Co-reporter:Mingliang Wang, Yanjia Fang, Shoulai Gu, Fangfang Chen, Zhengjiang Zhu, Xun Sun, Jidong Zhu
European Journal of Medicinal Chemistry 2017 Volume 132(Volume 132) pp:
Publication Date(Web):26 May 2017
DOI:10.1016/j.ejmech.2017.03.037
•Novel potent and selective CYP17 inhibitors have been discovered.•Compound 9c showed more potent inhibitory activity than abiraterone for rat and human CYP17.•Compound 9c reduced testosterone production in both NCI-H295R cells and male rat.The inhibition of CYP17 to block androgen biosynthesis is a well validated strategy for the treatment of prostate cancer. Herein we reported the design, synthesis and structure–activity relationship (SAR) study for a series of novel 1,2,3,4- tetrahydrobenzo[4,5]thieno[2,3-c]pyridine derivatives. Some analogs demonstrated a potent inhibition to both rat and human CYP17 protein and reduced testosterone production in human H295R cell line. Some analogs also showed high selectivity against other CYP enzymes such as 3A4, 1A2, 2C9, 2C19 and 2D6, which may limit side effects due to drug-drug interactions. Among these analogs, the most potent compound 9c showed 1.5 fold more potent against rat and human CYP17 protein than that of abiraterone (IC50 = 16 nM and 20 nM vs. 25 nM and 36 nM respectively). In NCI-H295R cells, the inhibitory effect of compound 9c on testosterone production (52± 2%) was also more potent than that of abiraterone (74± 15%) at the concentration of 1 μM. Further, it was shown that 9c reduced plasma testosterone level in a dose-dependent manner in Sprague-Dawley rats. Thus, analog 9c maybe a potential agent used for the treatment of prostate cancer.Download high-res image (152KB)Download full-size image
Co-reporter:Mei-Lin Tang, Lu Zhou, Jun Chang, Zhuo-Han Hu, Yan Qin, Xun Sun
European Journal of Medicinal Chemistry 2016 Volume 113() pp:81-91
Publication Date(Web):4 May 2016
DOI:10.1016/j.ejmech.2016.02.007
•The C–F for C–H replacement at the C3′ group of docetaxel blocks its metabolism.•The major metabolism site shifted from the C3′ group of docetaxel to the baccatin moiety of 3FDT.•The main P450 switched from CYP3A for docetaxel to CYP3A and CYP2E1 for 3FDT.3FDT, an analog of docetaxel with a blocked metabolism at its 3′-N-tert-butyloxyl group with three fluorine atoms, exhibits more potent cytotoxicity than docetaxel both with human cancer cell line SK-OV-3 in vitro and with human non-small cell lung cancer A549 xenografts in vivo. To further develop pharmacodynamically and pharmacokinetically favorable fluorinated docetaxel analogs as anticancer agents, we chose 3FDT as the model compound to identify the metabolites of 3FDT in RLMs, rats, and HLMs and the cytochrome P450 enzymes responsible for the metabolism of 3FDT. Our findings indicated that the major metabolic site switched from the C3′ appendage for docetaxel to the taxane ring for 3FDT, and the main metabolizing P450 enzymes switched from CYP3A to CYP3A4 and CYP2E1.Compared with docetaxel, the major oxidation site of 3FDT shifted from the C3′-N-tert-butyloxyl group to the baccatin moiety and the main metabolizing P450 shifted from CYP3A to CYP3A and CYP2E1.
Co-reporter:Mei-Lin Tang, Chen Zhong, Zheng-Yu Liu, Peng Peng, Xin-Hua Liu, Xun Sun
European Journal of Medicinal Chemistry 2016 Volume 113() pp:63-74
Publication Date(Web):4 May 2016
DOI:10.1016/j.ejmech.2016.02.021
•Compound 11k was more effective than resveratrol.•(±)-11k, (+)-11k and (−)-11k, do not have impacts on activity and toxicity.•(±)-11k could suppress iNOS, COX-2 expression and NO production.•The mechanism of (±)-11k was through TLR4/JNK/NF-κB signaling pathway.To develop novel anti-inflammatory agents with improved pharmaceutical profiles, twenty-eight novel sesquistilbene indanone analogues were synthesized and evaluated for anti-inflammatory activity using RAW264.7 cells. Among these compounds, compound 11k was found to be one of the most potent analogues in inhibiting NO production in LPS-stimulated RAW264.7 cells. Furthermore, it could also significantly suppress LPS-induced iNOS and COX-2 expression and NO production through TLR4/JNK/NF-κB signaling pathway in a concentration dependent manner.Sesquistilbene indanone analogue 11k was much more potent than resveratrol for its anti-inflammatory activity. It could also significantly suppress LPS-induced iNOS and COX-2 expression and NO production through TLR4/JNK/NF-κB signaling pathway in a concentration dependent manner.
Co-reporter:Mingliang Wang, Yimin Hu, Zhe Jiang, Hong C. Shen and Xun Sun
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 18) pp:4239-4246
Publication Date(Web):04 Apr 2016
DOI:10.1039/C6OB00392C
Convenient methods were developed for copper-mediated oxidative C–H activation of aminoquinoline benzamides. The reaction conditions can be tuned to give either hydroxylation or dimerization compounds as the major products efficiently. Preliminary mechanistic studies suggested that different coordination states of copper may lead to different reaction outcomes.
Co-reporter:Jiao Song, Peng Peng, Jun Chang, Ming-Ming Liu, Jian-Ming Yu, Lu Zhou, Xun Sun
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 9) pp:2174-2178
Publication Date(Web):1 May 2016
DOI:10.1016/j.bmcl.2016.03.064
Novel Ilomastat analogs with substituted benzamide groups, instead of hydroxamic acid groups, were designed, synthesized and evaluated against MMP-2 and MMP-9. Among these analogs, the most potent compound 10a exhibited potent inhibitory activity against MMP-2 with IC50 value of 0.19 nM, which is 5 times more potent than that of Ilomastat (IC50 = 0.94 nM). Importantly, 10a exhibited more than 8300 fold selectivity for MMP-2 versus MMP-9 (IC50 = 1.58 μM). Molecular docking studies showed that 10a bond to the catalytic active pocket of MMP-2 by a non-zinc-chelating mechanism which was different from that of Ilomastat. Furthermore, the invasion assay showed that 10a was effective in reducing HEY cells invasion at 84.6% in 50 μM concentration. For 10a, the pharmacokinetic properties had been improved and especially the more desirable t1/2z was achieved compared with these of the lead compound Ilomastat.
Co-reporter:Mingliang Wang, Xixi Liu, Lu Zhou, Jidong Zhu and Xun Sun
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 11) pp:3190-3193
Publication Date(Web):28 Jan 2015
DOI:10.1039/C4OB02691H
An efficient protocol was developed to access 3-fluoro-2-hydroxy-2-substituted benzo[b]furans with Selectfluor™ as the fluorinating reagent in MeCN and water. By utilizing SOCl2/Py as the dehydrating agent, the compounds above were readily converted to 3-fluorinated, 2-substituted benzo[b]furans in high yields.
Co-reporter:Xiao-Dong Hao, Jun Chang, Bo-Yin Qin, Chen Zhong, Zhi-Bo Chu, Jin Huang, Wen-Jiang Zhou, Xun Sun
European Journal of Medicinal Chemistry 2015 Volume 102() pp:26-38
Publication Date(Web):18 September 2015
DOI:10.1016/j.ejmech.2015.07.042
•Three series of novel resveratrol oligomer derivatives were synthesized.•Several compounds (2, 14f, 4, 4d) exhibited potent ERβ agonist activity.•Compound 2 exhibited a potent anti-osteoporosis effect in ovariectomized rats.Three series of resveratrol oligomer derivatives were synthesized, including the indenone-type, indene-type and octahydropentalene-type derivatives, among which ten derivatives were novel compounds. Compounds 2, 14f, and 4d were confirmed as ERβ agonists by yeast two-hybrid assay, and compound 2 (isopaucifloral F) was further chosen to evaluate its anti-osteoporosis activity in vivo. Compared with the sham-operated and the positive control groups, isopaucifloral F (10 μg/kg) showed a notable anti-osteoporosis effect in the ovariectomized (OVX) female rats based on a micro-CT analysis and the following measurements: bone mineral density, bone volume/tissue volume, trabecular thickness, trabecular separation/spacing, and the serum biochemical parameters. LD50 of isopaucifloral F was found to be greater than 5 mg/kg and its effective dose (ED) was found to be about 10 μg/kg. Therefore, isopaucifloral F may be a promising lead compound for the treatment of postmenopausal osteoporosis.
