Co-reporter:Sally Higson, Fabiana Subrizi, Tom D. Sheppard and Helen C. Hailes
Green Chemistry 2016 vol. 18(Issue 7) pp:1855-1858
Publication Date(Web):07 Jan 2016
DOI:10.1039/C5GC02935J
One-pot synthetic routes from furfurals to polysubstituted aromatic compounds have been developed in water, without the need for any organic solvents. The reaction proceeds via an uncatalysed, one-pot reaction cascade through formation of a hydrazone derivative, in situ cycloaddition with a dienophile, then aromatisation. A range of substituted phthalimides can be accessed with complete control over the substitution pattern. The reaction was also extended to other dienophiles and the diene 2-furylacrolein. The phthalimide products were further elaborated to produce a variety of polysubstituted benzenes including pharmaceutically relevant compounds.
Co-reporter:Rachel M. Lanigan, Valerija Karaluka, Marco T. Sabatini, Pavel Starkov, Matthew Badland, Lee Boulton and Tom D. Sheppard
Chemical Communications 2016 vol. 52(Issue 57) pp:8846-8849
Publication Date(Web):22 Jun 2016
DOI:10.1039/C6CC05147B
A commercially available borate ester, B(OCH2CF3)3, can be used to achieve protecting-group free direct amidation of α-amino acids with a range of amines in cyclopentyl methyl ether. The method can be applied to the synthesis of medicinally relevant compounds, and can be scaled up to obtain gram quantities of products.
Co-reporter:Paul M. Murray, Fiona Bellany, Laure Benhamou, Dejan-Krešimir Bučar, Alethea B. Tabor and Tom D. Sheppard
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 8) pp:2373-2384
Publication Date(Web):24 Dec 2015
DOI:10.1039/C5OB01892G
This article outlines the benefits of using ‘Design of Experiments’ (DoE) optimisation during the development of new synthetic methodology. A particularly important factor in the development of new chemical reactions is the choice of solvent which can often drastically alter the efficiency and selectivity of a process. Whilst solvent optimisation is usually done in a non-systematic way based upon a chemist's intuition and previous laboratory experience, we illustrate how optimisation of the solvent for a reaction can be carried out by using a ‘map of solvent space’ in a DoE optimisation. A new solvent map has been developed specifically for optimisation of new chemical reactions using principle component analysis (PCA) incorporating 136 solvents with a wide range of properties. The new solvent map has been used to identify safer alternatives to toxic/hazardous solvents, and also in the optimisation of an SNAr reaction.
Co-reporter:Samantha M. Gibson, Rachel M. Lanigan, Laure Benhamou, Abil E. Aliev and Tom D. Sheppard
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 34) pp:9050-9054
Publication Date(Web):29 Jul 2015
DOI:10.1039/C5OB01398D
In this paper we describe the use of a chiral aldehyde derived from lactate esters for determining the enantiopurity of primary amines, via the formation of diastereomeric imines. The method was shown to be suitable for reproducibly determining the enantiopurity of a diverse set of chiral amines. Both enantiomers of the aldehyde can be prepared in two steps from commercially available materials.
Co-reporter:Valerija Karaluka, Rachel M. Lanigan, Paul M. Murray, Matthew Badland and Tom D. Sheppard
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 44) pp:10888-10894
Publication Date(Web):14 Sep 2015
DOI:10.1039/C5OB01801C
The use of B(OCH2CF3)3 for mediating direct amidation reactions of a wide range of pharmaceutically relevant carboxylic acids and amines is described, including numerous heterocycle-containing examples. An initial screen of solvents for the direct amidation reaction suggested that cyclopentyl methyl ether, a solvent with a very good safety profile suitable for use over a wide temperature range, was an excellent replacement for the previously used solvent acetonitrile. Under these conditions amides could be prepared from 18 of the 21 carboxylic acids and 18 of the 21 amines examined. Further optimisation of one of the low yielding amidation reactions (36% yield) via a design of experiments approach enabled an 84% yield of the amide to be obtained.
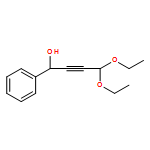
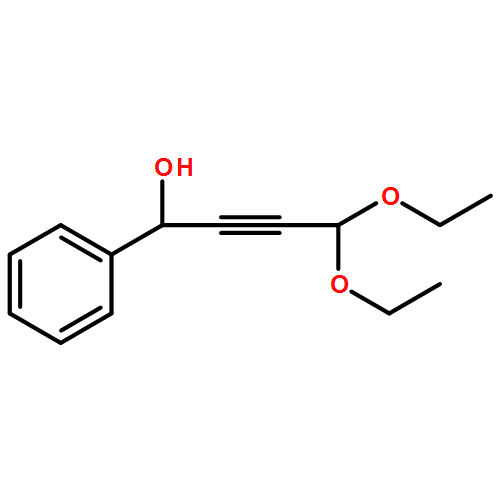
Co-reporter:Matthew N. Pennell, Robert W. Foster, Peter G. Turner, Helen C. Hailes, Christopher J. Tame and Tom D. Sheppard
Chemical Communications 2014 vol. 50(Issue 11) pp:1302-1304
Publication Date(Web):18 Dec 2013
DOI:10.1039/C3CC48290A
Synthetically important 3-alkoxyfurans can be prepared efficiently via treatment of acetal-containing propargylic alcohols (obtained from the addition of 3,3-diethoxypropyne to aldehydes) with 2 mol% gold catalyst in an alcohol solvent at room temperature. The resulting furans show useful reactivity in a variety of subsequent transformations.
Co-reporter:Dr. Persis Dhankher;Dr. Laure Benhamou ;Dr. Tom D. Sheppard
Chemistry - A European Journal 2014 Volume 20( Issue 41) pp:13375-13381
Publication Date(Web):
DOI:10.1002/chem.201403940
Abstract
Herein, we report the application of allyl acetate to the palladium-catalysed dearomatising diallylation of indoles. The reaction can be carried out by using a readily available palladium catalyst at room temperature, and can be applied to a wide range of substituted indoles to provide access to the corresponding 3,3-diallylindolinines. These compounds are versatile synthetic intermediates that readily undergo Ugi reactions or proline-catalysed asymmetric Mannich reactions. Alternatively, acylation of the 3,3-diallylindolinines with an acid chloride or a chloroformate, followed by treatment with aluminium chloride, enables 2,3-diallylindoles to be prepared. By using ring-closing metathesis, functionalised spirocyclic indoline scaffolds can be accessed from the Ugi products, and a dihydrocarbazole can be prepared from the corresponding 2,3-diallylindole.
