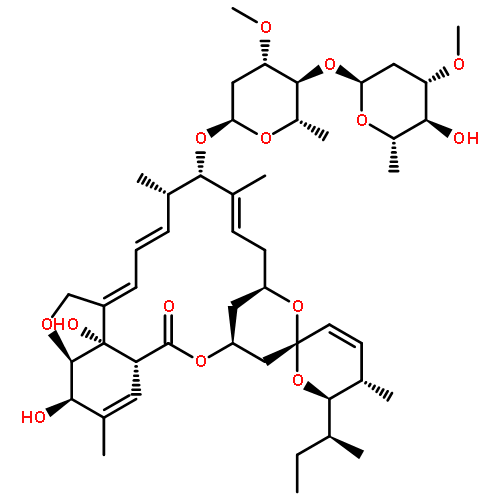
The total synthesis of 7,10-epimer of the proposed structure of amphidinolide N was accomplished. The requisite chiral C17–C29 subunit was assembled stereoselectively via Keck allylation, Shi epoxidation, diastereoselective 1,3-reduction, and a later oxidative synthesis of the THF framework. The C1–C13 and C17–C29 subunits were successfully coupled using a Enders RAMP “linchpin” as the C14–C16 three carbon unit, thereby controlling the chirality at C14 and C16. The labile allyl epoxy moiety was successfully constructed by Grieco–Nishizawa olefination at a final stage of the synthesis.
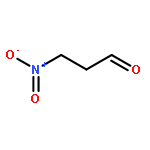
Recently, we developed a direct method to oxidatively convert primary nitroalkanes into amides that entailed mixing an iodonium source with an amine, base, and oxygen. Herein, we systematically investigated the mechanism and likely intermediates of such methods. We conclude that an amine–iodonium complex first forms through N−halogen bonding. This complex reacts with aci-nitronates to give both α-iodo- and α,α-diiodonitroalkanes, which can act as alternative sources of electrophilic iodine and also generate an extra equimolar amount of I+ under O2. In particular, evidence supports α,α-diiodonitroalkane intermediates reacting with molecular oxygen to form a peroxy adduct; alternatively, these tetrahedral intermediates rearrange anaerobically to form a cleavable nitrite ester. In either case, activated esters are proposed to form that eventually reacts with nucleophilic amines in a traditional fashion.
Organocatalyzed Michael, Mannich, and aldol reactions of aldehydes or ketones, as nucleophiles, have triggered several discussions regarding their reaction mechanism. H218O has been utilized to determine if the reaction proceeds through an enamine or enol mechanism by monitoring the ratio of 18O incorporated into the final product. In this communication, we describe the risk of H218O as an evaluation tool for this mechanistic investigation. We have demonstrated that exchange of 16O/18O occurs in the aldehyde or ketone starting material, caused by the presence of H218O and amine catalysts, before the Michael, Mannich, and aldol reactions proceed. Because the newly generated 18O starting aldehydes or ketones and 16O water affect the incorporation ratio of 18O in the final product, the use of H218O would not be appropriate to distinguish the mechanism of these organocatalyzed reactions.
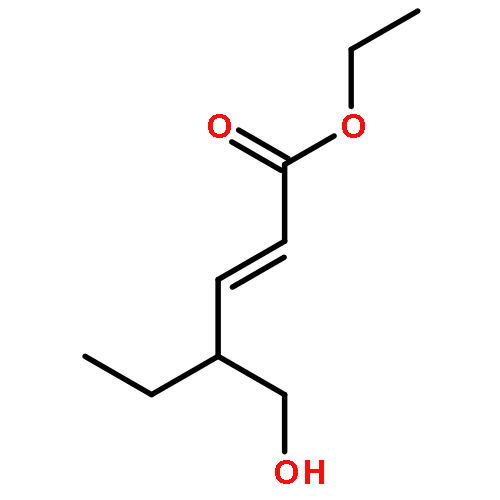
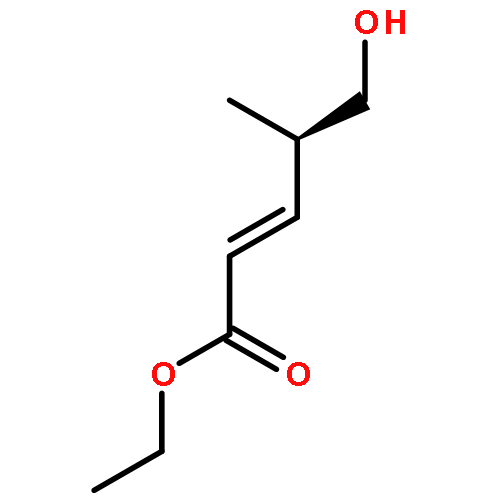
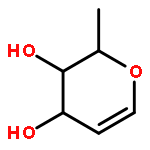
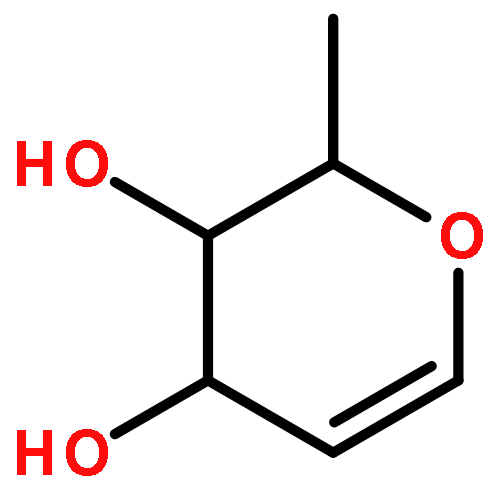
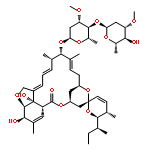
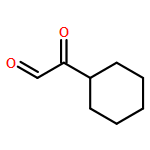
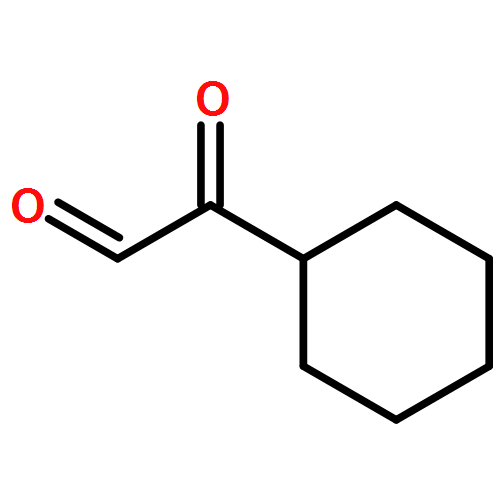
Amphidinolide N, the structure of which has been recently revised, is a 26-membered macrolide featuring allyl epoxide and tetrahydropyran moieties with 13 chiral centers. Due to its challenging structure and extraordinary potent cytotoxicity, amphidinolide N is a highly attractive target of total synthesis. During our total synthesis studies of the 7,10-epimer of the proposed structure of amphidinolide N, we have synthesized the C1–C13 subunit enantio- and diastereoselectively. Key reactions include an l-proline catalyzed enantioselective intramolecular aldol reaction, Evans aldol reaction, Sharpless asymmetric epoxidation and Tamao–Fleming oxidation. To aid late-stage manipulations, we also developed the 4-(N-benzyloxycarbonyl-N-methylamino)butyryl group as a novel ester protective group for the C9 alcohol.
The enantioselective domino Michael/Henry reaction of nitroalkenes with succinaldehyde was found to proceed efficiently upon using diphenylprolinol silyl ether as the organocatalyst. The reaction affords cis-disubstituted nitropentenes with excellent diastereoselectivities and enantioselectivities after treatment of the Michael product with Ac2O and pyridine.
The asymmetric cross-aldol reaction of α,α-disubstituted acetaldehydes with commercial ethyl glyoxylate polymer was successfully catalyzed by diphenylprolinol silyl ether 3 to generate all-carbon quaternary stereogenic centers with good enantioselectivity.
The diphenylprolinol silyl ether mediated asymmetric nitrocyclopropanation of α-substituted α,β-unsaturated aldehydes with bromonitromethane, followed by base-promoted isomerization was found to afford trans-nitrocyclopropanecarbaldehydes with all-carbon quaternary stereogenic centers with excellent diastereo- and enantioselectivities. DFT calculations indicated that the s-trans conformer of the iminium ion intermediate is more stable than the s-cis conformer. In addition, nucleophilic attack of the bromonitromethane anion to the iminium ion intermediate was calculated to occur preferentially from the opposite side of the bulky substituents of the pyrrolidine iminium intermediate.
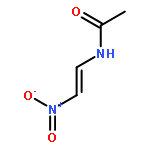
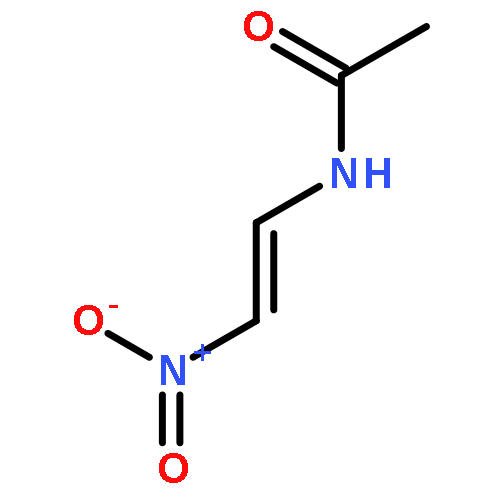
The formation of amides and peptides often necessitates powerful yet mild reagent systems. The reagents used, however, are often expensive and highly elaborate. New atom-economical and practical methods that achieve such goals are highly desirable. Ideally, the methods should start with substrates that are readily available in both chiral and non-chiral forms and utilize cheap reagents that are compatible with a wide variety of functional groups, steric encumberance, and epimerizable stereocenters. A direct oxidative method was developed to form amide and peptide bonds between amines and primary nitroalkanes simply by using I2 and K2CO3 under O2. Contrary to expectations, a 1:1 halogen-bonded complex forms between the iodonium source and the amine, which reacts with nitronates to form α-iodo nitroalkanes as precursors to the amides.
The asymmetric epoxidation of 2-oxoindoline-3-ylidene acetaldehydes, catalyzed by diarylprolinol silyl ether, has been developed. The reaction provides oxindole derivatives possessing chiral epoxides in good yield with good diastereoselectivity and excellent enantioselectivity.
The asymmetric cross-aldol reaction of chloral hydrate with aldehyde pronucleophiles catalyzed by a trifluoromethyl-substituted diarylprolinol was accomplished to afford γ-trichloro-β-hydroxy aldehydes in good yields with excellent enantioselectivities. The resulting aldehyde products were converted into chiral α-azido, α-(4-methyl)phenoxy, and α-fluoro esters without a loss in the diastereo- or enantioselectivities.
The formation of amides and peptides often necessitates powerful yet mild reagent systems. The reagents used, however, are often expensive and highly elaborate. New atom-economical and practical methods that achieve such goals are highly desirable. Ideally, the methods should start with substrates that are readily available in both chiral and non-chiral forms and utilize cheap reagents that are compatible with a wide variety of functional groups, steric encumberance, and epimerizable stereocenters. A direct oxidative method was developed to form amide and peptide bonds between amines and primary nitroalkanes simply by using I2 and K2CO3 under O2. Contrary to expectations, a 1:1 halogen-bonded complex forms between the iodonium source and the amine, which reacts with nitronates to form α-iodo nitroalkanes as precursors to the amides.
The reactions of α,β-unsaturated aldehydes with cyclopentadiene in the presence of diarylprolinol silyl ethers as catalyst proceed via iminium cations as intermediates, and can be divided into two types; one involving a Michael-type reaction (type A) and one involving a cycloaddition (type B). Diphenylprolinol silyl ethers and trifluoromethyl-substituted diarylprolinol silyl ethers, which are widely used proline-type organocatalysts, have been investigated in this study. As the LUMO of the iminium ion derived from trifluoromethyl-substituted diarylprolinol silyl ether is lower in energy than that derived from diphenylprolinol silyl ether, as supported by ab initio calculations, the trifluoromethyl-substituted catalyst is more reactive in a type B reaction. The iminium ion from an α,β-unsaturated aldehyde is generated more quickly with diphenylprolinol silyl ether than with the trifluoromethyl-substituted diarylprolinol silyl ether. When the generation of the iminium ion is the rate-determining step, the diphenylprolinol silyl ether catalyst is the more reactive. Because acid accelerates the generation of iminium ions and reduces the generation of anionic nucleophiles in the Michael-type reaction (type A), it is necessary to select the appropriate acid for specific reactions. In general, diphenylprolinol silyl ether is a superior catalyst for type A reactions, whereas the trifluoromethyl-substituted diarylprolinol silyl ether catalyst is preferred for type B reactions.
