Co-reporter:Yingzhan Tang, Chongguo Jiang, Xinhang Zhang, Chengjun Liu, Jingsheng Lin, Yanshi Wang, Chuan Du, Xiaoshi Peng, Wei Li, Yongxiang Liu, and Maosheng Cheng
The Journal of Organic Chemistry October 20, 2017 Volume 82(Issue 20) pp:11102-11102
Publication Date(Web):September 25, 2017
DOI:10.1021/acs.joc.7b02066
A cascade [3,3]-sigmatropic rearrangement/aromatization strategy to the synthesis of 2-(3-methylbenzofuran-2-yl)phenol derivatives was developed and applied to the collective syntheses of seven 2-arylbenzofuran-containing natural products, namely glycybenzofuran, glycyuralin E, lespedezol A1, puerariafuran, 7,2′,4′-trihydroxy-3-benzofurancarboxylic acid, coumestrol, and 4′-O-methylcoumestrol. Among them, the total syntheses of glycybenzofuran, glycyuralin E, puerariafuran, 7,2′,4′-trihydroxy-3-benzofurancarboxylic acid, and 4′-O-methylcoumestrol were reported for the first time. The practicality of this novel strategy in preparation of the key intermediates was demonstrated by performing the reaction on gram scale and by synthesizing a series of natural products with 2-(3-methylbenzofuran-2-yl)phenol scaffolds in a common strategy.
Co-reporter:Wenxi Ma;Qiaoling Tong;Jian Wang;Huali Yang;Meng Zhang;Hailun Jiang;Qinghe Wang;Yongxiang Liu;Maosheng Cheng
RSC Advances (2011-Present) 2017 vol. 7(Issue 11) pp:6720-6723
Publication Date(Web):2017/01/18
DOI:10.1039/C6RA27690C
A sustainable catalyst for the selective oxidation of benzyl alcohol (BnOH) to benzaldehyde (BzH) was developed by mineralizing Ti(SO4)2 on graphene oxide foam (GOF) surface. The Ti(SO4)2/GOF was characterized by scanning electron microscopy (SEM), energy disperse spectroscopy (EDS), X-ray diffraction (XRD), solid-state NMR (SSNMR), thermal gravimetric analysis (TGA), infrared (IR) and BET analysis. Recycling experiments proved that Ti(SO4)2/GOF possessed excellent reusability and durability. The industrial perspective of Ti(SO4)2/GOF was demonstrated by the preparation of BzH in a large-scale and solvent-free oxidation process.
Co-reporter:Jian Wang;Wei Li;Bo Wang;Baichun Hu;Hailun Jiang
Current Pharmacology Reports 2017 Volume 3( Issue 4) pp:184-195
Publication Date(Web):21 July 2017
DOI:10.1007/s40495-017-0094-1
Increasing insight into the area of molecular cancer biology and structural biology has resulted in the disclosure of an increasing number of chemopreventive targets, which can be exploited for rapid rational discovery of chemopreventive agents. In this article, we discuss the application of in silicon approaches including molecular docking, pharmacophore modeling, fragment-based drug design, homology modeling, and target prediction in facilitating the search and designing of chemopreventive agents. These computational strategies have shown promising potential in speeding drug discovery and is expected to contribute to intelligent chemopreventive agents.
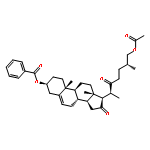
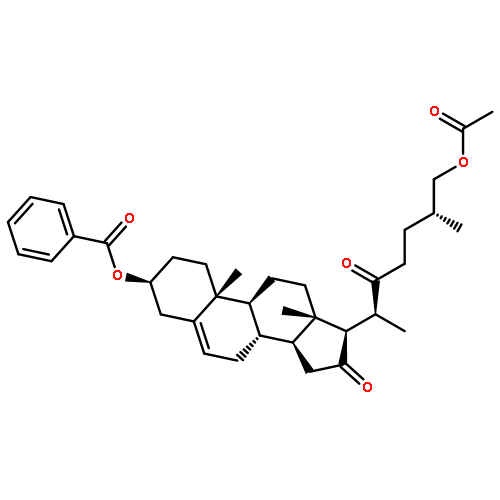
Co-reporter:Yongxiang Liu, Shengfei Jin, Yanshi Wang, Shanshan Cui, Xiaoshi Peng, Yuanyuan Niu, Chuan Du and Maosheng Cheng
Chemical Communications 2016 vol. 52(Issue 37) pp:6233-6236
Publication Date(Web):09 Mar 2016
DOI:10.1039/C6CC01770C
A gold(I)-catalyzed substituent-controlled strategy for the stereoselective synthesis of bicyclic furan and pyran derivatives has been developed. The mechanisms of the reactions have been studied thoroughly by deuterium labelling experiments. The applications of the methodologies were demonstrated by evaluating the preliminary anti-fungal activity of the synthetic compounds.
Co-reporter:Shengfei Jin, Chongguo Jiang, Xiaoshi Peng, Chunhui Shan, Shanshan Cui, Yuanyuan Niu, Yang Liu, Yu Lan, Yongxiang Liu, and Maosheng Cheng
Organic Letters 2016 Volume 18(Issue 4) pp:680-683
Publication Date(Web):February 3, 2016
DOI:10.1021/acs.orglett.5b03641
A unique strategy for the regiospecific synthesis of bicyclic furopyran derivatives has been developed via a gold(I)-catalyzed propargyl-Claisen rearrangement/6-endo-trig cyclization of propargyl vinyl ethers. The introduction of angle strain into the substrates significantly altered the reaction’s regioselectivity. Insight into the regioselectivity of the cycloisomerization was obtained with density functional theory calculations.
Co-reporter:Gaofei Wei, Shanshan Cui, Weijing Luan, Shuai Wang, Zhuang Hou, Yongxiang Liu, Yang Liu, Maosheng Cheng
European Journal of Medicinal Chemistry 2016 Volume 113() pp:92-101
Publication Date(Web):4 May 2016
DOI:10.1016/j.ejmech.2015.12.034
•A series of Albiziabioside A derivatives substituted with cinnamoyl groups were designed and synthesized.•The compound 8n was found with much less cytotoxicity towards normal cells lines.•The anti-proliferative activity of 8n could be attributed to the induction of cell cycle arrest and apoptosis in HCT116 cells.A series of Albiziabioside A coupled substituents of cinnamoyl derivatives were designed and synthesized. The synthesized compounds were screened for anticancer activity against a panel of six human cancer cell lines using a MTT assay. Synthetic derivatives showed excellent selectivity, as they were toxic against only HCT116 cell line. Some compounds exhibited better anti-cancer activity against HCT116 compared to positive controls, such as 5-fluorouracil and Albiziabioside A. Compound 8n was the most active derivative. Importantly, it was also found that the anti-proliferative activity of 8n could be attributed to the induction of cell cycle arrest and apoptosis in HCT116 cells.
Co-reporter:Fengran Li;Yang Liu;Shuai Wang;Gaofei Wei
Chemical Research in Chinese Universities 2016 Volume 32( Issue 6) pp:938-942
Publication Date(Web):2016 December
DOI:10.1007/s40242-016-6301-5
Fifteen novel 3-oxo-oleanolic acid esters bearing aryl substituted 1,2,3-triazolyl methyl moiety were synthesized via the method of Copper(I)-catalyzed Huisgen cycloaddition. The cytotoxicity evaluation results of these compounds against five human tumor cell lines show that most of these compounds presented potent activity and selectivity against A375-S2 and HT1080 cells. Compound 6c, with a p-NO2 at the bezene ring, possesses the best inhibitory activity against A375-S2(IC50=2.82 μmol/L) and HT1080(IC50=1.69 μmol/L).
Co-reporter:Yongxiang Liu;Xiaoyu Wang;Yanshi Wang;Chuan Du;Hui Shi;Shengfei Jin;Chongguo Jiang;Jianyong Xiao ;Maosheng Cheng
Advanced Synthesis & Catalysis 2015 Volume 357( Issue 5) pp:1029-1036
Publication Date(Web):
DOI:10.1002/adsc.201401097
Co-reporter:Gaofei Wei, Weijing Luan, Shuai Wang, Shanshan Cui, Fengran Li, Yongxiang Liu, Yang Liu and Maosheng Cheng
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 5) pp:1507-1514
Publication Date(Web):18 Nov 2014
DOI:10.1039/C4OB01605J
A series of novel oleanolic acid coupled 1,2,3-triazole derivatives have been designed and synthesized by employing a Cu(I) catalyzed Huisgen 1,3-dipolar cycloaddition reaction. The anti-proliferative evaluation indicated that some compounds exhibited excellent anti-cancer activity against the examined cancer cell lines. Among all derivatives, compound 3t possesses the best inhibitory activity against HT1080 cells. A series of pharmacology experiments show that compound 3t significantly induced HT1080 cell apoptosis. Therefore, this compound can serve as a promising lead candidate for further study.
Co-reporter:Ruijuan Li, Xiaolin Su, Zheng Chen, Wanxu Huang, Yali Wang, Kaibo Wang, Bin Lin, Jian Wang and Maosheng Cheng
RSC Advances 2015 vol. 5(Issue 30) pp:23202-23209
Publication Date(Web):25 Feb 2015
DOI:10.1039/C4RA16963H
A structure-based virtual screening approach to targeting p21-activated kinase 4 (PAK4) was performed to identify good chemical starting points for medicinal chemistry. A pre-filtrated database was screened against two designed PAK4 pharmacophores, and the pharmacophore search hits were docked into a PAK4 crystal structure. Twenty-seven compounds were then selected for in vitro PAK4 inhibition assay, and results showed three compounds exhibiting a micro-molar IC50 in a dose–response assay. Interactive modes of the three compounds were studied and showed good binding modes in the PAK4 active site. Calculated ADME/T properties of the three hits were also analyzed and showed good drug-like properties. The results of in vitro PAK4 inhibition assay, interactive mode study and ADME/T prediction revealed that the three compounds have potential PAK4 inhibitory activities and can be further optimized and developed as lead compounds.
Co-reporter:Xiaoyu Wang, Chuan Du, Hui Shi, Yadong Pang, Shengfei Jin, Yuqian Hou, Yanshi Wang, Xiaoshi Peng, Jianyong Xiao, Yang Liu, Yongxiang Liu, Maosheng Cheng
Tetrahedron 2015 Volume 71(Issue 38) pp:6744-6748
Publication Date(Web):23 September 2015
DOI:10.1016/j.tet.2015.07.040
During investigating water-compatible Lewis acids catalyzed etherifications using alcohols as alkylating reagents directly, we developed Fe(III)-catalyzed trityl benzyl ether formations irradiated by microwave. Then an in situ trityl benzyl ether formation and disproportionation cascade reaction was achieved to yield the benzaldehyde products with good functional group tolerances under neat conditions at relative higher temperatures. The substituent effects of the substrates on the etherification and disproportionation were explored by changing the substitutions on benzyl alcohols and triarylmethanols using chemical kinetic plots methods and the mechanism of the transformation was studied by crossover experiments. The etherification and disproportionation cascade process could be conveniently scaled up in laboratory without losing much efficiency.Fe(III)-catalyzed disproportionation of benzyl alcohols via a trityl ether intermediate was achieved under neat conditions irradiated by microwave. The substituent effects were examined by chemical kinetic plots and the mechanism of the transformation was studied by a crossover experiment.
Co-reporter:Yongxiang Liu, Jia Guo, Yang Liu, Xiaoyu Wang, Yanshi Wang, Xinyu Jia, Gaofei Wei, Lizhu Chen, Jianyong Xiao and Maosheng Cheng
Chemical Communications 2014 vol. 50(Issue 47) pp:6243-6245
Publication Date(Web):17 Mar 2014
DOI:10.1039/C4CC00464G
A novel strategy for the synthesis of multisubstituted naphthalenes was developed via a Au(I)-catalyzed alkyne alkoxylation/dienolether aromaticity-driven cascade cyclization using 1,5-enyne substrates. The functional group toleration was examined by synthesizing a series of substrates and the mechanism was also studied based on intermediates isolated through deuterium labeling experiments.
Co-reporter:Dong-Mei Zhao, Wen-Yan Li, Yu-Fang Shi, Xu-Qiong Xiong, Shuai Song, Chen-Zhou Hao, Mao-Sheng Cheng, Jing-Kang Shen
