Co-reporter:K. C. Nicolaou, Kiran Kumar Pulukuri, Stephan Rigol, Marek Buchman, Akshay A. Shah, Nicholas Cen, Megan D. McCurry, Kathryn Beabout, and Yousif Shamoo
Journal of the American Chemical Society November 8, 2017 Volume 139(Issue 44) pp:15868-15868
Publication Date(Web):October 24, 2017
DOI:10.1021/jacs.7b08749
An improved and enantioselective total synthesis of antibiotic CJ-16,264 through a practical kinetic resolution and an iodolactonization reaction to form the iodo pyrrolizidinone fragment of the molecule is described. A series of racemic and enantiopure analogues of CJ-16,264 was designed and synthesized through the developed synthetic technologies and tested against drug-resistant bacterial strains. These studies led to interesting structure–activity relationships and the identification of a number of simpler, and yet equipotent, or even more potent, antibacterial agents than the natural product, thereby setting the foundation for further investigations in the quest for new anti-infective drugs.
Co-reporter:K. C. Nicolaou, Gabriel Bellavance, Marek Buchman, and Kiran Kumar Pulukuri
Journal of the American Chemical Society November 8, 2017 Volume 139(Issue 44) pp:15636-15636
Publication Date(Web):October 24, 2017
DOI:10.1021/jacs.7b09843
Described herein are the first total syntheses of naturally occurring antitumor agents disorazoles A1 and B1 and the full structural assignment of the latter. The syntheses were achieved through convergent strategies employing enantioselective constructions of the required building blocks, including a novel Sharpless epoxidation/enzymatic kinetic resolution of stannane-containing substrates that led selectively to both enantiomeric forms of an epoxy vinyl stannane, and a series of coupling reactions, including a Wittig reaction, a Suzuki coupling, a Stille coupling, a Yamaguchi esterification and a Yamaguchi macrolactonization.
Co-reporter:K. C. Nicolaou, Pengxi Chen, Shugao Zhu, Quan Cai, Rohan D. Erande, Ruofan Li, Hongbao Sun, Kiran Kumar Pulukuri, Stephan Rigol, Monette Aujay, Joseph Sandoval, and Julia Gavrilyuk
Journal of the American Chemical Society November 1, 2017 Volume 139(Issue 43) pp:15467-15467
Publication Date(Web):October 20, 2017
DOI:10.1021/jacs.7b08820
A streamlined total synthesis of the naturally occurring antitumor agents trioxacarcins is described, along with its application to the construction of a series of designed analogues of these complex natural products. Biological evaluation of the synthesized compounds revealed a number of highly potent, and yet structurally simpler, compounds that are effective against certain cancer cell lines, including a drug-resistant line. A novel one-step synthesis of anthraquinones and chloro anthraquinones from simple ketone precursors and phenylselenyl chloride is also described. The reported work, featuring novel chemistry and cascade reactions, has potential applications in cancer therapy, including targeted approaches as in antibody–drug conjugates.
Co-reporter:K. C. Nicolaou, Derek Rhoades, Yanping Wang, Ruoli Bai, Ernest Hamel, Monette Aujay, Joseph Sandoval, and Julia Gavrilyuk
Journal of the American Chemical Society May 31, 2017 Volume 139(Issue 21) pp:7318-7318
Publication Date(Web):May 17, 2017
DOI:10.1021/jacs.7b02655
The synthesis and biological evaluation of a series of 12,13-aziridinyl epothilone B analogues is described. These compounds were accessed by a practical, general process that involved a 12,13-olefinic methyl ketone as a starting material obtained by ozonolytic cleavage of epothilone B followed by tungsten-induced deoxygenation of the epoxide moiety. The attachment of the aziridine structural motif was achieved by application of the Ess–Kürti–Falck aziridination, while the heterocyclic side chains were introduced via stereoselective phosphonate-based olefinations. In order to ensure high (E) selectivities for the latter reaction for electron-rich heterocycles, it became necessary to develop and apply an unprecedented modification of the venerable Horner–Wadsworth–Emmons reaction, employing 2-fluoroethoxyphosphonates that may prove to be of general value in organic synthesis. These studies resulted in the discovery of some of the most potent epothilones reported to date. Equipped with functional groups to accommodate modern drug delivery technologies, some of these compounds exhibited picomolar potencies that qualify them as payloads for antibody drug conjugates (ADCs), while a number of them revealed impressive activities against drug resistant human cancer cells, making them desirable for potential medical applications.
