Co-reporter:Mathew D. Anker, Christos E. Kefalidis, Yan Yang, Jian Fang, Michael S. Hill, Mary F. Mahon, and Laurent Maron
Journal of the American Chemical Society July 26, 2017 Volume 139(Issue 29) pp:10036-10036
Publication Date(Web):June 22, 2017
DOI:10.1021/jacs.7b04926
Reactions of β-diketiminato magnesium and calcium hydrides with 1 atm of CO result in a reductive coupling process to produce the corresponding derivatives of the cis-ethenediolate dianion. Computational (DFT) analysis of this process mediated by Ca, Sr, and Ba highlights a common mechanism and a facility for the reaction that is enhanced by increasing alkaline earth atomic weight. Reaction of CO with PhSiH3 in the presence of the magnesium or calcium hydrides results in catalytic reduction to methylsilane and methylene silyl ether products, respectively. These reactions are proposed to ensue via the interception of initially formed group 2 formyl intermediates, an inference which is confirmed by a DFT analysis of the magnesium-centered reaction. The computational results identify the rate-determining process, requiring traversal of a 33.9 kcal mol–1 barrier, as a Mg–H/C–O σ-bond metathesis reaction, associated with the ultimate cleavage of the C–O bond. The carbonylation reactivity is extended to a variety of magnesium and calcium amides. With primary amido complexes, which for calcium include a derivative of the parent [NH2]− anion, CO insertion is facile and ensues with subsequent nitrogen-to-carbon migration of hydrogen to yield a variety of dinuclear and, in one case, trinuclear formamidate species. The generation of initial carbenic carbamoyl intermediates is strongly implicated through the isolation of the CO insertion product of a magnesium N-methylanilide derivative. These observations are reinforced by a DFT analysis of the calcium-centered reaction with aniline, which confirms the exothermicity of the formamidate formation (ΔH = −67.7 kcal mol–1). Stoichiometric reduction of the resultant magnesium and calcium formamidates with pinacolborane results in the synthesis of the corresponding N-borylated methylamines. This takes place via a sequence of reactions initiated through the generation of amidatohydridoborate intermediates and a cascade of reactivity that is analogous to that previously reported for the deoxygenative hydroboration of organic isocyanates catalyzed by the same magnesium hydride precatalyst. Although a sequence of amine formylation and deoxygenation may be readily envisaged for the catalytic utilization of CO as a C1 source in the production of methylamines, our observations demonstrate that competitive amine–borane dehydrocoupling is too facile under the conditions of 1 atm of CO employed.
Co-reporter:Mathew D. Anker, Christos E. Kefalidis, Yan Yang, Jian Fang, Michael S. Hill, Mary F. Mahon, and Laurent Maron
Journal of the American Chemical Society July 26, 2017 Volume 139(Issue 29) pp:10036-10036
Publication Date(Web):June 22, 2017
DOI:10.1021/jacs.7b04926
Reactions of β-diketiminato magnesium and calcium hydrides with 1 atm of CO result in a reductive coupling process to produce the corresponding derivatives of the cis-ethenediolate dianion. Computational (DFT) analysis of this process mediated by Ca, Sr, and Ba highlights a common mechanism and a facility for the reaction that is enhanced by increasing alkaline earth atomic weight. Reaction of CO with PhSiH3 in the presence of the magnesium or calcium hydrides results in catalytic reduction to methylsilane and methylene silyl ether products, respectively. These reactions are proposed to ensue via the interception of initially formed group 2 formyl intermediates, an inference which is confirmed by a DFT analysis of the magnesium-centered reaction. The computational results identify the rate-determining process, requiring traversal of a 33.9 kcal mol–1 barrier, as a Mg–H/C–O σ-bond metathesis reaction, associated with the ultimate cleavage of the C–O bond. The carbonylation reactivity is extended to a variety of magnesium and calcium amides. With primary amido complexes, which for calcium include a derivative of the parent [NH2]− anion, CO insertion is facile and ensues with subsequent nitrogen-to-carbon migration of hydrogen to yield a variety of dinuclear and, in one case, trinuclear formamidate species. The generation of initial carbenic carbamoyl intermediates is strongly implicated through the isolation of the CO insertion product of a magnesium N-methylanilide derivative. These observations are reinforced by a DFT analysis of the calcium-centered reaction with aniline, which confirms the exothermicity of the formamidate formation (ΔH = −67.7 kcal mol–1). Stoichiometric reduction of the resultant magnesium and calcium formamidates with pinacolborane results in the synthesis of the corresponding N-borylated methylamines. This takes place via a sequence of reactions initiated through the generation of amidatohydridoborate intermediates and a cascade of reactivity that is analogous to that previously reported for the deoxygenative hydroboration of organic isocyanates catalyzed by the same magnesium hydride precatalyst. Although a sequence of amine formylation and deoxygenation may be readily envisaged for the catalytic utilization of CO as a C1 source in the production of methylamines, our observations demonstrate that competitive amine–borane dehydrocoupling is too facile under the conditions of 1 atm of CO employed.
Co-reporter:Kimberley J. Gallagher;Maialen Espinal-Viguri;Ruth L. Webster
Advanced Synthesis & Catalysis 2016 Volume 358( Issue 15) pp:2460-2468
Publication Date(Web):
DOI:10.1002/adsc.201501179
Co-reporter:William J. Gee, Laura K. Cadman, Harina Amer Hamzah, Mary F. Mahon, Paul R. Raithby, and Andrew D. Burrows
Inorganic Chemistry 2016 Volume 55(Issue 21) pp:10839
Publication Date(Web):October 17, 2016
DOI:10.1021/acs.inorgchem.6b01917
Postsynthetic modification (PSM) of amino-functionalized metal–organic frameworks (MOFs) to those bearing pendant β-amidoketone arms using diketene is herein reported. Three unique MOF families demonstrate the scope of this transformation, which both is atom-economical and yields high conversions. In each case, the crystallinity was retained, and instances of exceptional solid-state ordering were observed in the PSM products, which has allowed detailed crystallographic characterization in multiple instances.
Co-reporter:P. McKeown, M. G. Davidson, J. P. Lowe, M. F. Mahon, L. H. Thomas, T. J. Woodman and M. D. Jones
Dalton Transactions 2016 vol. 45(Issue 12) pp:5374-5387
Publication Date(Web):18 Feb 2016
DOI:10.1039/C5DT04695E
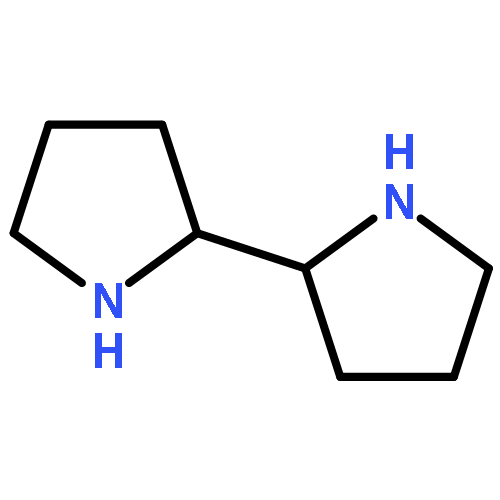
Herein we report the synthesis and characterisation of a series of salalen and salan ligands derived from 2-(aminomethyl)piperidine. Depending on the choice of starting salicylaldehyde, a bicyclic salan type ligand (1–3H2) or imino salalen type ligand (4–6H, 7–9H2) were prepared. The ligands were successfully complexed to group 4 metals and aluminium; with hafnium and zirconium octahedral complexes, M(1–3)2, were realised; whilst with aluminium tetrahedral and trigonal bipyramidal complexes, Al(1–9)Mex (x = 1,2), were isolated. The complexes have been characterised in solution via1H and 13C{1H} NMR spectroscopy and in the solid state by X-ray crystallography. The group 4 complexes were observed to have a fac–fac arrangement of ligands and there were two isomers present when 3H2 was ligated. The imino aluminium complexes Al(7–9)Me were isolated as a mixture of diastereoisomers. The resultant complexes were trialed in the ring opening polymerisation of rac-lactide with both heterotactic and isotactic PLA being demonstrated. Tacticity was found to be dependent on the nature of the ligand and metal used; the M(1–3)2 complexes were generally found to have a heterotactic preference (Pr = 0.67–0.76) and the aluminium polymerisation outcome was dictated more by the steric influence of the ligand, particularly for Al(4/6)Me2/Al(7/9)Me.
Co-reporter:Lee R. Collins, Louise A. Moffat, Mary F. Mahon, Matthew D. Jones, Michael K. Whittlesey
Polyhedron 2016 Volume 103(Part A) pp:121-125
Publication Date(Web):8 January 2016
DOI:10.1016/j.poly.2015.09.022
The reaction of the ring-expanded N-heterocyclic carbenes (RE-NHCs) with [ZnMe2] affords the structurally characterised complexes [(6-Mes)ZnMe2] (1) and [(7-Mes)ZnMe2] (2). The activity of the two complexes, along with that of the free carbenes, for the ring-opening polymerisation of rac-lactide has been probed. Even for such strongly σ-donating carbenes, dissociation from the metal centre cannot be ruled out.The activity of ring-expanded N-heterocyclic carbene complexes of zinc for the ring-opening polymerisation of rac-lactide has been probed alongside that of the free carbenes.
Co-reporter:Ian M. Riddlestone, David McKay, Matthias J. Gutmann, Stuart A. Macgregor, Mary F. Mahon, Hazel A. Sparkes, and Michael K. Whittlesey
Organometallics 2016 Volume 35(Issue 9) pp:1301-1312
Publication Date(Web):April 26, 2016
DOI:10.1021/acs.organomet.6b00173