Chinese Chemical Letters 2014 Volume 25(Issue 2) pp:299-304
Publication Date(Web):February 2014
DOI:10.1016/j.cclet.2013.11.033
Cholesteryl ester transfer protein (CETP) is a plasma glycoprotein that plays an important role in decreasing high-density lipoprotein cholesterol (HDL-C) levels and increasing low-density lipoprotein cholesterol (LDL-C) levels. Inhibition of CETP may be a new therapy for treating atherosclerosis. Herein, we report the development of a ligand-based pharmacophore model and pharmacophore-based virtual screening of the ZINC/big-n-greasy database, leading to the identification of compound H-10 as a potential CETP inhibitor in vitro. Based on H-10, a series of 3-((3,4-dichlorophenyl)(4-substituted benzyl)amino) propanamides were designed, synthesized, and evaluated against CETP. Compound 4l was found to have the best activity, resulting in 85.0% inhibition of CETP at 10 μmol/L.A ligand-based pharmacophore with 5 features was developed and used to database screening. A novel series of 3-((3,4-dichlorophenyl)(4-substituted benzyl)amino) propanamides were designed based on H-10 which is a potential CETP inhibitor obtained by pharmacophore-based database screening. Compound 4l was found to be a preferable CETP inhibitor with inhibition rate of 85.0%.
Co-reporter:Rui-Juan Li;Jian Wang;Zhen Xu;Wan-Xu Huang;Jia Li;Sheng-Fei Jin; Dong-Mei Zhao; Mao-Sheng Cheng
ChemMedChem 2014 Volume 9( Issue 5) pp:1012-1022
Publication Date(Web):
DOI:10.1002/cmdc.201400016
Abstract
p21-Activated kinase 4 (PAK4) is a serine/threonine protein kinase that plays important roles in a wide variety of human diseases including cancer. Targeting this kinase with specific inhibitors is of great interest in the treatment of cancer. In this study, PAK4 and its interaction with ATP-competitive inhibitors was investigated by a combined ligand- and structure-based approach. First, a ligand-based pharmacophore model was generated, consisting of five chemical features: a positive ionizable center, two hydrophobic groups, a hydrogen bond donor, and a hydrogen bond acceptor, which is consistent with available SAR information. The characteristics of the active site were then described as a topological region and used in docking of nine selected inhibitors. Combination of the pharmacophore model and results from the docking studies allowed us to weigh the various pharmacophore features and to identify the positive ionizable center as a spacer rather than an essential point. This research led to the proposal of an interaction model inside the PAK4 active site and provided guidance for the design of more potent PAK4 inhibitors.
Co-reporter:Li Ren;Yang Liu;Guihua Yu;Yuan Gao;Xin Liu
Chemical Research in Chinese Universities 2014 Volume 30( Issue 4) pp:639-643
Publication Date(Web):2014 August
DOI:10.1007/s40242-014-3522-3
Eleven novel triterpenoid saponins, N-substituted-β-D-glucosaminide derivatives of oleanolic acid, were designed and synthesized via a stepwise glycosylation strategy. These compounds were evaluated for in vitro cytotoxic activity against six different tumor cell lines. Most of the compounds inhibited the growth of, at least, one tumor cell line effectively at micromolar concentrations. Preliminary structure-activity relationships(SARs) indicate that acylation of the nitrogen of the glucosamine-bearing triterpenoid saponins affords the compounds that are highly cytotoxic towards specific tumor cell lines.
Co-reporter:Li Ren;Yang Liu;Gui-Hua Yu;Jing-Zheng Hao
Chemical Papers 2014 Volume 68( Issue 4) pp:525-530
Publication Date(Web):2014 April
DOI:10.2478/s11696-013-0482-x
I2 is an effective promoter for the synthesis of 2,3-unsaturated glycosides via Ferrier glycosylation. This reaction was used in the present work for the synthesis of O-glycosylated Fmoc amino acid building blocks. This metal-free reaction afforded the desired products with good to excellent yields with good α-selectivity.
Co-reporter:Fengrong Li, Dongmei Zhao, Jinhong Ren, Feiyue Hao, Guyue Liu, Shengfei Jin, Yongkui Jing, Maosheng Cheng
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 11) pp:3256-3261
Publication Date(Web):1 June 2013
DOI:10.1016/j.bmc.2013.03.044
To develop new CYP26A1 inhibitors, a three-cycle virtual screening was carried out based on the constructed homology model of human CYP26A1 using Dock, Fred, Gold and AutoDock. Twenty-two compounds exhibited high scores and reasonable binding modes in molecular docking were purchased from Specs Company. Eighteen compounds were tested their abilities to enhance ATRA-induced differentiation in human acute promyelocytic leukemia NB4 cells. Eight of them enhanced the ability of ATRA to induce differentiation at concentrations of 0.5 and 1 μM. Among these compounds, 2-(2-methylfuran-3-carboxamido)-3-phenylpropanoic acid (S8) is of most effective in blocking ATRA breaking down in NB4 cells based on the LC–MS/MS assay.
Co-reporter:Gang Wang, Jian Wang, Han Nie, Shen-Peng Tan, Wei-Qun Shi, Dong-Mei Zhao, Mao-Sheng Cheng
Journal of Molecular Structure 2013 Volume 1036() pp:372-379
Publication Date(Web):27 March 2013
DOI:10.1016/j.molstruc.2012.12.017
In the present study, 1-phenyl-2-thiobarbituric acid (1) was synthesized and the tautomerism of this compound was investigated by FT-IR spectroscopy, X-ray analysis and 1H NMR study as well as quantum chemical calculations. It is found that compound 1 exists in triketo form in the solid state and in CDCl3 solution while tautomerization was observed in DMSO-d6, DMF-d7 and CD3OD solution. The geometry optimization of eight possible tautomers of 1-phenyl-2-thiobarbituric acid was performed in gas phase and in different solvents using COSMO method. The calculated results are in good accordance with experimental data. Additionally, the methylation reaction of 1-phenyl-2-thiobarbituric acid was investigated for the first time by combination of experimental and theoretical methods. The methylation product might be varied with three possible isomers because of the structural features of 1-phenyl-2-thiobarbituric acid. We identified the obtained methylation product by X-ray diffraction and FT-IR spectroscopy, and transition state search and energy calculation were conducted to elucidate experimental data, the results revealed that the theoretical calculations are consistent with experimental data.Highlights► Synthesis and structural identification of thiobarbituric derivatives are presented. ► Tautomerism form was investigated by experimental methods and quantum calculations. ► The methylation site was identified experimentally and theoretically.
Co-reporter:Chao Ma;Anhui Wu;Yongqi Wu;Xuhong Ren;Maosheng Cheng
Archiv der Pharmazie 2013 Volume 346( Issue 12) pp:891-900
Publication Date(Web):
DOI:10.1002/ardp.201300276
To find new H+/K+-ATPase inhibitors for the treatment of peptic ulcer disease, a series of novel N-aryl isothiourea derivatives were synthesized and their structures were identified by 1H NMR and GC-MS. The effects of these compounds on inhibiting gastric acid secretion were evaluated by the guinea pig stomach mucous membrane study with pantoprazole magnesium as a positive control. The results showed that, of the 37 N-aryl isothiourea compounds synthesized, 20 compounds have comparable or stronger gastric acid inhibitory activities than that of pantoprazole magnesium. The quantitative structure–activity relationships (QSARs) of the N-aryl isothiourea compounds were also studied by comparative molecular field analysis (CoMFA) computation, and the model structure that was supposed to give more powerful bioactivities was finally predicted.
Co-reporter:Hong-Bo Xie, Jian Wang, Yu Sha, Mao-Sheng Cheng
Biophysical Chemistry 2013 s 180–181() pp: 1-9
Publication Date(Web):
DOI:10.1016/j.bpc.2013.05.004
Co-reporter:Na Yang;Jian Wang;Zhi-Wei Wang;Qing-He Wang;Hong-Guang Yang;Xiu-Jun Wang
Chemical Biology & Drug Design 2012 Volume 79( Issue 6) pp:1063-1071
Publication Date(Web):
DOI:10.1111/j.1747-0285.2012.01375.x
Inosine 5′-monophosphate dehydrogenase (IMPDH) is a key enzyme in the de novo synthesis of guanosine nucleotides. It is considered as an important target in the quest for drugs in the immunosuppressive, antiviral, antibacterial, and anticancer therapeutic areas. Herein, we report the 3D-QSAR analyses using comparative molecular field analysis (CoMFA), comparative molecular similarity indices analysis (CoMSIA) and docking on mycophenolic acid derivates for the first time. We obtained cross-validated q2 value of 0.805 for CoMFA and 0.620 for CoMSIA, while the non-cross-validated r2 values for them were 0.969 and 0.935, respectively. Based on the CoMFA and CoMSIA contour maps and docking analyses, some key structural factors responsible for inhibitory activity were revealed. The results obtained from this study could be used for the rational design of potent inhibitors against IMPDH.
Co-reporter:Hai Shang;Hong Chen;Dongmei Zhao;Xiaowei Tang;Yongfeng Liu;Li Pan;Maosheng Cheng
Archiv der Pharmazie 2012 Volume 345( Issue 1) pp:43-48
Publication Date(Web):
DOI:10.1002/ardp.201100094
Abstract
A series of 4α/4β-imidazolyl podophyllotoxin analogues have been designed and synthesized. All of the compounds were evaluated for their anticancer activity against a panel of three human cancer cell lines. Within the cell lines tested, some of the synthesized compounds showed promising anticancer activity. Compound 12, in particular, exhibited remarkable cytotoxicity, demonstrating effects against all tumor cell lines, including the K562/ADM cell line.
Co-reporter:Yu Tian;Chao Ma;Linyin Feng;Lei Zhang;Feiyue Hao;Li Pan;Maosheng Cheng
Archiv der Pharmazie 2012 Volume 345( Issue 6) pp:423-430
Publication Date(Web):
DOI:10.1002/ardp.201100424
Abstract
A series of novel (−)-1,2,3,9-tetrahydropyrrolo[2,1-b]quinazoline-1-carboxylic acid derivatives were designed and synthesized. All of the prepared compounds were screened for their neuroprotective effects using an in vitro oxygen glucose deprivation (OGD) model of ischemic stroke. Some of the target compounds exhibited moderate to excellent protective potency. In particular, compounds 9d, 9e, 9g, and 9h showed significant protective effects in the SH-SY5Y cell line at all three concentrations tested.
Co-reporter:Xiu-Jun Wang, Na-Ying Chu, Qin-He Wang, Chao Liu, Chun-guo Jiang, Xiao-Yu Wang, Takashi Ikejima, Mao-Sheng Cheng
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 19) pp:6297-6300
Publication Date(Web):1 October 2012
DOI:10.1016/j.bmcl.2012.06.102
