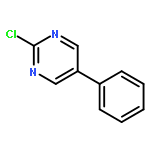
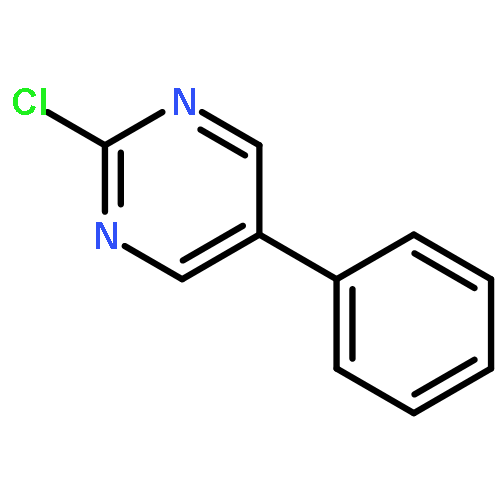
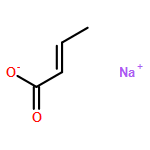
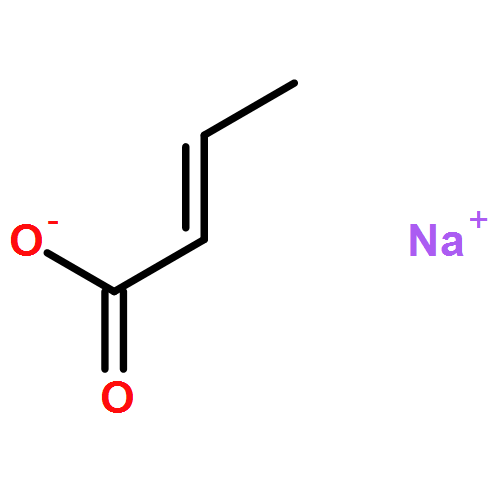
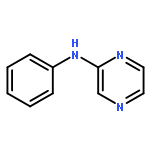
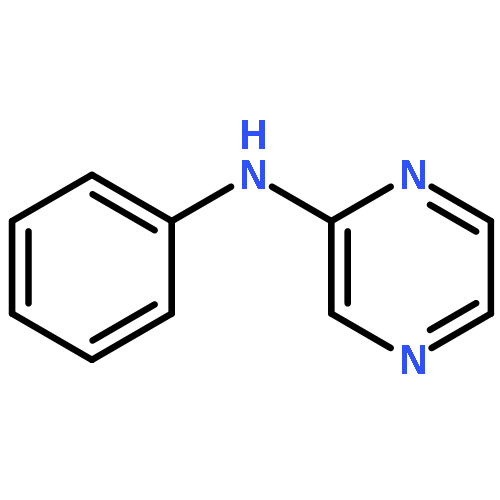
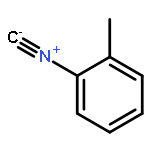
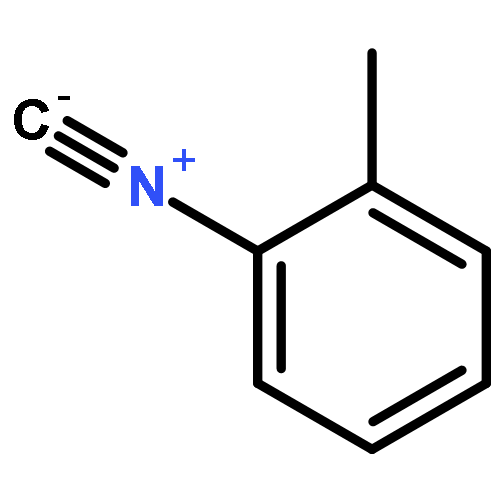
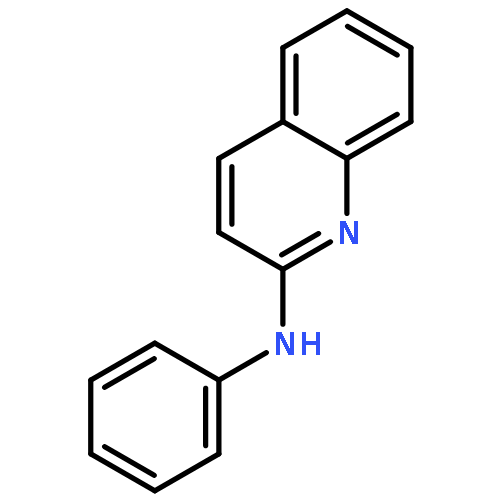
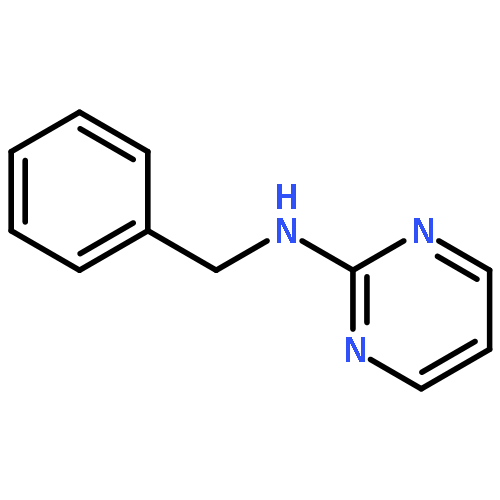
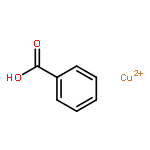
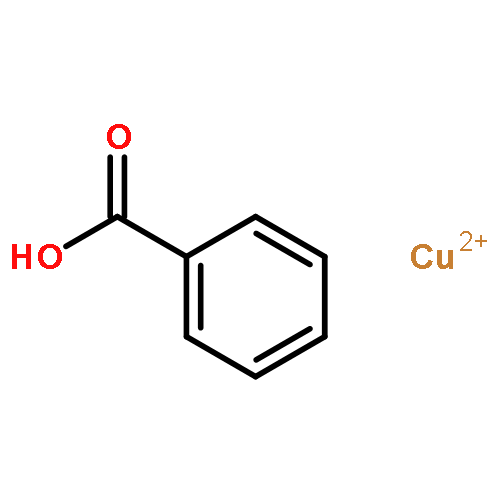
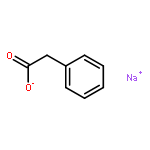
The addition reaction mechanism of OH radical with guanine-cytosine (G.C) base pair has been explored at the B3LYP/DZP++ level of density functional theory (DFT). Structures perturbations along the hydroxylation of G.C base pair cause strain in the pairing and double-strand breaks in DNA. Seven possible hydroxylation reactions are exothermic, and the reaction energy decreases in the order of G.CC4 > GC5.C > GC2.C > GC4.C > G.CC5 > G.CC6 > GC8.C. The hydroxylation reactions at G.CC5 and GC8.C sites appear to be barrierless, and the sequence of the barrier energy is G.CC4 > GC4.C > GC2.C > GC5.C > G.CC6 > G.CC5 ~ GC8.C. The results indicate that hydroxylation at GC8.C, G.CC5 and G.CC6 are more thermodynamically and kinetically favorable than other sites in G.C base pair. Considering the solvent effects by using the polarizable continuum model, the stabilities of all the compounds are increased significantly. Little change is taken place on the data of the reaction energies and barrier energies. Their sequences and the stability order follow the same trends like them in gas phase. The fluctuation of natural bond orbital charge further confirms that the hydroxylation reactions are exothermic. And transient spectra computed with the time-dependent density functional theory (TD-DFT) match well with the previous experimental and theoretical reports. Our deduced mechanism is in good agreement with the experimentally observed hydroxylated adducts. Copyright © 2015 John Wiley & Sons, Ltd.