Co-reporter:K. C. Nicolaou, Guodu Liu, Kathryn Beabout, Megan D. McCurry, and Yousif Shamoo
Journal of the American Chemical Society March 15, 2017 Volume 139(Issue 10) pp:3736-3736
Publication Date(Web):March 3, 2017
DOI:10.1021/jacs.6b12654
A phase transfer catalyzed asymmetric alkylation of anthrones with cyclic allylic bromides using quinidine- or quinine-derived catalysts is described. Utilizing mild basic conditions and as low as 0.5 mol % catalyst loading, and achieving up to >99:1 dr selectivity, this asymmetric reaction was successfully applied to produce enantioselectively (−)- and (+)-viridicatumtoxins B, and thus allowed assignment of the absolute configuration of this naturally occurring antibiotic. While the developed asymmetric synthesis of C10 substituted anthrones is anticipated to find wider applications in organic synthesis, its immediate application to the construction of a variety of designed enantiopure analogues of viridicatumtoxin B led to the discovery of highly potent, yet simpler analogues of the molecule. These studies are expected to facilitate drug discovery and development efforts toward new antibacterial agents.
Co-reporter:
Israel Journal of Chemistry 2017 Volume 57(Issue 3-4) pp:179-191
Publication Date(Web):2017/04/01
DOI:10.1002/ijch.201600087
AbstractThe advent of organic synthesis in the nineteenth century sparked a revolution in chemistry that led from a serendipitous discovery to the art and science that it is today. Its creative nature turned into enabling technologies that set in motion entire new industries such as the dye and pharmaceutical enterprises and helped elucidate and confirm structures of countless natural products. It also served as the locomotive for discovery and invention of new reactions, synthetic strategies and technologies, and delivered myriad valuable compounds, more or less complex, for research and applications in our everyday lives. Today it is a partner of biology in the quest of understanding living nature and applying the gathered intelligence to discover and develop newer and more effective drugs.
Co-reporter:K. C. Nicolaou; Kiran Kumar Pulukuri; Stephan Rigol; Philipp Heretsch; Ruocheng Yu; Charles I. Grove; Christopher R. H. Hale; Abdelatif ElMarrouni; Verena Fetz; Mark Brönstrup; Monette Aujay; Joseph Sandoval;Julia Gavrilyuk
Journal of the American Chemical Society 2016 Volume 138(Issue 20) pp:6550-6560
Publication Date(Web):May 12, 2016
DOI:10.1021/jacs.6b02075
A series of Δ12-prostaglandin J3 (Δ12-PGJ3) analogues and derivatives were synthesized employing an array of synthetic strategies developed specifically to render them readily available for biological investigations. The synthesized compounds were evaluated for their cytotoxicity against a number of cancer cell lines, revealing nanomolar potencies for a number of them against certain cancer cell lines. Four analogues (2, 11, 21, and 27) demonstrated inhibition of nuclear export through a covalent addition at Cys528 of the export receptor Crm1. One of these compounds (i.e., 11) is currently under evaluation as a potential drug candidate for the treatment of certain types of cancer. These studies culminated in useful and path-pointing structure–activity relationships (SARs) that provide guidance for further improvements in the biological/pharmacological profiles of compounds within this class.