In this study, a new series of bis-benzimidazole derivatives were designed and synthesized. Most of these new compounds showed significant anti-tumor activity in vitro compared to Hoechst 33258. Among them, the most potent compound 8 had the IC50 values of 0.56 μM for HL60 (Human promyelocytic leukemia cells) tumor cell line and 0.58 μM for U937 (Human leukemic monocyte lymphoma cells) tumor cell line. Subsequent toxicity study on human peripheral blood mononuclear cells (PBMC) showed that compound 8 exhibited less toxicity than 5-FU. We also found that apoptosis and autophagy were simultaneously induced by compound 8 in HL60 cells, and inhibition of autophagy by 3-MA decreased compound 8-induced apoptosis, indicating that they acted in synergy to exert tumor cell death.A new series of bis-benzimidazole derivatives were designed and synthesized. They showed significant anti-tumor activity in vitro compared to Hoechst 33258. Among them, the most potent compound 8 exhibited less toxicity than 5-FU on human PBMC. Compound 8 also induced autophagy and apoptosis in HL60 cells.
Co-reporter:Bo Wang, Yang Liu, Yanshi Wang, Xin Liu, Mao-Sheng Cheng
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 23) pp:7110-7113
Publication Date(Web):1 December 2012
DOI:10.1016/j.bmcl.2012.09.075
Twenty-four diosgenyl saponins bearing cinnamoyl, carbamido and thiosemicarbazone groups were synthesized concisely. The cytotoxicities of the synthetic compounds on six human caner cell lines were evaluated employing MTT method. Structure–activity relationship could be observed, and two of the synthesized compounds (5c and 5f) exhibited selective inhibition on HeLa and MCF-7 cells, while three of them (5d, 5f and 5h) showed strong inhibition against HT1080.
Co-reporter:Na Yang, Qing-He Wang, Wen-Qian Wang, Jian Wang, Feng Li, Shen-Peng Tan, Mao-Sheng Cheng
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 1) pp:53-56
Publication Date(Web):1 January 2012
DOI:10.1016/j.bmcl.2011.11.078
The synthesis and biological evaluation of a series of novel isobenzofuran-based compounds are described. The compounds were evaluated for their immunosuppressive effects of T-cell proliferation and IMPDH type II inhibitor activity in vitro, as well as their structure–activity relationships were assessed. Several compounds demonstrated highly efficacious immunosuppressive properties, especially compounds 2d, 2e, 2h and 2j, which were superior to MPA, while compounds 2k, 2m, 2n, 4c and 5d exhibited an equipotent inhibitory activity compared to MPA. Generally, it was obviously demonstrated that α,β-unsaturated amides proved more potent than the diamide and urea series. The present study provides a guide for further research on development of safe and effective immunosuppressive agents.
Co-reporter:Jian Wang;Shao-Jie Wang;Dan-Dan Song;Yong-Kui Jing
Medicinal Chemistry Research 2012 Volume 21( Issue 10) pp:2823-2826
Publication Date(Web):2012 October
DOI:10.1007/s00044-011-9806-y
Glutathione S-transferase P1-1 (GSTP1-1, EC 2.5.1.18) is generally conceived to be an important player in the acquired resistance of tumor to anticancer agents, thus a series of quinolinoneacetic acid derivatives were synthesized and their inhibitory effects on GSTP-1 were tested in this study. Interestingly, it was found that some compounds showed remarkable inhibitory effect on GSTP-1 activity. Furthermore, molecular docking study was conducted to provide the binding modes of quinolinoneacetic acid derivatives on GSTP1-1 activity. In summary, we present the discovery of a series of quinolinoneacetic acid compounds as inhibitors of GSTP1-1 and their interaction modes toward GSTP1-1 binding site.
Co-reporter:Na Yang;Jian Wang;Jian Li;Qing-He Wang;Yu Wang
Chemical Biology & Drug Design 2011 Volume 78( Issue 1) pp:175-182
Publication Date(Web):
DOI:10.1111/j.1747-0285.2011.01128.x
Inosine 5′-monophosphate dehydrogenase (IMPDH) is a key enzyme in the de novo synthesis of guanosine nucleotides. It is considered an important target in the quest for drugs in the immunosuppressive, antiviral, antibacterial and anticancer therapeutic areas. In this study, a chemical feature-based pharmacophore model of IMPDH inhibitors has been firstly developed with the aid of the HypoRefine protocol within Discovery Studio 2.5. The best model for IMPDH inhibitors, Hypo1-1, was characterized by the best correlation coefficient (0.97595) and the lowest RMSD (0.582058). It consisted of one hydrogen-bond donor, one hydrogen-bond acceptor, one aromatic ring and one hydrophobic feature, as well as two excluded volumes. The model was validated using a wide range of test molecules and a cross-validation. Furthermore, the pharmacophore features were confirmed by molecular docking studies. The pharmacophore model could quantitatively predict inhibitor activity and identify highly potent molecules. Therefore, the present results could be valuable for the discovery and development of specific IMPDH inhibitors.
Co-reporter:Yan-Hui Yang, Mao-Sheng Cheng, Qing-He Wang, Han Nie, Na Liao, Jian Wang, Hong Chen
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 4) pp:1808-1812
Publication Date(Web):April 2009
DOI:10.1016/j.ejmech.2008.07.021
A novel symmetrical bis-benzimidazole was designed as DNA minor groove binder. Molecular modeling study showed that it could dock into the minor groove of DNA. Several derivatives were synthesized and confirmed by IR, MS, and 1H NMR. All these novel compounds were screened for cytotoxic activity on SKOV-3, HeLa, and BGC-823 cell lines in vitro. Some compounds showed IC50s in the single-digit micromolar range for cytotoxicity in several tumor cell lines.2,2′-Di-[(substituted-pyrid-2-yl)-methylenethio]-5,5′-bis-1H,1′H-benzimidazole was designed and compound 16 showed micromolar cytotoxicity on SKOV-3, HeLa, and BGC-823 cell lines in preliminary in vitro anti-tumor test.
Co-reporter:Jian Wang;Shaojie Wang;Dan Song;Dongmei Zhao;Yu Sha;Yuting Jiang;Yongkui Jing;Maosheng Cheng
Chemical Biology & Drug Design 2009 Volume 73( Issue 5) pp:511-514
Publication Date(Web):
DOI:10.1111/j.1747-0285.2009.00807.x
Resistance to chemotherapeutic drugs has long been a considerable barrier to successful treatment of many cancers and over-expression of glutathione S-transferase P1-1 is correlated to carcinogenesis and resistance of cancer cells against chemotherapeutic agents. This study throws light on the role of chalcone derivatives, a new class of glutathione S-transferase P1-1 inhibitors potentially to overcome glutathione S-transferase P1-1-mediated chemotherapy resistance. Nineteen α-substituted chalcone derivatives were synthesized and their in vitro inhibitory effects on glutathione S-transferase P1-1 were determined. We interestingly found that most of these compounds showed inhibitory effect on glutathione S-transferase P1-1 activity. In addition, molecular field-based similarity analysis provides the necessary three-dimensional molecular field properties of α, β-unsaturated carbonyl derivatives to inhibit glutathione S-transferase P1-1 activity. Thus, these compounds have great potential to be developed into novel chemotherapeutic sensitizers.
Co-reporter:Yang Liu, Dong-Mei Zhao, Xue-Hua Lu, Hui Wang, Hong Chen, Ying Ke, Ling Leng, Mao-Sheng Cheng
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 1) pp:156-160
Publication Date(Web):1 January 2007
DOI:10.1016/j.bmcl.2006.09.071
A bisdesmosidic steroidal saponins library, composed of 16 novel kryptogenin glycosides, was set up via six random glycosylation procedures, wherein two compounds showed their antitumor activity against HeLa cell in the preliminary pharmacological research.A library of 16 novel bisdesmosidic kryptogenyl saponins was built for investigation of antitumor activity, wherein two compounds showed inhibition against HeLa cells (IC50 = 4.25 μM for 7a and 8.36 μM for 7i).
Co-reporter:Mao-Cai Yan, Yang Liu, Hong Chen, Ying Ke, Qing-Chun Xu, Mao-Sheng Cheng
Bioorganic & Medicinal Chemistry Letters 2006 Volume 16(Issue 16) pp:4200-4204
Publication Date(Web):15 August 2006
DOI:10.1016/j.bmcl.2006.05.086
Two natural triterpenoid saponins bearing N-acetylglucosamine, lotoidoside D and lotoidoside E, which had been available only from Glinus lotoides growing in Egyptian desert, were facilely synthesized from readily available oleanolic acid. Preliminary pharmacological research showed their antitumor activity against HeLa cell.Two antiproliferative triterpenoid saponins bearing N-acetylglucosamine were facilely synthesized. Antitumor activity was preliminarily investigated (IC50 = 2.74 μM against HeLa cell).
Co-reporter:Yongxiang Liu, Shengfei Jin, Yanshi Wang, Shanshan Cui, Xiaoshi Peng, Yuanyuan Niu, Chuan Du and Maosheng Cheng
Chemical Communications 2016 - vol. 52(Issue 37) pp:NaN6236-6236
Publication Date(Web):2016/03/09
DOI:10.1039/C6CC01770C
A gold(I)-catalyzed substituent-controlled strategy for the stereoselective synthesis of bicyclic furan and pyran derivatives has been developed. The mechanisms of the reactions have been studied thoroughly by deuterium labelling experiments. The applications of the methodologies were demonstrated by evaluating the preliminary anti-fungal activity of the synthetic compounds.
Co-reporter:Gaofei Wei, Weijing Luan, Shuai Wang, Shanshan Cui, Fengran Li, Yongxiang Liu, Yang Liu and Maosheng Cheng
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 5) pp:NaN1514-1514
Publication Date(Web):2014/11/18
DOI:10.1039/C4OB01605J
A series of novel oleanolic acid coupled 1,2,3-triazole derivatives have been designed and synthesized by employing a Cu(I) catalyzed Huisgen 1,3-dipolar cycloaddition reaction. The anti-proliferative evaluation indicated that some compounds exhibited excellent anti-cancer activity against the examined cancer cell lines. Among all derivatives, compound 3t possesses the best inhibitory activity against HT1080 cells. A series of pharmacology experiments show that compound 3t significantly induced HT1080 cell apoptosis. Therefore, this compound can serve as a promising lead candidate for further study.
Co-reporter:Yongxiang Liu, Jia Guo, Yang Liu, Xiaoyu Wang, Yanshi Wang, Xinyu Jia, Gaofei Wei, Lizhu Chen, Jianyong Xiao and Maosheng Cheng
Chemical Communications 2014 - vol. 50(Issue 47) pp:NaN6245-6245
Publication Date(Web):2014/03/17
DOI:10.1039/C4CC00464G
A novel strategy for the synthesis of multisubstituted naphthalenes was developed via a Au(I)-catalyzed alkyne alkoxylation/dienolether aromaticity-driven cascade cyclization using 1,5-enyne substrates. The functional group toleration was examined by synthesizing a series of substrates and the mechanism was also studied based on intermediates isolated through deuterium labeling experiments.