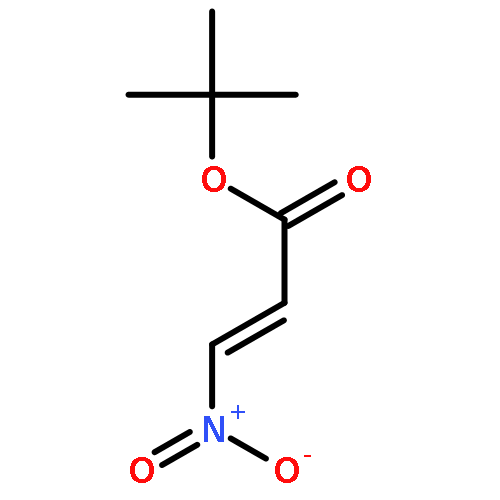
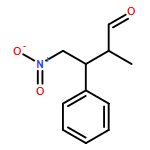
The asymmetric Michael reaction of nitroalkanes and β,β-disubstituted α,β-unsaturated aldehydes was catalyzed by diphenylprolinol silyl ether to afford 1,4-addition products with an all-carbon quaternary stereogenic center with excellent enantioselectivity. The reaction is general for β-substituents such as β-aryl and β-alkyl groups, and both nitromethane and nitroethane can be employed. The addition of nitroethane is considered a synthetic equivalent of the asymmetric Michael reaction of ethyl and acetyl substituents by means of radical denitration and Nef reaction, respectively. The short asymmetric synthesis of (S)-ethosuximide with a quaternary carbon center was accomplished by using the present asymmetric Michael reaction as the key step. The reaction mechanism that involves the E/Z isomerization of α,β-unsaturated aldehydes, the retro-Michael reaction, and the different reactivity between nitromethane and nitroethane is discussed.
(−)-Horsfiline and (−)-coerulescine were synthesized through three one-pot operations in 33 and 46 % overall yield, respectively. Key to the success was the efficient use of a diarylprolinol silyl ether to catalyze the asymmetric Michael addition of nitromethane to a 2-oxoindoline-3-ylidene acetaldehyde. This allowed the all-carbon quaternary, spirocyclic carbon stereocenter to be constructed in good yield with excellent enantioselectivity.
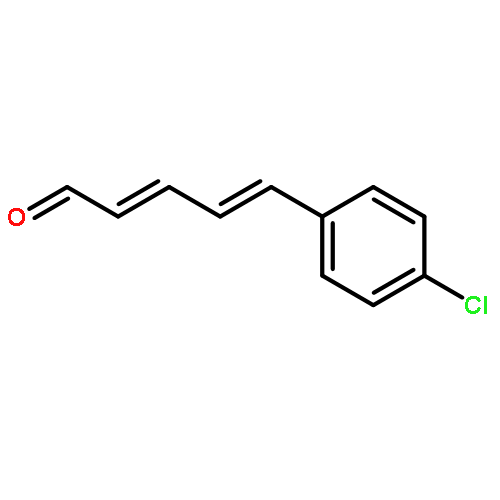
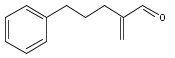
A Nef reaction has been developed that is conducted under mildly basic conditions with molecular oxygen as an oxidant, without the need for metal additives. Whereas nitroalkanes are converted into ketones in good yield, nitroalkenes are transformed into α,β-unsaturated ketones in one-pot by double-bond isomerization followed by the oxygen-mediated Nef reaction. The reaction protocol is both mild and general, and tolerates acid- and base-labile functionality or protecting groups. When oxygen-saturated solvents are employed, the reaction completes within 20 min. Mechanistically, the addition of nitronate ion and molecular oxygen is proposed to proceed initially through a single-electron transfer event, as indicated by radical clock experiments. This ultimately generates a putative 1,1-dioxirane, which reacts further with another nitronate ion to generate the ketone. Involvement of a 1,1-dioxirane is supported by intramolecular trapping experiments with sulfide at the γ-position of the nitro-moiety.
The effect of silyl substituents in diphenylprolinol silyl ether catalysts was investigated. Mechanistically, reactions catalyzed by diphenylprolinol silyl ether can be categorized into three types: two that involve an iminium ion intermediate, such as for the Michael-type reaction (type A) and the cycloaddition reaction (type B), and one that proceeds via an enamine intermediate (type C). In the Michael-type reaction via iminium ions (type A), excellent enantioselectivity is realized when the catalyst with a bulky silyl moiety is employed, in which efficient shielding of a diastereotopic face of the iminium ion is directed by the bulky silyl moiety. In the cycloaddition reaction of iminium ions (type B) and reactions via enamines (type C), excellent enantioselectivity is obtained even when the silyl group is less bulky and, in this case, too much bulk reduces the reaction rate. In other cases, the yield increases when diphenylprolinol silyl ethers with bulky substituents are employed, presumably by suppressing side reactions between the nucleophilic catalyst and the reagent. The conformational behaviors of the iminium and enamine species have been determined by theoretical calculations. These data explain the effect of the bulkiness of the silyl substituent on the enantioselectivity and reactivity of the catalysts.
The stoichiometric reactions of enamines prepared from aldehydes and diphenyl-prolinol silyl ethers (intermediates of numerous organocatalytic processes) with nitro olefins have been investigated. As reported in the last century for simple achiral and chiral enamines, the products are cyclobutanes (4 with monosubstituted nitro-ethenes), dihydro-oxazine N-oxide derivatives (5 with disubstituted nitro-ethenes), and nitro enamines derived from γ-nitro aldehydes (6, often formed after longer reaction times). The same types of products were shown to be formed, when the reactions were carried out with peptides H-Pro-Pro-Xaa-OMe that lack an acidic H-atom. Functionalized components such as alkoxy enamines, nitro-acrylates, acetamido-nitro-ethylene, or hydroxylated nitro olefins also form products carrying the diphenyl-prolinol silyl ether as a substituent. All of these products must be considered intermediates in the corresponding catalytic reactions; the investigation of their chemical properties provided useful hints about the rates, the conditions, the catalyst resting states or irreversible traps, and/or the limitations of the corresponding organocatalytic processes. High-level DFT and MP2 computations of the structures of alkoxy enamines and thermodynamic data of a cyclobutane dissociation are also described. Some results obtained with the stoichiometrically prepared intermediates are not compatible with previous mechanistic proposals and assumptions.
The one-pot sequential synthesis of (−)-oseltamivir has been achieved without evaporation or solvent exchange in 36 % yield over seven reactions. The key step was the asymmetric Michael reaction of pentan-3-yloxyacetaldehyde with (Z)-N-2-nitroethenylacetamide, catalyzed by a diphenylprolinol silyl ether. The use of a bulky O-silyl-substituted diphenylprolinol catalyst, chlorobenzene as a solvent, and HCO2H as an acid additive, were key to produce the first Michael adduct in both excellent yield and excellent diastereo- and enantioselectivity. Investigation into the effect of acid demonstrated that an acid additive accelerates not only the E–Z isomerization of the enamines derived from pentan-3-yloxyacetaldehyde with diphenylprolinol silyl ether, but also ring opening of the cyclobutane intermediate and the addition reaction of the enamine to (Z)-N-2-nitroethenylacetamide. The transition-state model for the Michael reaction of pentan-3-yloxyacetaldehyde with (Z)-N-2-nitroethenylacetamide was proposed by consideration of the absolute configuration of the major and minor isomers of the Michael product with the results of the Michael reaction of pentan-3-yloxyacetaldehyde with phenylmaleimide and naphthoquinone.
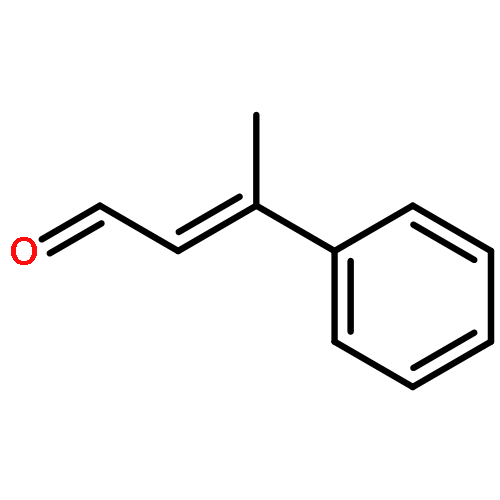
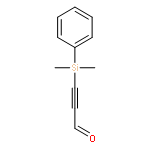
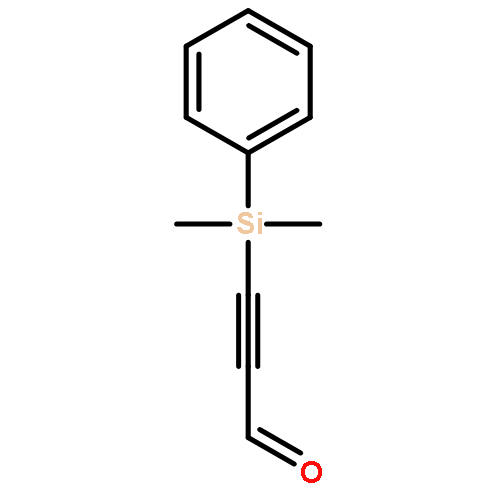
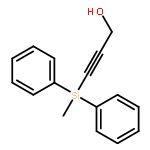
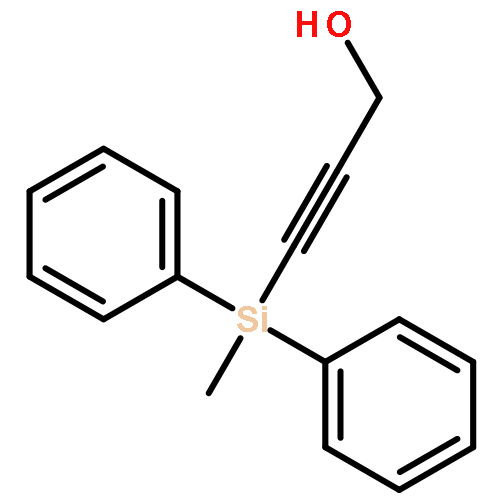
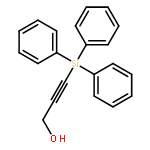
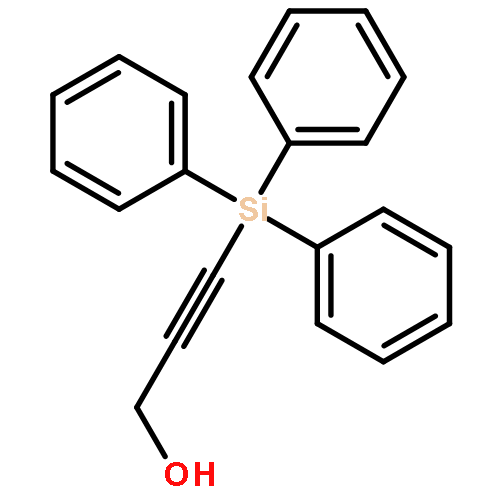
The direct cross-aldol reaction of alkynyl aldehydes catalyzed by a trifluoromethylated diarylprolinol provides a practical route for the highly enantioselective synthesis of chiral β-alkynyl-β-hydroxy aldehydes. Good anti selectivity and excellent enantioselectivity were obtained in the reactions of silylpropynals, which afford synthetically useful chiral building blocks.
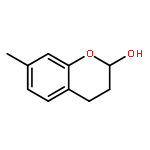
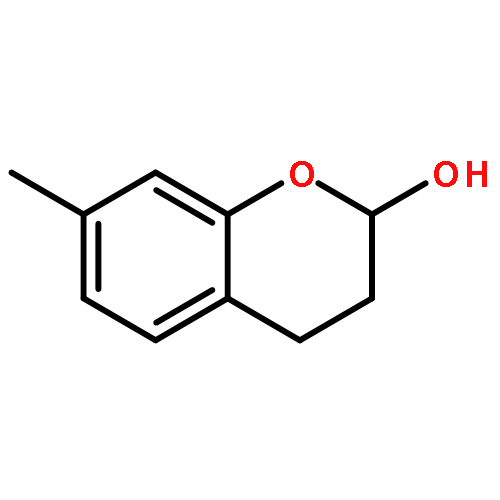
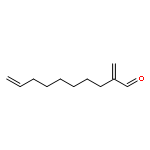
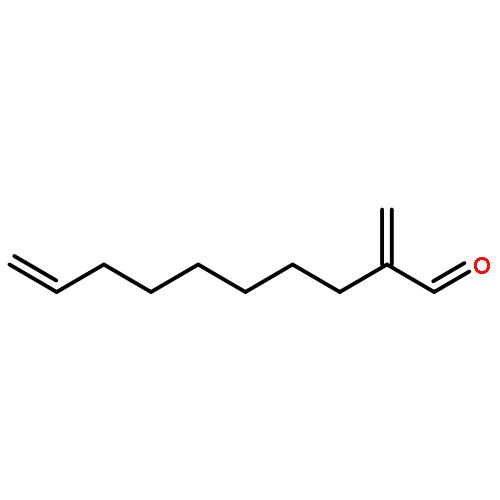