Co-reporter:K. C. Nicolaou; Derek Rhoades; Manjunath Lamani; Manas R. Pattanayak;S. Mothish Kumar
Journal of the American Chemical Society 2016 Volume 138(Issue 24) pp:7532-7535
Publication Date(Web):June 8, 2016
DOI:10.1021/jacs.6b04781
The total synthesis of the spliceosome inhibitor thailanstatin A has been achieved in a longest linear sequence of nine steps from readily available starting materials. A key feature of the developed synthetic strategy is the implementation of a unique, biomimetic asymmetric intramolecular oxa-Michael reaction/hydrogenation sequence that allows diastereodivergent access to highly functionalized tetrahydropyrans, which can be used for the synthesis of designed analogues of this bioactive molecule.
Co-reporter:K. C. Nicolaou; Yanping Wang; Min Lu; Debashis Mandal; Manas R. Pattanayak; Ruocheng Yu; Akshay A. Shah; Jason S. Chen; Hongjun Zhang; James J. Crawford; Laxman Pasunoori; Yam B. Poudel; Naidu S. Chowdari; Chin Pan; Ayesha Nazeer; Sanjeev Gangwar; Gregory Vite;Emmanuel N. Pitsinos
Journal of the American Chemical Society 2016 Volume 138(Issue 26) pp:8235-8246
Publication Date(Web):June 7, 2016
DOI:10.1021/jacs.6b04339
From the enediyne class of antitumor antibiotics, uncialamycin is among the rarest and most potent, yet one of the structurally simpler, making it attractive for chemical synthesis and potential applications in biology and medicine. In this article we describe a streamlined and practical enantioselective total synthesis of uncialamycin that is amenable to the synthesis of novel analogues and renders the natural product readily available for biological and drug development studies. Starting from hydroxy- or methoxyisatin, the synthesis features a Noyori enantioselective reduction, a Yamaguchi acetylide-pyridinium coupling, a stereoselective acetylide-aldehyde cyclization, and a newly developed annulation reaction that allows efficient coupling of a cyanophthalide and a p-methoxy semiquinone aminal to forge the anthraquinone moiety of the molecule. Overall, the developed streamlined synthesis proceeds in 22 linear steps (14 chromatographic separations) and 11% overall yield. The developed synthetic strategies and technologies were applied to the synthesis of a series of designed uncialamycin analogues equipped with suitable functional groups for conjugation to antibodies and other delivery systems. Biological evaluation of a select number of these analogues led to the identification of compounds with low picomolar potencies against certain cancer cell lines. These compounds and others like them may serve as powerful payloads for the development of antibody drug conjugates (ADCs) intended for personalized targeted cancer therapy.
Co-reporter:K. C. Nicolaou; Jun Yin; Debashis Mandal; Rohan D. Erande; Philipp Klahn; Michael Jin; Monette Aujay; Joseph Sandoval; Julia Gavrilyuk;Dionisios Vourloumis
Journal of the American Chemical Society 2016 Volume 138(Issue 5) pp:1698-1708
Publication Date(Web):February 1, 2016
DOI:10.1021/jacs.5b12557
A streamlined total synthesis of N14-desacetoxytubulysin H (Tb1) based on a C–H activation strategy and a short total synthesis of pretubulysin D (PTb-D43) are described. Applications of the developed synthetic strategies and technologies to the synthesis of a series of tubulysin analogues (Tb2–Tb41 and PTb-D42) are also reported. Biological evaluation of the synthesized compounds against an array of cancer cells revealed a number of novel analogues (e.g., Tb14), some with exceptional potencies against certain cell lines [e.g., Tb32 with IC50 = 12 pM against MES SA (uterine sarcoma) cell line and 2 pM against HEK 293T (human embryonic kidney) cell line], and a set of valuable structure–activity relationships. The highly potent cytotoxic compounds discovered in this study are highly desirable as payloads for antibody–drug conjugates and other drug delivery systems for personalized targeted cancer chemotherapies.
