





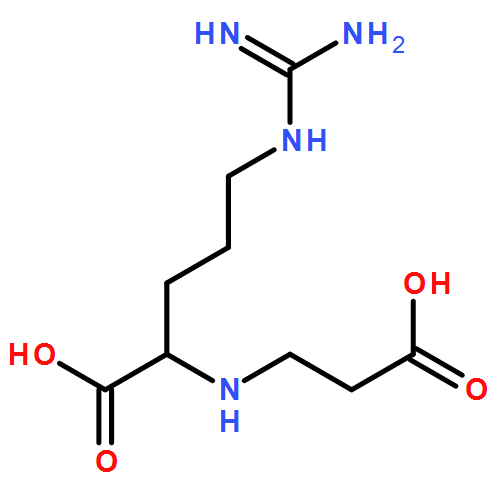
![L-Aspartic acid,N-[[[(4S)-4-amino-4-carboxybutyl]amino]iminomethyl]-](http://img.cochemist.com/ccimg/2400/2387-71-5.png)
![L-Aspartic acid,N-[[[(4S)-4-amino-4-carboxybutyl]amino]iminomethyl]-](http://img.cochemist.com/ccimg/2400/2387-71-5_b.png)



![2-Propenamide,N-[(1S)-1-(hydroxymethyl)-2-[(R)-[(methylthio)methyl]sulfinyl]ethyl]-3-(1,2,3,4-tetrahydro-6-methyl-2,4-dioxo-5-pyrimidinyl)-,(2E)-](http://img.cochemist.com/ccimg/1500/1404-64-4.png)
![2-Propenamide,N-[(1S)-1-(hydroxymethyl)-2-[(R)-[(methylthio)methyl]sulfinyl]ethyl]-3-(1,2,3,4-tetrahydro-6-methyl-2,4-dioxo-5-pyrimidinyl)-,(2E)-](http://img.cochemist.com/ccimg/1500/1404-64-4_b.png)


![L-Glutamic acid,N-[(5S)-5-amino-5-carboxypentyl]-](http://img.cochemist.com/ccimg/1000/997-68-2.png)
![L-Glutamic acid,N-[(5S)-5-amino-5-carboxypentyl]-](http://img.cochemist.com/ccimg/1000/997-68-2_b.png)






![(1AR,4R,4AR,7S,7AS,7BR)-1,1,4,7-TETRAMETHYLDECAHYDRO-1H-CYCLOPROP<WBR />A[E]AZULENE-4,7-DIOL](http://img.cochemist.com/ccimg/800/763-10-0.png)
![(1AR,4R,4AR,7S,7AS,7BR)-1,1,4,7-TETRAMETHYLDECAHYDRO-1H-CYCLOPROP<WBR />A[E]AZULENE-4,7-DIOL](http://img.cochemist.com/ccimg/800/763-10-0_b.png)





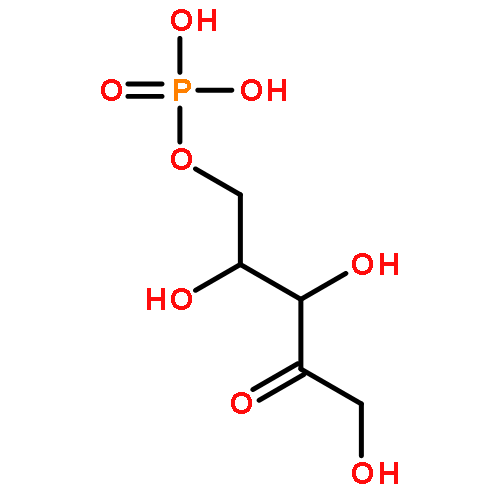

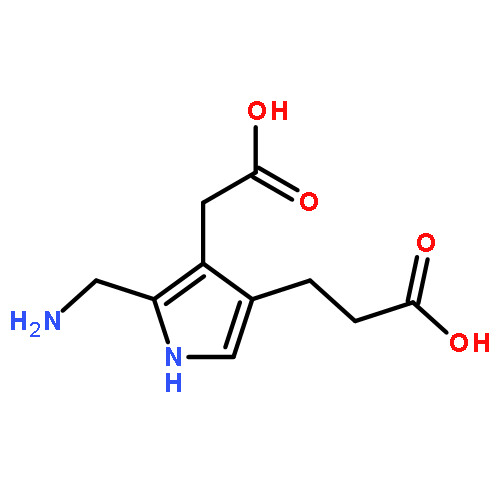
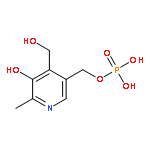
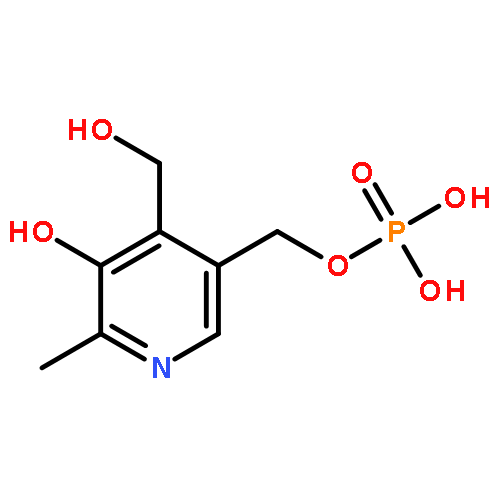












![(1R,2R,4S)-2-(6-chloropyridin-3-yl)-7-aza-bicyclo[2.2.1]heptane](http://img.cochemist.com/ccimg/140200/140111-52-0.png)
![(1R,2R,4S)-2-(6-chloropyridin-3-yl)-7-aza-bicyclo[2.2.1]heptane](http://img.cochemist.com/ccimg/140200/140111-52-0_b.png)