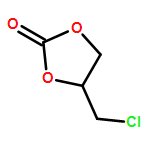
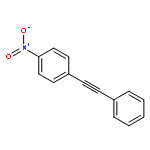
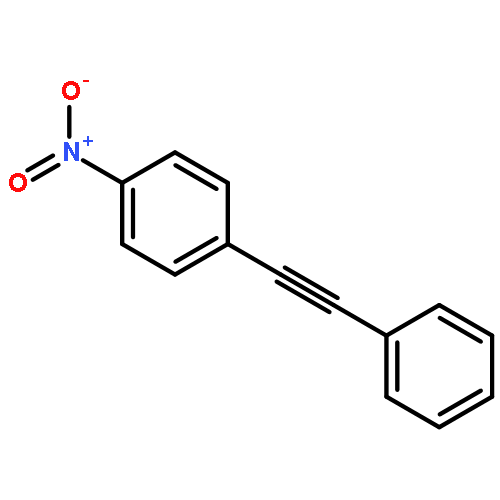
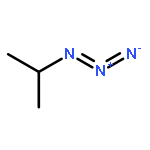
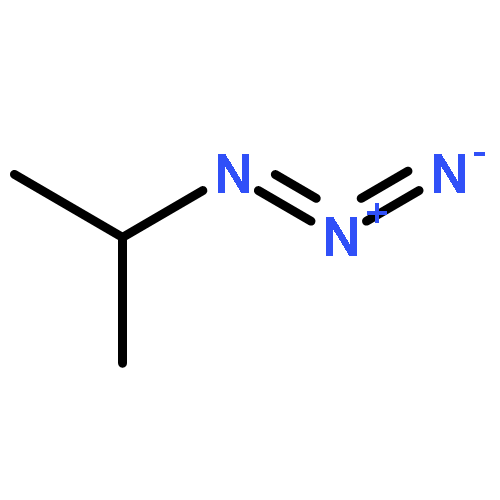
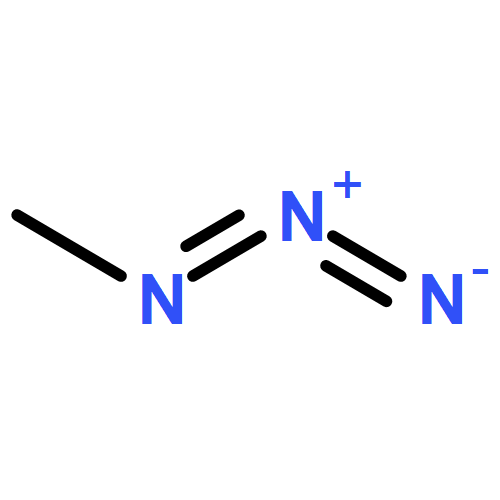
Co-reporter:Kui Zhou, Somboon Chaemchuen, Zhaoxuan Wu, Francis Verpoort
Microporous and Mesoporous Materials 2017 Volume 239() pp:28-33
Publication Date(Web):February 2017
DOI:10.1016/j.micromeso.2016.09.038
•Straight and energy efficient synthesis of mixed ligand MOFs under mild conditions generating Kagomé pillar net frameworks.•Unified synthesis of Kagomé pillared net frameworks using a variance of metal clusters in M2(bdc)2(dabco) (M = Zn, Ni, Co).•The metal cluster species in M2(bdc)2(dabco) determine the physical properties of the 3D pillared structures.•The advantage of the room temperature procedure is the low cost and easy accessibility of the MOFs synthesis.A direct synthesis of pillared metal-organic frameworks with Kagomé net topology under mild conditions (room temperature and ambient pressure) was developed which provides a straightforward and energy-saving procedure to produce MOFs. A series of metals (M = Zn, Ni and Co) bridging with ligands 1,4-benzene dicarboxylate (bdc) and 1,4-diazabicyclo[2.2.2]octane (dabco) were used to construct MOF frameworks denoted as M2(bdc)2(dabco). The Kagomé net pillared structures were confirmed via crystal phase analysis (XRD). The use of different metals resulted in diverse crystal morphologies and particle shapes as observed from scanning electron microscope analysis (SEM). The obtained Kagomé net pillared structure influences the physical properties such as surface area, pore size, N2/CO2/CH4 gas adsorption and also thermal stability. Furthermore, modeling was applied to determine the behavior of the isosteric heat of adsorption on CO2 and gas selectivity of CO2/CH4 and CO2/N2.
Co-reporter:Somboon Chaemchuen, Zhixiong Luo, Kui Zhou, Bibimaryam Mousavi, Suphot Phatanasri, Mietek Jaroniec, Francis Verpoort
Journal of Catalysis 2017 Volume 354(Volume 354) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.jcat.2017.08.012
•Defects (nature & amount) are determined by the crystal growth rate of the procedure.•Identification & quantification of defects via probe gas techniques was demonstrated.•The defect structures related catalytic performance was demonstrated.•A new strategy for defect creation in MOFs can be applied to design catalysts.The defect formation in metal–organic frameworks (MOFs) initiated by the crystal growth-rate of different synthesis procedures affects the structure, morphology and other properties of MOFs such as particle shape, gas adsorption capacity and specifically catalytic performance etc. Although the design of defect structures and the defect structures itself are well known, comparatively little is known regarding defect creation introduced by the synthesis procedure (different from mixed-linker approach, the use of acid modulators, post-synthetic treatment, etc.) and characterization of the resulting naturally occurring abnormalities (defects) in the material. Moreover, high performance of MOFs in various catalytic reactions can be correlated to the higher amount of defect structures because defects can possibly exhibit acid and/or basic properties. While studies of MOF crystallinity confirmed that for a given type of MOF different synthesis methods generate samples of similar crystal structures, their morphologies are often different due to the differences in the crystallization rates associated with these methods.Download high-res image (97KB)Download full-size image
Co-reporter:Jeet Chakraborty, Ipsita Nath, Francis Verpoort
Coordination Chemistry Reviews 2016 Volume 326() pp:135-163
Publication Date(Web):1 November 2016
DOI:10.1016/j.ccr.2016.08.006
•Porphyrinoids and heme enzymes entrapped in metal-organic materials are discussed.•Immobilization techniques and coordination between hosts and guests are described.•Catalytic activities based on the similarities of mechanistic pathways are given.Literature sources on metal-organic materials encapsulating versatile porphyrinoids and heme enzymes have been discussed in this review. General fundamentals and structural and chemical requisites of these architectures are discussed in introduction followed by historical perspectives of the initial composites. We have categorized the structural aspects of this class of compounds according to the host and guest skeletons as well as encapsulating strategies in a sequential manner. Heme mimetic, as well as some novel applications of the materials including oxidation, electron transfer, carbene transfer and bio-molecule sensing, are then discussed and compared with each other and their homogeneous analogues based on mechanistic similarities, wherever possible.
Co-reporter:Md. Ali Asraf, Hussein A. Younus, Chizoba I. Ezugwu, Akshay Mehta and Francis Verpoort
Catalysis Science & Technology 2016 vol. 6(Issue 12) pp:4271-4282
Publication Date(Web):19 Jan 2016
DOI:10.1039/C5CY02157J
Earth-abundant molecular complexes have been found to be excellent catalysts for the light-driven water oxidation reaction. Here, we demonstrate the photochemical water oxidation catalysed by three cobalt salophen complexes with different axial ligands in the presence of [Ru(bpy)3]2+ as a photosensitizer and Na2S2O8 as an electron acceptor in phosphate buffer of pH 9 and 7. The electrochemical investigation of the complexes in pH 9 including cyclic voltammetry (CV) and linear sweep voltammetry (LSV) verify the deposition of catalytic films on the surface of the working electrode. The deposited film characterization using scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) confirms that the complexes decomposed to form nanoparticles. Electrospray ionization mass spectrometry (ESI-MS) studies along with dynamic light-scattering (DLS) measurements of the catalyst solution during the course of photochemical water oxidation suggest the catalyst decomposition and the formation of nanoparticles. The XPS measurements of the produced nanoparticles suggest that the surface of the particles is composed of Co(II) and OH species. In contrast, multiple experiments argue that light-driven water oxidation using the same complexes in pH 7 is homogeneous. We thus conclude that cobalt salophen complexes act as precatalysts that decompose under basic conditions to form cobalt hydroxide nanoparticles which act as a real catalyst for the light-driven water oxidation reaction, whereas the same complexes act as a homogeneous catalyst in the photochemical water oxidation reaction under neutral conditions.