Halide abstraction from the ruthenium N-heterocyclic carbene complex Ru(IPr)2(CO)HCl (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) with NaBAr4F (BAr4F = B{C6H3(3,5-CF3)2}4) gave the salt [Ru(IPr)2(CO)H]BAr4F (2), which was shown through a combined X-ray/neutron structure refinement and quantum theory of atoms in molecules (QTAIM) study to contain a bifurcated Ru···η3-H2C ξ-agostic interaction involving one iPr substituent of the IPr ligand. This system complements the previously reported [Ru(IMes)2(CO)H]+ cation (IMes =1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene), where a non-agostic form is favored. Treatment of 2 with CO, H2, and the amine–boranes H3B·NR2H (R = Me, H) gave [Ru(IPr)2(CO)3H]BAr4F (3), [Ru(IPr)2(CO)(η2-H2)H]BAr4F (4), and [Ru(IPr)2(CO)(κ2-H2BH·NR2H)H]BAr4F (R = Me, 5; R = H, 6), respectively. Heating 5 in the presence of Me3SiCH═CH2 led to alkene hydroboration and formation of the C–H activated product [Ru(IPr)(IPr)′(CO)]BAr4F (7). X-ray characterization of 3 and 5–7 was complemented by DFT calculations, and the mechanism of H2/H exchange in 4 was also elucidated. Treatment of 2 with HBcat resulted in Ru–H abstraction to form the boryl complex [Ru(IPr)2(CO)(Bcat)] BAr4F (8), which proved to be competent in the catalytic hydroboration of 1-hexene. In 8, a combined X-ray/neutron structure refinement and QTAIM analysis suggested the presence of a single Ru···η2-HC ξ-agostic interaction.
Co-reporter:Lee R. Collins;Dr. Ian M. Riddlestone;Dr. Mary F. Mahon; Michael K. Whittlesey
Chemistry - A European Journal 2015 Volume 21( Issue 40) pp:14075-14084
Publication Date(Web):
DOI:10.1002/chem.201502476
Abstract
The mononuclear N-heterocyclic carbene (NHC) copper alkoxide complexes [(6-NHC)CuOtBu] (6-NHC=6-MesDAC (1), 6-Mes (2)) have been prepared by addition of the free carbenes to the tetrameric tert-butoxide precursor [Cu(OtBu)]4, or by protonolysis of [(6-NHC)CuMes] (6-NHC=6-MesDAC (3), 6-Mes (4)) with tBuOH. In contrast to the relatively stable diaminocarbene complex 2, the diamidocarbene derivative 1 proved susceptible to both thermal and hydrolytic ring-opening reactions, the latter affording [(6-MesDAC)Cu(OC(O)CMe2C(O)N(H)Mes)(CNMes)] (6). The intermediacy of [(6-MesDAC)Cu(OH)] in this reaction was supported by the generation of Cu2O as an additional product. Attempts to generate an isolable copper hydride complex of the type [(6-MesDAC)CuH] by reaction of 1 with Et3SiH resulted instead in migratory insertion to generate [(6-MesDAC-H)Cu(P(p-tolyl)3)] (9) upon trapping by P(p-tolyl)3. Migratory insertion was also observed during attempts to prepare [(6-Mes)CuH], with [(6-Mes-H)Cu(6-Mes)] (10) isolated, following a reaction that was significantly slower than in the 6-MesDAC case. The longer lifetime of [(6-Mes)CuH] allowed it to be trapped stoichiometrically by alkyne, and also employed in the catalytic semi-reduction of alkynes and hydrosilylation of ketones.
Co-reporter:Matthew D. Jones, Stuart L. Hancock, Paul McKeown, Pascal M. Schäfer, Antoine Buchard, Lynne H. Thomas, Mary F. Mahon and John P. Lowe
Chemical Communications 2014 vol. 50(Issue 100) pp:15967-15970
Publication Date(Web):04 Nov 2014
DOI:10.1039/C4CC07871C
Herein we report the synthesis and characterisation of a series of Zr(IV) 2,2′-bipyrrolidine–salan derived complexes and their exploitation for the ring opening polymerisation of rac-lactide to afford highly isotactically enriched polymers.
Co-reporter:Catherine L. Lyall, Mario Uosis-Martin, John P. Lowe, Mary F. Mahon, G. Dan Pantoş and Simon E. Lewis
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 9) pp:1468-1475
Publication Date(Web):21 Dec 2012
DOI:10.1039/C2OB26765A
The functionalisation of decalin by means of an “aliphatic Friedel–Crafts” reaction was reported over fifty years ago by Baddeley et al. This protocol is of current relevance in the context of C–H activation and here we demonstrate its applicability to a range of other saturated hydrocarbons. Structural elucidation of the products is described and a mechanistic rationale for their formation is presented. The “aliphatic Friedel–Crafts” procedure allows for production of novel oxygenated building blocks from abundant hydrocarbons and as such can be considered to add significant synthetic value in a single step.
Co-reporter:Dr. Michael J. Page;Wei Y. Lu;Rebecca C. Poulten;Dr. Emma Carter;Dr. Andrés G. Algarra;Dr. Benson M. Kariuki; Stuart A. Macgregor;Dr. Mary F. Mahon; Kingsley J. Cavell;Dr. Damien M. Murphy; Michael K. Whittlesey
Chemistry - A European Journal 2013 Volume 19( Issue 6) pp:2158-2167
Publication Date(Web):
DOI:10.1002/chem.201202950
Abstract
Comproportionation of [Ni(cod)2] (cod=cyclooctadiene) and [Ni(PPh3)2X2] (X=Br, Cl) in the presence of six-, seven- and eight-membered ring N-aryl-substituted heterocyclic carbenes (NHCs) provides a route to a series of isostructural three-coordinate NiI complexes [Ni(NHC)(PPh3)X] (X=Br, Cl; NHC=6-Mes 1, 6-Anis 2, 6-AnisMes 3, 7-o-Tol 4, 8-Mes 5, 8-o-Tol 6, O-8-o-Tol 7). Continuous wave (CW) and pulsed EPR measurements on 1, 4, 5, 6 and 7 reveal that the spin Hamiltonian parameters are particularly sensitive to changes in NHC ring size, N substituents and halide. In combination with DFT calculations, a mixed SOMO of ∣3d〉 and ∣3d〉 character, which was found to be dependent on the complex geometry, was observed and this was compared to the experimental g values obtained from the EPR spectra. A pronounced 31P superhyperfine coupling to the PPh3 group was also identified, consistent with the large spin density on the phosphorus, along with partially resolved bromine couplings. The use of 1, 4, 5 and 6 as pre-catalysts for the Kumada coupling of aryl chlorides and fluorides with ArMgY (Ar=Ph, Mes) showed the highest activity for the smaller ring systems and/or smaller substituents (i.e., 1>4≈6≫5).
Co-reporter:Andrew D. Burrows, Mary F. Mahon, Catherine L. Renouf, Christopher Richardson, Anna J. Warren and John E. Warren
Dalton Transactions 2012 vol. 41(Issue 14) pp:4153-4163
Publication Date(Web):26 Jan 2012
DOI:10.1039/C2DT12115H
The iron(III) and aluminium(III) complexes of 1,3-di(4-pyridyl)propane-1,3-dionato (dppd) and 1,3-di(3-pyridyl)propane-1,3-dionato (dmppd), [Fe(dppd)3] 1, [Fe(dmppd)3] 2, [Al(dppd)3] 3 and [Al(dmppd)3] 4 have been prepared. These complexes adopt molecular structures in which the metal centres contain distorted octahedral geometries. In contrast, the copper(II) and zinc(II) complexes [Cu(dppd)2] 5 and [Zn(dmppd)2] 6 both form polymeric structures in which coordination of the pyridyl groups into the axial positions of neighbouring metal centres links discrete square-planar complexes into two-dimensional networks. The europium complex [Eu(dmppd)2(H2O)4]Cl·2EtOH·0.5H2O 7 forms a structure containing discrete cations that are linked into sheets through hydrogen bonds, whereas the lanthanum complex [La(dmppd)3(H2O)]·2H2O 8 adopts a one-dimensional network structure, connected into sheets by hydrogen bonds. The iron complexes 1 and 2 act as metalloligands in reactions with silver(I) salts, with the nature of the product depending on the counter-ions present. Thus, the reaction between 1 and AgBF4 gave [AgFe(dppd)3]BF4·DMSO 9, in which the silver centres link the metalloligands into discrete nanotubes, whereas reactions with AgPF6 and AgSbF6 gave [AgFe(dppd)3]PF6·3.28DMSO 10 and [AgFe(dppd)3]SbF6·1.25DMSO 11, in which the metalloligands are linked into sheets. In all three cases, only four of the six pyridyl groups present on the metalloligands are coordinated. The reaction between 2 and AgNO3 gave [Ag2Fe(dmppd)3(ONO2)]NO3·MeCN·CH2Cl212. Compound 12 adopts a layer structure in which all pyridyl groups are coordinated to silver centres and, in addition, a nitrate ion bridges between two silver centres. A similar structure is adopted by [Ag2Fe(dmppd)3(O2CCF3)]CF3CO2·2MeCN·0.25CH2Cl213, with a bridging trifluoroacetate ion playing the same role as the nitrate ion in 12.
Co-reporter:Ravi Shankar ; Archana Jain ; Gabriele Kociok-Köhn ; Mary F. Mahon ;Kieran C. Molloy
Inorganic Chemistry 2010 Volume 49(Issue 10) pp:4708-4715
Publication Date(Web):April 21, 2010
DOI:10.1021/ic100465u
Hydrolysis of the mixed-ligand dimethyltin(ethoxy)ethanesulfonate, [Me2Sn(OEt)(OSO2Et)]n (1a) in moist hexane proceeds via disproportionation and partial cleavage of Sn−C and S−C bonds to afford a novel oxo-/hydroxo- organotin cluster of the composition [(Me2Sn)(MeSn)4(OSO2Et)2(OH)4(O)2(SO3)2] (1) bearing both mono- and dimethyltin fragments and in situ generated sulfite (SO32−) anion in the structural framework. On the other hand, similar reactions with analogous mixed ligand diorganotin precursors, [R2Sn(OR1)(OSO2R1)]n (R = n-Bu, R1 = Et (2a); R = Et, R1 = Me (3a)), result in the formation of tetranuclear diorganotin clusters, [{(n-Bu2Sn)2(OH)(OSO2Et)}O]2 (2) and [(Et2Sn)4(OH)(O)2(OSO2Me)3] (3), respectively. The activation of the Sn−C or S−C bond is not observed in these cases. These findings provide a preliminary insight into the unusual reactivity of 1a under hydrolytic conditions.
Co-reporter:Carlo Di Iulio, Matthew D. Jones, Mary F. Mahon, and David C. Apperley
Inorganic Chemistry 2010 Volume 49(Issue 22) pp:10232-10234
Publication Date(Web):October 12, 2010
DOI:10.1021/ic101809r
In this Communication, we report the unprecedented solid-state structures for a series of zinc(II) silsesquioxane complexes. Initial catalytic data for the ring-opening polymerization of rac-lactide are also presented together with analogous heterogeneous species.