Co-reporter:Jun Chang, Zhi-Bo Chu, Jiao Song, Lin Jin, Xun Sun
Tetrahedron Letters 2015 Volume 56(Issue 1) pp:225-228
Publication Date(Web):1 January 2015
DOI:10.1016/j.tetlet.2014.11.077
Two new isoquinoline alkaloids, decumbensine (1) and decumbendine (2), together with nine known compounds including tetrahydropalmatine (3), bicuculline (4), allocryptopine (5), bulbocapine (6), protopine (7), kikemanine (8), decumbenine B (9), decumbenine C (10), and muramine (11) were isolated from the seedling of Corydalis decumbens (Thunb.) Pers. (Papaveraceae). Their structures were decisively elucidated by spectroscopic analysis including 1D and 2D NMR techniques. Furthermore, the biogenetic pathways of two new compounds were proposed, which would provide valuable information for further understanding of the isoquinoline alkaloids of C. decumbens.
Co-reporter:Ge Xu ; Binhua Lv ; Jacques Y. Roberge ; Baihua Xu ; Jiyan Du ; Jiajia Dong ; Yuanwei Chen ; Kun Peng ; Lili Zhang ; Xinxing Tang ; Yan Feng ; Min Xu ; Wei Fu ; Wenbin Zhang ; Liangcheng Zhu ; Zhongping Deng ; Zelin Sheng ; Ajith Welihinda
Journal of Medicinal Chemistry 2014 Volume 57(Issue 4) pp:1236-1251
Publication Date(Web):January 23, 2014
DOI:10.1021/jm401780b
SGLT2 inhibitors deuterated at sites susceptible to oxidative metabolism were found to have a slightly longer tmax and half-life (t1/2), dose-dependent increase in urinary glucose excretion (UGE) in rats, and slightly superior effects on UGE in dogs while retaining similar in vitro inhibitory activities against hSGLT2. In particular, deuterated compound 41 has the potential to be a robust long-acting antidiabetic agent.
Co-reporter:Chen Zhong, Xin-Hua Liu, Xiao-Dong Hao, Jun Chang, Xun Sun
European Journal of Medicinal Chemistry 2013 Volume 62() pp:187-198
Publication Date(Web):April 2013
DOI:10.1016/j.ejmech.2012.12.037
Three types of resveratrol analogues were designed, synthesized and evaluated in vitro against paraquat-induced apoptosis in SH-SY5Y cells. The results showed that some compounds, especially those containing an indene core, exhibit good activities. Analogue 3′a showed a potent neuroprotective effect at a low concentration (10 μM). Further investigation showed that compound 3′a could attenuate paraquat-induced nuclear morphological changes, significantly decrease paraquat-induced ROS (reactive oxygen species) generation in SH-SY5Y cells and elevate the expression of SOD (Superoxide Dismutase) and GPx (glutathione peroxidase) in a dose-dependent manner. Furthermore, analogue 3′a could decrease Bax protein levels in a concentration-dependent manner and increase Bcl-2 protein expression, which was accompanied by increasing chances of cell survival.Graphical abstractThree types of novel neuroprotective agents were designed, synthesized and evaluated in vitro in human neuronal SH-SY5Y cells.Highlights► Three types of novel neuroprotective agents were designed and synthesized. ► Analogues exhibited activities in vitro against paraquat-induced apoptosis. ► The most potent compound 3′a was chosen for further investigation.
Co-reporter:Chen Zhang;Hongrui Liu;Qing Yang;Jun Chang
Chinese Journal of Chemistry 2013 Volume 31( Issue 1) pp:154-158
Publication Date(Web):
DOI:10.1002/cjoc.201201168
Abstract
A series of new platinum(II) complexes with C2-asymmetric and C2-symmetric 1,2-diamines were designed and synthesized by convenient methods, involving samarium diiodide induced reductive coupling as the key step. The results of cytotoxicity showed that compounds (R,R)-11a and (S,S)-11a, two novel platinum(II) complexes with asymmetric 1,2-diamines, exhibited more potent cytotoxicity than that of oxaliplatin against all leukemia cell lines. Interestingly, (R,R)-11a and (S,S)-11a demonstrated less potent activity against three solid cancer cell lines than that of oxaliplatin, which indicated that these two compounds may only selectively inhibit the leukemia cell lines. In contrast, (R,R)-15a and (S,S)-15a, two platinum(II) complexes with symmetric 1,2-diamines, showed similar cytotoxicity to that of oxaliplatin against all leukemia cell lines and more potent activity against solid cancer cell lines. Further flow cytometry data indicated that (R,R)-11a could obviously arrest leukemia K562 cells in G2/M phases.
Co-reporter:Zhao-Lin Que, Wen-Jiang Zhou, Jun Chang, Xin-Hua Liu, Jian-Ming Yu, Xun Sun
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 6) pp:1793-1796
Publication Date(Web):15 March 2013
DOI:10.1016/j.bmcl.2013.01.038
A series of mercaptoethylleonurine and mercaptoethylguanidine derivatives were designed and synthesized. Their neuroprotective effects toward H2O2-induced apoptosis were investigated in human SH-SY5Y cells. The results from these studies identified several potent compounds, with compound 8k emerging as the most effective. Further investigation demonstrated that 8k reduced H2O2-induced activation of mitochondrial apoptosis by inhibiting the expression of Bax and elevating the expression of Bcl-2. Moreover, the molecular mechanism underlying the observed neuroprotective effects of 8k was exerted via the Akt and JNK pathways. Compound 8k can be a lead compound for further discovery of neuroprotective medicine.Novel mercaptoethylleonurine and mercaptoethylguanidine derivatives were designed, synthesized, and evaluated for their neuroprotective effects on H2O2-induced apoptosis in human SH-SY5Y cells, and the mechanism of compound 8k was studied.
Co-reporter:Qing Yang, Jun Chang, Jiao Song, Meng-Ting Qian, Jian-Ming Yu, Xun Sun
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 16) pp:4602-4607
Publication Date(Web):15 August 2013
DOI:10.1016/j.bmcl.2013.06.024
Four novel iridium(III) complexes with enantiopure C2-symmetrical vicinal diamine ligands were designed, synthesized, and characterized by FT-IR, NMR, and MS. The cytotoxicities of all of the complexes against the human solid tumor cell lines A2780, A549, KB, and MDA-MB-231 were evaluated. Both R,R-configured complexes (R,R)-5a and (R,R)-5b exhibited more potent or similar activity compared with oxaliplatin, whereas their corresponding (S,S)-isomers (S,S)-5a and (S,S)-5b were found to be mostly inactive. As indicated by the activation of caspase-3, the cleavage of PARP, and the upregulation of p53, the preliminary mechanism studies revealed that the mode of cell death initiated by (R,R)-5a in A2780 cells was predominantly p53-mediated apoptosis. In addition, the structure of (R,R)-5a was unambiguously confirmed through single crystal X-ray structure determination.Iridium(III) complexes with enantiopure C2-symmetrical 1,2-diarylethane-1,2-diamine ligands were designed, synthesized, and biologically evaluated against human solid tumor cell lines.
Co-reporter:Chen Zhong, Xin-Hua Liu, Jun Chang, Jian-Ming Yu, Xun Sun
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 15) pp:4413-4418
Publication Date(Web):1 August 2013
DOI:10.1016/j.bmcl.2013.05.058
Four types of resveratrol dimerized analogues were synthesized and evaluated in vitro on LPS-induced NO production in RAW 264.7 cells. The results showed that several compounds, especially those containing 1,2-diphenyl-2,3-dihydro-1H-indene core (type I), exhibited good inhibitory activities. Among 25 analogues, 12b showed a significant inhibitory activity (49% NO production at 10 μM, IC50 = 3.38 μM). Further study revealed that compound 12b could suppress LPS-induced iNOS expression, NO production, and IL-1β release in a concentration-dependently manner. The mechanism of action (MOA) involved for its anti-inflammatory responses was through signaling pathways of p38 MAPK and JNK1/2, but not ERK1/2.Twenty five (four types) of resveratrol dimerized analogues were synthesized and evaluated in vitro on nitric oxide (NO) production in lipopolysaccharide (LPS)-induced RAW 264.7 cells.