Co-reporter: Kyriacos C. Nicolaou;Dr. Dionisios Vourloumis;Dr. Sotirios Totokotsopoulos;Dr. Athanasios Papakyriakou;Dr. Holger Karsunky;Hanan Ferno;Dr. Julia Gavrilyuk;Dr. Damien Webb;Dr. Antonia F. Stepan
ChemMedChem 2016 Volume 11( Issue 1) pp:31-37
Publication Date(Web):
DOI:10.1002/cmdc.201500510
Abstract
A convenient synthesis of imatinib, a potent inhibitor of ABL1 kinase and widely prescribed drug for the treatment of a variety of leukemias, was devised and applied to the construction of a series of novel imatinib analogues featuring a number of non-aromatic structural motifs in place of the parent molecule's phenyl moiety. These analogues were subsequently evaluated for their biopharmaceutical properties (e.g., ABL1 kinase inhibitory activity, cytotoxicity). The bicyclo[1.1.1]pentane- and cubane-containing analogues were found to possess higher themodynamic solubility, whereas cubane- and cyclohexyl-containing analogues exhibited the highest inhibitory activity against ABL1 kinase and the most potent cytotoxicity values against cancer cell lines K562 and SUP-B15. Molecular modeling was employed to rationalize the weak activity of the compounds against ABL1 kinase, and it is likely that the observed cytotoxicity of these agents arises through off-target effects.
Co-reporter:Dr. K. C. Nicolaou;Dr. Kiran Kumar Pulukuri;Ruocheng Yu;Dr. Stephan Rigol;Dr. Philipp Heretsch;Dr. Charles I. Grove;Christopher R. H. Hale;Dr. Abdelatif ElMarrouni
Chemistry - A European Journal 2016 Volume 22( Issue 25) pp:8559-8570
Publication Date(Web):
DOI:10.1002/chem.201601449
Abstract
The total synthesis of Δ12-prostaglandin J3 (Δ12-PGJ3, 1), a reported leukemia stem cell ablator, through a number of strategies and tactics is described. The signature cross-conjugated dienone structural motif of 1 was forged by an aldol reaction/dehydration sequence from key building blocks enone 13 and aldehyde 14, whose lone stereocenters were generated by an asymmetric Tsuji–Trost reaction and an asymmetric Mukaiyama aldol reaction, respectively. During this program, a substituent-governed regioselectivity pattern for the Rh-catalyzed C−H functionalization of cyclopentenes and related olefins was discovered. The evolution of the synthesis of 1 from the original strategy to the final streamlined process proceeded through improvements in the construction of both fragments 13 and 14, exploration of the chemistry of the hitherto underutilized chiral lactone synthon 57, and a diastereoselective alkylation of a cyclopentenone intermediate. The described chemistry sets the stage for large-scale production of Δ12-PGJ3 and designed analogues for further biological and pharmacological studies.
Co-reporter:K. C. Nicolaou; Christian Nilewski; Christopher R. H. Hale; Christopher F. Ahles; Chiao An Chiu; Christian Ebner; Abdelatif ElMarrouni; Lifeng Yang; Katherine Stiles;Deepak Nagrath
Journal of the American Chemical Society 2015 Volume 137(Issue 14) pp:4766-4770
Publication Date(Web):April 1, 2015
DOI:10.1021/jacs.5b00141
A recently developed dimerization/macrocyclization was employed to synthesize a series of macroheterocycles which were biologically evaluated, leading to the discovery of a number of potent cytotoxic agents (e.g., 27: GI50 = 51 nM against leukemia CCRF-CEM cell line; 29: GI50 = 99 nM against melanoma MDA-MB-435 cell line). Further biological studies support an apoptosis mechanism of action for these compounds involving deregulation of the tricarboxylic acid cycle activity and suppression of mitochondrial function in cancer cells.