Co-reporter:Hussein A. Younus;Wei Su;Nazir Ahmad;Si Chen
Advanced Synthesis & Catalysis 2015 Volume 357( Issue 2-3) pp:283-330
Publication Date(Web):
DOI:10.1002/adsc.201400777
Co-reporter:Md. Ali Asraf, Hussein A. Younus, Mekhman Yusubov and Francis Verpoort
Catalysis Science & Technology 2015 vol. 5(Issue 11) pp:4901-4925
Publication Date(Web):13 Aug 2015
DOI:10.1039/C5CY01251A
Recent developments in the oxidation of water to dioxygen using metal complexes as catalysts or precatalysts are presented, as well as the conversion of homogeneous catalysts into nanoparticles or heterogeneous catalysts during the oxidation of water. Homogeneous catalysts are advantageous in the careful design and elucidation of the mechanisms for catalytic water oxidation. In distinction, the use of heterogeneous catalysts in water oxidation is advantageous for applications as a result of their robust catalytic activity. Although detailed studies on homogeneous and heterogeneous water oxidation reactions are performed rather often, understanding the link between homogeneous catalysts and heterogeneous ones is becoming an additional requirement in order to produce economically attractive water oxidation catalysts (WOCs). This minireview focuses on the aspects that determine whether particular catalysts for the oxidation of water are homogeneous or heterogeneous.
Co-reporter:Somboon Chaemchuen, Kui Zhou, Nawsad Alam Kabir, Yao Chen, Xiaoxing Ke, Gustaaf Van Tendeloo, Francis Verpoort
Microporous and Mesoporous Materials 2015 Volume 201() pp:277-285
Publication Date(Web):1 January 2015
DOI:10.1016/j.micromeso.2014.09.038
•A 3-D pillared-layer M-DABCO MOF (M = Co, Ni, Cu, Zn) series is synthesized.•The metal atom has an impact on stability and adsorptive properties.•Ni-DABCO MOF exhibits the highest CO2 adsorption and CO2/CH4 selectivity.•Saturated metal sites can improve adsorption and selectivity at ambient conditions.•Modeling demonstrates the effect of the metal on CO2 and CH4 adsorption.Metal–organic frameworks (MOFs) have emerged as new porous materials for capture and separation of binary gas mixtures. Tuning the metal sites in MOF structures has an impact on properties, which enhance affinity of gas adsorption and selectivity (e.g., surface area, cavity, electric field, etc.). The synthesis and characterization of a M-DABCO series (M = Ni, Co, Cu, Zn) of MOFs are described in this study. The experiments were conducted using multicomponent gas mixtures and the Ideal Adsorbed Solution Theory (IAST) was applied to determine the CO2/CH4 selectivity. Experimental adsorption isotherms were fitted with a model equation to evaluate the characteristic adsorption energy (Isosteric, Qst) of this series. The Ni metal in the M-DABCO series reveals the best performance concerning CO2 adsorption and CH4/CO2 selectivity at ambient conditions based on IAST calculations. The combination of characterizations, calculations and adsorption experiments were used to discuss the metal impact on the adsorption sites in the M-DABCO series at ambient conditions.
Co-reporter:Hussein A. Younus, Nazir Ahmad, Wei Su, Francis Verpoort
Coordination Chemistry Reviews 2014 Volume 276() pp:112-152
Publication Date(Web):15 September 2014
DOI:10.1016/j.ccr.2014.06.016
•Definitions of pincer ligands and their nomenclature are presented.•Modification of the pincer backbone to control steric and electronic properties.•Design of pincer ligands based on pyridine, benzene, etc. for ruthenium pincer complexes (RPCs).•Synthesis of RPCs via direct metallation, C/SiH bond activation, trans-metallation, and trans-cyclometallation.•Key RPC structures affect the catalyst behavior in catalytic hydrogenation reactions.Active transition–metal complexes based on relatively inexpensive metals are considered to be a desirable method for sustainable human industrial growth. Considering their cost efficiency, ruthenium complexes are gaining increasing attraction, instead of palladium, rhodium, and iridium. Among the ruthenium complexes, ruthenium pincer complexes (RPCs) have received much attention due to their outstanding performance. Various strategies have been developed for pincer ligand design and RPC synthesis, which indicate that ligand design is a key feature of pincer chemistry. In addition, electronic and steric effects, hemilability, non-innocent behavior, and the flexibility of the pincer ligand have significant effects on the catalytic performance of RPCs in hydrogenation and dehydrogenation reactions.
Co-reporter:Nazir Ahmad, Adeel H. Chughtai, Hussein A. Younus, Francis Verpoort
Coordination Chemistry Reviews 2014 280() pp: 1-27
Publication Date(Web):
DOI:10.1016/j.ccr.2014.07.005
Co-reporter:Zhi-Zhong Xie, Wen-Juan Liao, Jun Cao, Li-Ping Guo, Francis Verpoort, and Weihai Fang
Organometallics 2014 Volume 33(Issue 10) pp:2448-2456
Publication Date(Web):May 15, 2014
DOI:10.1021/om401092h