![Benzene, 1-[(R)-ethynylsulfinyl]-4-methyl-](http://img.cochemist.com/ccimg/100000/99930-83-3.png)
![Benzene, 1-[(R)-ethynylsulfinyl]-4-methyl-](http://img.cochemist.com/ccimg/100000/99930-83-3_b.png)










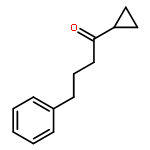
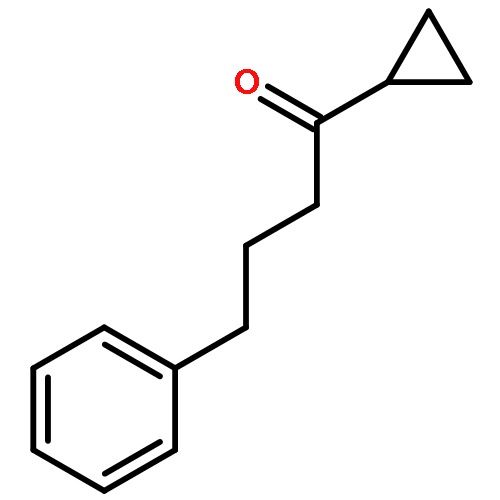
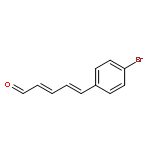
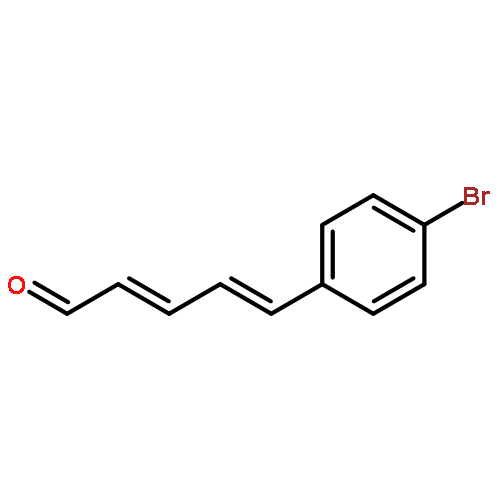