Co-reporter:Nanhai Singh, Abhinav Kumar, Rajendra Prasad, Kieran C. Molloy and Mary F. Mahon
Dalton Transactions 2010 vol. 39(Issue 10) pp:2667-2675
Publication Date(Web):20 Jan 2010
DOI:10.1039/B917871F
A series of new heterobimetallic phenylmercury(II) dithiocarbamate complexes incorporating the ferrocenyl moiety (C5H5)Fe(C5H4) (Fc), namely PhHgS2CN(CH2Fc)CH2C6H5, (1), PhHgS2CN(CH2Fc)CH(CH3)2, (2), PhHgS2CN(CH2Fc)(CH2)3CH3, (3) and [PhHgS2CN(CH2Fc)]2(CH2C6H4CH2), (4) have been prepared and characterized by elemental analysis, UV-Vis, IR, 1H and 13C NMR spectroscopies. The crystal structures of 1, 2 and 4 showed a linear core at the Hg(II) centre of the molecule, bound by the sulfur atom of the dithiocarbamate ligand and carbon atom of the aromatic ring. Weak intermolecular Hg⋯S interactions form “head-to-tail” dimers in the cases of 1 and 2. 4 forms a similar dimeric structure, forming two pairs of Hg⋯S interactions to generate a tetrametallic unit. The observed quasi-reversible cyclic voltammograms of the complexes have been corroborated by calculating gross electron population at each atom for the neutral as well its oxidized species obtained at the density functional level (DFT) of theory, which suggests an electron withdrawing effect from the organomercury(II)-dithiocarbamate group. The electronic absorption bands of all the four complexes were assigned with the help of time dependent density functional theory (TD-DFT) calculations. Upon excitation at ∼ 440 nm 1, 3 and 4 exhibited a medium strong photoluminescence emission at ∼ 500 nm as a consequence of MLCT intraligand charge transfer. 1, when excited at 256 nm exhibits photoluminescence emission at 398 nm.
Co-reporter:Emma L. Whitelaw, Matthew D. Jones, Mary F. Mahon and Gabriele Kociok-Kohn
Dalton Transactions 2009 (Issue 41) pp:9020-9025
Publication Date(Web):11 Aug 2009
DOI:10.1039/B911545E
A series of group 4 amine tris(phenolate) complexes have been prepared and characterised by single crystal X-ray diffraction and multinuclear NMR spectroscopy. It was found that the ligands afforded monomeric titanium complexes and dimeric zirconium structures in the solid-state. The complexes have been tested for the ring-opening polymerisation of rac-lactide under both solution and melt conditions, which showed varying degrees of selectivity and control, with PDIs in the range of 1.09–2.07. The initiators were also tested for the production of copolymers containing rac-lactide and isosorbide. From NMR spectroscopic analysis and MALDI-TOF mass spectrometry the isosorbide is incorporated into the polymer. The complexes were also screened for the ring-opening polymerisation of 1,3-dioxan-2-one to produce a polycarbonate with good conversions (24–99%).
Co-reporter:Mary F. Mahon, John McGinley, A. Denise Rooney, John M.D. Walsh
Inorganica Chimica Acta 2009 Volume 362(Issue 7) pp:2353-2360
Publication Date(Web):15 May 2009
DOI:10.1016/j.ica.2008.10.029
The Schiff-base ligands (1–3) formed from the reaction of 3-hydroxybenzaldehyde with three alkyl diamines have been synthesised and characterised. They are stable both in solution and in the solid state for several weeks, with no degradation to the starting amines and aldehyde observed. The reactions of the three Schiff-base ligands with various MX2 salts (M = Cu, Ni or Zn; X = chloride, perchlorate or acetate) resulted in the cleavage of the imine bond and formation of metal–amine complexes, as well as the entrapment of a two coordinate CuCl2 molecule within the lattice in one particular case.The reactions of the three Schiff-base ligands with various MX2 salts (M = Cu, Ni or Zn; X = chloride, perchlorate or acetate) resulted in the cleavage of the imine bond and formation of metal–amine complexes, as well as the entrapment of a two coordinate CuCl2 molecule within the lattice in one particular case.
Co-reporter:Nanhai Singh, Abhinav Kumar, Kieran C. Molloy and Mary F. Mahon
Dalton Transactions 2008 (Issue 37) pp:4999-5007
Publication Date(Web):01 Aug 2008
DOI:10.1039/B804635B
A series of new phenylmercury(II) dithio complexes [PhHg(Bun2dtc)] (1; Bun2dtc− = di-n-butyldithiocarbamate), [PhHg(morphdtc)] (2; morphdtc− = morpholinedithiocarbamate), [PhHg(Bz2dtc)] (3; Bz2dtc− = dibenzyldithiocarbamate), [PhHg(methoxethxant)] (4; methoxethxant− = 2-methoxyethylxanthate) [(PhHg)2NED] (5; NED2− = 1-nitroethylene-2,2-dithiolate) and [(PhHg)2CDC] (6; CDC2− = cyanodithioimidocarbonate) have been prepared and characterized by elemental analysis, UV-Vis, IR, 1H and 13C NMR spectra and mass spectrometry. The crystal structures of 1, 2 and 3 showed a linear Hg(II) core at the center of the molecules. The weak intra- and intermolecular Hg⋯S interactions provide a molecular chain framework. The reaction of PhHgO2CCH3 with Bun2dtcH gave the known dimeric complex Hg(Bun2dtc)2 while the Ni(O2CCH3)2 mediated reaction gave 1 instead of the expected heterobimetallic complex [PhHgNi(Bun2CS2)2]O2CCH3 which has been corroborated by natural charges at each atom obtained at the density functional level (DFT) of theory. Upon excitation at 358 nm 3 exhibited a medium strong photoluminescence emission at 420 nm as a consequence of intraligand π → π* transitions. The electronic absorption bands of 3 were assigned from time dependent density functional theory (TD-DFT) calculations. Geometrical configurations of 4, 5 and 6 have been optimized using the DFT method. All of the complexes are weakly conducting (σrt ∼ 10−12 S cm−1). However 2 and 6 exhibited semiconductivity with band gaps of 0.39 and 0.94 eV respectively.
Co-reporter:Andrew D. Burrows, Kevin Cassar, Tina Düren, Richard M. W. Friend, Mary F. Mahon, Sean P. Rigby and Teresa L. Savarese
Dalton Transactions 2008 (Issue 18) pp:2465-2474
Publication Date(Web):19 Mar 2008
DOI:10.1039/B718947H
The products isolated from the reaction between Cd(NO3)2·4H2O and 1,4-benzenedicarboxylic acid (H2bdc) in DMF are very dependent on the conditions. At 115 °C, the reaction gives [Cd(bdc)(DMF)]∞1, which has a three-dimensional network structure, whereas at 95 °C, 1 is formed alongside [Cd3(bdc)3(DMF)4]∞2, which has a two-dimensional network structure. When the reaction is carried out under pressure, it yields [Cd3(bdc)3(DMF)4]∞3, which is a supramolecular isomer of 2. The structure of 3 differs from that of 2 regarding the way the Cd3(O2CR)6 units are interlinked to form layers. When the reaction was carried out in DMF that had undergone partial hydrolysis, the only isolated product was {(NMe2H2)2[Cd(bdc)2]·2DMF}∞4. Compound 4 has a three-dimensional triply-interpenetrated diamondoid structure, with dimethylammonium cations and DMF molecules included within the pores. The reaction between Cd(NO3)2·4H2O and H2bdc in DEF gave [Cd(bdc)(DEF)]∞5, regardless of the solvent quality. Compound 5 has a three-dimensional network structure. The reaction of Cd(NO3)2·4H2O and 1,3-benzenedicarboxylic acid (H2mbdc) in DMF gave [Cd(mbdc)(DMF)]∞6 which has a bilayer structure. The thermal properties of the new materials have been investigated, and the coordinated DEF molecules from 5 can be removed on heating to 400 °C without any change in the powder X-ray diffraction pattern. The H2 sorption isotherm for the desolvated material shows marked hysteresis between adsorption and desorption, and less adsorption than predicted by simulations. Kinetic data indicate that the hysteresis is not due to mass transfer limitations, and the most likely explanation for this behaviour lies in partial collapse of the framework to an amorphous phase under the conditions of activation.
Co-reporter:Andrew L. Johnson, Matthew G. Davidson and Mary F. Mahon
Dalton Transactions 2007 (Issue 46) pp:5405-5411
Publication Date(Web):27 Sep 2007
DOI:10.1039/B708378E
Treatment of the titanium(IV) alkoxide complex [Ti(OiPr)(OC6Me2H2CH2)3N] (2) with BH3·THF, as part of a study into the utility and reactivity of (2) in the metal mediated borane reduction of acetophenone, results in alkoxide–hydride exchange and formation of the structurally characterised titanium(IV) tetrahydroborate complex [Ti{BH4}(OC6Me2H2CH2)3N] (3). Complex (3) readily undergoes reduction to form the isolable titanium(III) species [Ti(OC6Me2H2CH2)3N]2 (4). Reaction of (2) with B(C6F5)3 results in formation of the Lewis acid adduct [Ti(OC6Me2H2CH2)3N][HO·B(C6F5)3] (5). In comparison, treatment of the less sterically encumbered alkoxide Ti(OiPr)4 with B(C6F5)3 results in alkoxide–aryl exchange and formation of the organometallic titanium complex [Ti(OiPr)3(C6F5)]2 (6). The molecular structures of 3, 4, 5 and 6 have been determined by X-ray diffraction.
Co-reporter:Andrew D. Burrows, Kevin Cassar, Mary F. Mahon and John E. Warren
Dalton Transactions 2007 (Issue 24) pp:2499-2509
Publication Date(Web):23 Apr 2007
DOI:10.1039/B702074K