Co-reporter:Jarryl M. D'Oyley;Dr. Abil E. Aliev ;Dr. Tom D. Sheppard
Angewandte Chemie International Edition 2014 Volume 53( Issue 40) pp:10747-10750
Publication Date(Web):
DOI:10.1002/anie.201405348
Abstract
The regioselective conversion of propargylic alcohols into previously unreported α,α-diiodo-β-hydroxyketones was achieved by treatment with N-iodosuccinimide in the presence of a gold catalyst. The corresponding α,α-dichloro-β-hydroxyketones were obtained by treatment with trichloroisocyanuric acid in the absence of a catalyst. The latter reaction can be extended to other alkynols. These transformations can be used to prepare potentially useful halogenated building blocks. Preliminary mechanistic studies suggest that the reaction involves participation of the acetonitrile solvent in the formation of a 5-halo-1,3-oxazine intermediate.
Co-reporter:Jarryl M. D'Oyley;Dr. Abil E. Aliev ;Dr. Tom D. Sheppard
Angewandte Chemie 2014 Volume 126( Issue 40) pp:10923-10926
Publication Date(Web):
DOI:10.1002/ange.201405348
Abstract
The regioselective conversion of propargylic alcohols into previously unreported α,α-diiodo-β-hydroxyketones was achieved by treatment with N-iodosuccinimide in the presence of a gold catalyst. The corresponding α,α-dichloro-β-hydroxyketones were obtained by treatment with trichloroisocyanuric acid in the absence of a catalyst. The latter reaction can be extended to other alkynols. These transformations can be used to prepare potentially useful halogenated building blocks. Preliminary mechanistic studies suggest that the reaction involves participation of the acetonitrile solvent in the formation of a 5-halo-1,3-oxazine intermediate.
Co-reporter:Robert W. Foster;Christopher J. Tame;Helen C. Hailes
Advanced Synthesis & Catalysis 2013 Volume 355( Issue 11-12) pp:2353-2360
Publication Date(Web):
DOI:10.1002/adsc.201300055
Co-reporter:Rachel M. Lanigan
European Journal of Organic Chemistry 2013 Volume 2013( Issue 33) pp:7453-7465
Publication Date(Web):
DOI:10.1002/ejoc.201300573
Abstract
The synthesis of amides is of huge importance in a wide variety of industrial and academic fields and is of particular significance in the synthesis of pharmaceuticals. Many of the well established methods for amide synthesis involve reagents that are difficult to handle and lead to the generation of large quantities of waste products. As a consequence, there has been a considerable amount of interest in the development of new approaches to amide synthesis. Over the past few years a wide range of new reagents and catalysts for direct amidation of carboxylic acids have been reported. In addition, the interconversion of amide derivatives through transamidation is emerging as a potential alternative strategy for accessing certain amides. This microreview covers recent developments in the direct amidation of carboxylic acids and the interconversion of amides through transamidation. The advantages and disadvantages of the various methods are discussed, as well as the possible mechanisms of the reactions.
Co-reporter:Rachel M. Lanigan, Pavel Starkov, and Tom D. Sheppard
The Journal of Organic Chemistry 2013 Volume 78(Issue 9) pp:4512-4523
Publication Date(Web):April 16, 2013
DOI:10.1021/jo400509n
B(OCH2CF3)3, prepared from readily available B2O3 and 2,2,2-trifluoroethanol, is as an effective reagent for the direct amidation of a variety of carboxylic acids with a broad range of amines. In most cases, the amide products can be purified by a simple filtration procedure using commercially available resins, with no need for aqueous workup or chromatography. The amidation of N-protected amino acids with both primary and secondary amines proceeds effectively, with very low levels of racemization. B(OCH2CF3)3 can also be used for the formylation of a range of amines in good to excellent yield, via transamidation of dimethylformamide.
Co-reporter:Pavel Starkov;Filippo Rota;Jarryl M. D'Oyley
Advanced Synthesis & Catalysis 2012 Volume 354( Issue 17) pp:3217-3224
Publication Date(Web):
DOI:10.1002/adsc.201200491
Abstract
In the presence of a cationic gold(I) catalyst and N-halosuccinimide, both trimethylsilyl-protected and terminal alkynes are converted into alkynyl halides. Further experiments showed that silyl-protected alkynes undergo electrophilic iodination and bromination under Brønsted acid catalysis, whilst terminal alkynes require a cationic gold catalyst. The former reactions probably proceed via activation of the electrophile, whilst the latter reactions proceed via a gold(I) acetylide intermediate. Gold-catalysed halogenation was further combined with gold-catalysed hydration and subsequent annulation to provide convenient routes to iodomethyl ketones and five-membered aromatic heterocycles.
Co-reporter:Martin Bachman, Sam E. Mann and Tom D. Sheppard
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 1) pp:162-170
Publication Date(Web):10 Oct 2011
DOI:10.1039/C1OB06534C
Four-component reactions between amino alcohols, aldehydes, isocyanides and thiols proceed rapidly under microwave or conventional heating at 60 °C in methanol. The reaction is successful with a wide range of components and gives access to potentially drug-like products containing amine, amide and thioether functionality in moderate to excellent yield. The reaction conditions are also applicable to the synthesis of a range of 8–10 membered medium ring lactonesvia three-component reactions of amino alcohols, isocyanides and acid-aldehydes. Incorporation of L-prolinol as the amino alcohol component in each case gives access to multicomponent products with moderate to high diastereoselectivity.
