Co-reporter:Feng-Hua Zhang, Bikash Debnath, Zhong-Liang Xu, Liu-Meng Yang, Li-Rui Song, Yong-Tang Zheng, Nouri Neamati, Ya-Qiu Long
European Journal of Medicinal Chemistry 2017 Volume 125() pp:1051-1063
Publication Date(Web):5 January 2017
DOI:10.1016/j.ejmech.2016.10.045
•A new class of selective HIV-1 IN-LEDGF/p75 inhibitors without carboxylic acid/bioisostere are identified through scaffold hopping.•The 3-hydroxypicolinamides function as dual HIV-1 IN inhibitors targeting IN-LEDGF/p75 interaction and IN dimerization with anti-HIV effect.•The binding mode of the novel IN-LEDGF/p75 inhibitors was established through shape-based ROCS pharmacophore model.Currently, three HIV-1 integrase (IN) active site-directed inhibitors are in clinical use for the treatment of HIV infection. However, emergence of drug resistance mutations have limited the promise of a long-term cure. As an alternative, allosteric inhibition of IN activity has drawn great attention and several of such inhibitors are under early stage clinical development. Specifically, inhibitors of IN and the cellular cofactor LEDGF/p75 remarkably diminish proviral integration in cells and deliver a potent reduction in viral replicative capacity. Distinct from the extensively studied 2-(quinolin-3-yl) acetic acid or 1H-indol-3-yl-2-hydroxy-4-oxobut-2-enoic acid chemotypes, this study discloses a new class of selective IN-LEDGF/p75 inhibitors without the carboxylic acid functionality. More significantly, 3-hydroxypicolinamides also show low micromolar inhibition against IN dimerization, providing novel dual IN inhibitors with in vitro therapeutically selective antiviral effect for further development. Finally, our shape-based ROCS pharmacophore model of the 3-hydroxypicolinamide class of compounds provides a new insight into the binding mode of these novel IN-LEDGF/p75 inhibitors.A novel class of HIV-1 integrase (IN) allosteric inhibitors targeting both IN-LEDGF/p75 interaction and IN dimerization were discovered bearing 3-hydroxypicolinamide scaffold, with low micromolar IC50 values and therapeutically selective antiviral effect.
Co-reporter:Gang Liu, Ding Xue, Jun Yang, Juan Wang, Xiaohua Liu, Wenjing Huang, Jie Li, Ya-Qiu Long, Wenfu Tan, and Ao Zhang
Journal of Medicinal Chemistry 2016 Volume 59(Issue 24) pp:11050-11068
Publication Date(Web):October 13, 2016
DOI:10.1021/acs.jmedchem.6b01247
A series of novel Smo antagonists were developed either by directly incorporating the basic skeleton of the natural product artemisinin or by first breaking artemisinin into structurally simpler and stable intermediates and then reconstructing into diversified heterocyclic derivatives, equipped with a Smo-targeting bullet. 2-(2,5-Dimethyl-5,6,7,8-tetrahydroquinolin-8-yl)-N-arylpropanamide 65 was identified as the most potent, with an IC50 value of 9.53 nM against the Hh signaling pathway. Complementary mechanism studies confirmed that 65 inhibits Hh signaling pathway by targeting Smo and shares the same binding site as that of the tool drug cyclopamine. Meanwhile, 65 has a good plasma exposure and an acceptable oral bioavailability. Dose-dependent antiproliferative effects were observed in ptch+/–;p53–/– medulloblastoma cells, and significant tumor growth inhibitions were achieved for 65 in the ptch+/–;p53–/– medulloblastoma allograft model.
Co-reporter:Yu Cheng, Jian Shen, Run-Ze Peng, Gui-Feng Wang, Jian-Ping Zuo, Ya-Qiu Long
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 12) pp:2900-2906
Publication Date(Web):15 June 2016
DOI:10.1016/j.bmcl.2016.04.042