The complexes [Cu(L1)2] 1, [Fe(L1)3] 3 and [Al(L1)3] 4 [L1 = CH3C(O)C(CN)C(O)CH3] have been prepared for use as metallo-ligands in mixed-metal coordination networks. Surprisingly, the nature of the copper precursor is important in the synthesis of 1, with the reaction between Cu(NO3)2·3H2O, HL1 and NEt3 giving [Cu6(µ3-OMe)4(µ-OMe)2(L1)6] 2 instead of the anticipated 1, which was obtained with CuCl2·2H2O under the same conditions. Compound 1 reacts with AgNO3 to form [Cu(L1)2·AgNO3]∞5, the structure of which contains one-dimensional chains in which Ag+ ions bridge between molecules of 1. These chains are cross-linked into ladders by bridging nitrates. The product obtained from the reaction of 3 and AgNO3 is crucially dependent on the solvent used. The reaction in methanol–acetone gives [Fe(L1)3·AgNO3]∞6, {[Fe2(µ-OMe)2(L1)4·2AgNO3]·CH3C(O)CH3}∞7 and [Fe2(µ-OMe)2(L1)4·AgNO3]∞8. Compounds 6 and 8 both have one-dimensional chain structures, whereas 7 has a two-dimensional layer structure. The reaction in methanol gives 6 and 8 as the major products and, in addition, small quantities of {[AgFe2(µ-OMe)2(L1)4]OH·0.4H2O]∞9. Compound 9 has a three-dimensional structure based on doubly interpenetrated PtS nets. Compounds 7–9 contain Fe2(µ-OMe)2(L1)4 dimers, but the coordination properties of the dimers differ, with all the cyanides coordinated in 7 and 9 but one uncoordinated in 8. The orientation of the cyanide groups depends on the relative chirality of the iron centres. A transmetallation reaction occurs between 4 and AgNO3 to give [Ag(L1)]∞10, which has a two-dimensional layer structure. Compounds 2, 3 and 5–10 have been characterised by X-ray crystallography.
Co-reporter:Christopher J. Chapman, Christopher G. Frost and Mary F. Mahon
Dalton Transactions 2006 (Issue 18) pp:2251-2262
Publication Date(Web):09 Feb 2006
DOI:10.1039/B513390D
New heterofunctional phosphine ligands have been synthesised, incorporating the substitutionally labile sulfone and sulfonamide moieties as chelating groups, which display activity in the palladium-catalysed Suzuki and amination cross-coupling reactions. Single-crystal X-ray diffraction studies of the complexes formed with [Pd(µ-Cl)(dmba)] (dmba-H = N,N-dimethylbenzylamine) highlight the coordinating nature of these ligands; showing the formation of a bis chelate complex through a six-membered Pd–P–C–C–S–O ring with the sulfonamide class of ligands.
Co-reporter:Nichola J. Burke, Andrew D. Burrows, Mary F. Mahon, John E. Warren
Inorganica Chimica Acta 2006 Volume 359(Issue 11) pp:3497-3506
Publication Date(Web):1 August 2006
DOI:10.1016/j.ica.2006.01.008
Interactions between guanidinium cations and the sulfonate groups on the phosphine [PPh2C6H4-m-SO3]− have been exploited to incorporate iridium(I) centres into hydrogen-bonded networks. The crystal structure of [C(NH2)3]2{trans-[IrCl(CO)(PPh2C6H4-m-SO3)2]} (4) contains hexagonal guanidinium sulfonate (GS) sheets in which both of the sulfonate groups from each complex anion form hydrogen bonds within the same sheet. The crystal structures of [C(NH2)2(NHMe)][PPh2C6H4-m-SO3] (5) and [C(NH2)2(NHEt)][PPh2C6H4-m-SO3] (6) reveal that the GS sheets can tolerate the loss of one hydrogen bond donor, though twisting occurs to accommodate the alkyl group. However, the crystal structure of [C(NH2)2(NMe2)][PPh2C6H4-m-SO3] (7) shows that ribbon structures are formed instead of sheets when two hydrogen bond donors are lost. The compound [C(NH2)2(NHMe)]2{trans-[IrCl(CO)(PPh2C6H4-m-SO3)2]} · 3/8H2O (8) contains hydrogen-bonded cylinders as opposed to sheets. This is a likely consequence of a mismatch between the intramolecular S⋯S distance present in the anion, and the closer S⋯S distance present in a twisted GS sheet such as that in 5. The crystal structures of [C(NH2)2(NHEt)][P(O)Ph2C6H4-m-SO3] (9) and [C(NH2)2(NMe2)][P(O)Ph2C6H4-m-SO3] · H2O (10) show that the phosphine oxide group successfully competes with the sulfonate as a hydrogen bond acceptor. The crystal structure of 9 contains hydrogen-bonded ribbons that are interlinked through the anions which act as pillars to form a layer structure. In contrast, the crystal structure of 10 contains hydrogen-bonded sheets that involve cations, sulfonate groups, phosphine oxides and the included water molecule. These sheets are linked into a three-dimensional network through the anion pillars.The crystal structures of seven compounds containing guanidinium or substituted guanidinium cations and derivatives of the sulfonated phosphine PPh2C6H4-m-SO3- are reported. The effects on the supramolecular structure of inclusion of the trans-[IrCl(CO)(PPh2C6H4-m-SO3)2]2− anion is investigated, along with those of substitution on the cation and incorporation of a phosphine oxide group which is a competing hydrogen bond acceptor.
Co-reporter:Catherine L. Lyall, Mario Uosis-Martin, John P. Lowe, Mary F. Mahon, G. Dan Pantoş and Simon E. Lewis
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 9) pp:NaN1475-1475
Publication Date(Web):2012/12/21
DOI:10.1039/C2OB26765A
The functionalisation of decalin by means of an “aliphatic Friedel–Crafts” reaction was reported over fifty years ago by Baddeley et al. This protocol is of current relevance in the context of C–H activation and here we demonstrate its applicability to a range of other saturated hydrocarbons. Structural elucidation of the products is described and a mechanistic rationale for their formation is presented. The “aliphatic Friedel–Crafts” procedure allows for production of novel oxygenated building blocks from abundant hydrocarbons and as such can be considered to add significant synthetic value in a single step.
Co-reporter:Andrew D. Burrows, Kevin Cassar, Tina Düren, Richard M. W. Friend, Mary F. Mahon, Sean P. Rigby and Teresa L. Savarese
Dalton Transactions 2008(Issue 18) pp:NaN2474-2474
Publication Date(Web):2008/03/19
DOI:10.1039/B718947H
The products isolated from the reaction between Cd(NO3)2·4H2O and 1,4-benzenedicarboxylic acid (H2bdc) in DMF are very dependent on the conditions. At 115 °C, the reaction gives [Cd(bdc)(DMF)]∞1, which has a three-dimensional network structure, whereas at 95 °C, 1 is formed alongside [Cd3(bdc)3(DMF)4]∞2, which has a two-dimensional network structure. When the reaction is carried out under pressure, it yields [Cd3(bdc)3(DMF)4]∞3, which is a supramolecular isomer of 2. The structure of 3 differs from that of 2 regarding the way the Cd3(O2CR)6 units are interlinked to form layers. When the reaction was carried out in DMF that had undergone partial hydrolysis, the only isolated product was {(NMe2H2)2[Cd(bdc)2]·2DMF}∞4. Compound 4 has a three-dimensional triply-interpenetrated diamondoid structure, with dimethylammonium cations and DMF molecules included within the pores. The reaction between Cd(NO3)2·4H2O and H2bdc in DEF gave [Cd(bdc)(DEF)]∞5, regardless of the solvent quality. Compound 5 has a three-dimensional network structure. The reaction of Cd(NO3)2·4H2O and 1,3-benzenedicarboxylic acid (H2mbdc) in DMF gave [Cd(mbdc)(DMF)]∞6 which has a bilayer structure. The thermal properties of the new materials have been investigated, and the coordinated DEF molecules from 5 can be removed on heating to 400 °C without any change in the powder X-ray diffraction pattern. The H2 sorption isotherm for the desolvated material shows marked hysteresis between adsorption and desorption, and less adsorption than predicted by simulations. Kinetic data indicate that the hysteresis is not due to mass transfer limitations, and the most likely explanation for this behaviour lies in partial collapse of the framework to an amorphous phase under the conditions of activation.
Co-reporter:Andrew D. Burrows, Kevin Cassar, Mary F. Mahon and John E. Warren
Dalton Transactions 2007(Issue 24) pp:NaN2509-2509
Publication Date(Web):2007/04/23
DOI:10.1039/B702074K
The complexes [Cu(L1)2] 1, [Fe(L1)3] 3 and [Al(L1)3] 4 [L1 = CH3C(O)C(CN)C(O)CH3] have been prepared for use as metallo-ligands in mixed-metal coordination networks. Surprisingly, the nature of the copper precursor is important in the synthesis of 1, with the reaction between Cu(NO3)2·3H2O, HL1 and NEt3 giving [Cu6(µ3-OMe)4(µ-OMe)2(L1)6] 2 instead of the anticipated 1, which was obtained with CuCl2·2H2O under the same conditions. Compound 1 reacts with AgNO3 to form [Cu(L1)2·AgNO3]∞5, the structure of which contains one-dimensional chains in which Ag+ ions bridge between molecules of 1. These chains are cross-linked into ladders by bridging nitrates. The product obtained from the reaction of 3 and AgNO3 is crucially dependent on the solvent used. The reaction in methanol–acetone gives [Fe(L1)3·AgNO3]∞6, {[Fe2(µ-OMe)2(L1)4·2AgNO3]·CH3C(O)CH3}∞7 and [Fe2(µ-OMe)2(L1)4·AgNO3]∞8. Compounds 6 and 8 both have one-dimensional chain structures, whereas 7 has a two-dimensional layer structure. The reaction in methanol gives 6 and 8 as the major products and, in addition, small quantities of {[AgFe2(µ-OMe)2(L1)4]OH·0.4H2O]∞9. Compound 9 has a three-dimensional structure based on doubly interpenetrated PtS nets. Compounds 7–9 contain Fe2(µ-OMe)2(L1)4 dimers, but the coordination properties of the dimers differ, with all the cyanides coordinated in 7 and 9 but one uncoordinated in 8. The orientation of the cyanide groups depends on the relative chirality of the iron centres. A transmetallation reaction occurs between 4 and AgNO3 to give [Ag(L1)]∞10, which has a two-dimensional layer structure. Compounds 2, 3 and 5–10 have been characterised by X-ray crystallography.
Co-reporter:Emma L. Whitelaw, Matthew D. Jones, Mary F. Mahon and Gabriele Kociok-Kohn
Dalton Transactions 2009(Issue 41) pp:NaN9025-9025
Publication Date(Web):2009/08/11
DOI:10.1039/B911545E