Co-reporter:Matthew N. Pennell;Dr. Peter G. Turner;Dr. Tom D. Sheppard
Chemistry - A European Journal 2012 Volume 18( Issue 15) pp:4748-4758
Publication Date(Web):
DOI:10.1002/chem.201102830
Abstract
A wide range of primary, secondary and tertiary propargylic alcohols undergo a Meyer–Schuster rearrangement to give enones at room temperature in the presence of a gold(I) catalyst and small quantities of MeOH or 4-methoxyphenylboronic acid. The syntheses of the enone natural products isoegomaketone and daphenone were achieved using this reaction as the key step. The rearrangement of primary propargylic alcohols can readily be combined in a one-pot procedure with the addition of a nucleophile to the resulting terminal enone, to give β-aryl, β-alkoxy, β-amino or β-sulfido ketones. Propargylic alcohols bearing an adjacent electron-rich aryl group can also undergo silver-catalyzed substitution of the alcohol with oxygen, nitrogen and carbon nucleophiles. This latter reaction was initially observed with a batch of gold catalyst that was probably contaminated with small quantities of silver salt.
Co-reporter:Pavel Starkov and Tom D. Sheppard
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 5) pp:1320-1323
Publication Date(Web):16 Dec 2010
DOI:10.1039/C0OB01069C
Simple borates serve as effective promoters for amide bond formation with a variety of carboxylic acids and amines. With trimethyl or tris(2,2,2-trifluoroethyl) borate, amides are obtained in good to excellent yield and high purity after a simple work-up procedure. Tris(2,2,2-trifluoroethyl) borate can also be used for the straightforward conversion of primary amides to secondary amidesvia transamidation.
Co-reporter:Sam E. Mann, Abil E. Aliev, Graham J. Tizzard, and Tom D. Sheppard
Organometallics 2011 Volume 30(Issue 7) pp:1772-1775
Publication Date(Web):March 9, 2011
DOI:10.1021/om2000585
The Pd-mediated oxidation of unsaturated thioacetals gives either allyl or vinyl esters, depending on the substrate structure. We report the characterization of a range of sulfur-stabilized palladium intermediates via a combined computational and experimental NMR approach, demonstrating that the oxidation proceeds via two divergent reaction mechanisms. We were also able to synthesize an unusual σ-bound Pd complex, via acetoxypalladation of an unsaturated dithiane, which was characterized by X-ray crystallography.
Co-reporter:Matthew N. Pennell, Matthew G. Unthank, Peter Turner, and Tom D. Sheppard
The Journal of Organic Chemistry 2011 Volume 76(Issue 5) pp:1479-1482
Publication Date(Web):January 25, 2011
DOI:10.1021/jo102263t
Meyer−Schuster rearrangements of propargylic alcohols take place readily at room temperature in toluene with 1−2 mol % PPh3AuNTf2, in the presence of 0.2 equiv of 4-methoxyphenylboronic acid or 1 equiv of methanol. Good to excellent yields of enones can be obtained from secondary and tertiary alcohols, with high selectivity for the E-alkene in most cases. A one-pot procedure for the conversion of primary propargylic alcohols into β-arylketones was also developed, via Meyer-Schuster rearrangement followed by Pd-catalayzed addition of a boronic acid.
Co-reporter:Cindy Körner ; Pavel Starkov
Journal of the American Chemical Society 2010 Volume 132(Issue 17) pp:5968-5969
Publication Date(Web):April 9, 2010
DOI:10.1021/ja102129c
A new method for enolate generation via the gold-catalyzed addition of boronic acids to alkynes is reported. The formation of boron enolates from readily accessible ortho-alkynylbenzeneboronic acids proceeds rapidly with 2 mol % PPh3AuNTf2 at ambient temperature. The enolates undergo aldol reaction with an aldehyde present in the reaction mixture to give cyclic boronate esters, which can be subsequently transformed into phenols, biaryls, or dihydrobenzofurans via oxidation, Suzuki−Miyaura, or intramolecular Chan−Lam coupling, respectively. A combined gold/boronic acid catalyzed aldol condensation reaction of an alkynyl aldehyde was also successfully achieved.
Co-reporter:Robert W. Waller, Louis J. Diorazio, Brian A. Taylor, William B. Motherwell, Tom D. Sheppard
Tetrahedron 2010 66(33) pp: 6496-6507
Publication Date(Web):
DOI:10.1016/j.tet.2010.05.083
Co-reporter:Tom D. Sheppard
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 6) pp:1043-1052
Publication Date(Web):30 Jan 2009
DOI:10.1039/B818155A
Aryl halides are common synthetic targets themselves, and also highly versatile synthetic intermediates. Aryl chlorides are much more widely available and easier to synthesise than the other halide derivatives, so the development of effective methods for interconverting aryl halide derivatives would therefore be extremely useful. This article outlines which transformations are particularly desirable, and describes the progress that has been made on developing methods for carrying out those transformations using copper, nickel or palladium catalysts. The possible mechanisms of these reactions are discussed, with a view to identifying areas for future investigation.
Co-reporter:Samantha M. Gibson, Rachel M. Lanigan, Laure Benhamou, Abil E. Aliev and Tom D. Sheppard
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 34) pp:NaN9054-9054
Publication Date(Web):2015/07/29
DOI:10.1039/C5OB01398D
In this paper we describe the use of a chiral aldehyde derived from lactate esters for determining the enantiopurity of primary amines, via the formation of diastereomeric imines. The method was shown to be suitable for reproducibly determining the enantiopurity of a diverse set of chiral amines. Both enantiomers of the aldehyde can be prepared in two steps from commercially available materials.