HCV NS5B polymerase is an attractive and validated target for anti-HCV therapy. Starting from our previously identified 2-aryl quinolones as novel non-nucleoside NS5B polymerase inhibitors, structure-based optimization furnished 2-alkyl-N-benzyl quinolones with improved antiviral potency by employing privileged fragment hybridization strategy. The N-(4-chlorobenzyl)-2-(methoxymethyl)quinolone derivative 5f proved to be the best compound of this series, exhibiting a selective sub-micromolar antiviral effect (EC50 = 0.4 μM, SI = 10.8) in Huh7.5.1 cells carrying a HCV genotype 2a. Considering the undesirable pharmacokinetic property of the highly substituted quinolones, a novel chemotype of 1,6-naphthyridine-4,5-diones were evolved via scaffold hopping, affording brand new structure HCV inhibitors with compound 6h (EC50 (gt2a) = 2.5 μM, SI = 7.2) as a promising hit. Molecular modeling studies suggest that both of 2-alkyl quinolones and 1,6-naphthyridine-4,5-diones function as HCV NS5B thumb pocket II inhibitors.Based on the binding mode of quinolones with HCV NS5B, 2-alkyl-N-arylquinolones and 1,6-naphthyridine-4,5-diones were designed and discovered as novel HCV inhibitors with submicromolar cellular replican potency.
Co-reporter:Xiao-De An, Hongyan Liu, Zhong-Liang Xu, Yi Jin, Xia Peng, Ying-Ming Yao, Meiyu Geng, Ya-Qiu Long
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 3) pp:708-716
Publication Date(Web):1 February 2015
DOI:10.1016/j.bmcl.2014.11.070
Starting from our previously identified novel c-Met kinase inhibitors bearing 1H-imidazo[4,5-h][1,6]naphthyridin-2(3H)-one scaffold, a global structural exploration was conducted to furnish an optimal binding motif for further development, directed by the enzyme inhibitory mechanism. First round SAR study picked two imidazonaphthyridinone frameworks with 1,8- and 3,5-disubstitution pattern as class I and class II c-Met kinase inhibitors, respectively. Further structural optimization on type II inhibitors by truncation of the imidazonaphthyridinone core and incorporation of an N-phenyl cyclopropane-1,1-dicarboxamide pharmacophore led to the discovery of novel imidazopyridine-based c-Met kinase inhibitors, displaying nanomolar enzyme inhibitory activity and improved Met kinase selectivity. More significantly, the new chemotype c-Met kinase inhibitors effectively inhibited Met phosphorylation and its downstream signaling as well as the proliferation of Met-dependent EBC-1 human lung cancer cells at submicromolar concentrations.A new class of imidazopyridine-based c-Met kinase inhibitors exhibited nanomolar inhibition against the enzyme and the growth of Met-driven EBC-1 human lung cancer cells.
Co-reporter:Jian-Ping Lin, Feng-Hua Zhang, and Ya-Qiu Long
Organic Letters 2014 Volume 16(Issue 11) pp:2822-2825
Publication Date(Web):May 9, 2014
DOI:10.1021/ol500864r
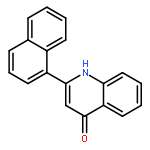
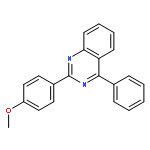
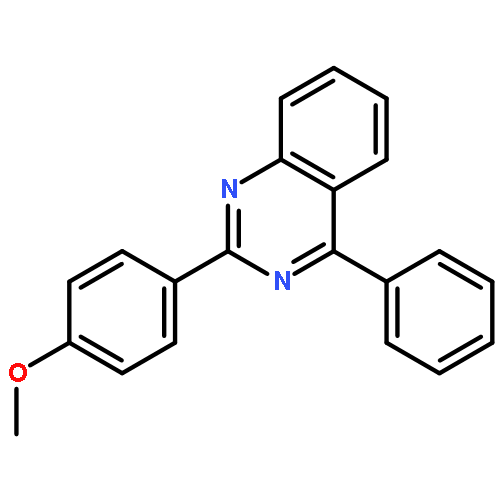
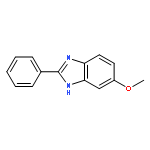
A fast and simple divergent synthesis of multisubstituted quinazolines and benzimidazoles was developed from readily available amidines, via iodine(III)-promoted oxidative C(sp3)–C(sp2) and C(sp2)–N bond formation in nonpolar and polar solvents, respectively. Further selective synthesis of quinazolines in polar solvent was realized by TEMPO-catalyzed sp3C–H/sp2C–H direct coupling of the amidine with K2S2O8 as the oxidant. No metal, base, or other additives were needed.
Co-reporter:Cheng Chen, He Li, Ya-Qiu Long
Bioorganic & Medicinal Chemistry Letters (1 December 2016) Volume 26(Issue 23) pp:5603-5612
Publication Date(Web):1 December 2016
DOI:10.1016/j.bmcl.2016.10.074
GPR40 belongs to the GPCR family and the activation of GPR40 has been shown to induce glucose-stimulated insulin secretion (GSIS) from pancreatic beta cells as well as incretin secretion from intestinal endocrine cells. Therefore, GPR40 has emerged as a viable and promising therapeutic target for type 2 diabetes mellitus (T2DM) without the risk of hypoglycemia. However, the termination of TAK-875 in phase III clinical trials for the hepatotoxicity issue threw doubt over the long-term safety of targeting GPR40. Herein, we summarized the newly disclosed biological characteristics and the druglikeness-based structural evolution of GPR40 agonists to advance the development of GPR40-based anti-diabetic drugs.Figure optionsDownload full-size imageDownload high-quality image (77 K)Download as PowerPoint slide
Co-reporter:Cheng Chen, He Li, Ya-Qiu Long
Bioorganic & Medicinal Chemistry Letters (1 December 2016) Volume 26(Issue 23) pp:5603-5612
Publication Date(Web):1 December 2016
DOI:10.1016/j.bmcl.2016.10.074