![PYRAZINECARBOXYLIC ACID, 3-[[(5-CHLORO-2-PYRIDINYL)AMINO]CARBONYL]-](http://img.cochemist.com/ccimg/43300/43200-83-5.png)
![PYRAZINECARBOXYLIC ACID, 3-[[(5-CHLORO-2-PYRIDINYL)AMINO]CARBONYL]-](http://img.cochemist.com/ccimg/43300/43200-83-5_b.png)
![6-(5-Chloropyridin-2-yl)-7-hydroxy-6,7-dihydro-5H-pyrrolo[3,4-b]pyrazin-5-one](http://img.cochemist.com/ccimg/43300/43200-81-3.png)
![6-(5-Chloropyridin-2-yl)-7-hydroxy-6,7-dihydro-5H-pyrrolo[3,4-b]pyrazin-5-one](http://img.cochemist.com/ccimg/43300/43200-81-3_b.png)






![BENZENE, 1,1',1''-[(1-PHENYLETHOXY)METHYLIDYNE]TRIS-](http://img.cochemist.com/ccimg/39900/39834-50-9.png)
![BENZENE, 1,1',1''-[(1-PHENYLETHOXY)METHYLIDYNE]TRIS-](http://img.cochemist.com/ccimg/39900/39834-50-9_b.png)
![BENZENE, 1-METHYL-4-[(TRIPHENYLMETHOXY)METHYL]-](http://img.cochemist.com/ccimg/39900/39834-48-5.png)
![BENZENE, 1-METHYL-4-[(TRIPHENYLMETHOXY)METHYL]-](http://img.cochemist.com/ccimg/39900/39834-48-5_b.png)