Co-reporter:Valerija Karaluka, Rachel M. Lanigan, Paul M. Murray, Matthew Badland and Tom D. Sheppard
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 44) pp:NaN10894-10894
Publication Date(Web):2015/09/14
DOI:10.1039/C5OB01801C
The use of B(OCH2CF3)3 for mediating direct amidation reactions of a wide range of pharmaceutically relevant carboxylic acids and amines is described, including numerous heterocycle-containing examples. An initial screen of solvents for the direct amidation reaction suggested that cyclopentyl methyl ether, a solvent with a very good safety profile suitable for use over a wide temperature range, was an excellent replacement for the previously used solvent acetonitrile. Under these conditions amides could be prepared from 18 of the 21 carboxylic acids and 18 of the 21 amines examined. Further optimisation of one of the low yielding amidation reactions (36% yield) via a design of experiments approach enabled an 84% yield of the amide to be obtained.
Co-reporter:Pavel Starkov and Tom D. Sheppard
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 5) pp:NaN1323-1323
Publication Date(Web):2010/12/16
DOI:10.1039/C0OB01069C
Simple borates serve as effective promoters for amide bond formation with a variety of carboxylic acids and amines. With trimethyl or tris(2,2,2-trifluoroethyl) borate, amides are obtained in good to excellent yield and high purity after a simple work-up procedure. Tris(2,2,2-trifluoroethyl) borate can also be used for the straightforward conversion of primary amides to secondary amidesvia transamidation.
Co-reporter:Martin Bachman, Sam E. Mann and Tom D. Sheppard
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 1) pp:NaN170-170
Publication Date(Web):2011/10/10
DOI:10.1039/C1OB06534C
Four-component reactions between amino alcohols, aldehydes, isocyanides and thiols proceed rapidly under microwave or conventional heating at 60 °C in methanol. The reaction is successful with a wide range of components and gives access to potentially drug-like products containing amine, amide and thioether functionality in moderate to excellent yield. The reaction conditions are also applicable to the synthesis of a range of 8–10 membered medium ring lactonesvia three-component reactions of amino alcohols, isocyanides and acid-aldehydes. Incorporation of L-prolinol as the amino alcohol component in each case gives access to multicomponent products with moderate to high diastereoselectivity.
Co-reporter:Rachel M. Lanigan, Valerija Karaluka, Marco T. Sabatini, Pavel Starkov, Matthew Badland, Lee Boulton and Tom D. Sheppard
Chemical Communications 2016 - vol. 52(Issue 57) pp:NaN8849-8849
Publication Date(Web):2016/06/22
DOI:10.1039/C6CC05147B
A commercially available borate ester, B(OCH2CF3)3, can be used to achieve protecting-group free direct amidation of α-amino acids with a range of amines in cyclopentyl methyl ether. The method can be applied to the synthesis of medicinally relevant compounds, and can be scaled up to obtain gram quantities of products.
Co-reporter:Matthew N. Pennell, Robert W. Foster, Peter G. Turner, Helen C. Hailes, Christopher J. Tame and Tom D. Sheppard
Chemical Communications 2014 - vol. 50(Issue 11) pp:NaN1304-1304
Publication Date(Web):2013/12/18
DOI:10.1039/C3CC48290A
Synthetically important 3-alkoxyfurans can be prepared efficiently via treatment of acetal-containing propargylic alcohols (obtained from the addition of 3,3-diethoxypropyne to aldehydes) with 2 mol% gold catalyst in an alcohol solvent at room temperature. The resulting furans show useful reactivity in a variety of subsequent transformations.
Co-reporter:Paul M. Murray, Fiona Bellany, Laure Benhamou, Dejan-Krešimir Bučar, Alethea B. Tabor and Tom D. Sheppard
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 8) pp:NaN2384-2384
Publication Date(Web):2015/12/24
DOI:10.1039/C5OB01892G
This article outlines the benefits of using ‘Design of Experiments’ (DoE) optimisation during the development of new synthetic methodology. A particularly important factor in the development of new chemical reactions is the choice of solvent which can often drastically alter the efficiency and selectivity of a process. Whilst solvent optimisation is usually done in a non-systematic way based upon a chemist's intuition and previous laboratory experience, we illustrate how optimisation of the solvent for a reaction can be carried out by using a ‘map of solvent space’ in a DoE optimisation. A new solvent map has been developed specifically for optimisation of new chemical reactions using principle component analysis (PCA) incorporating 136 solvents with a wide range of properties. The new solvent map has been used to identify safer alternatives to toxic/hazardous solvents, and also in the optimisation of an SNAr reaction.
Co-reporter:Tom D. Sheppard
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 6) pp:NaN1052-1052
Publication Date(Web):2009/01/30
DOI:10.1039/B818155A
Aryl halides are common synthetic targets themselves, and also highly versatile synthetic intermediates. Aryl chlorides are much more widely available and easier to synthesise than the other halide derivatives, so the development of effective methods for interconverting aryl halide derivatives would therefore be extremely useful. This article outlines which transformations are particularly desirable, and describes the progress that has been made on developing methods for carrying out those transformations using copper, nickel or palladium catalysts. The possible mechanisms of these reactions are discussed, with a view to identifying areas for future investigation.