Co-reporter:K. C. Nicolaou; Zhaoyong Lu; Ruofan Li; James R. Woods;Te-ik Sohn
Journal of the American Chemical Society 2015 Volume 137(Issue 27) pp:8716-8719
Publication Date(Web):July 2, 2015
DOI:10.1021/jacs.5b05575
The total synthesis of the rare but extremely potent antitumor agent shishijimicin A has been achieved via a convergent strategy involving carboline disaccharide 3 and hydroxy enediyne thioacetate 4.
Co-reporter:Dr. K. C. Nicolaou;Dr. Quan Cai;Dr. Bo Qin;Mette T. Petersen;Remi J. T. Mikkelsen ;Dr. Philipp Heretsch
Angewandte Chemie International Edition 2015 Volume 54( Issue 10) pp:3074-3078
Publication Date(Web):
DOI:10.1002/anie.201410369
Abstract
An enantioselective total synthesis of trioxacarcin DC-45-A2 (1) featuring a novel Lewis acid-induced cascade rearrangement of epoxyketone 6 to forge the polyoxygenated 2,7-dioxabicyclo[2.2.1]heptane core of the molecule is described.
Co-reporter:Dr. K. C. Nicolaou;Dr. Quan Cai;Dr. Bo Qin;Mette T. Petersen;Remi J. T. Mikkelsen ;Dr. Philipp Heretsch
Angewandte Chemie 2015 Volume 127( Issue 10) pp:3117-3121
Publication Date(Web):
DOI:10.1002/ange.201410369
Abstract
An enantioselective total synthesis of trioxacarcin DC-45-A2 (1) featuring a novel Lewis acid-induced cascade rearrangement of epoxyketone 6 to forge the polyoxygenated 2,7-dioxabicyclo[2.2.1]heptane core of the molecule is described.
Co-reporter:Dr. K. C. Nicolaou;Dr. Min Lu;Pengxi Chen ;Dr. Akshay A. Shah
Angewandte Chemie 2015 Volume 127( Issue 43) pp:12878-12882
Publication Date(Web):
DOI:10.1002/ange.201507007
Abstract
New versatile and selective methods for the syntheses of substituted amino- and methoxyphenolic anthraquinones (I–IV) based on fusion of cyanophthalides (V) and semiquinone aminals (VI, VII) under basic conditions are described.
Co-reporter:Dr. K. C. Nicolaou;Derek Rhoades;Dr. Yanping Wang;Dr. Sotirios Totokotsopoulos;Dr. Ruoli Bai;Dr. Ernest Hamel
ChemMedChem 2015 Volume 10( Issue 12) pp:1974-1979
Publication Date(Web):
DOI:10.1002/cmdc.201500401
Abstract
The design, synthesis, and biological evaluation of a series of epothilone analogues with novel side chains equipped with an amino group are described. Their design facilitates potential conjugation to selective drug delivery systems such as antibodies. Their synthesis proceeded efficiently via Stille coupling of a readily available vinyl iodide and heterocyclic stannanes. Cytotoxicity studies and tubulin binding assays revealed two of these analogues to be more potent than epothilones A–D and the anticancer agent ixabepilone, currently in clinical use.
Co-reporter:Dr. K. C. Nicolaou;Dr. Akshay A. Shah;Dr. Henry Korman;Dr. Tabrez Khan;Dr. Lei Shi;Dr. Wisuttaya Worawalai;Dr. Emmanuel A. Theodorakis
Angewandte Chemie International Edition 2015 Volume 54( Issue 32) pp:9203-9208
Publication Date(Web):
DOI:10.1002/anie.201504337
Abstract
The total synthesis and structural revision of antibiotic CJ-16,264 is described. Starting with citronellal, the quest for the target molecule featured a novel bis-transannular Diels–Alder reaction that casted stereoselectively the decalin system and included the synthesis of six isomers before demystification of its true structure.