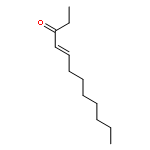

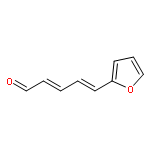
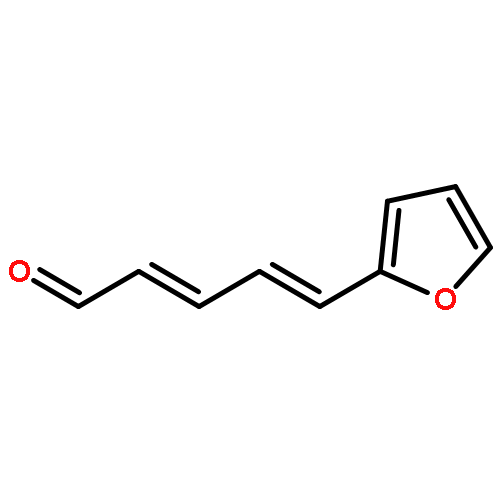
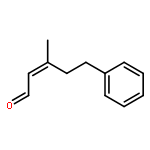
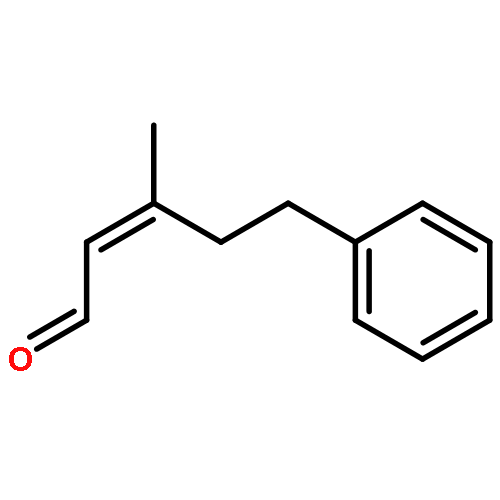
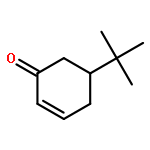
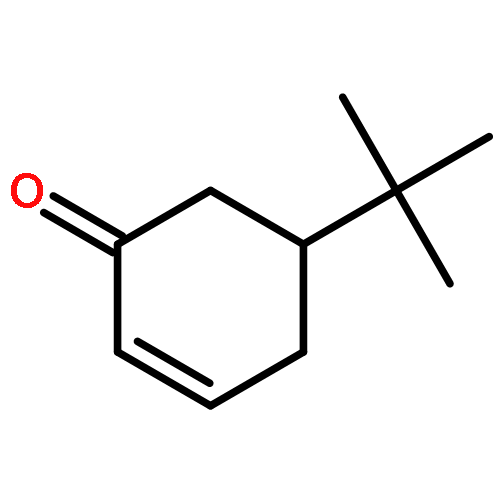
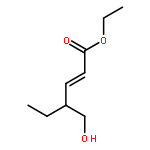

![2,4-Pentadienal, 5-[4-(trifluoromethyl)phenyl]-, (2E,4E)-](http://img.cochemist.com/ccimg/183900/183800-82-0.png)
![2,4-Pentadienal, 5-[4-(trifluoromethyl)phenyl]-, (2E,4E)-](http://img.cochemist.com/ccimg/183900/183800-82-0_b.png)
![SILANE, (1,1-DIMETHYLETHYL)DIPHENYL[[(2S)-2-(PHENYLMETHYL)-3-BUTYNYL]OXY]-](http://img.cochemist.com/ccimg/866500/866404-25-3.png)
![SILANE, (1,1-DIMETHYLETHYL)DIPHENYL[[(2S)-2-(PHENYLMETHYL)-3-BUTYNYL]OXY]-](http://img.cochemist.com/ccimg/866500/866404-25-3_b.png)