A DFT study on the reaction of diazoacetate with primary allyl alcohol mediated by dirhodium catalyst has been carried out in detail. Calculations indicate that the major O–H insertion product can be obtained via either a [1,3]-proton shift of the free enol or a [1,2]-proton shift of the free oxonium ylide, which are regulated by the orientation of the ester group. In the case of a [1,3]-proton shift the reaction begins with the nucleophilic attack of the alcohol at the carbenoid, generating a metal-associated oxonium ylide followed by a [1,4]-proton shift to the adjacent carbonyl oxygen atom of the ester group, resulting in a metal-associated enol. Subsequently, its decomposition liberates a free enol intermediate. The whole process requires an overall barrier of 4.2 kcal/mol and is exergonic by 6.4 kcal/mol. The [1,3]-proton shift of the enol also readily provides the final O–H insertion product, which has a barrier of 11.7 kcal/mol using a three-alcohol cluster as catalyst. For the free oxonium ylide pathway, formation of an alternative metal-associated oxonium ylide is also straightforward, having an overall barrier of 4.5 kcal/mol. In the presence of extra alcohol molecules, the decomposition of the metal-associated oxonium ylide can generate an alcohol-stabilized free oxonium ylide (endergonic by only 4.1 kcal/mol). Afterward, it undergoes a [1,2]-proton shift, resulting in the O–H insertion product, which requires an energy barrier of 4.7 kcal/mol. In comparison, the competitive [2,3]-sigmatropic rearrangement for the metal-associated oxonium ylides is not sensitive to the orientation of the ester, which has a similar activation free energy around 14.0 kcal/mol. Accordingly, it is always disfavored over the O–H insertion, which kinetically agrees well with the experimental observations, in which traces of [2,3]-sigmatropic rearrangement product were obtained for the primary allyl alcohol.
Co-reporter:Xu-Chao Wang, Xian-Shuang Song, Li-Ping Guo, Deyu Qu, Zhi-Zhong Xie, Francis Verpoort, and Jun Cao
Organometallics 2014 Volume 33(Issue 15) pp:4042-4050
Publication Date(Web):July 29, 2014
DOI:10.1021/om5005572
The insertion of a carbenoid into an N–H bond of an amine cooperatively catalyzed by a dirhodium catalyst and a spiro chiral phosphoric acid has been investigated in detail using density functional theory methods. Calculations indicate that the reaction begins with the nucleophilic amine attacking at the carbenoid, forming a metal-associated ammonium ylide first followed by a rapid proton transfer to afford a metal-associated enamine intermediate. Subsequently, the enamine intermediate dissociates from the metal and yield a more stable seven-membered-ring conformation via an intramolecular hydrogen-bond exchange. Formation of the enamine intermediate requires an overall barrier of 5.7 kcal/mol and is exergonic by 5.1 kcal/mol. Calculations demonstrated that, although the conversion of the achiral enamine into the N–H insertion product can be facilitated efficiently by the dirhodium catalyst through a two-step process, it can be compressed to a large extent. This is due to the more competitive decomposition of the diazoacetate catalyzed by the dirhodium catalyst, which can give a carbenoid for the next catalytic cycle. Meanwhile, formation of the carbenoid is considerably exergonic, which can promote the direct [1,3]-proton shift of enamine. However, in the presence of the spiro chiral phosphoric acid, the asymmetric proton induction of enamine is greatly favored, requiring an activation free energy of 6.0 kcal/mol to afford the major R product. This agrees well with the experimental observation.
Co-reporter:Md. Ali Asraf, Hussein A. Younus, Chizoba I. Ezugwu, Akshay Mehta and Francis Verpoort
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 12) pp:NaN4282-4282
Publication Date(Web):2016/01/19
DOI:10.1039/C5CY02157J
Earth-abundant molecular complexes have been found to be excellent catalysts for the light-driven water oxidation reaction. Here, we demonstrate the photochemical water oxidation catalysed by three cobalt salophen complexes with different axial ligands in the presence of [Ru(bpy)3]2+ as a photosensitizer and Na2S2O8 as an electron acceptor in phosphate buffer of pH 9 and 7. The electrochemical investigation of the complexes in pH 9 including cyclic voltammetry (CV) and linear sweep voltammetry (LSV) verify the deposition of catalytic films on the surface of the working electrode. The deposited film characterization using scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) confirms that the complexes decomposed to form nanoparticles. Electrospray ionization mass spectrometry (ESI-MS) studies along with dynamic light-scattering (DLS) measurements of the catalyst solution during the course of photochemical water oxidation suggest the catalyst decomposition and the formation of nanoparticles. The XPS measurements of the produced nanoparticles suggest that the surface of the particles is composed of Co(II) and OH species. In contrast, multiple experiments argue that light-driven water oxidation using the same complexes in pH 7 is homogeneous. We thus conclude that cobalt salophen complexes act as precatalysts that decompose under basic conditions to form cobalt hydroxide nanoparticles which act as a real catalyst for the light-driven water oxidation reaction, whereas the same complexes act as a homogeneous catalyst in the photochemical water oxidation reaction under neutral conditions.
Co-reporter:Md. Ali Asraf, Hussein A. Younus, Mekhman Yusubov and Francis Verpoort
Catalysis Science & Technology (2011-Present) 2015 - vol. 5(Issue 11) pp:NaN4925-4925
Publication Date(Web):2015/08/13
DOI:10.1039/C5CY01251A
Recent developments in the oxidation of water to dioxygen using metal complexes as catalysts or precatalysts are presented, as well as the conversion of homogeneous catalysts into nanoparticles or heterogeneous catalysts during the oxidation of water. Homogeneous catalysts are advantageous in the careful design and elucidation of the mechanisms for catalytic water oxidation. In distinction, the use of heterogeneous catalysts in water oxidation is advantageous for applications as a result of their robust catalytic activity. Although detailed studies on homogeneous and heterogeneous water oxidation reactions are performed rather often, understanding the link between homogeneous catalysts and heterogeneous ones is becoming an additional requirement in order to produce economically attractive water oxidation catalysts (WOCs). This minireview focuses on the aspects that determine whether particular catalysts for the oxidation of water are homogeneous or heterogeneous.