![8-(4-(4-(Pyrimidin-2-yl)piperazin-1-yl)butyl)-8-azaspiro[4.5]decane-7,9-dione](http://img.cochemist.com/ccimg/36600/36505-84-7.png)
![8-(4-(4-(Pyrimidin-2-yl)piperazin-1-yl)butyl)-8-azaspiro[4.5]decane-7,9-dione](http://img.cochemist.com/ccimg/36600/36505-84-7_b.png)









![Tetracosanamide,N-[(1S,2S,3R)-2,3-dihydroxy-1-(hydroxymethyl)heptadecyl]-](http://img.cochemist.com/ccimg/34500/34437-74-6.png)
![Tetracosanamide,N-[(1S,2S,3R)-2,3-dihydroxy-1-(hydroxymethyl)heptadecyl]-](http://img.cochemist.com/ccimg/34500/34437-74-6_b.png)


![(3WEI )-3-{[6-DEOXY-WEI -L-MANNOPYRANOSYL-(1->2)-[WEI -D-XYLOPYRANOSYL-(1->3)]-WEI -L-ARABINOPYRANOSYL]OXY}-20-HYDROXY-19-OXODAMMAR-24-EN-21-YL WEI -D-GLUCOPYRANOSIDE](http://img.cochemist.com/ccimg/33300/33228-65-8.png)
![(3WEI )-3-{[6-DEOXY-WEI -L-MANNOPYRANOSYL-(1->2)-[WEI -D-XYLOPYRANOSYL-(1->3)]-WEI -L-ARABINOPYRANOSYL]OXY}-20-HYDROXY-19-OXODAMMAR-24-EN-21-YL WEI -D-GLUCOPYRANOSIDE](http://img.cochemist.com/ccimg/33300/33228-65-8_b.png)







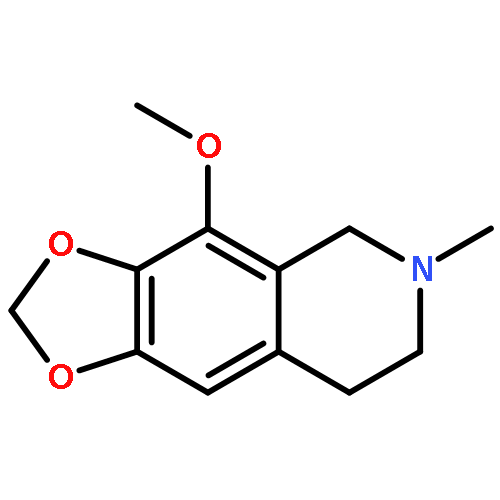
![1,3-Dioxolo[4,5-g]isoquinolinium,7,8-dihydro-4-methoxy-6-methyl-, iodide (1:1)](http://img.cochemist.com/ccimg/31000/30936-27-7.png)
![1,3-Dioxolo[4,5-g]isoquinolinium,7,8-dihydro-4-methoxy-6-methyl-, iodide (1:1)](http://img.cochemist.com/ccimg/31000/30936-27-7_b.png)













![Benzene, 1,1'-[(phenylmethoxy)methylene]bis-](http://img.cochemist.com/ccimg/26100/26059-49-4.png)
![Benzene, 1,1'-[(phenylmethoxy)methylene]bis-](http://img.cochemist.com/ccimg/26100/26059-49-4_b.png)






![Acetic acid, [(4-bromophenyl)amino]oxo-, ethyl ester](http://img.cochemist.com/ccimg/24500/24451-15-8.png)
![Acetic acid, [(4-bromophenyl)amino]oxo-, ethyl ester](http://img.cochemist.com/ccimg/24500/24451-15-8_b.png)




![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 6-bromo-3,3-dimethyl-7-oxo-, (2S,5R,6S)-](http://img.cochemist.com/ccimg/24200/24138-28-1.png)
![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 6-bromo-3,3-dimethyl-7-oxo-, (2S,5R,6S)-](http://img.cochemist.com/ccimg/24200/24138-28-1_b.png)
![1H-Benzimidazole,2-[[(4-chlorophenyl)methyl]thio]-](http://img.cochemist.com/ccimg/24000/23976-76-3.png)
![1H-Benzimidazole,2-[[(4-chlorophenyl)methyl]thio]-](http://img.cochemist.com/ccimg/24000/23976-76-3_b.png)
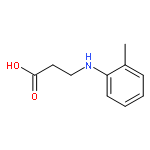
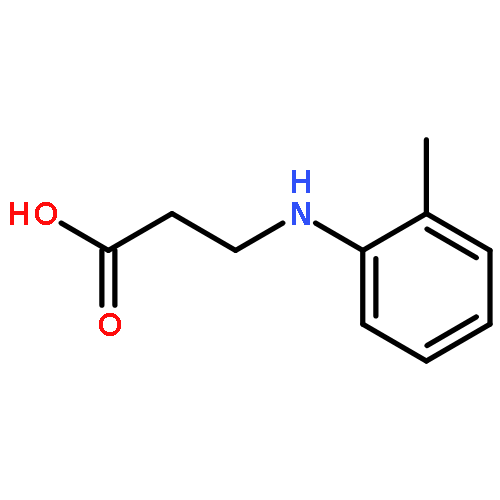