![2-Pyrrolidinone, 1-[(1R,6S,7R)-1-methylbicyclo[4.1.0]hept-7-yl]-, rel-](http://img.cochemist.com/ccimg/645500/645412-68-6.png)
![2-Pyrrolidinone, 1-[(1R,6S,7R)-1-methylbicyclo[4.1.0]hept-7-yl]-, rel-](http://img.cochemist.com/ccimg/645500/645412-68-6_b.png)
![2-Pyrrolidinone, 1-[(1R,2S)-2-(1,1-dimethylethyl)cyclopropyl]-, rel-](http://img.cochemist.com/ccimg/645500/645412-66-4.png)
![2-Pyrrolidinone, 1-[(1R,2S)-2-(1,1-dimethylethyl)cyclopropyl]-, rel-](http://img.cochemist.com/ccimg/645500/645412-66-4_b.png)
![Carbamic acid, [(1R,2R)-2-hydroxycyclopropyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/876200/876125-09-6.png)
![Carbamic acid, [(1R,2R)-2-hydroxycyclopropyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/876200/876125-09-6_b.png)


![2-Pyrrolidinone, 1-[(1R,2R)-2-(phenylmethyl)cyclopropyl]-, rel-](http://img.cochemist.com/ccimg/645500/645412-64-2.png)
![2-Pyrrolidinone, 1-[(1R,2R)-2-(phenylmethyl)cyclopropyl]-, rel-](http://img.cochemist.com/ccimg/645500/645412-64-2_b.png)