![2H-Furo[2,3-c]pyran-2-one](http://img.cochemist.com/ccimg/857100/857054-03-6.png)
![2H-Furo[2,3-c]pyran-2-one](http://img.cochemist.com/ccimg/857100/857054-03-6_b.png)
![3-Methyl 2H-Furo[2,3-c]pyran-2-one](http://img.cochemist.com/ccimg/857100/857054-02-5.png)
![3-Methyl 2H-Furo[2,3-c]pyran-2-one](http://img.cochemist.com/ccimg/857100/857054-02-5_b.png)
![2-Propynoic acid, 3-[4-(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/220100/220006-53-1.png)
![2-Propynoic acid, 3-[4-(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/220100/220006-53-1_b.png)






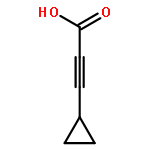
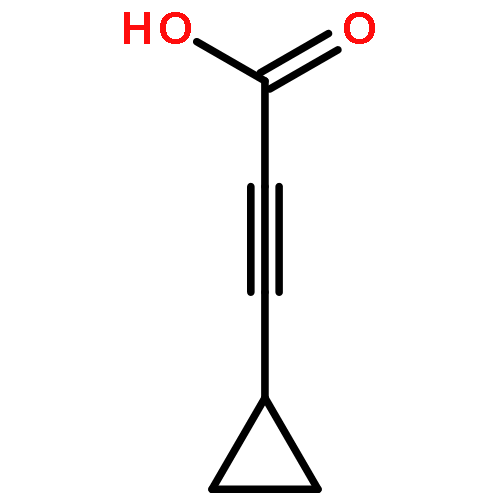


