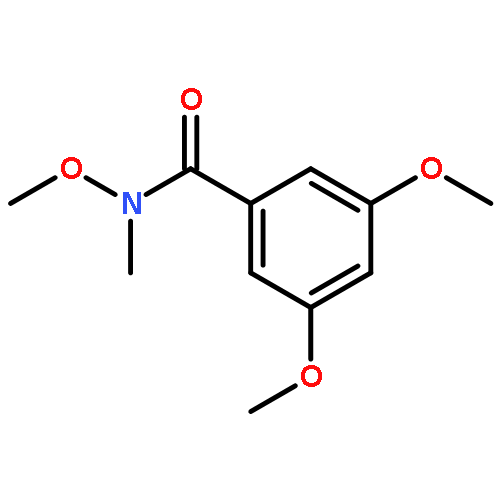
β-secretase (BACE-1) is a potential target for the treatment of Alzheimer's disease (AD). Despite its potential, only few compounds targeting BACE have entered the clinical trials. Herein, we describe the identification of Gefitinib as a potential lead compound for BACE through an integrated approach of structural bioinformatics analysis, experimental assessment and computational analysis. In particular, we performed ELISA and western analysis to assess the effect of Gefitinib using N2a human APP695 cells. In addition, we investigated the binding mechanism of Gefitinib with BACE through molecular docking coupled with molecular dynamics simulations. The computational analyses revealed that hydrophobic contact is a major contributing factor to the binding of Gefitinib with BACE. The results obtained in the study have rendered Gefitinib as a putative lead compound for BACE. Further optimization studies are warranted to improve its potency and pharmacological properties against BACE for potential AD treatment.
Four novel compounds were designed by “tailoring” 3,3′-dihydroxyisorenieratene (a natural carotenoid) based on an isoprene unit retention truncation strategy. Among them, the smallest molecule 1 (2,3,6,2′,3′,6′-hexamethyl-4,4′-dihydroxy-trans-stilbene) was concisely synthesized in a one-pot Stille–Heck tandem sequence, and surfaced as a promising lead molecule in terms of its selective antiproliferative activity mediated by blocking the NCI-H460 cell cycle in G1 phase. Additionally, theoretical calculations and cell uptake experiments indicate that the unique polymethylation pattern of compound 1 significantly induces a conformational change shift out of planarity and increases its cell uptake and metabolic stability. The observation should be helpful to rationally design resveratrol-inspired antiproliferative agents.




![N-{2-[(3-CYANO-5,7-DIMETHYL-2-QUINOLINYL)AMINO]ETHYL}-3-METHOXYBE<WBR />NZAMIDE](http://img.cochemist.com/ccimg/321700/321661-62-5.png)
![N-{2-[(3-CYANO-5,7-DIMETHYL-2-QUINOLINYL)AMINO]ETHYL}-3-METHOXYBE<WBR />NZAMIDE](http://img.cochemist.com/ccimg/321700/321661-62-5_b.png)