![1-Propanol, 2-amino-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2S)-](http://img.cochemist.com/ccimg/566200/566199-97-1.png)
![1-Propanol, 2-amino-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2S)-](http://img.cochemist.com/ccimg/566200/566199-97-1_b.png)






![Benzeneacetamide, N-[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/52200/52195-00-3.png)
![Benzeneacetamide, N-[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/52200/52195-00-3_b.png)




















![Boronic acid, [2-(phenylethynyl)phenyl]-](http://img.cochemist.com/ccimg/124200/124160-41-4.png)
![Boronic acid, [2-(phenylethynyl)phenyl]-](http://img.cochemist.com/ccimg/124200/124160-41-4_b.png)







![BICYCLO[3.1.1]HEPTANE-2-METHANAMINE, 6,6-DIMETHYL-, (1S,2R,5S)-](http://img.cochemist.com/ccimg/73600/73522-42-6.png)
![BICYCLO[3.1.1]HEPTANE-2-METHANAMINE, 6,6-DIMETHYL-, (1S,2R,5S)-](http://img.cochemist.com/ccimg/73600/73522-42-6_b.png)





















![2-Oxazolidinone, 4-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-, (4S)-](http://img.cochemist.com/ccimg/155400/155398-62-2.png)
![2-Oxazolidinone, 4-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-, (4S)-](http://img.cochemist.com/ccimg/155400/155398-62-2_b.png)
![ACETAMIDE, 2-HYDROXY-N-[2-(1H-INDOL-3-YL)ETHYL]-](http://img.cochemist.com/ccimg/645500/645405-32-9.png)
![ACETAMIDE, 2-HYDROXY-N-[2-(1H-INDOL-3-YL)ETHYL]-](http://img.cochemist.com/ccimg/645500/645405-32-9_b.png)

























![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-L-alanyl-, methyl ester](http://img.cochemist.com/ccimg/2300/2280-66-2.png)
![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-L-alanyl-, methyl ester](http://img.cochemist.com/ccimg/2300/2280-66-2_b.png)






























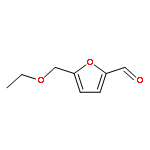
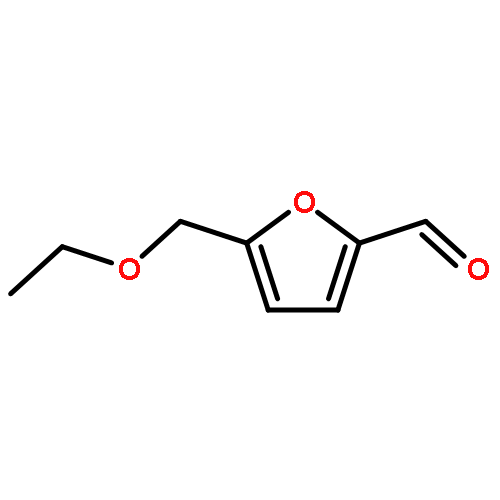