![2-Propynoic acid, 3-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/3800/3792-88-9.png)
![2-Propynoic acid, 3-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/3800/3792-88-9_b.png)
![Propanedinitrile,2-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/2900/2826-26-8.png)
![Propanedinitrile,2-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/2900/2826-26-8_b.png)
![2-[(4-methylphenyl)methylidene]propanedinitrile](http://img.cochemist.com/ccimg/2900/2826-25-7.png)
![2-[(4-methylphenyl)methylidene]propanedinitrile](http://img.cochemist.com/ccimg/2900/2826-25-7_b.png)
![Propanedinitrile, [(4-fluorophenyl)methylene]-](http://img.cochemist.com/ccimg/2900/2826-22-4.png)
![Propanedinitrile, [(4-fluorophenyl)methylene]-](http://img.cochemist.com/ccimg/2900/2826-22-4_b.png)
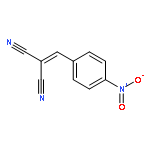

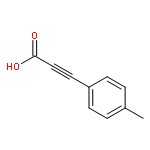
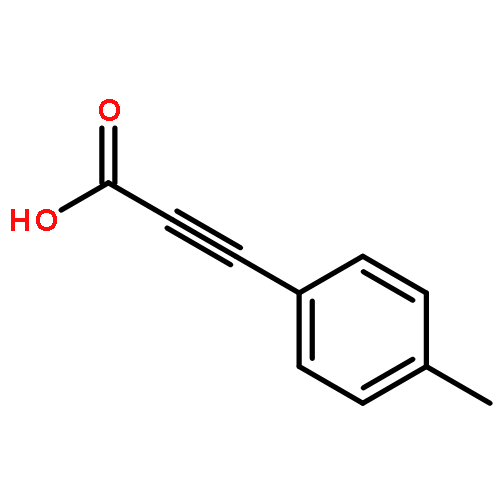
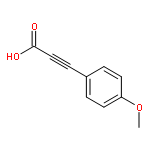


![Propanedinitrile,2-[(4-chlorophenyl)methylene]-](http://img.cochemist.com/ccimg/1900/1867-38-5.png)
![Propanedinitrile,2-[(4-chlorophenyl)methylene]-](http://img.cochemist.com/ccimg/1900/1867-38-5_b.png)




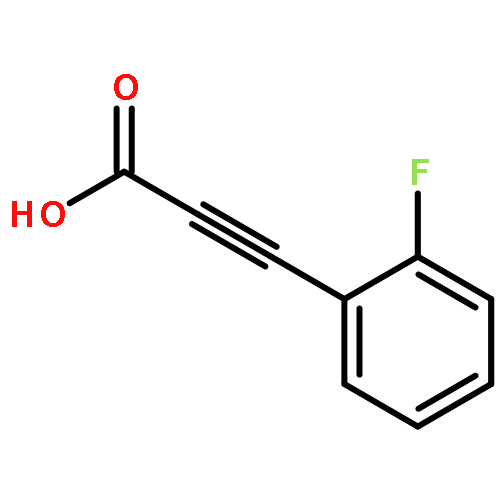




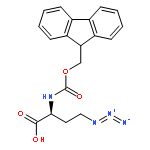
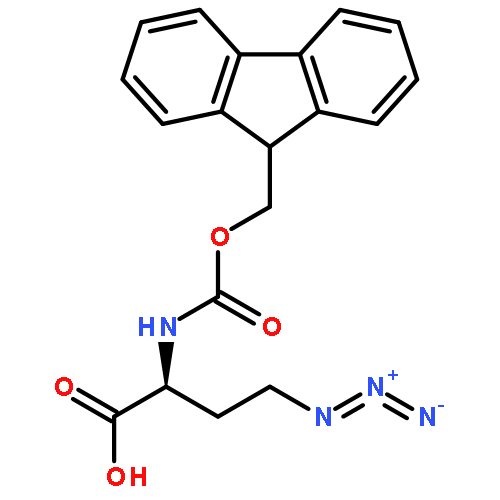