Co-reporter:Jun Chang, Xiao-Dong Hao, Yun-Peng Hao, Hong-Fu Lu, Jian-Ming Yu, Xun Sun
Bioorganic & Medicinal Chemistry Letters 2013 23(24) pp: 6834-6837
Publication Date(Web):
DOI:10.1016/j.bmcl.2013.10.007
Co-reporter:Dr. Jun Chang;Si-Ji Zhang;Yong-Wei Jiang;Dr. Liang Xu;Dr. Jian-Ming Yu; Wen-Jiang Zhou;Dr. Xun Sun
ChemMedChem 2013 Volume 8( Issue 6) pp:976-984
Publication Date(Web):
DOI:10.1002/cmdc.201300011
Abstract
Five novel N-substituted demethylvancomycin derivatives were rationally designed and synthesized by using a structure-based approach. The in vitro antibacterial activities against methicillin-resistant Staphylococcus aureus (MRSA), gentamicin-resistant Enterococcus faecalis (GRE), methicillin-resistant Streptococcus pneumoniae (MRS), and vancomycin-resistant Enterococcus faecalis (VRE) were evaluated. One of the compounds, N-(6-phenylheptyl)demethylvancomycin (12 a), was found to exhibit more potent antibacterial activity than vancomycin and demethylvancomycin. Compound 12 a was also found to be ∼18-fold more efficacious than vancomycin against MRSA; however, the two compounds were found to have similar efficacy against MRS. Furthermore, compound 12 a exhibited a favorable pharmacokinetic profile with a half-life of 5.11±0.52 h, which is longer than that of vancomycin (4.3±1.9 h). These results suggest that 12 a is a promising antibacterial drug candidate for further preclinical evaluation.
Co-reporter:Dr. Jun Chang;Si-Ji Zhang;Yong-Wei Jiang;Dr. Liang Xu;Dr. Jian-Ming Yu; Wen-Jiang Zhou;Dr. Xun Sun
ChemMedChem 2013 Volume 8( Issue 6) pp:
Publication Date(Web):
DOI:10.1002/cmdc.201390021
Co-reporter:Si-Ji Zhang, Qing Yang, Liang Xu, Jun Chang, Xun Sun
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 15) pp:4942-4945
Publication Date(Web):1 August 2012
DOI:10.1016/j.bmcl.2012.06.039
To explore the structure–activity relationships (SAR) of demethylvancomycin (2) and find more effective new chemical entities than known glycopeptides for the treatment of Clostridium difficile (C. difficile), 17 novel N-substituted (N-arylmethylene or -aliphatic substituents) demethylvancomycin derivatives were prepared. These analogues have been evaluated in vitro for their antibacterial activities against C. difficile and Enterococcus faecium (E. faecium). Compounds 5d, 5h, and 5i with N-arylmethylene substituents, structurally similar to Oritavancin, showed more potent antibacterial activity against C. difficile than vancomycin (1) or demethylvancomycin (2). Meanwhile, compound 5k with an undecyl side chain showed the most potent antibacterial activity against E. faecium (vancomycin-resistant strain).Seventeen novel N-substituted demethylvancomycin derivatives were prepared and evaluated in vitro for their antibacterial activities against C. difficile and E. faecium. Compounds 5d, 5h, and 5i showed more potent antibacterial activity against C. difficile, while 5k showed the most potent antibacterial activity against E. faecium.
Co-reporter:Hong-Fu Lu, Cheng Xie, Jun Chang, Guo-Qiang Lin, Xun Sun
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 5) pp:1743-1748
Publication Date(Web):May 2011
DOI:10.1016/j.ejmech.2011.02.027
Three novel fluorinated docetaxel analogs, along with six previous reported, were evaluated for their cytotoxicity against five tumor cell lines. The results indicated that these analogs maintained similar/more potent activity than docetaxel against these tumor cell lines. They were also evaluated for their metabolic stability and pharmacokinetics, which demonstrated that these analogs showed better profiles of metabolic stability and pharmacokinetics than that of docetaxel.Three novel fluorinated docetaxel analogs, along with six previous reported, were evaluated for their cytotoxicity, metabolic stability and pharmacokinetics, which demonstrated that these analogs showed better profiles than docetaxel.Highlights► Three novel fluorinated docetaxel analogs were synthesized. ► These analogs maintained similar/more potent cytotoxicity than docetaxel. ► These analogs also showed better profiles of metabolic stability and pharmacokinetics.
Co-reporter:Chen Zhong, Jun Zhu, Jun Chang, Xun Sun
Tetrahedron Letters 2011 Volume 52(Issue 22) pp:2815-2817
Publication Date(Web):1 June 2011
DOI:10.1016/j.tetlet.2011.03.002
Concise total syntheses of (±)isopaucifloral F, (±)quadrangularin A, and (±)pallidol, starting from commercially available 3,5-dimethoxybenzoic acid, have been achieved by a sequential process. The overall synthetic strategy involves Nazarov cyclization, Ramberg–Backlund olefination, and Friedel–Crafts alkylation.
Co-reporter:Hong-Fu Lu, Xun Sun, Liang Xu, Li-Guang Lou, Guo-Qiang Lin
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 2) pp:482-491
Publication Date(Web):February 2009
DOI:10.1016/j.ejmech.2008.04.004
A series of novel fluorinated docetaxel analogues have been synthesized and evaluated in vitro and in vivo. Incorporated one, two or three fluorine atom(s) either at both meta position on C-2 benzolate and 3′-N-tert-butyloxyl group or only at 3′-N-tert-butyloxyl group has resulted in potent analogues which have comparable or superior in vitro and in vivo cytotoxicity to docetaxel. Among them, compounds 14d and 14e have displayed more potent cytotoxicity than docetaxel both in human cancer cell line SK-OV-3 in vitro and in human non-small cell lung cancer A549 xenografts in vivo. Preliminary data show that compound 14a has reduced acute animal toxicity in mice compared with docetaxel.Novel fluorinated docetaxel analogues have been synthesized and evaluated in vitro and in vivo. Some of them have displayed comparable or superior cytotoxicity to docetaxel and reduced acute animal toxicity in mice compared with docetaxel.
Co-reporter:Xun SUN;Jun ZHU;Chen ZHONG;Ken-Ji IZUMI;Chen ZHANG
Chinese Journal of Chemistry 2007 Volume 25(Issue 12) pp:
Publication Date(Web):12 DEC 2007
DOI:10.1002/cjoc.200790344
A new and convenient synthetic method has been developed for the construction of stilbenes bearing electron-withdrawing group(s) by using benzils and arylmethyldiphenylphosphine oxides via sequences involving Wittig-Horner reaction and a rearrangement in the presence of t-BuOK in toluene under mild conditions. This approach could be readily applied to a facile synthesis of biologically important natural products, resveratrol and its derivatives, such as trimethoxystilbenes 1 (Z) and 2 (E).
Co-reporter:Mingliang Wang, Yimin Hu, Zhe Jiang, Hong C. Shen and Xun Sun
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 18) pp:NaN4246-4246
Publication Date(Web):2016/04/04
DOI:10.1039/C6OB00392C
Convenient methods were developed for copper-mediated oxidative C–H activation of aminoquinoline benzamides. The reaction conditions can be tuned to give either hydroxylation or dimerization compounds as the major products efficiently. Preliminary mechanistic studies suggested that different coordination states of copper may lead to different reaction outcomes.
Co-reporter:Mingliang Wang, Xixi Liu, Lu Zhou, Jidong Zhu and Xun Sun
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 11) pp:NaN3193-3193
Publication Date(Web):2015/01/28
DOI:10.1039/C4OB02691H
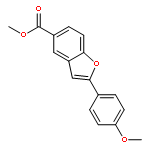
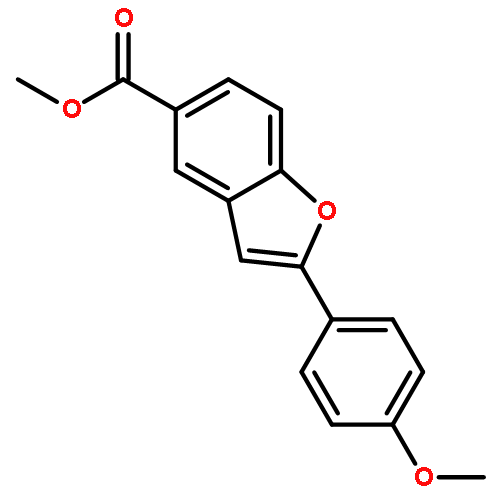
An efficient protocol was developed to access 3-fluoro-2-hydroxy-2-substituted benzo[b]furans with Selectfluor™ as the fluorinating reagent in MeCN and water. By utilizing SOCl2/Py as the dehydrating agent, the compounds above were readily converted to 3-fluorinated, 2-substituted benzo[b]furans in high yields.