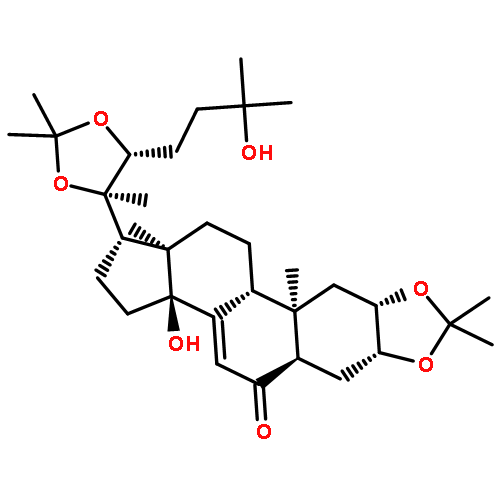
![Cholest-7-en-6-one,2,3,14,25-tetrahydroxy-20,22-[(1-methylethylidene)bis(oxy)]-, (2b,3b,5b,22R)-](http://img.cochemist.com/ccimg/22800/22798-96-5.png)
![Cholest-7-en-6-one,2,3,14,25-tetrahydroxy-20,22-[(1-methylethylidene)bis(oxy)]-, (2b,3b,5b,22R)-](http://img.cochemist.com/ccimg/22800/22798-96-5_b.png)
![Tricyclo[3.3.1.13,7]decan-1-ol, acetate](http://img.cochemist.com/ccimg/22700/22635-62-7.png)
![Tricyclo[3.3.1.13,7]decan-1-ol, acetate](http://img.cochemist.com/ccimg/22700/22635-62-7_b.png)


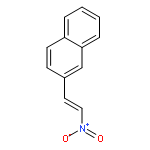
![Benzamide, N-[[(4-methoxyphenyl)amino]thioxomethyl]-](http://img.cochemist.com/ccimg/21300/21258-08-2.png)
![Benzamide, N-[[(4-methoxyphenyl)amino]thioxomethyl]-](http://img.cochemist.com/ccimg/21300/21258-08-2_b.png)
![N-[(2-chlorophenyl)carbamothioyl]benzamide](http://img.cochemist.com/ccimg/21300/21258-05-9.png)
![N-[(2-chlorophenyl)carbamothioyl]benzamide](http://img.cochemist.com/ccimg/21300/21258-05-9_b.png)

![5-Methyl-1-phenyl-1H-[1,2,3]triazole-4-carboxylic acid](http://img.cochemist.com/ccimg/20800/20725-32-0.png)
![5-Methyl-1-phenyl-1H-[1,2,3]triazole-4-carboxylic acid](http://img.cochemist.com/ccimg/20800/20725-32-0_b.png)
![Benzene, 1,1',1''-[(cyclohexyloxy)methylidyne]tris-](http://img.cochemist.com/ccimg/20800/20705-40-2.png)
![Benzene, 1,1',1''-[(cyclohexyloxy)methylidyne]tris-](http://img.cochemist.com/ccimg/20800/20705-40-2_b.png)




![Benzamide, N-[[(4-bromophenyl)amino]thioxomethyl]-](http://img.cochemist.com/ccimg/19300/19249-89-9.png)
![Benzamide, N-[[(4-bromophenyl)amino]thioxomethyl]-](http://img.cochemist.com/ccimg/19300/19249-89-9_b.png)


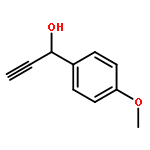
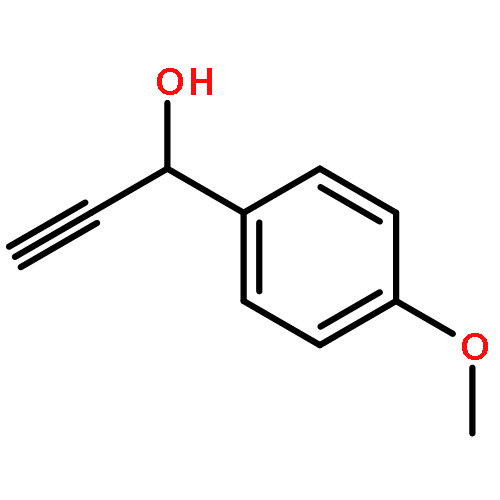




![Acetic acid,2-[(4-methylphenyl)amino]-2-oxo-, ethyl ester](http://img.cochemist.com/ccimg/18600/18522-98-0.png)
![Acetic acid,2-[(4-methylphenyl)amino]-2-oxo-, ethyl ester](http://img.cochemist.com/ccimg/18600/18522-98-0_b.png)
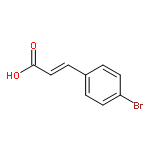
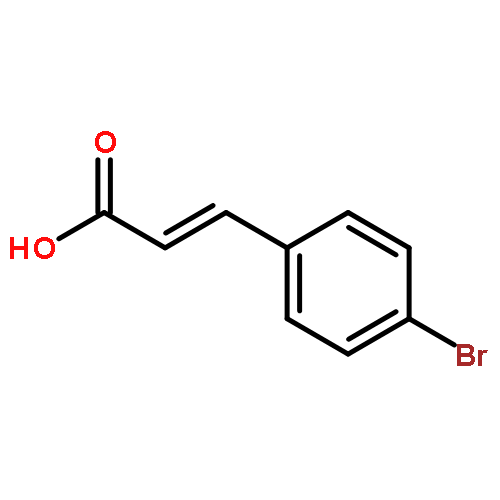






![[(cyclohexyloxy)methyl]benzene](http://img.cochemist.com/ccimg/16300/16224-09-2.png)
![[(cyclohexyloxy)methyl]benzene](http://img.cochemist.com/ccimg/16300/16224-09-2_b.png)















![4H-1-Benzopyran-4-one,3,5-dihydroxy-2-[3-hydroxy-4-(phenylmethoxy)phenyl]-7-(phenylmethoxy)-](http://img.cochemist.com/ccimg/13500/13459-13-7.png)
![4H-1-Benzopyran-4-one,3,5-dihydroxy-2-[3-hydroxy-4-(phenylmethoxy)phenyl]-7-(phenylmethoxy)-](http://img.cochemist.com/ccimg/13500/13459-13-7_b.png)

![Urea, [2-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/13200/13114-85-7.png)
![Urea, [2-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/13200/13114-85-7_b.png)
![[1,1'-Biphenyl]-4-carboxylicacid, 4'-fluoro-, ethyl ester](http://img.cochemist.com/ccimg/10600/10540-36-0.png)
![[1,1'-Biphenyl]-4-carboxylicacid, 4'-fluoro-, ethyl ester](http://img.cochemist.com/ccimg/10600/10540-36-0_b.png)









![N-[(4-methylphenyl)carbamothioyl]benzamide](http://img.cochemist.com/ccimg/6300/6281-61-4.png)
![N-[(4-methylphenyl)carbamothioyl]benzamide](http://img.cochemist.com/ccimg/6300/6281-61-4_b.png)