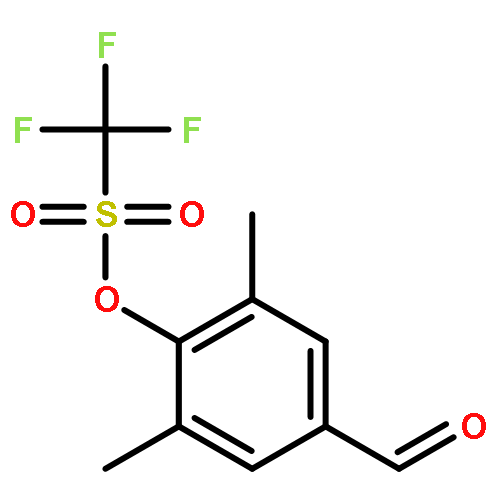
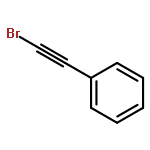















![(2s)-2-[tert-butyl(dimethyl)silyl]oxypropanal](http://img.cochemist.com/ccimg/87800/87727-28-4.png)
![(2s)-2-[tert-butyl(dimethyl)silyl]oxypropanal](http://img.cochemist.com/ccimg/87800/87727-28-4_b.png)
![tert-butyl [(1S)-2-(benzylamino)-1-methyl-2-oxoethyl]carbamate](http://img.cochemist.com/ccimg/51900/51814-55-2.png)
![tert-butyl [(1S)-2-(benzylamino)-1-methyl-2-oxoethyl]carbamate](http://img.cochemist.com/ccimg/51900/51814-55-2_b.png)



















![1-Pyrrolidinecarboxylic acid, 2-[[(phenylmethyl)amino]carbonyl]-,1,1-dimethylethyl ester, (2S)-](http://img.cochemist.com/ccimg/92300/92235-33-1.png)
![1-Pyrrolidinecarboxylic acid, 2-[[(phenylmethyl)amino]carbonyl]-,1,1-dimethylethyl ester, (2S)-](http://img.cochemist.com/ccimg/92300/92235-33-1_b.png)


























![Formamide, N-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/17100/17061-63-1.png)
![Formamide, N-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/17100/17061-63-1_b.png)







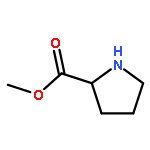
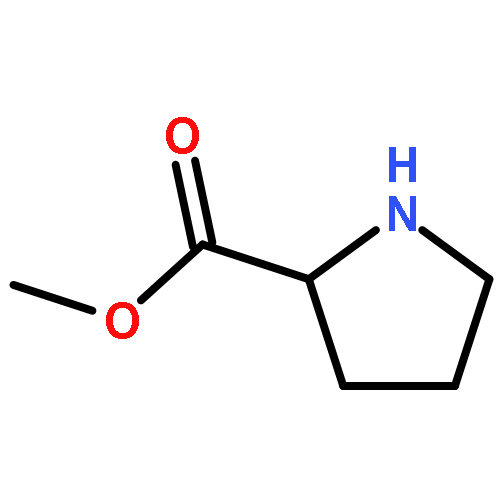
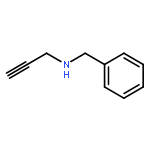
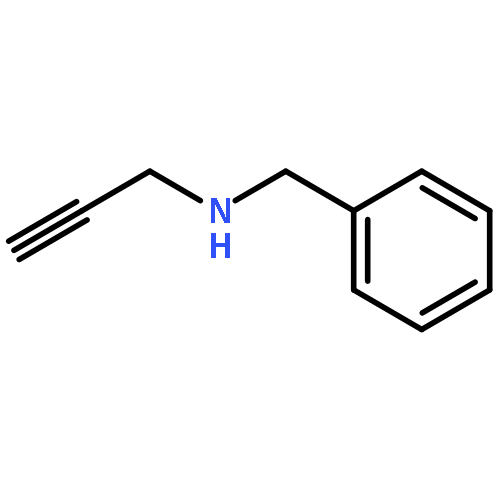





![Methyl 2-[(2-phenylacetyl)amino]acetate](http://img.cochemist.com/ccimg/5300/5259-87-0.png)
![Methyl 2-[(2-phenylacetyl)amino]acetate](http://img.cochemist.com/ccimg/5300/5259-87-0_b.png)










![Formamide, N-[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/31600/31502-34-8.png)
![Formamide, N-[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/31600/31502-34-8_b.png)
![Benzeneacetamide, N-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/305900/305849-49-4.png)
![Benzeneacetamide, N-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/305900/305849-49-4_b.png)