Co-reporter:Dr. K. C. Nicolaou;Dr. Akshay A. Shah;Dr. Henry Korman;Dr. Tabrez Khan;Dr. Lei Shi;Dr. Wisuttaya Worawalai;Dr. Emmanuel A. Theodorakis
Angewandte Chemie 2015 Volume 127( Issue 32) pp:9335-9340
Publication Date(Web):
DOI:10.1002/ange.201504337
Abstract
The total synthesis and structural revision of antibiotic CJ-16,264 is described. Starting with citronellal, the quest for the target molecule featured a novel bis-transannular Diels–Alder reaction that casted stereoselectively the decalin system and included the synthesis of six isomers before demystification of its true structure.
Co-reporter:Dr. K. C. Nicolaou;Dr. Min Lu;Pengxi Chen ;Dr. Akshay A. Shah
Angewandte Chemie International Edition 2015 Volume 54( Issue 43) pp:12687-12691
Publication Date(Web):
DOI:10.1002/anie.201507007
Abstract
New versatile and selective methods for the syntheses of substituted amino- and methoxyphenolic anthraquinones (I–IV) based on fusion of cyanophthalides (V) and semiquinone aminals (VI, VII) under basic conditions are described.
Co-reporter:K. C. Nicolaou ; Christopher R. H. Hale ; Christian Nilewski ; Heraklidia A. Ioannidou ; Abdelatif ElMarrouni ; Lizanne G. Nilewski ; Kathryn Beabout ; Tim T. Wang ;Yousif Shamoo
Journal of the American Chemical Society 2014 Volume 136(Issue 34) pp:12137-12160
Publication Date(Web):August 15, 2014
DOI:10.1021/ja506472u
The details of the total synthesis of viridicatumtoxin B (1) are described. Initial synthetic strategies toward this intriguing tetracycline antibiotic resulted in the development of key alkylation and Lewis acid-mediated spirocyclization reactions to form the hindered EF spirojunction, as well as Michael–Dieckmann reactions to set the A and C rings. The use of an aromatic A-ring substrate, however, was found to be unsuitable for the introduction of the requisite hydroxyl groups at carbons 4a and 12a. Applying these previous tactics, we developed stepwise approaches to oxidize carbons 12a and 4a based on enol- and enolate-based oxidations, respectively, the latter of which was accomplished after systematic investigations that revealed critical reactivity patterns. The herein described synthetic strategy resulted in the total synthesis of viridicatumtoxin B (1), which, in turn, formed the basis for the revision of its originally assigned structure. The developed chemistry facilitated the synthesis of a series of viridicatumtoxin analogues, which were evaluated against Gram-positive and Gram-negative bacterial strains, including drug-resistant pathogens, revealing the first structure–activity relationships within this structural type.
Co-reporter:K. C. Nicolaou, Philipp Heretsch, Tsuyoshi Nakamura, Anna Rudo, Michio Murata, and Keiichi Konoki
Journal of the American Chemical Society 2014 Volume 136(Issue 46) pp:16444-16451
Publication Date(Web):November 6, 2014
DOI:10.1021/ja509829e
The synthesis of QRSTUVWXYZA′ domains 7, 8, and 9 of the highly potent marine neurotoxin maitotoxin (1), the largest secondary metabolite isolated to date, is described. The devised synthetic strategy entailed a cascade Takai–Utimoto ester olefination/ring closing metathesis to construct ring Y, a hydroxydithioketal cyclization/methylation sequence to cast ring X, a Horner–Wadsworth–Emmons coupling of WXYZA′ ketophosphonate 11 with QRSTU aldehyde 12 to form enone 10, and a reductive hydroxyketone ring closure to forge ring V. 2D NMR spectroscopic analysis and comparison of 13C chemical shifts with those of the corresponding carbons of maitotoxin revealed close similarities supporting the originally assigned structure of this region of the natural product. Biological evaluations of various synthesized domains of maitotoxin in this and previous studies from these laboratories led to fragment structure–activity relationships regarding their ability to inhibit maitotoxin-elicited Ca2+ influx in rat C6 glioma cells.