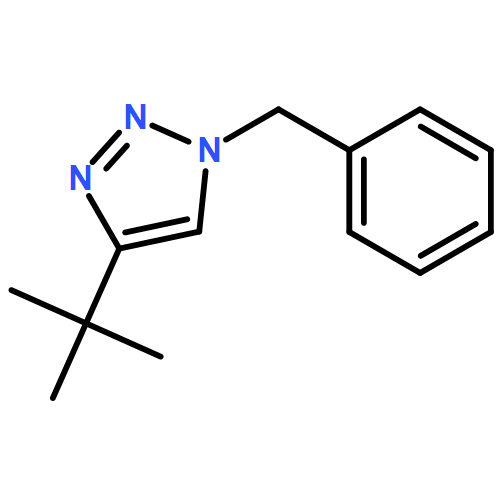
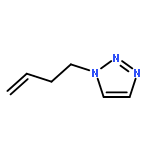
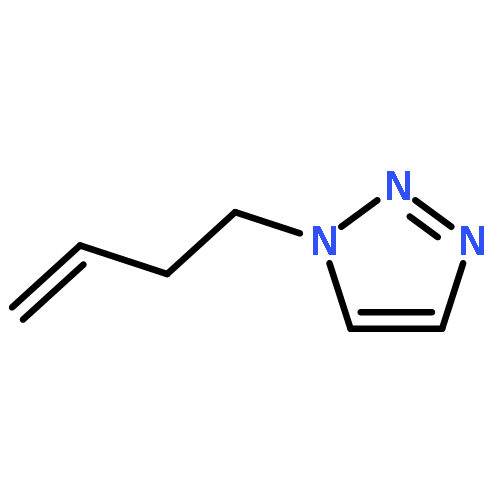
![Phenol,4,4'-[(1S,2S)-1,2-bis[[[3,5-bis(phenylmethoxy)phenyl]methyl]amino]-1,2-ethanediyl]bis-](http://img.cochemist.com/ccimg/863200/863117-86-6.png)
![Phenol,4,4'-[(1S,2S)-1,2-bis[[[3,5-bis(phenylmethoxy)phenyl]methyl]amino]-1,2-ethanediyl]bis-](http://img.cochemist.com/ccimg/863200/863117-86-6_b.png)



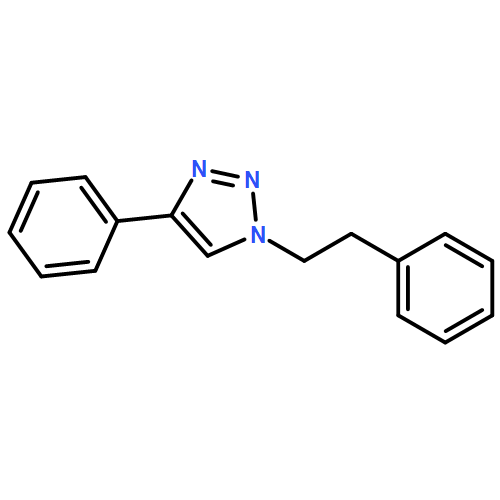








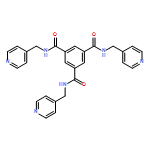
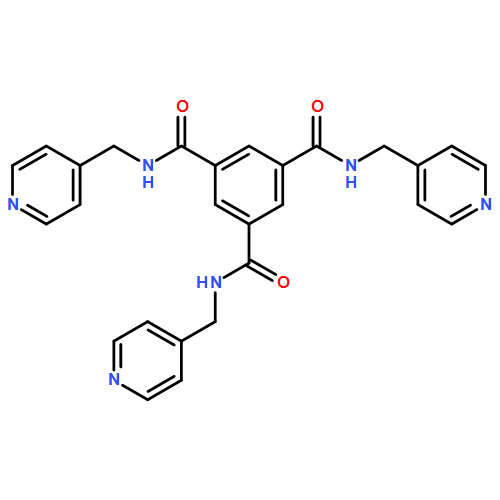
![[1,1'-BIPHENYL]-4,4'-DICARBOXAMIDE, N,N'-BIS(4-PYRIDINYLMETHYL)-](http://img.cochemist.com/ccimg/637400/637324-69-7.png)
![[1,1'-BIPHENYL]-4,4'-DICARBOXAMIDE, N,N'-BIS(4-PYRIDINYLMETHYL)-](http://img.cochemist.com/ccimg/637400/637324-69-7_b.png)
![Phosphonic acid, [2-methyl-1-(phenylamino)propyl]-, diethyl ester](http://img.cochemist.com/ccimg/476100/476014-41-2.png)
![Phosphonic acid, [2-methyl-1-(phenylamino)propyl]-, diethyl ester](http://img.cochemist.com/ccimg/476100/476014-41-2_b.png)

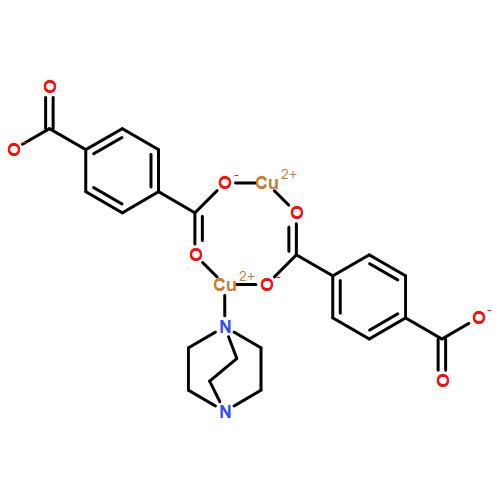
![Phosphonic acid, [(2-methylphenyl)(phenylamino)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/374900/374808-40-9.png)
![Phosphonic acid, [(2-methylphenyl)(phenylamino)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/374900/374808-40-9_b.png)