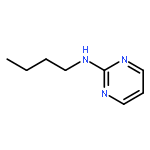
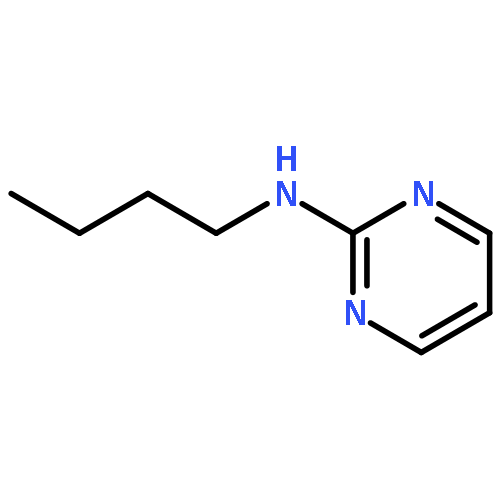
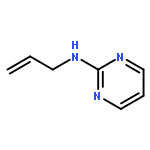
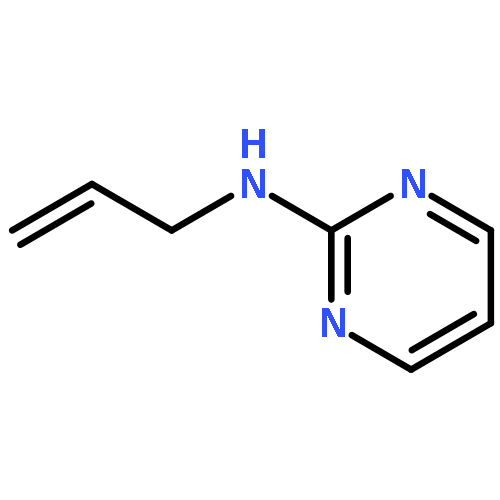
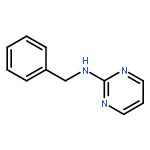
The hydrogen abstraction and addition reactions of OH radical with A·U base pair have been explored by using density functional theory (DFT) both in gas phase and in aqueous solution. Solvent effects were taken into consideration by using the polarized continuum model. All the reaction pathways are exothermic in energy, and the compounds in aqueous phase are more favorable than those in gas phase. The relative free energies of adducts in the addition reaction are lower than those obtained for products in hydrogen abstraction reaction. Among dehydrogenation reaction, the hydrogen abstractions from AC2·U and AN6·U sites are more favorable than those from AC8·U, A·UC5, and A·UC6 sites. In addition, hydroxylation at AC8·U, A·UC5, and A·UC6 sites are more probable than other investigated positions. The hydroxylation at AH8·U site is most favorable, and hydroxylation at A·UC5 site is more preference than that at A·UC6 site controlled by the kinetics factors. The data in both gas phase and water solution demonstrated that addition of OH radical to A·UC5 and A·UC6 sites are more thermodynamically and kinetically favorable than abstracting the hydrogen atom form A·UC5 and A·UC6 sites. The phenomena are in agreement with the experimental observations. Copyright © 2015 John Wiley & Sons, Ltd.