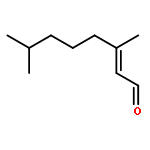
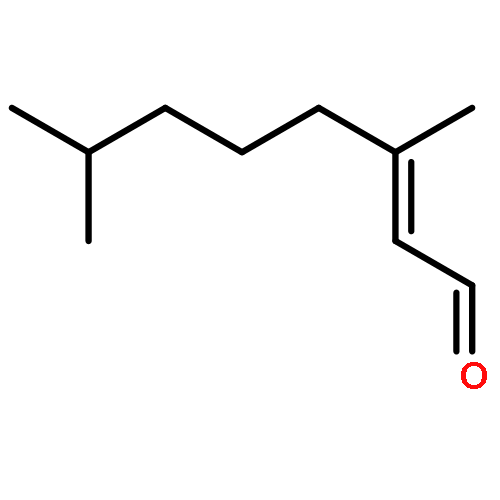
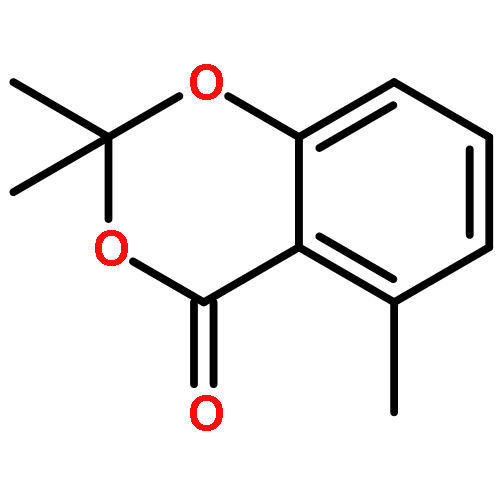
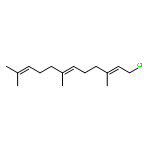
![OXIRANE, 3-[(3E)-5-CHLORO-3-METHYL-3-PENTENYL]-2,2-DIMETHYL-](http://img.cochemist.com/ccimg/43200/43119-82-0.png)
![OXIRANE, 3-[(3E)-5-CHLORO-3-METHYL-3-PENTENYL]-2,2-DIMETHYL-](http://img.cochemist.com/ccimg/43200/43119-82-0_b.png)
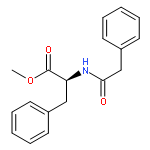
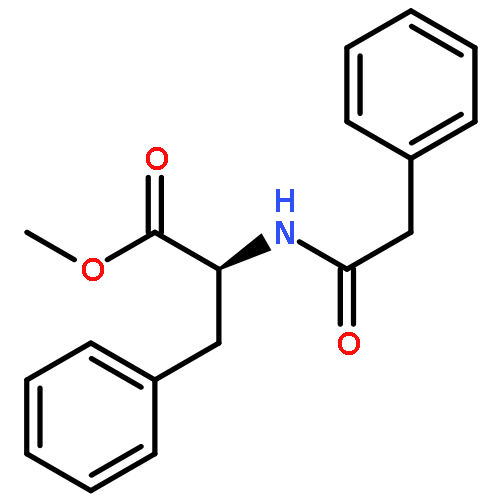
![Benzene, 1-[(S)-ethynylsulfinyl]-4-methyl-](http://img.cochemist.com/ccimg/195100/195063-98-0.png)
![Benzene, 1-[(S)-ethynylsulfinyl]-4-methyl-](http://img.cochemist.com/ccimg/195100/195063-98-0_b.png)

![Tricyclo[3.3.1.13,7]decane-1-carboxamide, N-methyl-N-(phenylmethyl)-](http://img.cochemist.com/ccimg/113500/113435-65-7.png)
![Tricyclo[3.3.1.13,7]decane-1-carboxamide, N-methyl-N-(phenylmethyl)-](http://img.cochemist.com/ccimg/113500/113435-65-7_b.png)


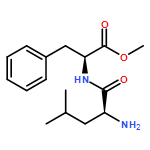
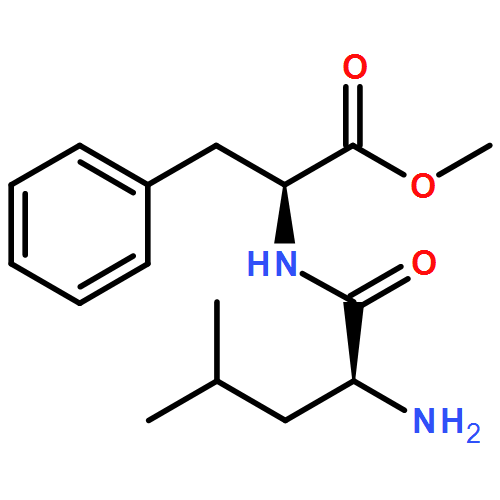


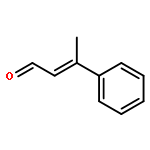

![S-2-[bis[3,5-bis(trifluoroMethyl)phenyl] [(triethylsilyl)oxy]Methyl]-Pyrrolidine](/data/chemimg/2074200/946074-05-1.png)
![S-2-[bis[3,5-bis(trifluoroMethyl)phenyl] [(triethylsilyl)oxy]Methyl]-Pyrrolidine](/data/chemimg/2074200/946074-05-1_b.png)












![3-OCTEN-2-ONE, 8-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-, (3E)-](http://img.cochemist.com/ccimg/479000/478943-07-6.png)
![3-OCTEN-2-ONE, 8-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-, (3E)-](http://img.cochemist.com/ccimg/479000/478943-07-6_b.png)
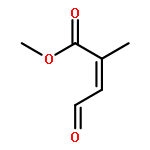
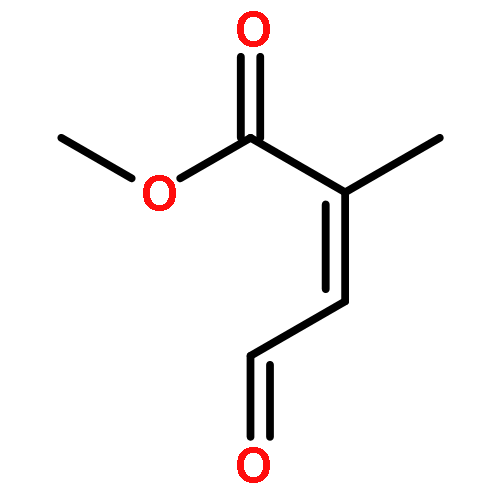
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-methyl-, (1S,2R,4S)-](http://img.cochemist.com/ccimg/72300/72203-36-2.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-methyl-, (1S,2R,4S)-](http://img.cochemist.com/ccimg/72300/72203-36-2_b.png)




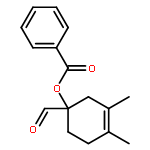
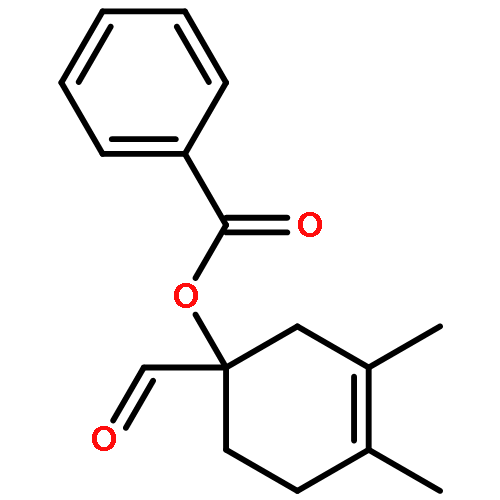
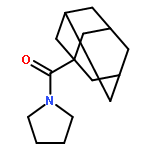
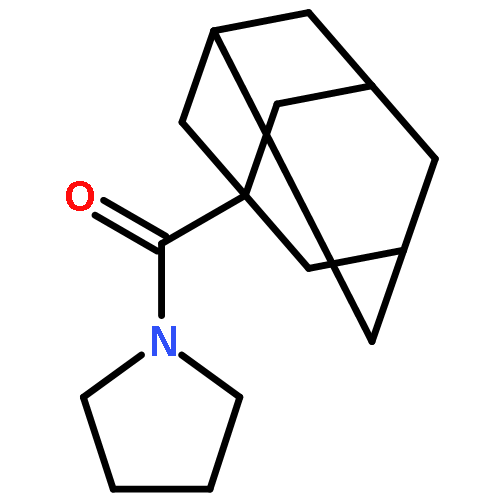


![Benzene, [(1-nitro-2-phenylethyl)sulfonyl]-](http://img.cochemist.com/ccimg/74800/74737-94-3.png)
![Benzene, [(1-nitro-2-phenylethyl)sulfonyl]-](http://img.cochemist.com/ccimg/74800/74737-94-3_b.png)




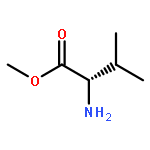
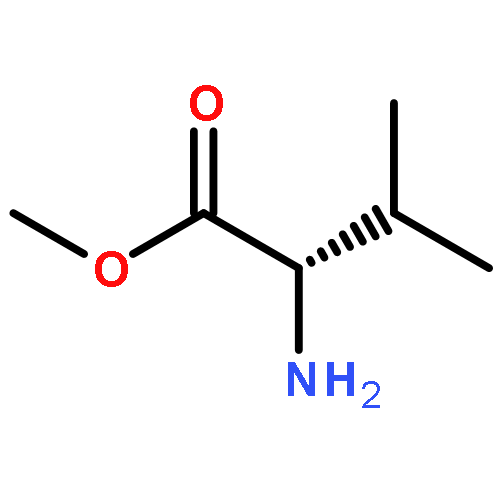