![Phosphonic acid, [(butylamino)phenylmethyl]-, diethyl ester](http://img.cochemist.com/ccimg/157900/157841-12-8.png)
![Phosphonic acid, [(butylamino)phenylmethyl]-, diethyl ester](http://img.cochemist.com/ccimg/157900/157841-12-8_b.png)
![Benzene, 1,3-bis[[3,5-bis[[3,5-bis(phenylmethoxy)phenyl]methoxy]phenyl]methoxy]-5-(bromomethyl)-](/data/chemimg/107700/137472-17-4.png)
![Benzene, 1,3-bis[[3,5-bis[[3,5-bis(phenylmethoxy)phenyl]methoxy]phenyl]methoxy]-5-(bromomethyl)-](/data/chemimg/107700/137472-17-4_b.png)



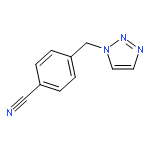
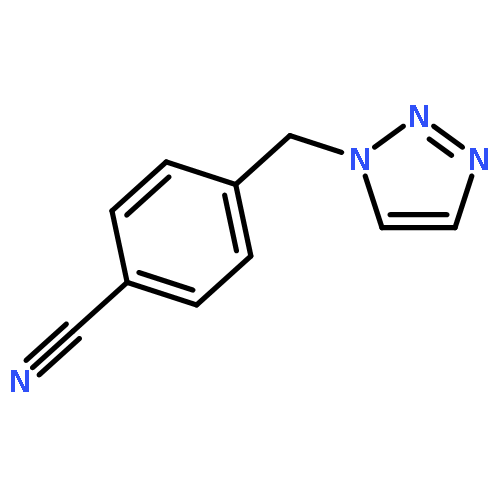



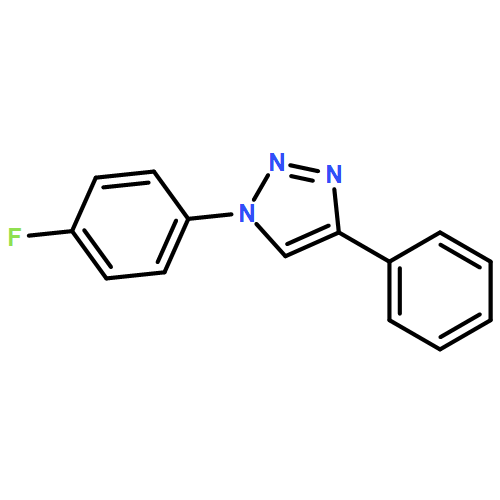

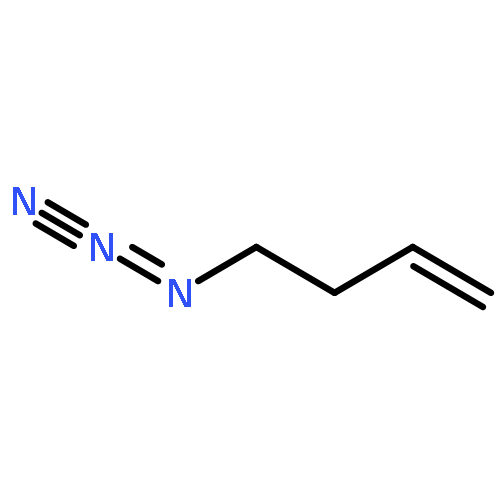
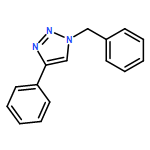
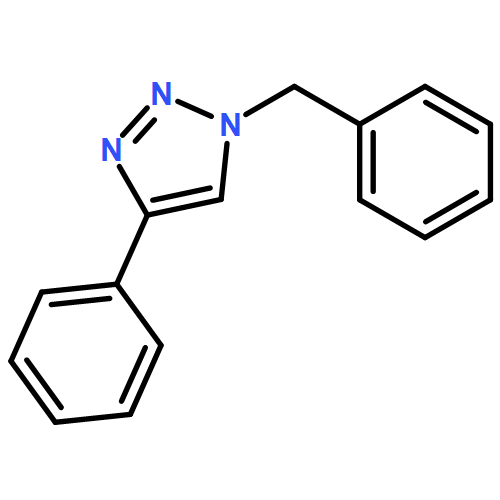
![PHOSPHONIC ACID, [(METHYLPHENYLAMINO)PHENYLMETHYL]-, DIETHYL ESTER](http://img.cochemist.com/ccimg/104600/104541-79-9.png)
![PHOSPHONIC ACID, [(METHYLPHENYLAMINO)PHENYLMETHYL]-, DIETHYL ESTER](http://img.cochemist.com/ccimg/104600/104541-79-9_b.png)
![1H-1,2,3-Triazole, 1-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/99600/99590-25-7.png)
![1H-1,2,3-Triazole, 1-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/99600/99590-25-7_b.png)