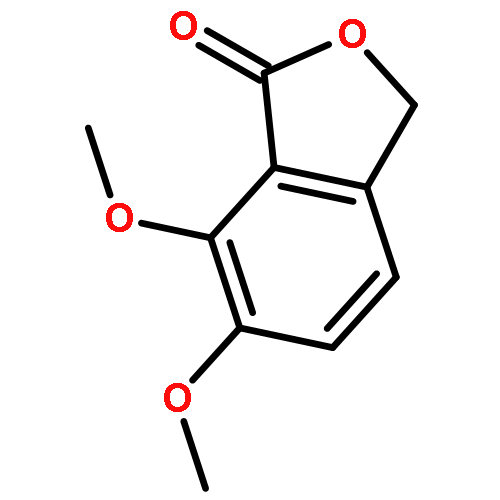
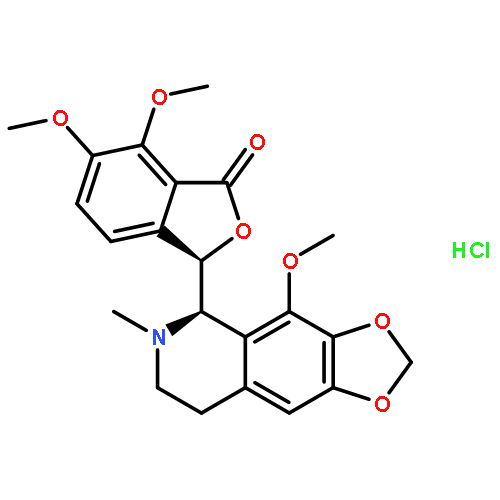
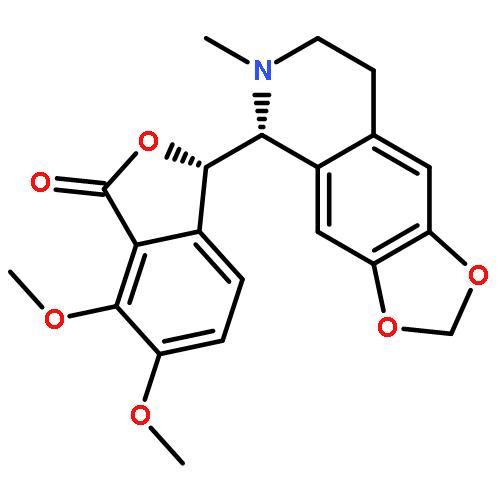
![(3R)-6,7-dimethoxy-3-[(5S)-4-methoxy-6-methyl-5,6,7,8-tetrahydro[1,3]dioxolo[4,5-g]isoquinolin-5-yl]-2-benzofuran-1(3H)-one](http://img.cochemist.com/ccimg/6100/6035-40-1.png)
![(3R)-6,7-dimethoxy-3-[(5S)-4-methoxy-6-methyl-5,6,7,8-tetrahydro[1,3]dioxolo[4,5-g]isoquinolin-5-yl]-2-benzofuran-1(3H)-one](http://img.cochemist.com/ccimg/6100/6035-40-1_b.png)




![Acetic acid,2-[(4-chlorophenyl)amino]-2-oxo-, ethyl ester](http://img.cochemist.com/ccimg/5400/5397-14-8.png)
![Acetic acid,2-[(4-chlorophenyl)amino]-2-oxo-, ethyl ester](http://img.cochemist.com/ccimg/5400/5397-14-8_b.png)
![N-[(2-bromophenyl)carbamothioyl]benzamide](http://img.cochemist.com/ccimg/5400/5391-29-7.png)
![N-[(2-bromophenyl)carbamothioyl]benzamide](http://img.cochemist.com/ccimg/5400/5391-29-7_b.png)




![N-[(2-methylphenyl)carbamothioyl]benzamide](http://img.cochemist.com/ccimg/5000/4949-88-6.png)
![N-[(2-methylphenyl)carbamothioyl]benzamide](http://img.cochemist.com/ccimg/5000/4949-88-6_b.png)
![BENZAMIDE, N-[[(3-METHYLPHENYL)AMINO]THIOXOMETHYL]-](http://img.cochemist.com/ccimg/5000/4949-87-5.png)
![BENZAMIDE, N-[[(3-METHYLPHENYL)AMINO]THIOXOMETHYL]-](http://img.cochemist.com/ccimg/5000/4949-87-5_b.png)
![N-[(4-chlorophenyl)carbamothioyl]benzamide](http://img.cochemist.com/ccimg/5000/4921-83-9.png)
![N-[(4-chlorophenyl)carbamothioyl]benzamide](http://img.cochemist.com/ccimg/5000/4921-83-9_b.png)
![2-Chloro-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/4900/4857-06-1.png)
![2-Chloro-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/4900/4857-06-1_b.png)

![Furo[3',4':6,7]naphtho[2,3-d]-1,3-dioxol-6(5aH)-one,5,8,8a,9-tetrahydro-9-hydroxy-5-(3,4,5-trimethoxyphenyl)-, (5R,5aR,8aR,9S)-](http://img.cochemist.com/ccimg/4400/4375-07-9.png)
![Furo[3',4':6,7]naphtho[2,3-d]-1,3-dioxol-6(5aH)-one,5,8,8a,9-tetrahydro-9-hydroxy-5-(3,4,5-trimethoxyphenyl)-, (5R,5aR,8aR,9S)-](http://img.cochemist.com/ccimg/4400/4375-07-9_b.png)




![4-(4,4,5,5-TETRAMETHYL-1,3,2-DIOXABOROLAN-2-YL)-2-{[4-(TRIFLUOROM<WBR />ETHYL)BENZYL]OXY}PYRIDINE](http://img.cochemist.com/ccimg/3700/3622-03-5.png)
![4-(4,4,5,5-TETRAMETHYL-1,3,2-DIOXABOROLAN-2-YL)-2-{[4-(TRIFLUOROM<WBR />ETHYL)BENZYL]OXY}PYRIDINE](http://img.cochemist.com/ccimg/3700/3622-03-5_b.png)



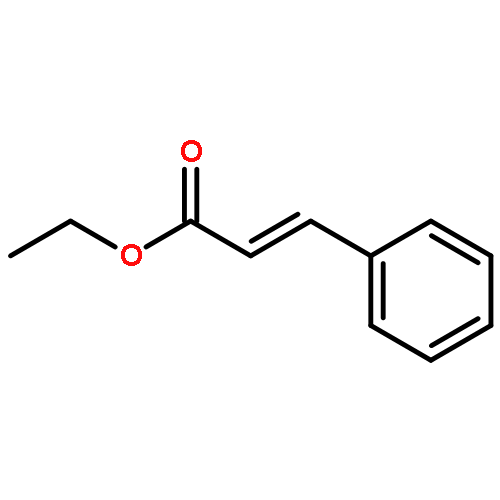
![Benzene,1-methoxy-4-[(phenylmethoxy)methyl]-](http://img.cochemist.com/ccimg/3700/3613-53-4.png)
![Benzene,1-methoxy-4-[(phenylmethoxy)methyl]-](http://img.cochemist.com/ccimg/3700/3613-53-4_b.png)


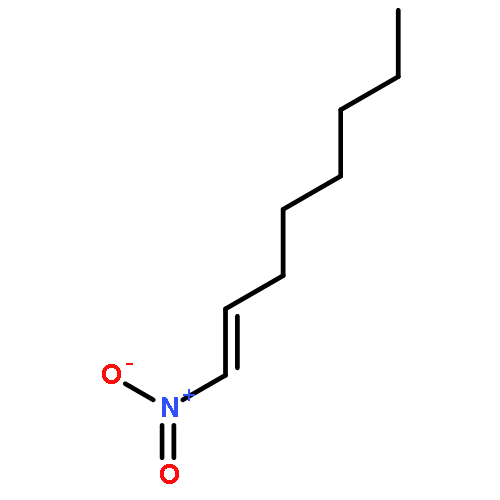
![Benzene, [(1,1-dimethylethoxy)methyl]-](http://img.cochemist.com/ccimg/3500/3459-80-1.png)
![Benzene, [(1,1-dimethylethoxy)methyl]-](http://img.cochemist.com/ccimg/3500/3459-80-1_b.png)
![2-[(2-METHYLPROPAN-2-YL)OXY]ETHYLBENZENE](http://img.cochemist.com/ccimg/3400/3354-66-3.png)
![2-[(2-METHYLPROPAN-2-YL)OXY]ETHYLBENZENE](http://img.cochemist.com/ccimg/3400/3354-66-3_b.png)



















![4H-1-Benzopyran-4-one,3,5,7-tris(acetyloxy)-2-[3,4-bis(acetyloxy)phenyl]-](http://img.cochemist.com/ccimg/1100/1064-06-8.png)
![4H-1-Benzopyran-4-one,3,5,7-tris(acetyloxy)-2-[3,4-bis(acetyloxy)phenyl]-](http://img.cochemist.com/ccimg/1100/1064-06-8_b.png)