A series of group 4 amine tris(phenolate) complexes have been prepared and characterised by single crystal X-ray diffraction and multinuclear NMR spectroscopy. It was found that the ligands afforded monomeric titanium complexes and dimeric zirconium structures in the solid-state. The complexes have been tested for the ring-opening polymerisation of rac-lactide under both solution and melt conditions, which showed varying degrees of selectivity and control, with PDIs in the range of 1.09–2.07. The initiators were also tested for the production of copolymers containing rac-lactide and isosorbide. From NMR spectroscopic analysis and MALDI-TOF mass spectrometry the isosorbide is incorporated into the polymer. The complexes were also screened for the ring-opening polymerisation of 1,3-dioxan-2-one to produce a polycarbonate with good conversions (24–99%).
Co-reporter:Andrew D. Burrows, Mary F. Mahon, Catherine L. Renouf, Christopher Richardson, Anna J. Warren and John E. Warren
Dalton Transactions 2012 - vol. 41(Issue 14) pp:NaN4163-4163
Publication Date(Web):2012/01/26
DOI:10.1039/C2DT12115H
The iron(III) and aluminium(III) complexes of 1,3-di(4-pyridyl)propane-1,3-dionato (dppd) and 1,3-di(3-pyridyl)propane-1,3-dionato (dmppd), [Fe(dppd)3] 1, [Fe(dmppd)3] 2, [Al(dppd)3] 3 and [Al(dmppd)3] 4 have been prepared. These complexes adopt molecular structures in which the metal centres contain distorted octahedral geometries. In contrast, the copper(II) and zinc(II) complexes [Cu(dppd)2] 5 and [Zn(dmppd)2] 6 both form polymeric structures in which coordination of the pyridyl groups into the axial positions of neighbouring metal centres links discrete square-planar complexes into two-dimensional networks. The europium complex [Eu(dmppd)2(H2O)4]Cl·2EtOH·0.5H2O 7 forms a structure containing discrete cations that are linked into sheets through hydrogen bonds, whereas the lanthanum complex [La(dmppd)3(H2O)]·2H2O 8 adopts a one-dimensional network structure, connected into sheets by hydrogen bonds. The iron complexes 1 and 2 act as metalloligands in reactions with silver(I) salts, with the nature of the product depending on the counter-ions present. Thus, the reaction between 1 and AgBF4 gave [AgFe(dppd)3]BF4·DMSO 9, in which the silver centres link the metalloligands into discrete nanotubes, whereas reactions with AgPF6 and AgSbF6 gave [AgFe(dppd)3]PF6·3.28DMSO 10 and [AgFe(dppd)3]SbF6·1.25DMSO 11, in which the metalloligands are linked into sheets. In all three cases, only four of the six pyridyl groups present on the metalloligands are coordinated. The reaction between 2 and AgNO3 gave [Ag2Fe(dmppd)3(ONO2)]NO3·MeCN·CH2Cl212. Compound 12 adopts a layer structure in which all pyridyl groups are coordinated to silver centres and, in addition, a nitrate ion bridges between two silver centres. A similar structure is adopted by [Ag2Fe(dmppd)3(O2CCF3)]CF3CO2·2MeCN·0.25CH2Cl213, with a bridging trifluoroacetate ion playing the same role as the nitrate ion in 12.
Co-reporter:Nanhai Singh, Abhinav Kumar, Kieran C. Molloy and Mary F. Mahon
Dalton Transactions 2008(Issue 37) pp:NaN5007-5007
Publication Date(Web):2008/08/01
DOI:10.1039/B804635B
A series of new phenylmercury(II) dithio complexes [PhHg(Bun2dtc)] (1; Bun2dtc− = di-n-butyldithiocarbamate), [PhHg(morphdtc)] (2; morphdtc− = morpholinedithiocarbamate), [PhHg(Bz2dtc)] (3; Bz2dtc− = dibenzyldithiocarbamate), [PhHg(methoxethxant)] (4; methoxethxant− = 2-methoxyethylxanthate) [(PhHg)2NED] (5; NED2− = 1-nitroethylene-2,2-dithiolate) and [(PhHg)2CDC] (6; CDC2− = cyanodithioimidocarbonate) have been prepared and characterized by elemental analysis, UV-Vis, IR, 1H and 13C NMR spectra and mass spectrometry. The crystal structures of 1, 2 and 3 showed a linear Hg(II) core at the center of the molecules. The weak intra- and intermolecular Hg⋯S interactions provide a molecular chain framework. The reaction of PhHgO2CCH3 with Bun2dtcH gave the known dimeric complex Hg(Bun2dtc)2 while the Ni(O2CCH3)2 mediated reaction gave 1 instead of the expected heterobimetallic complex [PhHgNi(Bun2CS2)2]O2CCH3 which has been corroborated by natural charges at each atom obtained at the density functional level (DFT) of theory. Upon excitation at 358 nm 3 exhibited a medium strong photoluminescence emission at 420 nm as a consequence of intraligand π → π* transitions. The electronic absorption bands of 3 were assigned from time dependent density functional theory (TD-DFT) calculations. Geometrical configurations of 4, 5 and 6 have been optimized using the DFT method. All of the complexes are weakly conducting (σrt ∼ 10−12 S cm−1). However 2 and 6 exhibited semiconductivity with band gaps of 0.39 and 0.94 eV respectively.
Co-reporter:Andrew L. Johnson, Matthew G. Davidson and Mary F. Mahon
Dalton Transactions 2007(Issue 46) pp:NaN5411-5411
Publication Date(Web):2007/09/27
DOI:10.1039/B708378E
Treatment of the titanium(IV) alkoxide complex [Ti(OiPr)(OC6Me2H2CH2)3N] (2) with BH3·THF, as part of a study into the utility and reactivity of (2) in the metal mediated borane reduction of acetophenone, results in alkoxide–hydride exchange and formation of the structurally characterised titanium(IV) tetrahydroborate complex [Ti{BH4}(OC6Me2H2CH2)3N] (3). Complex (3) readily undergoes reduction to form the isolable titanium(III) species [Ti(OC6Me2H2CH2)3N]2 (4). Reaction of (2) with B(C6F5)3 results in formation of the Lewis acid adduct [Ti(OC6Me2H2CH2)3N][HO·B(C6F5)3] (5). In comparison, treatment of the less sterically encumbered alkoxide Ti(OiPr)4 with B(C6F5)3 results in alkoxide–aryl exchange and formation of the organometallic titanium complex [Ti(OiPr)3(C6F5)]2 (6). The molecular structures of 3, 4, 5 and 6 have been determined by X-ray diffraction.
Co-reporter:Matthew D. Jones, Stuart L. Hancock, Paul McKeown, Pascal M. Schäfer, Antoine Buchard, Lynne H. Thomas, Mary F. Mahon and John P. Lowe
Chemical Communications 2014 - vol. 50(Issue 100) pp:NaN15970-15970
Publication Date(Web):2014/11/04
DOI:10.1039/C4CC07871C
Herein we report the synthesis and characterisation of a series of Zr(IV) 2,2′-bipyrrolidine–salan derived complexes and their exploitation for the ring opening polymerisation of rac-lactide to afford highly isotactically enriched polymers.
Co-reporter:P. McKeown, M. G. Davidson, J. P. Lowe, M. F. Mahon, L. H. Thomas, T. J. Woodman and M. D. Jones
Dalton Transactions 2016 - vol. 45(Issue 12) pp:NaN5387-5387
Publication Date(Web):2016/02/18
DOI:10.1039/C5DT04695E
Herein we report the synthesis and characterisation of a series of salalen and salan ligands derived from 2-(aminomethyl)piperidine. Depending on the choice of starting salicylaldehyde, a bicyclic salan type ligand (1–3H2) or imino salalen type ligand (4–6H, 7–9H2) were prepared. The ligands were successfully complexed to group 4 metals and aluminium; with hafnium and zirconium octahedral complexes, M(1–3)2, were realised; whilst with aluminium tetrahedral and trigonal bipyramidal complexes, Al(1–9)Mex (x = 1,2), were isolated. The complexes have been characterised in solution via1H and 13C{1H} NMR spectroscopy and in the solid state by X-ray crystallography. The group 4 complexes were observed to have a fac–fac arrangement of ligands and there were two isomers present when 3H2 was ligated. The imino aluminium complexes Al(7–9)Me were isolated as a mixture of diastereoisomers. The resultant complexes were trialed in the ring opening polymerisation of rac-lactide with both heterotactic and isotactic PLA being demonstrated. Tacticity was found to be dependent on the nature of the ligand and metal used; the M(1–3)2 complexes were generally found to have a heterotactic preference (Pr = 0.67–0.76) and the aluminium polymerisation outcome was dictated more by the steric influence of the ligand, particularly for Al(4/6)Me2/Al(7/9)Me.
Co-reporter:Nanhai Singh, Abhinav Kumar, Rajendra Prasad, Kieran C. Molloy and Mary F. Mahon
Dalton Transactions 2010 - vol. 39(Issue 10) pp:NaN2675-2675
Publication Date(Web):2010/01/20
DOI:10.1039/B917871F
A series of new heterobimetallic phenylmercury(II) dithiocarbamate complexes incorporating the ferrocenyl moiety (C5H5)Fe(C5H4) (Fc), namely PhHgS2CN(CH2Fc)CH2C6H5, (1), PhHgS2CN(CH2Fc)CH(CH3)2, (2), PhHgS2CN(CH2Fc)(CH2)3CH3, (3) and [PhHgS2CN(CH2Fc)]2(CH2C6H4CH2), (4) have been prepared and characterized by elemental analysis, UV-Vis, IR, 1H and 13C NMR spectroscopies. The crystal structures of 1, 2 and 4 showed a linear core at the Hg(II) centre of the molecule, bound by the sulfur atom of the dithiocarbamate ligand and carbon atom of the aromatic ring. Weak intermolecular Hg⋯S interactions form “head-to-tail” dimers in the cases of 1 and 2. 4 forms a similar dimeric structure, forming two pairs of Hg⋯S interactions to generate a tetrametallic unit. The observed quasi-reversible cyclic voltammograms of the complexes have been corroborated by calculating gross electron population at each atom for the neutral as well its oxidized species obtained at the density functional level (DFT) of theory, which suggests an electron withdrawing effect from the organomercury(II)-dithiocarbamate group. The electronic absorption bands of all the four complexes were assigned with the help of time dependent density functional theory (TD-DFT) calculations. Upon excitation at ∼ 440 nm 1, 3 and 4 exhibited a medium strong photoluminescence emission at ∼ 500 nm as a consequence of MLCT intraligand charge transfer. 1, when excited at 256 nm exhibits photoluminescence emission at 398 nm.