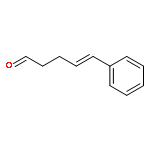
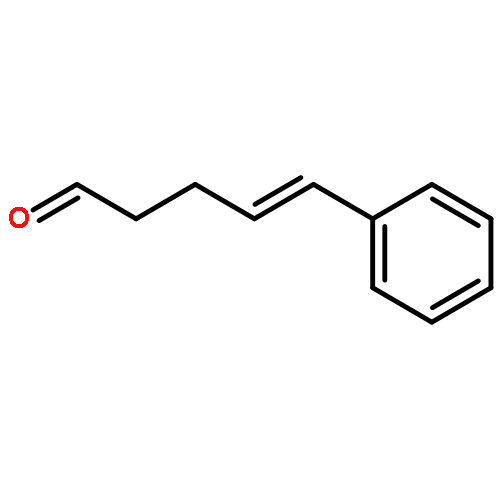
![Phenol,2,4-bis(1,1-dimethylethyl)-6-[(E)-[[(1S)-1-(hydroxymethyl)-2-methylpropyl]imino]phenylmethyl]-](http://img.cochemist.com/ccimg/210700/210626-68-9.png)
![Phenol,2,4-bis(1,1-dimethylethyl)-6-[(E)-[[(1S)-1-(hydroxymethyl)-2-methylpropyl]imino]phenylmethyl]-](http://img.cochemist.com/ccimg/210700/210626-68-9_b.png)
![Carbamic acid, [(1R)-2-nitro-1-phenylethyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/685200/685132-79-0.png)
![Carbamic acid, [(1R)-2-nitro-1-phenylethyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/685200/685132-79-0_b.png)



















![2-Butyn-1-ol, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/92900/92808-80-5.png)
![2-Butyn-1-ol, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/92900/92808-80-5_b.png)

![(2R,2'R,3a'S,4a'R,5'S,6'S,6a'S,9'S,10'S,10a'R,11a'S,12a'R,13a'S,14a'R,15a'S,17'E,19a'R,20a'S,22a'R,23a'S,24'R,24a'R,26'R,29a'S,30a'R,31a'S,32a'R,34a'S,35a'R,37'R,37a'S,38a'R)-26'-[(1E,3S)-3,4-dihydroxybut-1-en-1-yl]-2',5',9',10',37a'-pentamethyl-1',3',3a'](http://img.cochemist.com/ccimg/139400/139341-09-6.png)
![(2R,2'R,3a'S,4a'R,5'S,6'S,6a'S,9'S,10'S,10a'R,11a'S,12a'R,13a'S,14a'R,15a'S,17'E,19a'R,20a'S,22a'R,23a'S,24'R,24a'R,26'R,29a'S,30a'R,31a'S,32a'R,34a'S,35a'R,37'R,37a'S,38a'R)-26'-[(1E,3S)-3,4-dihydroxybut-1-en-1-yl]-2',5',9',10',37a'-pentamethyl-1',3',3a'](http://img.cochemist.com/ccimg/139400/139341-09-6_b.png)


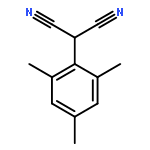
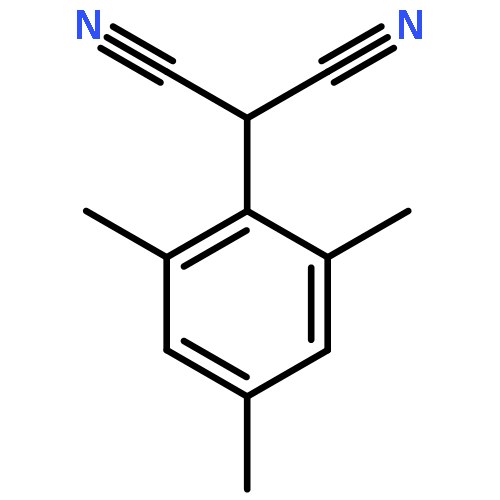

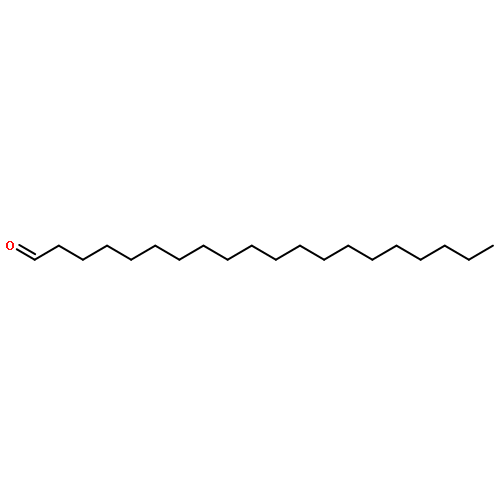
![2-Butynal, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/112300/112213-42-0.png)
![2-Butynal, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/112300/112213-42-0_b.png)

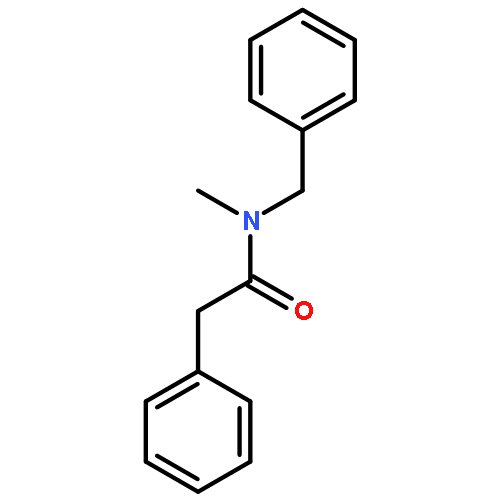








![(2E)-3-[(4S)-2,2-DiMethyl-1,3-dioxolan-4-yl]-2-Methyl-2-propenoic acid ethyl ester](http://img.cochemist.com/ccimg/82000/81997-76-4.png)
![(2E)-3-[(4S)-2,2-DiMethyl-1,3-dioxolan-4-yl]-2-Methyl-2-propenoic acid ethyl ester](http://img.cochemist.com/ccimg/82000/81997-76-4_b.png)