![Benzenepropanoic acid, β-cyclopropyl-3-[[2-(5,5-dimethyl-1-cyclopenten-1-yl)-2'-fluoro-5'-methoxy[1,1'-biphenyl]-4-yl]methoxy]-, (βS)-](/data/chemimg/345800/1142214-62-7.png)
![Benzenepropanoic acid, β-cyclopropyl-3-[[2-(5,5-dimethyl-1-cyclopenten-1-yl)-2'-fluoro-5'-methoxy[1,1'-biphenyl]-4-yl]methoxy]-, (βS)-](/data/chemimg/345800/1142214-62-7_b.png)
![Benzenepropanoic acid, β-1-propyn-1-yl-4-[[4'-(trifluoromethyl)[1,1'-biphenyl]-3-yl]methoxy]-, (βS)-](/data/chemimg/1015800/865231-46-5.png)
![Benzenepropanoic acid, β-1-propyn-1-yl-4-[[4'-(trifluoromethyl)[1,1'-biphenyl]-3-yl]methoxy]-, (βS)-](/data/chemimg/1015800/865231-46-5_b.png)




![2-PYRIDINECARBOXAMIDE, N-[(4-FLUOROPHENYL)METHYL]-](http://img.cochemist.com/ccimg/803700/803694-10-2.png)
![2-PYRIDINECARBOXAMIDE, N-[(4-FLUOROPHENYL)METHYL]-](http://img.cochemist.com/ccimg/803700/803694-10-2_b.png)
![2-PYRIDINECARBOXAMIDE, 3-HYDROXY-N-[[4-(TRIFLUOROMETHYL)PHENYL]METHYL]-](http://img.cochemist.com/ccimg/628800/628702-04-5.png)
![2-PYRIDINECARBOXAMIDE, 3-HYDROXY-N-[[4-(TRIFLUOROMETHYL)PHENYL]METHYL]-](http://img.cochemist.com/ccimg/628800/628702-04-5_b.png)
![2-PYRIDINECARBOXAMIDE, N-[(4-FLUOROPHENYL)METHYL]-3-HYDROXY-](http://img.cochemist.com/ccimg/628800/628702-02-3.png)
![2-PYRIDINECARBOXAMIDE, N-[(4-FLUOROPHENYL)METHYL]-3-HYDROXY-](http://img.cochemist.com/ccimg/628800/628702-02-3_b.png)