Co-reporter:K.C. Nicolaou
Chemistry & Biology 2014 Volume 21(Issue 9) pp:1039-1045
Publication Date(Web):18 September 2014
DOI:10.1016/j.chembiol.2014.07.020
Admirable as it is, the drug discovery and development process is continuously undergoing changes and adjustments in search of further improvements in efficiency, productivity, and profitability. Recent trends in academic-industrial partnerships promise to provide new opportunities for advancements of this process through transdisciplinary collaborations along the entire spectrum of activities involved in this complex process. This perspective discusses ways to promote the emerging academic paradigm of the chemistry-biology-medicine continuum as a means to advance the drug discovery and development process.
Co-reporter:Dr. K. C. Nicolaou;Dr. Philipp Heretsch;Dr. Abdelatif ElMarrouni;Christopher R. H. Hale;Dr. Kiran K. Pulukuri;Dr. Avinash K. Kudva;Dr. Vivek Narayan;Dr. K. Seep Prabhu
Angewandte Chemie 2014 Volume 126( Issue 39) pp:10611-10615
Publication Date(Web):
DOI:10.1002/ange.201404917
Abstract
A catalytic asymmetric total synthesis of the potent and selective antileukemic Δ12-prostaglandin J3 (Δ12-PGJ3) is described. The convergent synthesis proceeded through intermediates 2 and 3, formed enantioselectively from readily available starting materials and coupled through an aldol reaction followed by dehydration to afford stereoselectively the cyclopentenone alkylidene structural motif of the molecule.
Co-reporter:Dr. K. C. Nicolaou;Dr. Philipp Heretsch;Dr. Abdelatif ElMarrouni;Christopher R. H. Hale;Dr. Kiran K. Pulukuri;Dr. Avinash K. Kudva;Dr. Vivek Narayan;Dr. K. Seep Prabhu
Angewandte Chemie International Edition 2014 Volume 53( Issue 39) pp:10443-10447
Publication Date(Web):
DOI:10.1002/anie.201404917
Abstract
A catalytic asymmetric total synthesis of the potent and selective antileukemic Δ12-prostaglandin J3 (Δ12-PGJ3) is described. The convergent synthesis proceeded through intermediates 2 and 3, formed enantioselectively from readily available starting materials and coupled through an aldol reaction followed by dehydration to afford stereoselectively the cyclopentenone alkylidene structural motif of the molecule.
Co-reporter: K. C. Nicolaou
Angewandte Chemie International Edition 2014 Volume 53( Issue 19) pp:
Publication Date(Web):
DOI:10.1002/anie.201402816
Co-reporter: K. C. Nicolaou
Angewandte Chemie International Edition 2014 Volume 53( Issue 35) pp:9128-9140
Publication Date(Web):
DOI:10.1002/anie.201404761
Abstract
The current state of affairs in the drug discovery and development process is briefly summarized and then ways to take advantage of the ever-increasing fundamental knowledge and technical knowhow in chemistry and biology and related disciplines are discussed. The primary motivation of this Essay is to celebrate the great achievements of chemistry, biology, and medicine and to inform and inspire students and academics to enter the field of drug discovery and development while, at the same time, continue to advance the fundamentals of their disciplines. It is also meant to encourage and catalyze multidisciplinary partnerships between academia and industry as scientists attempt to merge their often complementary interests and expertise to achieve new improvements and breakthroughs in their respective fields, and the common goal of applying them to the discovery and invention of new and better medicines, especially in areas of unmet needs.
Co-reporter:Dr. K. C. Nicolaou;Dr. Lei Shi;Dr. Min Lu;Dr. Manas R. Pattanayak;Dr. Akshay A. Shah;Dr. Heraklidia A. Ioannidou ;Dr. Manjunath Lamani
Angewandte Chemie International Edition 2014 Volume 53( Issue 41) pp:10970-10974
Publication Date(Web):
DOI:10.1002/anie.201406815
Abstract
The total synthesis of cytotoxic polyketides myceliothermophins E (1), C (2), and D (3) through a cascade-based cyclization to form the trans-fused decalin system is described. The convergent synthesis delivered all three natural products through late-stage divergence and facilitated unambiguous C21 structural assignments for 2 and 3 through X-ray crystallographic analysis, which revealed an interesting dimeric structure between its enantiomeric forms.