![1H-Pyrrole, 1,1'-[[(diphenylphosphino)methyl]phosphinidene]bis-](http://img.cochemist.com/ccimg/623600/623561-06-8.png)
![1H-Pyrrole, 1,1'-[[(diphenylphosphino)methyl]phosphinidene]bis-](http://img.cochemist.com/ccimg/623600/623561-06-8_b.png)



![Ethanone, 1-[1-(di-1H-pyrrol-1-ylphosphino)-1H-pyrrol-2-yl]-](http://img.cochemist.com/ccimg/420100/420087-01-0.png)
![Ethanone, 1-[1-(di-1H-pyrrol-1-ylphosphino)-1H-pyrrol-2-yl]-](http://img.cochemist.com/ccimg/420100/420087-01-0_b.png)








![Benzo[d]carbazole, 11-(1,1-dimethylethyl)-1,2,3,4,4a,5,6,7-octahydro-](http://img.cochemist.com/ccimg/193000/192991-63-2.png)
![Benzo[d]carbazole, 11-(1,1-dimethylethyl)-1,2,3,4,4a,5,6,7-octahydro-](http://img.cochemist.com/ccimg/193000/192991-63-2_b.png)
![Benzo[d]carbazole, 1,2,3,4,4a,5,6,7-octahydro-11-(1-methylethyl)-](http://img.cochemist.com/ccimg/193000/192991-59-6.png)
![Benzo[d]carbazole, 1,2,3,4,4a,5,6,7-octahydro-11-(1-methylethyl)-](http://img.cochemist.com/ccimg/193000/192991-59-6_b.png)
![COPPER, [(4-METHOXYPHENYL)ETHYNYL]-](http://img.cochemist.com/ccimg/48200/48121-13-7.png)
![COPPER, [(4-METHOXYPHENYL)ETHYNYL]-](http://img.cochemist.com/ccimg/48200/48121-13-7_b.png)
![Phosphine, [2-(4-methoxyphenyl)ethyl]diphenyl-](http://img.cochemist.com/ccimg/152100/152076-32-9.png)
![Phosphine, [2-(4-methoxyphenyl)ethyl]diphenyl-](http://img.cochemist.com/ccimg/152100/152076-32-9_b.png)