![Benzene,[(ethenyldimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/18100/18001-46-2.png)
![Benzene,[(ethenyldimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/18100/18001-46-2_b.png)


![tert-butyl [(1S)-1-benzyl-2-oxo-2-pyrrolidin-1-ylethyl]carbamate](http://img.cochemist.com/ccimg/125200/125139-05-1.png)
![tert-butyl [(1S)-1-benzyl-2-oxo-2-pyrrolidin-1-ylethyl]carbamate](http://img.cochemist.com/ccimg/125200/125139-05-1_b.png)
![Benzeneacetamide,N-[2-(1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/19500/19462-24-9.png)
![Benzeneacetamide,N-[2-(1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/19500/19462-24-9_b.png)








![(1R,2R,4S)-Bicyclo[2.2.1]heptan-2-amine](http://img.cochemist.com/ccimg/130500/130463-97-7.png)
![(1R,2R,4S)-Bicyclo[2.2.1]heptan-2-amine](http://img.cochemist.com/ccimg/130500/130463-97-7_b.png)








![Ruthenium,chloro[(1,2,5,6-h)-1,5-cyclooctadiene][(1,2,3,4,5-h)-1,2,3,4,5-pentamethyl-2,4-cyclopentadien-1-yl]-](http://img.cochemist.com/ccimg/92400/92390-26-6.png)
![Ruthenium,chloro[(1,2,5,6-h)-1,5-cyclooctadiene][(1,2,3,4,5-h)-1,2,3,4,5-pentamethyl-2,4-cyclopentadien-1-yl]-](http://img.cochemist.com/ccimg/92400/92390-26-6_b.png)


![Benzene, 1-bromo-2-[2-(4-methylphenyl)ethenyl]-](http://img.cochemist.com/ccimg/92500/92497-32-0.png)
![Benzene, 1-bromo-2-[2-(4-methylphenyl)ethenyl]-](http://img.cochemist.com/ccimg/92500/92497-32-0_b.png)



![Bicyclo[2.2.1]heptan-2-amine](http://img.cochemist.com/ccimg/900/822-98-0.png)
![Bicyclo[2.2.1]heptan-2-amine](http://img.cochemist.com/ccimg/900/822-98-0_b.png)


![4-METHYL-1-OXA-4-AZASPIRO[4.5]DECANE](http://img.cochemist.com/ccimg/40000/39931-24-3.png)
![4-METHYL-1-OXA-4-AZASPIRO[4.5]DECANE](http://img.cochemist.com/ccimg/40000/39931-24-3_b.png)










































![4-[(2E)-3-CHLORO-2-PROPEN-1-YL]-1-DODECANAMINE](http://img.cochemist.com/ccimg/400/300-54-9.png)
![4-[(2E)-3-CHLORO-2-PROPEN-1-YL]-1-DODECANAMINE](http://img.cochemist.com/ccimg/400/300-54-9_b.png)




![Bicyclo[3.1.1]heptan-3-amine,2,6,6-trimethyl-, (1S,2S,3S,5R)-](http://img.cochemist.com/ccimg/13300/13293-47-5.png)
![Bicyclo[3.1.1]heptan-3-amine,2,6,6-trimethyl-, (1S,2S,3S,5R)-](http://img.cochemist.com/ccimg/13300/13293-47-5_b.png)






![Propanal, 2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (R)-](http://img.cochemist.com/ccimg/111900/111819-71-7.png)
![Propanal, 2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (R)-](http://img.cochemist.com/ccimg/111900/111819-71-7_b.png)




![2-Phenylimidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/4200/4105-21-9.png)
![2-Phenylimidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/4200/4105-21-9_b.png)








![Furan,3-[2-(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/465600/465521-19-1.png)
![Furan,3-[2-(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/465600/465521-19-1_b.png)


![1H-Pyrrole-2,5-dione,1-[(4-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/42900/42867-34-5.png)
![1H-Pyrrole-2,5-dione,1-[(4-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/42900/42867-34-5_b.png)


























![(1R,2S,4S)-Bicyclo[2.2.1]heptan-2-amine hydrochloride](http://img.cochemist.com/ccimg/121100/121055-07-0.png)
![(1R,2S,4S)-Bicyclo[2.2.1]heptan-2-amine hydrochloride](http://img.cochemist.com/ccimg/121100/121055-07-0_b.png)




![3,3-Dimethylbenzo[c][1,2]oxaborol-1(3H)-ol](/data/chemimg/19800/221352-10-9.png)
![3,3-Dimethylbenzo[c][1,2]oxaborol-1(3H)-ol](/data/chemimg/19800/221352-10-9_b.png)