![BUTANOIC ACID, 4-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-3-METHYL-, (3S)-](http://img.cochemist.com/ccimg/862400/862387-31-3.png)
![BUTANOIC ACID, 4-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-3-METHYL-, (3S)-](http://img.cochemist.com/ccimg/862400/862387-31-3_b.png)
![L-Isoleucine, N-[[(2R)-1-methyl-2-piperidinyl]carbonyl]-](http://img.cochemist.com/ccimg/645000/644960-70-3.png)
![L-Isoleucine, N-[[(2R)-1-methyl-2-piperidinyl]carbonyl]-](http://img.cochemist.com/ccimg/645000/644960-70-3_b.png)
![Butanal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3-methyl-, (3S)-](http://img.cochemist.com/ccimg/205000/204917-42-0.png)
![Butanal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3-methyl-, (3S)-](http://img.cochemist.com/ccimg/205000/204917-42-0_b.png)





![[(6,7-dichloro-2,3-dihydro-2-methyl-1-oxo-2-phenyl-1H-inden-5-yl)oxy]acetic acid](http://img.cochemist.com/ccimg/56100/56049-88-8.png)
![[(6,7-dichloro-2,3-dihydro-2-methyl-1-oxo-2-phenyl-1H-inden-5-yl)oxy]acetic acid](http://img.cochemist.com/ccimg/56100/56049-88-8_b.png)