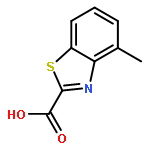
![6-Methoxybenzo[d]thiazole-2-carboxylic acid](http://img.cochemist.com/ccimg/1000/946-13-4.png)
![6-Methoxybenzo[d]thiazole-2-carboxylic acid](http://img.cochemist.com/ccimg/1000/946-13-4_b.png)











![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6_b.png)



![2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2R,3S,4R,5S,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydropyran-2-yl]oxy-chromen-4-one](http://img.cochemist.com/ccimg/500/482-35-9.png)
![2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2R,3S,4R,5S,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydropyran-2-yl]oxy-chromen-4-one](http://img.cochemist.com/ccimg/500/482-35-9_b.png)




![2-METHYL-2-PROPANYL 1-OXO-3-PHENYL-2-OXA-5-AZASPIRO[3.4]OCTANE-5-<WBR />CARBOXYLATE](http://img.cochemist.com/ccimg/400/366-69-8.png)
![2-METHYL-2-PROPANYL 1-OXO-3-PHENYL-2-OXA-5-AZASPIRO[3.4]OCTANE-5-<WBR />CARBOXYLATE](http://img.cochemist.com/ccimg/400/366-69-8_b.png)
![N-[BIS(4-FLUOROPHENYL)METHYLIDENE]HYDROXYLAMINE](http://img.cochemist.com/ccimg/400/363-02-0.png)
![N-[BIS(4-FLUOROPHENYL)METHYLIDENE]HYDROXYLAMINE](http://img.cochemist.com/ccimg/400/363-02-0_b.png)

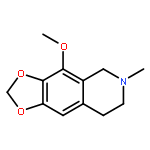
![1,3-Dioxolo[4,5-g]isoquinolin-5-ol,5,6,7,8-tetrahydro-4-methoxy-6-methyl-](http://img.cochemist.com/ccimg/100/82-54-2.png)
![1,3-Dioxolo[4,5-g]isoquinolin-5-ol,5,6,7,8-tetrahydro-4-methoxy-6-methyl-](http://img.cochemist.com/ccimg/100/82-54-2_b.png)












![QUINOLINE, 4-CHLORO-6-METHOXY-7-[3-(1-PIPERIDINYL)PROPOXY]-](http://img.cochemist.com/ccimg/861900/861881-05-2.png)
![QUINOLINE, 4-CHLORO-6-METHOXY-7-[3-(1-PIPERIDINYL)PROPOXY]-](http://img.cochemist.com/ccimg/861900/861881-05-2_b.png)


![2-Propenoic acid, 3-[4-(phenylmethoxy)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/227200/227105-11-5.png)
![2-Propenoic acid, 3-[4-(phenylmethoxy)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/227200/227105-11-5_b.png)
![Quinoline, 4-chloro-6-methoxy-7-[3-(4-morpholinyl)propoxy]-](http://img.cochemist.com/ccimg/205500/205448-32-4.png)
![Quinoline, 4-chloro-6-methoxy-7-[3-(4-morpholinyl)propoxy]-](http://img.cochemist.com/ccimg/205500/205448-32-4_b.png)
![L-Proline, 1-[(2-nitrophenyl)methyl]-5-oxo-](http://img.cochemist.com/ccimg/182900/182816-05-3.png)
![L-Proline, 1-[(2-nitrophenyl)methyl]-5-oxo-](http://img.cochemist.com/ccimg/182900/182816-05-3_b.png)



















![3-nitro-4-(phenylmethoxy)phenyl]-oxirane](http://img.cochemist.com/ccimg/51600/51582-41-3.png)
![3-nitro-4-(phenylmethoxy)phenyl]-oxirane](http://img.cochemist.com/ccimg/51600/51582-41-3_b.png)
![Benzenemethanol,4-amino-3,5-dichloro-a-[[(1,1-dimethylethyl)amino]methyl]-, (aS)-](http://img.cochemist.com/ccimg/50500/50499-60-0.png)
![Benzenemethanol,4-amino-3,5-dichloro-a-[[(1,1-dimethylethyl)amino]methyl]-, (aS)-](http://img.cochemist.com/ccimg/50500/50499-60-0_b.png)


![[(2s)-2-(3-formamido-4-hydroxyphenyl)-2-hydroxyethyl]-[(2s)-1-(4-methoxyphenyl)propan-2-yl]azanium](http://img.cochemist.com/ccimg/43300/43229-80-7.png)
![[(2s)-2-(3-formamido-4-hydroxyphenyl)-2-hydroxyethyl]-[(2s)-1-(4-methoxyphenyl)propan-2-yl]azanium](http://img.cochemist.com/ccimg/43300/43229-80-7_b.png)









![4'-Fluoro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/5800/5731-10-2.png)
![4'-Fluoro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/5800/5731-10-2_b.png)





![4H-PYRAZOLO[1,5-C][1,3]THIAZOLE-2-CARBALDEHYDE](http://img.cochemist.com/ccimg/3300/3296-03-5.png)
![4H-PYRAZOLO[1,5-C][1,3]THIAZOLE-2-CARBALDEHYDE](http://img.cochemist.com/ccimg/3300/3296-03-5_b.png)







![PYRROLO[2,1-B]QUINAZOLINE-1-CARBOXYLIC ACID, 1,2,3,9-TETRAHYDRO-, (-)-](http://img.cochemist.com/ccimg/481800/481710-98-9.png)
![PYRROLO[2,1-B]QUINAZOLINE-1-CARBOXYLIC ACID, 1,2,3,9-TETRAHYDRO-, (-)-](http://img.cochemist.com/ccimg/481800/481710-98-9_b.png)



![2-(3,5-DIMETHOXYPHENYL)-6-ETHYL-5-METHYLPYRAZOLO[1,5-A]PYRIMIDIN-7-OL](http://img.cochemist.com/ccimg/149800/149707-75-5.png)
![2-(3,5-DIMETHOXYPHENYL)-6-ETHYL-5-METHYLPYRAZOLO[1,5-A]PYRIMIDIN-7-OL](http://img.cochemist.com/ccimg/149800/149707-75-5_b.png)
![Benzenemethanol, 4-amino-3,5-dichloro-α-[[(1,1-dimethylethyl)amino]methyl]-](/data/chemimg/568900/37148-27-9.png)
![Benzenemethanol, 4-amino-3,5-dichloro-α-[[(1,1-dimethylethyl)amino]methyl]-](/data/chemimg/568900/37148-27-9_b.png)
![(3R,4R)-3-(5,6-Dihydro-4H-pyrrolo[3,2,1-ij]quinolin-1-yl)-4-(1H-indol-3-yl)pyrrolidine-2,5-dione](http://img.cochemist.com/ccimg/905900/905854-02-6.png)
![(3R,4R)-3-(5,6-Dihydro-4H-pyrrolo[3,2,1-ij]quinolin-1-yl)-4-(1H-indol-3-yl)pyrrolidine-2,5-dione](http://img.cochemist.com/ccimg/905900/905854-02-6_b.png)
![Pyrrolo[3,4-c]pyrazole-5(1H)-carboxamide, N-[(1S)-2-(dimethylamino)-1-phenylethyl]-4,6-dihydro-6,6-dimethyl-3-[(2-methylthieno[3,2-d]pyrimidin-4-](/data/chemimg/2242900/898044-15-0.png)
![Pyrrolo[3,4-c]pyrazole-5(1H)-carboxamide, N-[(1S)-2-(dimethylamino)-1-phenylethyl]-4,6-dihydro-6,6-dimethyl-3-[(2-methylthieno[3,2-d]pyrimidin-4-](/data/chemimg/2242900/898044-15-0_b.png)










![L-Tyrosine, N-[(9H-fluoren-9-ylmethoxy)carbonyl]-, phenylmethyl ester](http://img.cochemist.com/ccimg/83000/82911-77-1.png)
![L-Tyrosine, N-[(9H-fluoren-9-ylmethoxy)carbonyl]-, phenylmethyl ester](http://img.cochemist.com/ccimg/83000/82911-77-1_b.png)