![Spiro[indene-1,4'-piperidine]](http://img.cochemist.com/ccimg/33100/33042-66-9.png)
![Spiro[indene-1,4'-piperidine]](http://img.cochemist.com/ccimg/33100/33042-66-9_b.png)










![2-Pyridinecarboxamide, 3-hydroxy-N-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/1100/1032-85-5.png)
![2-Pyridinecarboxamide, 3-hydroxy-N-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/1100/1032-85-5_b.png)
































![Ethanone, 1-[2-[(phenylmethyl)amino]phenyl]-](http://img.cochemist.com/ccimg/104100/104014-46-2.png)
![Ethanone, 1-[2-[(phenylmethyl)amino]phenyl]-](http://img.cochemist.com/ccimg/104100/104014-46-2_b.png)








![BENZENECARBOXIMIDOYL CHLORIDE, N-[4-(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/80900/80819-62-1.png)
![BENZENECARBOXIMIDOYL CHLORIDE, N-[4-(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/80900/80819-62-1_b.png)
![ETHANONE, 1-[2-(2-PROPENYLAMINO)PHENYL]-](http://img.cochemist.com/ccimg/80300/80217-67-0.png)
![ETHANONE, 1-[2-(2-PROPENYLAMINO)PHENYL]-](http://img.cochemist.com/ccimg/80300/80217-67-0_b.png)




![Methanone, [3-amino-4-[(phenylmethyl)amino]phenyl]phenyl-](http://img.cochemist.com/ccimg/65400/65303-90-4.png)
![Methanone, [3-amino-4-[(phenylmethyl)amino]phenyl]phenyl-](http://img.cochemist.com/ccimg/65400/65303-90-4_b.png)
![1H-Benzimidazole, 2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/58800/58758-09-1.png)
![1H-Benzimidazole, 2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/58800/58758-09-1_b.png)

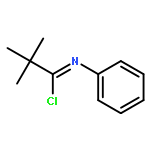
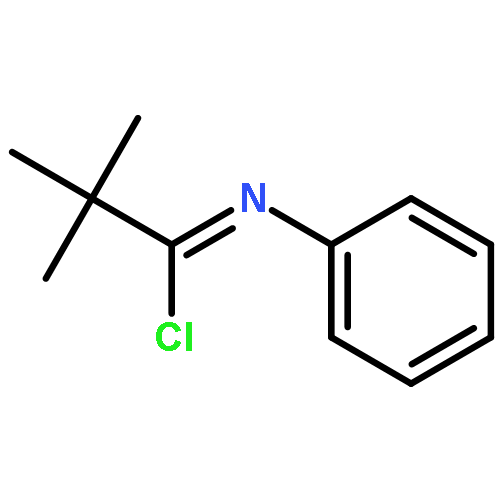








![2-{[2-(4-FLUOROPHENYL)-8-METHYL-4-QUINAZOLINYL]SULFANYL}-N-PROPYL<WBR />ACETAMIDE](http://img.cochemist.com/ccimg/37000/36947-70-3.png)
![2-{[2-(4-FLUOROPHENYL)-8-METHYL-4-QUINAZOLINYL]SULFANYL}-N-PROPYL<WBR />ACETAMIDE](http://img.cochemist.com/ccimg/37000/36947-70-3_b.png)