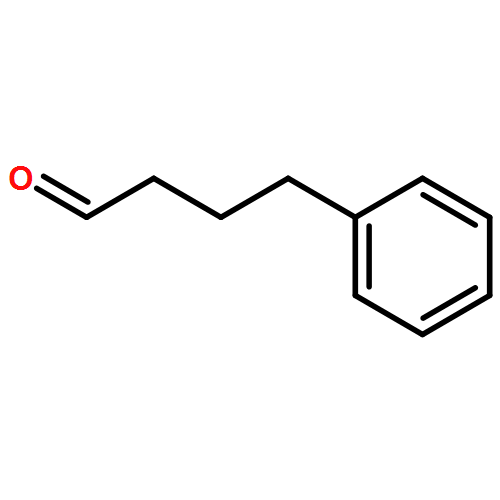
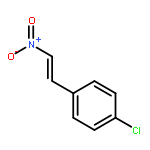
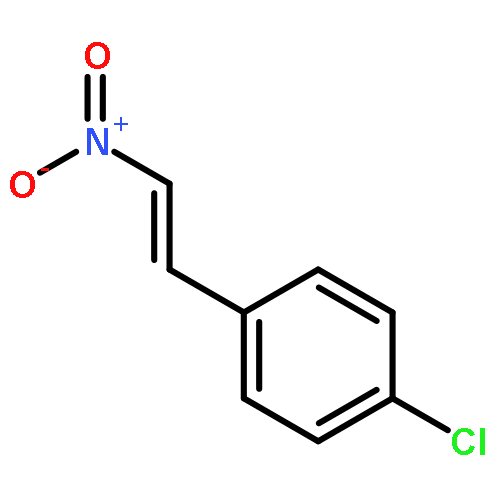