![Phenol, 4,4'-[(1S,2S)-1,2-diamino-1,2-ethanediyl]bis-](http://img.cochemist.com/ccimg/91600/91548-22-0.png)
![Phenol, 4,4'-[(1S,2S)-1,2-diamino-1,2-ethanediyl]bis-](http://img.cochemist.com/ccimg/91600/91548-22-0_b.png)
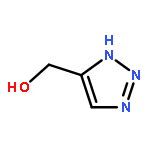
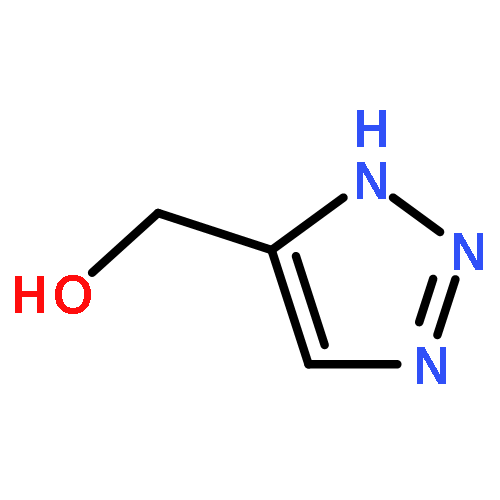
![Cyclohexanol, 1-[1-(phenylmethyl)-1H-1,2,3-triazol-4-yl]-](/data/chemimg/3722400/83027-66-1.png)
![Cyclohexanol, 1-[1-(phenylmethyl)-1H-1,2,3-triazol-4-yl]-](/data/chemimg/3722400/83027-66-1_b.png)
![PHOSPHONIC ACID, [PHENYL(PHENYLTHIO)METHYL]-, DIETHYL ESTER](http://img.cochemist.com/ccimg/73800/73778-49-1.png)
![PHOSPHONIC ACID, [PHENYL(PHENYLTHIO)METHYL]-, DIETHYL ESTER](http://img.cochemist.com/ccimg/73800/73778-49-1_b.png)


![Benzene, 1-[(4-methoxyphenyl)ethynyl]-2-methyl-](http://img.cochemist.com/ccimg/59200/59105-21-4.png)
![Benzene, 1-[(4-methoxyphenyl)ethynyl]-2-methyl-](http://img.cochemist.com/ccimg/59200/59105-21-4_b.png)



![Phosphonic acid, [[(3-nitrophenyl)amino]phenylmethyl]-, diethyl ester](http://img.cochemist.com/ccimg/37000/36934-13-1.png)
![Phosphonic acid, [[(3-nitrophenyl)amino]phenylmethyl]-, diethyl ester](http://img.cochemist.com/ccimg/37000/36934-13-1_b.png)



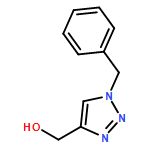

![Phosphonic acid, [(4-chlorophenyl)(phenylamino)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/26400/26321-54-0.png)
![Phosphonic acid, [(4-chlorophenyl)(phenylamino)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/26400/26321-54-0_b.png)
![Phosphonic acid, [(4-methylphenyl)(phenylamino)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/26400/26321-53-9.png)
![Phosphonic acid, [(4-methylphenyl)(phenylamino)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/26400/26321-53-9_b.png)


![Pyridine, 2-[(4-methoxyphenyl)ethynyl]-](http://img.cochemist.com/ccimg/16400/16344-79-9.png)
![Pyridine, 2-[(4-methoxyphenyl)ethynyl]-](http://img.cochemist.com/ccimg/16400/16344-79-9_b.png)
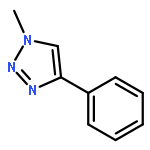



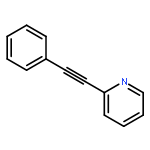
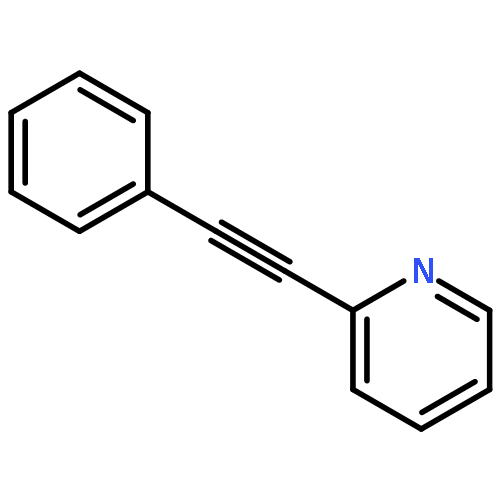


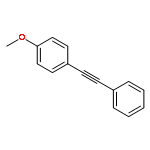
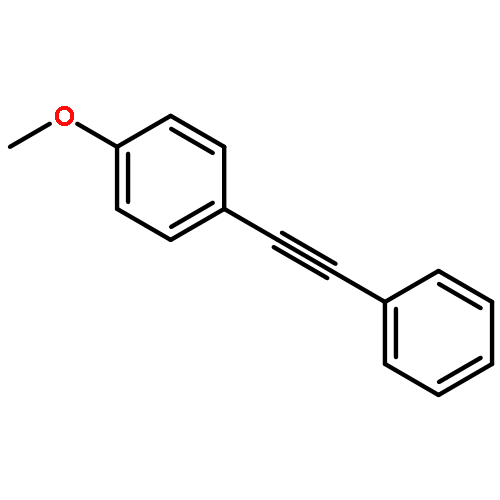
![Phosphonic acid, P-[phenyl(phenylamino)methyl]-, diethyl ester](/data/chemimg/1951600/7236-88-6.png)
![Phosphonic acid, P-[phenyl(phenylamino)methyl]-, diethyl ester](/data/chemimg/1951600/7236-88-6_b.png)