![3,3'-(5'-(3-(Pyridin-3-yl)phenyl)-[1,1':3',1''-terphenyl]-3,3''-diyl)dipyridine](http://img.cochemist.com/ccimg/921300/921205-03-0.png)
![3,3'-(5'-(3-(Pyridin-3-yl)phenyl)-[1,1':3',1''-terphenyl]-3,3''-diyl)dipyridine](http://img.cochemist.com/ccimg/921300/921205-03-0_b.png)


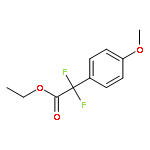
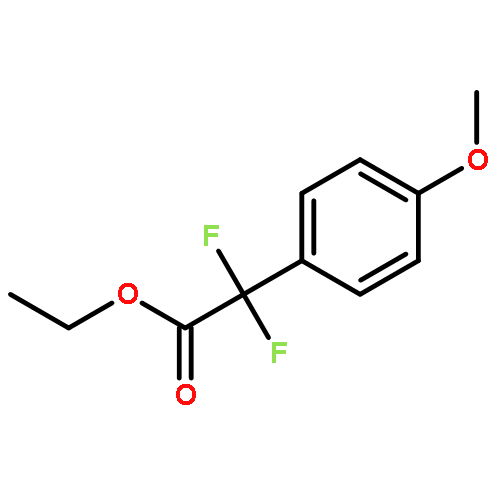


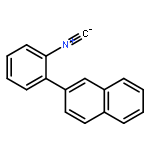
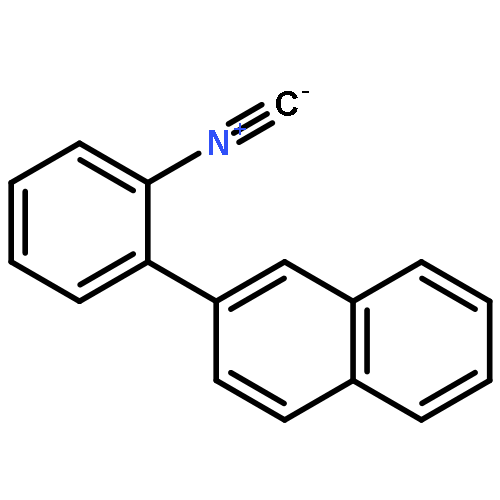




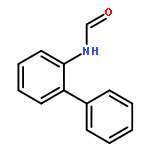
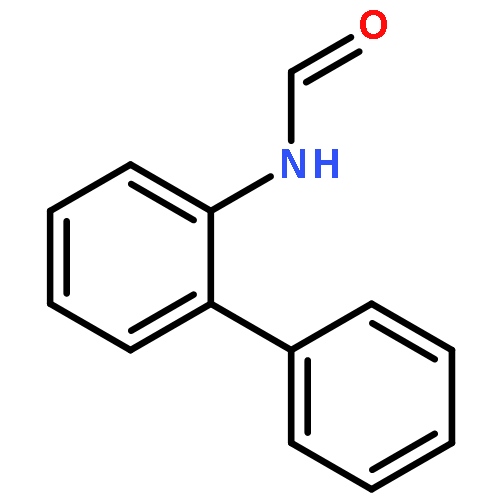
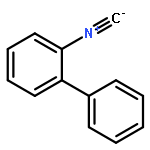
![5H-Pyrido[3,2-b]indole](http://img.cochemist.com/ccimg/300/245-08-9.png)
![5H-Pyrido[3,2-b]indole](http://img.cochemist.com/ccimg/300/245-08-9_b.png)
![1,3-dihydro-1,1,3,3-tetramethylnaphth[2,3-c][1,2,5]oxadisilole](http://img.cochemist.com/ccimg/443000/442912-20-1.png)
![1,3-dihydro-1,1,3,3-tetramethylnaphth[2,3-c][1,2,5]oxadisilole](http://img.cochemist.com/ccimg/443000/442912-20-1_b.png)