![[2H5]-Dapagliflozin](http://img.cochemist.com/ccimg/1204300/1204219-80-6.png)
![[2H5]-Dapagliflozin](http://img.cochemist.com/ccimg/1204300/1204219-80-6_b.png)
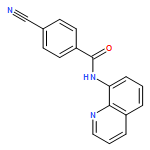
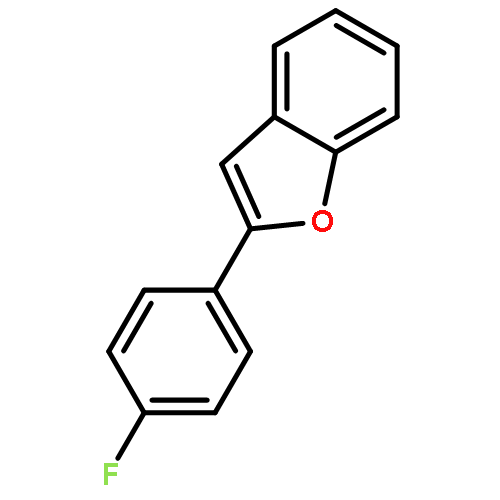
![Benzofuran, 2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/904400/904325-87-7.png)
![Benzofuran, 2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/904400/904325-87-7_b.png)
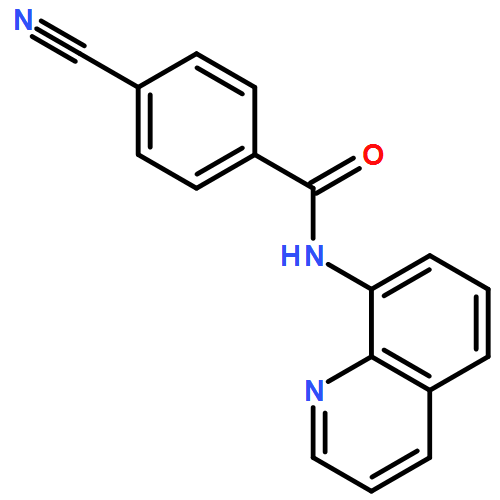
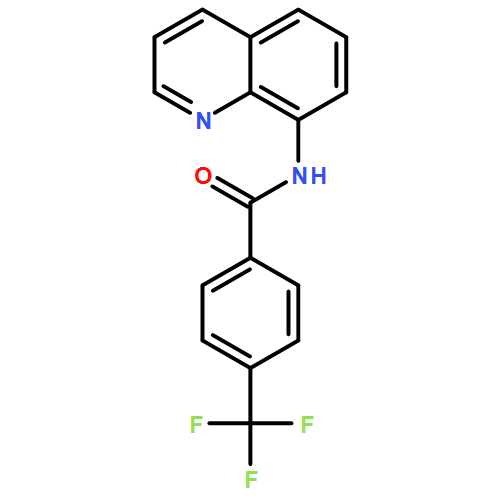
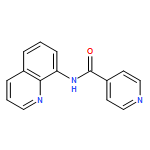
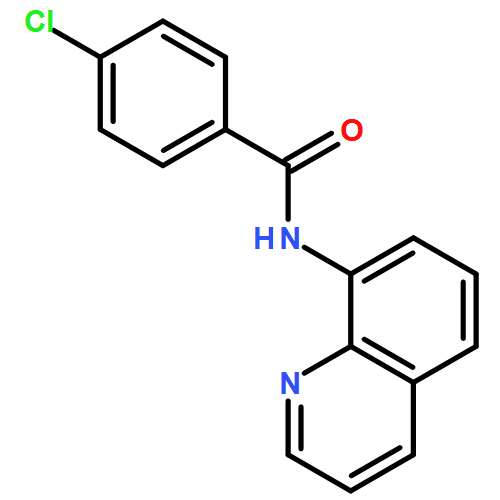
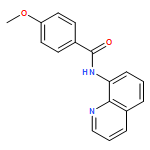
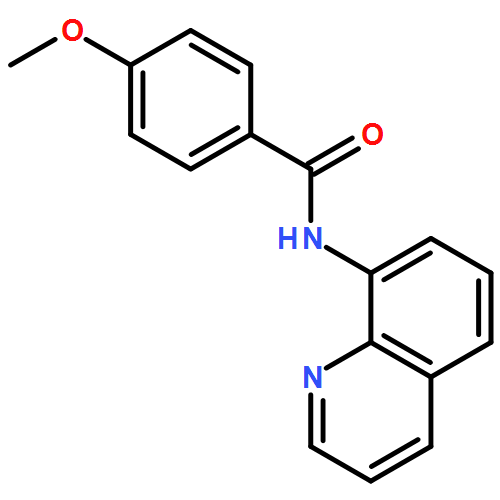
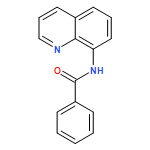
![Benzoic acid, 4-[(8-quinolinylamino)carbonyl]-, methyl ester](/data/chemimg/3735000/878687-85-5.png)
![Benzoic acid, 4-[(8-quinolinylamino)carbonyl]-, methyl ester](/data/chemimg/3735000/878687-85-5_b.png)