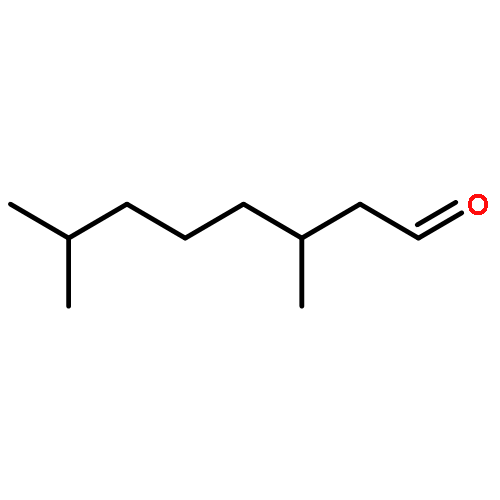


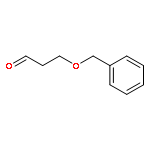
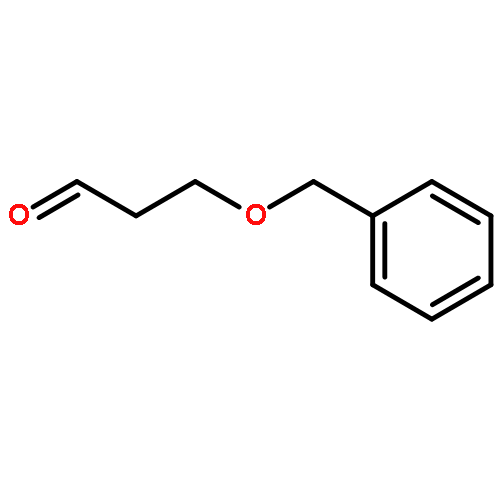
![Hexanamide,N-[4-[(hydroxyamino)sulfonyl]phenyl]-](http://img.cochemist.com/ccimg/7000/6937-32-2.png)
![Hexanamide,N-[4-[(hydroxyamino)sulfonyl]phenyl]-](http://img.cochemist.com/ccimg/7000/6937-32-2_b.png)
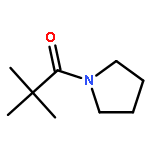
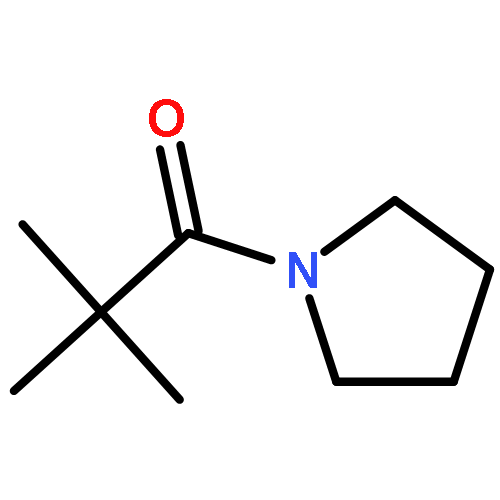
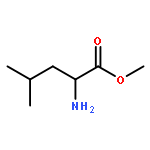
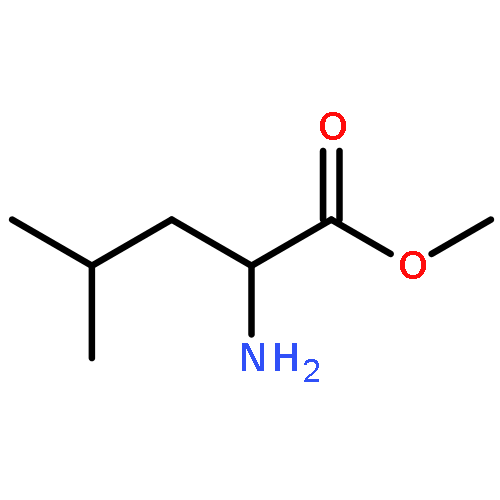


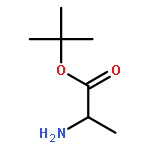
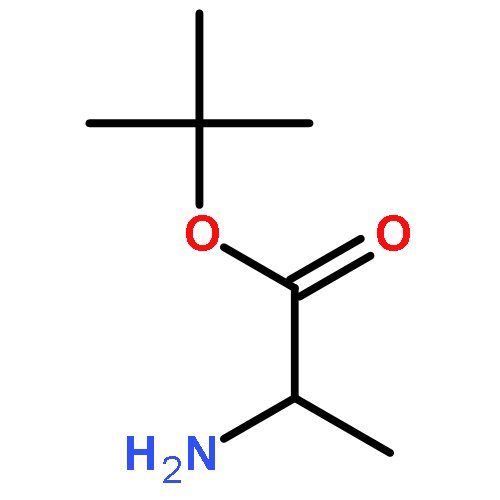
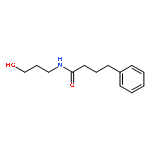
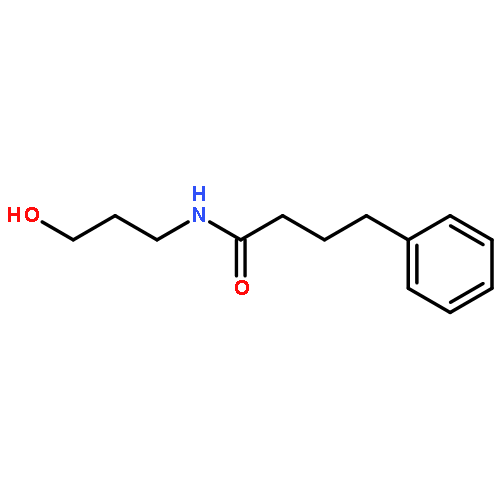
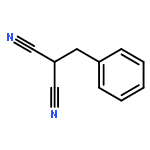

![Benzoic acid, 4-[(1E)-3-oxo-1-propen-1-yl]-, methyl ester](/data/chemimg/709300/78024-60-9.png)
![Benzoic acid, 4-[(1E)-3-oxo-1-propen-1-yl]-, methyl ester](/data/chemimg/709300/78024-60-9_b.png)






![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde,2-ethyl-, (1S,2R,4S)-](http://img.cochemist.com/ccimg/154300/154279-11-5.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde,2-ethyl-, (1S,2R,4S)-](http://img.cochemist.com/ccimg/154300/154279-11-5_b.png)






![L-Serine, O-[(1,1-dimethylethyl)dimethylsilyl]-, methyl ester](http://img.cochemist.com/ccimg/98700/98642-61-6.png)
![L-Serine, O-[(1,1-dimethylethyl)dimethylsilyl]-, methyl ester](http://img.cochemist.com/ccimg/98700/98642-61-6_b.png)


![2-Propenal, 3-[4-(trifluoromethyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/95200/95123-61-8.png)
![2-Propenal, 3-[4-(trifluoromethyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/95200/95123-61-8_b.png)
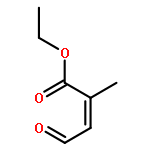
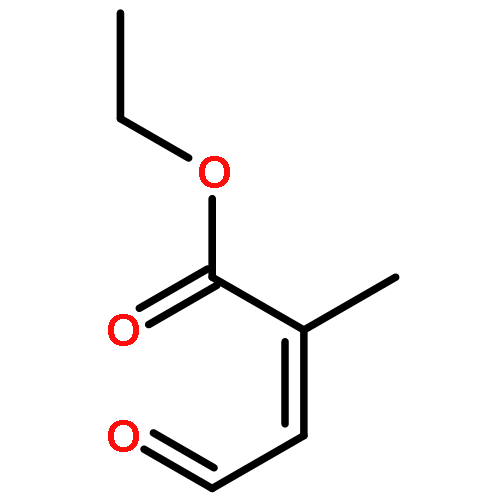
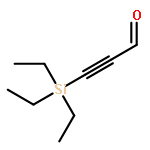


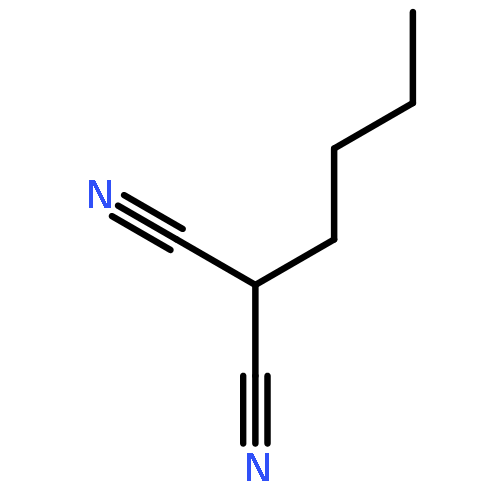
![Pentanal, 5-[(tetrahydro-2H-pyran-2-yl)oxy]-](http://img.cochemist.com/ccimg/14200/14194-86-6.png)
![Pentanal, 5-[(tetrahydro-2H-pyran-2-yl)oxy]-](http://img.cochemist.com/ccimg/14200/14194-86-6_b.png)















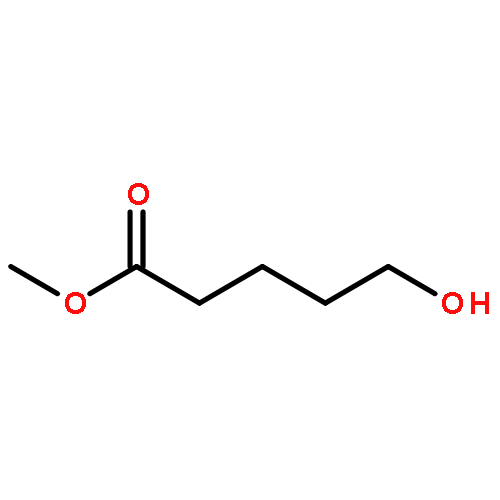
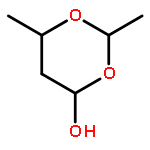