![PHOSPHINE, [2-(4-CHLOROPHENYL)ETHYL]DIPHENYL-](http://img.cochemist.com/ccimg/152100/152076-31-8.png)
![PHOSPHINE, [2-(4-CHLOROPHENYL)ETHYL]DIPHENYL-](http://img.cochemist.com/ccimg/152100/152076-31-8_b.png)

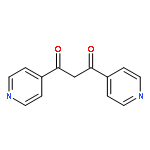
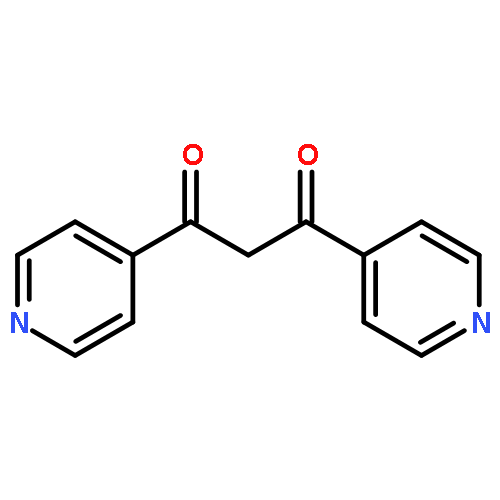
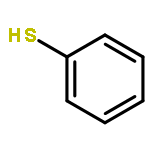
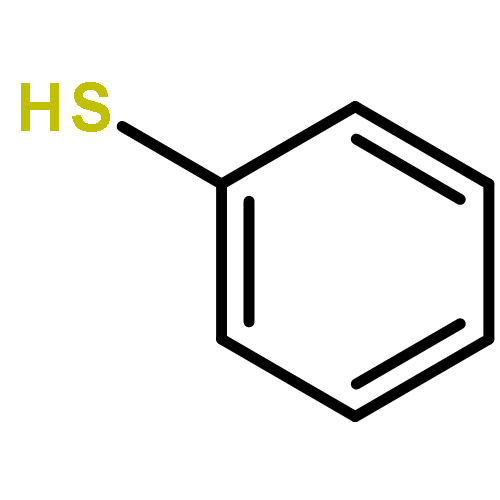
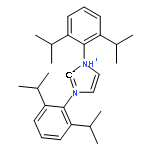
![Imidazolidine,1,3-bis[2,6-bis(1-methylethyl)phenyl]-2-(pentafluorophenyl)-](http://img.cochemist.com/ccimg/865700/865620-21-9.png)
![Imidazolidine,1,3-bis[2,6-bis(1-methylethyl)phenyl]-2-(pentafluorophenyl)-](http://img.cochemist.com/ccimg/865700/865620-21-9_b.png)