![Phenol, 4-[(5-bromo-2-chlorophenyl)methyl]-](/data/chemimg/964500/864070-18-8.png)
![Phenol, 4-[(5-bromo-2-chlorophenyl)methyl]-](/data/chemimg/964500/864070-18-8_b.png)


![Phosphonium, triphenyl[[4-(phenylmethoxy)phenyl]methyl]-, bromide](http://img.cochemist.com/ccimg/383200/383189-06-8.png)
![Phosphonium, triphenyl[[4-(phenylmethoxy)phenyl]methyl]-, bromide](http://img.cochemist.com/ccimg/383200/383189-06-8_b.png)
![Benzene, 4-bromo-1-chloro-2-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/333400/333361-51-6.png)
![Benzene, 4-bromo-1-chloro-2-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/333400/333361-51-6_b.png)










![(1E,2R,3R)-2-(3,5-dihydroxyphenyl)-3-(4-hydroxyphenyl)-1-[(4-hydroxyphenyl)methylidene]-2,3-dihydro-1H-indene-4,6-diol](http://img.cochemist.com/ccimg/252600/252557-25-8.png)
![(1E,2R,3R)-2-(3,5-dihydroxyphenyl)-3-(4-hydroxyphenyl)-1-[(4-hydroxyphenyl)methylidene]-2,3-dihydro-1H-indene-4,6-diol](http://img.cochemist.com/ccimg/252600/252557-25-8_b.png)
![1,3-Benzodioxole-4-methanol, 5-(1,3-dioxolo[4,5-f]isoquinolin-8-yl)-](http://img.cochemist.com/ccimg/165000/164991-68-8.png)
![1,3-Benzodioxole-4-methanol, 5-(1,3-dioxolo[4,5-f]isoquinolin-8-yl)-](http://img.cochemist.com/ccimg/165000/164991-68-8_b.png)








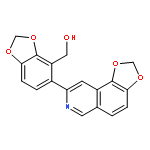
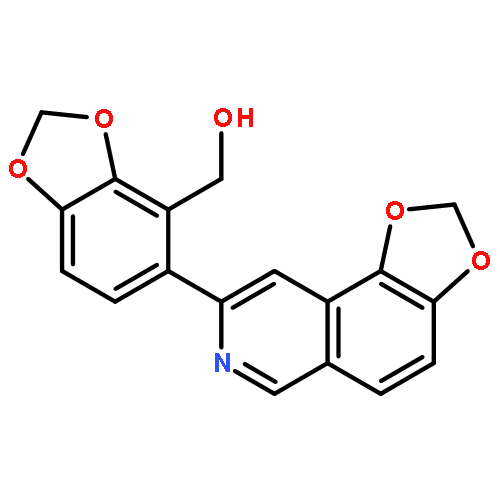

![Indeno[2,1-a]indene-1,3,6,8-tetrol,4b,5,9b,10-tetrahydro-5,10-bis(4-hydroxyphenyl)-, (4bR,5R,9bR,10R)-](http://img.cochemist.com/ccimg/105100/105037-88-5.png)
![Indeno[2,1-a]indene-1,3,6,8-tetrol,4b,5,9b,10-tetrahydro-5,10-bis(4-hydroxyphenyl)-, (4bR,5R,9bR,10R)-](http://img.cochemist.com/ccimg/105100/105037-88-5_b.png)
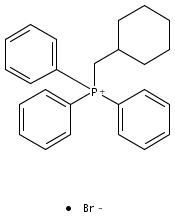
![PHOSPHONIUM, [(4-HYDROXYPHENYL)METHYL]TRIPHENYL-, BROMIDE](http://img.cochemist.com/ccimg/98900/98834-25-4.png)
![PHOSPHONIUM, [(4-HYDROXYPHENYL)METHYL]TRIPHENYL-, BROMIDE](http://img.cochemist.com/ccimg/98900/98834-25-4_b.png)
![Phosphonium, [(3-fluorophenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/89400/89302-81-8.png)
![Phosphonium, [(3-fluorophenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/89400/89302-81-8_b.png)