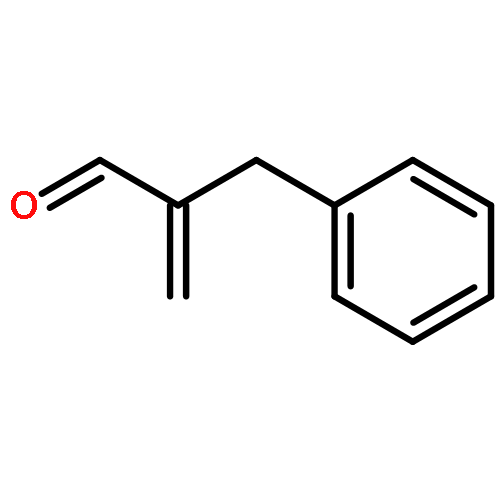


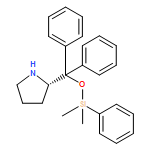

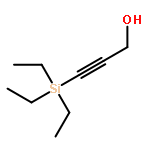
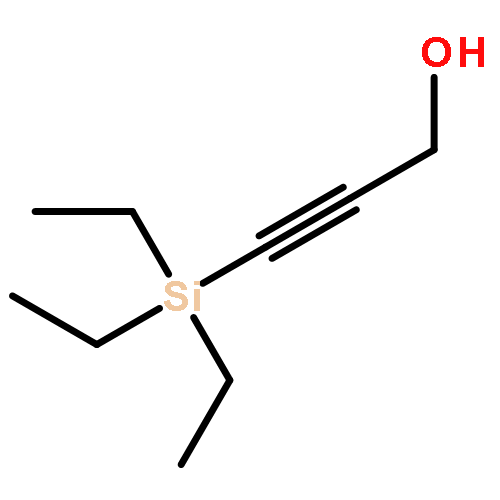





![3-[DIMETHYL(PHENYL)SILYL]PROP-2-YN-1-OL](/data/chemimg/64200/43019-63-2.png)
![3-[DIMETHYL(PHENYL)SILYL]PROP-2-YN-1-OL](/data/chemimg/64200/43019-63-2_b.png)






![Phosphonothioic acid,P-methyl-, S-[2-(diethylamino)ethyl] ester](http://img.cochemist.com/ccimg/21100/21068-51-9.png)
![Phosphonothioic acid,P-methyl-, S-[2-(diethylamino)ethyl] ester](http://img.cochemist.com/ccimg/21100/21068-51-9_b.png)


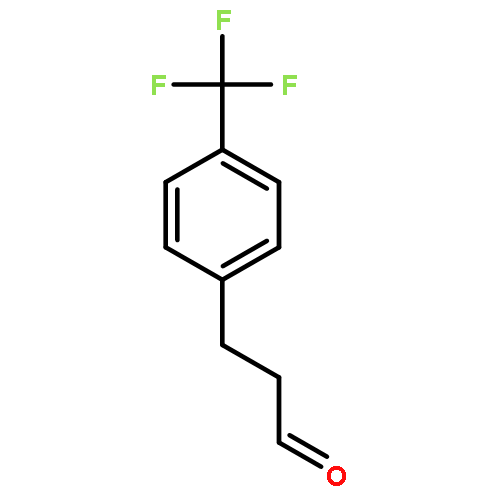


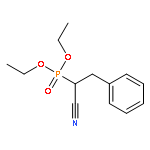
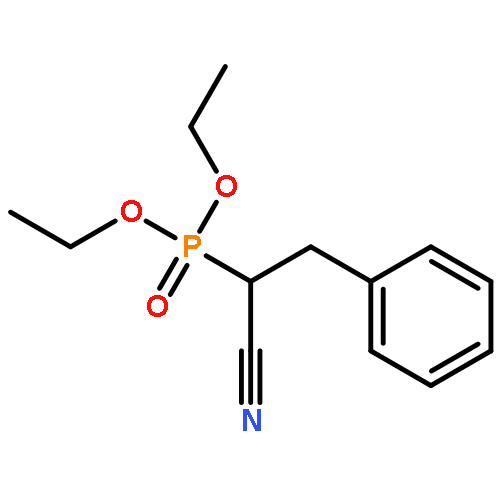
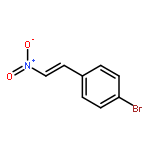
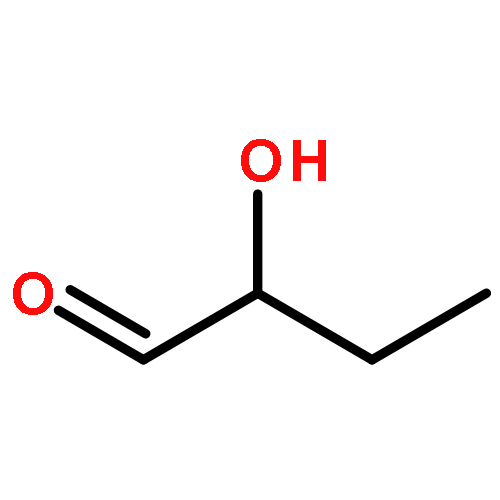


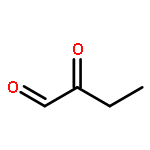

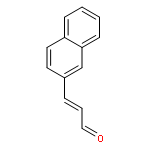
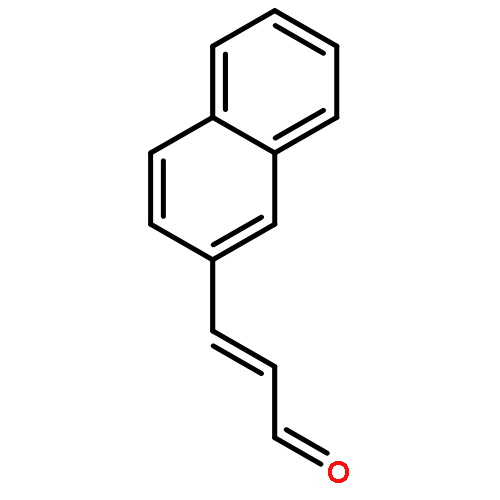



![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 3-phenyl-, (1S,2S,3S,4R)-](http://img.cochemist.com/ccimg/279700/279685-68-6.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 3-phenyl-, (1S,2S,3S,4R)-](http://img.cochemist.com/ccimg/279700/279685-68-6_b.png)







![Propanal, 3-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/128500/128461-65-4.png)
![Propanal, 3-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/128500/128461-65-4_b.png)


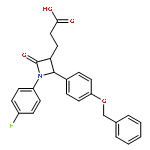

![3-Penten-2-one,5-(3,3-dimethylbicyclo[2.2.1]hept-2-ylidene)-](http://img.cochemist.com/ccimg/2300/2226-11-1.png)
![3-Penten-2-one,5-(3,3-dimethylbicyclo[2.2.1]hept-2-ylidene)-](http://img.cochemist.com/ccimg/2300/2226-11-1_b.png)
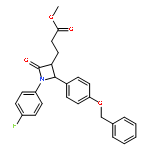
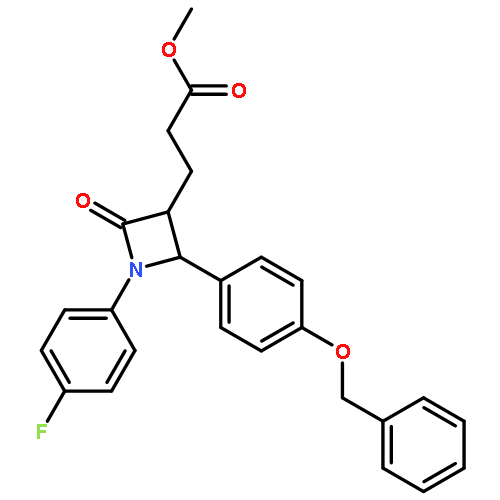


![1,3-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea](/data/chemimg/315700/1060-92-0.png)
![1,3-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea](/data/chemimg/315700/1060-92-0_b.png)
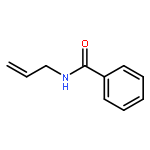






![Pyrrolidine, 2-[diphenyl[(triethylsilyl)oxy]methyl]-, (2S)-](/data/chemimg/102900/864466-70-6.png)
![Pyrrolidine, 2-[diphenyl[(triethylsilyl)oxy]methyl]-, (2S)-](/data/chemimg/102900/864466-70-6_b.png)