![[1,1'-Biphenyl]-4,4'-dicarboxylic acid, disodium salt](/data/chemimg/1152900/17274-14-5.png)
![[1,1'-Biphenyl]-4,4'-dicarboxylic acid, disodium salt](/data/chemimg/1152900/17274-14-5_b.png)









![(S)-6-((4-Chlorophenyl)(1H-1,2,4-triazol-1-yl)methyl)-1-methyl-1H-benzo[d][1,2,3]triazole](http://img.cochemist.com/ccimg/129800/129731-10-8.png)
![(S)-6-((4-Chlorophenyl)(1H-1,2,4-triazol-1-yl)methyl)-1-methyl-1H-benzo[d][1,2,3]triazole](http://img.cochemist.com/ccimg/129800/129731-10-8_b.png)








![Phenol, 2,2',2''-[nitrilotris(methylene)]tris[4,6-dimethyl-](http://img.cochemist.com/ccimg/58000/57914-22-4.png)
![Phenol, 2,2',2''-[nitrilotris(methylene)]tris[4,6-dimethyl-](http://img.cochemist.com/ccimg/58000/57914-22-4_b.png)



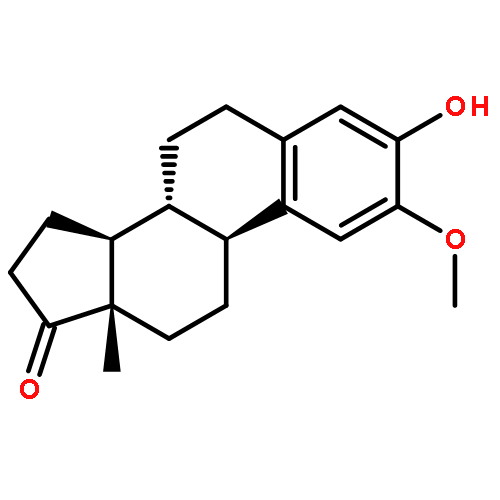
![1'H-Estra-1,3,5(10)-trieno[17,16-c]pyrazol-3-ol](http://img.cochemist.com/ccimg/3500/3460-91-1.png)
![1'H-Estra-1,3,5(10)-trieno[17,16-c]pyrazol-3-ol](http://img.cochemist.com/ccimg/3500/3460-91-1_b.png)
![N-[(3,4-DIMETHOXYPHENYL)METHYL]-1-PHENYLMETHANIMINE](http://img.cochemist.com/ccimg/71800/71703-43-0.png)
![N-[(3,4-DIMETHOXYPHENYL)METHYL]-1-PHENYLMETHANIMINE](http://img.cochemist.com/ccimg/71800/71703-43-0_b.png)

![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2.png)
![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2_b.png)
![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4.png)
![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4_b.png)
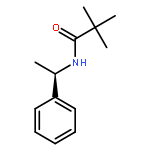
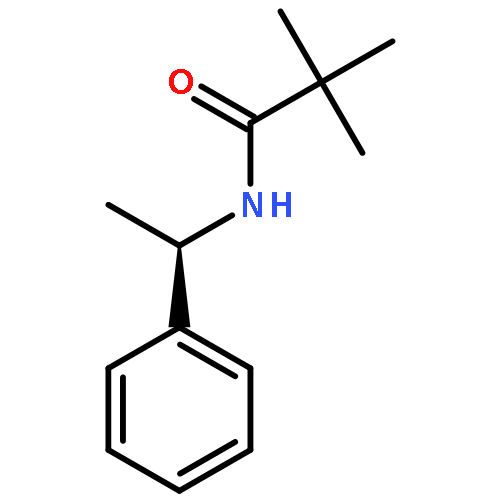
![Benzenesulfinic acid, 2-[1-[(2,2-dimethyl-1-oxopropyl)amino]ethyl]-, (R)-](/data/chemimg/1083600/130973-54-5.png)
![Benzenesulfinic acid, 2-[1-[(2,2-dimethyl-1-oxopropyl)amino]ethyl]-, (R)-](/data/chemimg/1083600/130973-54-5_b.png)















![Estra-1,3,5(10)-trien-17-one,3-[(aminosulfonyl)oxy]-](http://img.cochemist.com/ccimg/148700/148672-09-7.png)
![Estra-1,3,5(10)-trien-17-one,3-[(aminosulfonyl)oxy]-](http://img.cochemist.com/ccimg/148700/148672-09-7_b.png)


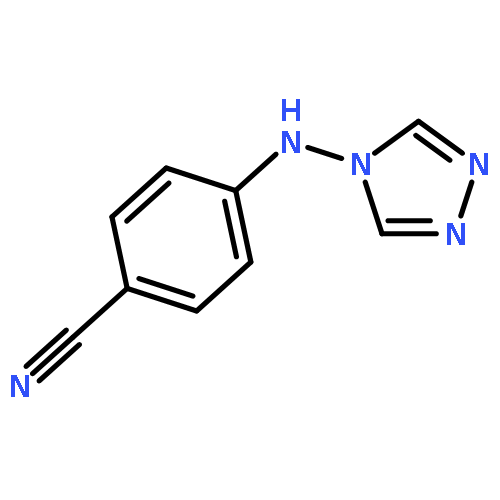
![Benzonitrile,4-[[(4-bromophenyl)methyl]-4H-1,2,4-triazol-4-ylamino]-](http://img.cochemist.com/ccimg/148900/148869-05-0.png)
![Benzonitrile,4-[[(4-bromophenyl)methyl]-4H-1,2,4-triazol-4-ylamino]-](http://img.cochemist.com/ccimg/148900/148869-05-0_b.png)











![Sulfamic acid,6,7,8,9,10,11-hexahydro-6-oxobenzo[b]cyclohepta[d]pyran-3-yl ester](http://img.cochemist.com/ccimg/288700/288628-05-7.png)
![Sulfamic acid,6,7,8,9,10,11-hexahydro-6-oxobenzo[b]cyclohepta[d]pyran-3-yl ester](http://img.cochemist.com/ccimg/288700/288628-05-7_b.png)







![Indeno[1,2-b]indole, 5,10-dihydro-](/data/chemimg/529900/3254-91-9.png)
![Indeno[1,2-b]indole, 5,10-dihydro-](/data/chemimg/529900/3254-91-9_b.png)




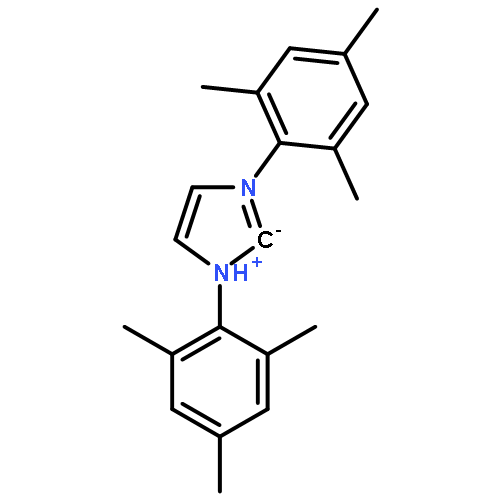
![2H-Imidazol-2-ylidene,1,3-dihydro-1,3-bis(tricyclo[3.3.1.13,7]dec-1-yl)-](http://img.cochemist.com/ccimg/131100/131042-77-8.png)
![2H-Imidazol-2-ylidene,1,3-dihydro-1,3-bis(tricyclo[3.3.1.13,7]dec-1-yl)-](http://img.cochemist.com/ccimg/131100/131042-77-8_b.png)









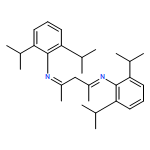
![Benzenamine, N-[3-[[2,6-bis(1-methylethyl)phenyl]amino]-1-methyl-2-buten-1-ylidene]-2,6-bis(1-methylethyl)-](/data/chemimg/120200/181708-81-6.png)
![Benzenamine, N-[3-[[2,6-bis(1-methylethyl)phenyl]amino]-1-methyl-2-buten-1-ylidene]-2,6-bis(1-methylethyl)-](/data/chemimg/120200/181708-81-6_b.png)





![Phenol, 2-[[[2,6-bis(1-methylethyl)phenyl]imino]methyl]-](http://img.cochemist.com/ccimg/187700/187605-57-8.png)
![Phenol, 2-[[[2,6-bis(1-methylethyl)phenyl]imino]methyl]-](http://img.cochemist.com/ccimg/187700/187605-57-8_b.png)