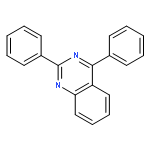
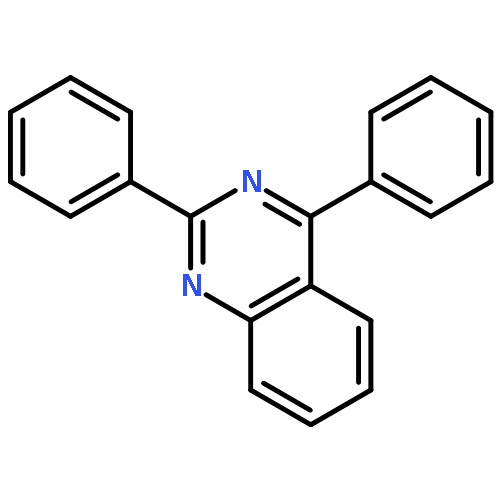
![1H-Naphth[2,3-d]imidazole, 2-phenyl-](http://img.cochemist.com/ccimg/30400/30384-79-3.png)
![1H-Naphth[2,3-d]imidazole, 2-phenyl-](http://img.cochemist.com/ccimg/30400/30384-79-3_b.png)












![1,2-Benzenediamine, N-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/5800/5729-18-0.png)
![1,2-Benzenediamine, N-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/5800/5729-18-0_b.png)













![1,2-Diphenyl-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/2700/2622-67-5.png)
![1,2-Diphenyl-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/2700/2622-67-5_b.png)







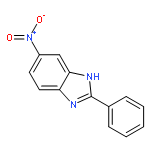
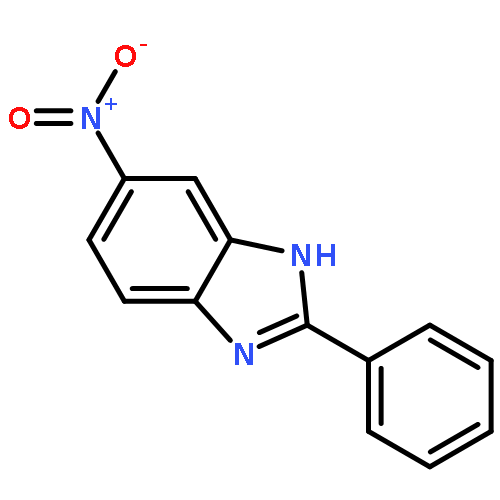


![N-[2-(CYCLOPENTYLAMINO)-2-OXOETHYL]-N-(4-FLUOROBENZYL)-1,2,3-THIA<WBR />DIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/800/739-88-8.png)
![N-[2-(CYCLOPENTYLAMINO)-2-OXOETHYL]-N-(4-FLUOROBENZYL)-1,2,3-THIA<WBR />DIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/800/739-88-8_b.png)
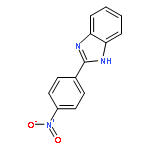
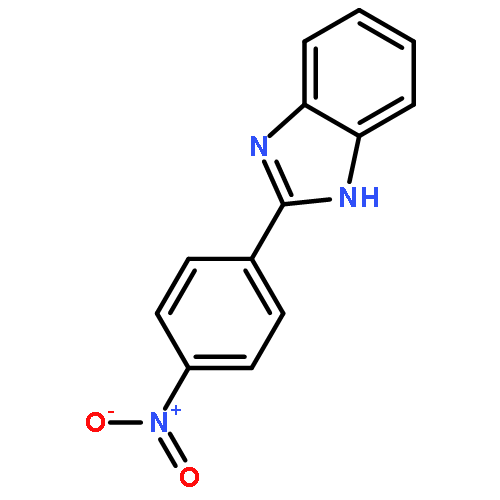
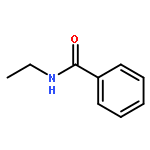

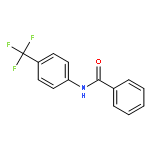

![11H-Isoindolo[2,1-a]benzimidazole](http://img.cochemist.com/ccimg/300/248-72-6.png)
![11H-Isoindolo[2,1-a]benzimidazole](http://img.cochemist.com/ccimg/300/248-72-6_b.png)