![TRIPHENYL-[[4-(TRIFLUOROMETHYL)PHENYL]METHYL]PHOSPHANIUM;BROMIDE](http://img.cochemist.com/ccimg/38800/38733-98-1.png)
![TRIPHENYL-[[4-(TRIFLUOROMETHYL)PHENYL]METHYL]PHOSPHANIUM;BROMIDE](http://img.cochemist.com/ccimg/38800/38733-98-1_b.png)




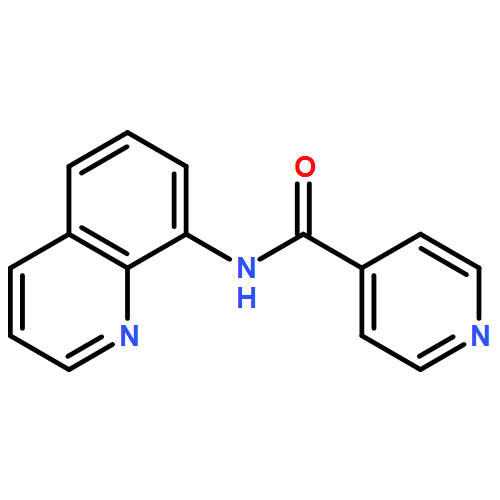
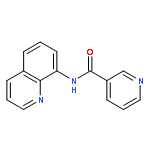
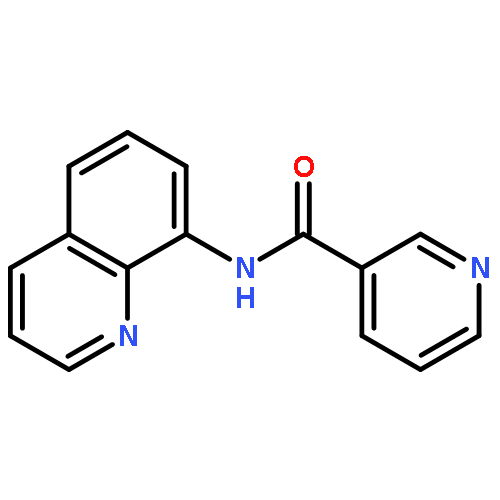
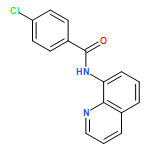













![Phosphine oxide, [(4-methoxyphenyl)methyl]diphenyl-](http://img.cochemist.com/ccimg/16200/16114-90-2.png)
![Phosphine oxide, [(4-methoxyphenyl)methyl]diphenyl-](http://img.cochemist.com/ccimg/16200/16114-90-2_b.png)
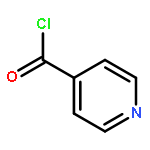
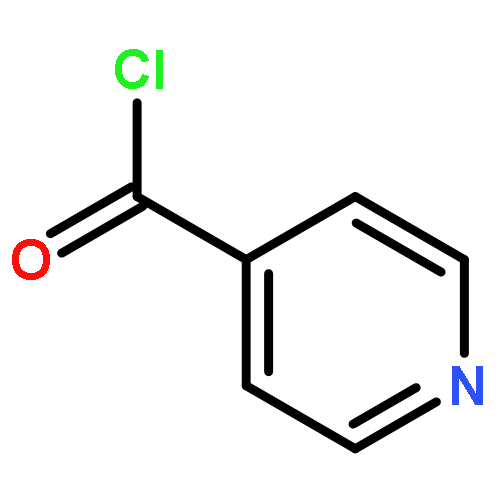





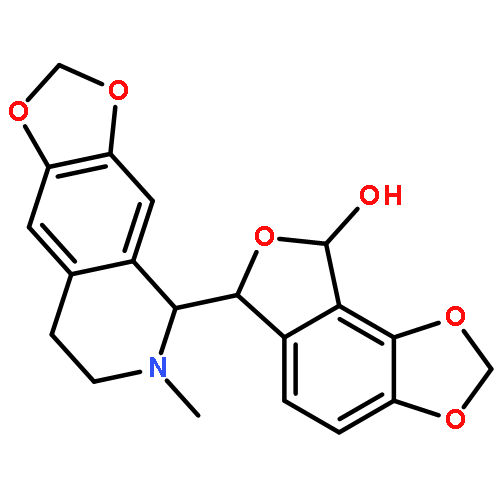
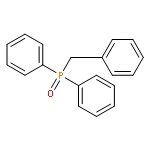

![Dibenz[c,g]azecin-13(6H)-one,5,7,8,14-tetrahydro-3,4,10,11-tetramethoxy-6-methyl-](http://img.cochemist.com/ccimg/2300/2292-20-8.png)
![Dibenz[c,g]azecin-13(6H)-one,5,7,8,14-tetrahydro-3,4,10,11-tetramethoxy-6-methyl-](http://img.cochemist.com/ccimg/2300/2292-20-8_b.png)



![5H-Benzo[g]-1,3-benzodioxolo[6,5,4-de]quinolin-12-ol,6,7,7a,8-tetrahydro-11-methoxy-7-methyl-, (7aS)-](http://img.cochemist.com/ccimg/300/298-45-3.png)
![5H-Benzo[g]-1,3-benzodioxolo[6,5,4-de]quinolin-12-ol,6,7,7a,8-tetrahydro-11-methoxy-7-methyl-, (7aS)-](http://img.cochemist.com/ccimg/300/298-45-3_b.png)