Co-reporter: K. C. Nicolaou
Angewandte Chemie 2014 Volume 126( Issue 19) pp:
Publication Date(Web):
DOI:10.1002/ange.201402816
Co-reporter: K. C. Nicolaou
Angewandte Chemie 2014 Volume 126( Issue 35) pp:9280-9292
Publication Date(Web):
DOI:10.1002/ange.201404761
Abstract
The current state of affairs in the drug discovery and development process is briefly summarized and then ways to take advantage of the ever-increasing fundamental knowledge and technical knowhow in chemistry and biology and related disciplines are discussed. The primary motivation of this Essay is to celebrate the great achievements of chemistry, biology, and medicine and to inform and inspire students and academics to enter the field of drug discovery and development while, at the same time, continue to advance the fundamentals of their disciplines. It is also meant to encourage and catalyze multidisciplinary partnerships between academia and industry as scientists attempt to merge their often complementary interests and expertise to achieve new improvements and breakthroughs in their respective fields, and the common goal of applying them to the discovery and invention of new and better medicines, especially in areas of unmet needs.
Co-reporter:Dr. K. C. Nicolaou;Dr. Lei Shi;Dr. Min Lu;Dr. Manas R. Pattanayak;Dr. Akshay A. Shah;Dr. Heraklidia A. Ioannidou ;Dr. Manjunath Lamani
Angewandte Chemie 2014 Volume 126( Issue 41) pp:11150-11154
Publication Date(Web):
DOI:10.1002/ange.201406815
Abstract
The total synthesis of cytotoxic polyketides myceliothermophins E (1), C (2), and D (3) through a cascade-based cyclization to form the trans-fused decalin system is described. The convergent synthesis delivered all three natural products through late-stage divergence and facilitated unambiguous C21 structural assignments for 2 and 3 through X-ray crystallographic analysis, which revealed an interesting dimeric structure between its enantiomeric forms.
Co-reporter:Kyriacos C. Nicolaou and Roman A. Valiulin
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 25) pp:4154-4163
Publication Date(Web):17 May 2013
DOI:10.1039/C3OB40654G
Reaction of 10-deacetylbaccatin III (III) and its 7-TES derivative (IV) with DAST under various conditions resulted in the formation of an array of new fluorinated and non-fluorinated 13-keto taxoid compounds (2a–4a) through a vinylogous pinacol–pinacolone rearrangement. Further fluorination of some of these products (2a, 3a) with NFSi or Selectfluor gave additional derivatives. Sodium borohydride reduction of the 13-keto group of these products (2a, 2b, 3a, 3b, 4a, 8, 9, 11–14) led to a series of 9α-hydroxy taxoid derivatives, which were esterified using the docetaxel side chain employing the corresponding protected β-lactam, followed by deprotection to furnish a library of docetaxel analogs and related compounds. A selected number of synthesized compounds (7, 10, 19a, 19b, 21a, 21b, 23, 27, 29, 34–36) were submitted to the National Cancer Institute (NCI) 60 cell line screening program and tested for cytotoxic properties. Taxoids 19a, 19b, 21a, 21b, 23, 27, 29, 34 and 35 were found to exhibit significant anticancer activity against various cancerous cell lines with 23, 27, and 29 being the most potent compounds, demonstrating GI50 values of ≤5 nM in several assays.
Co-reporter:Dr. K. C. Nicolaou;Dr. Christian Nilewski;Christopher R. H. Hale;Dr. Heraklidia A. Ioannidou;Dr. Abdelatif ElMarrouni;Lizanne G. Koch
Angewandte Chemie International Edition 2013 Volume 52( Issue 33) pp:8736-8741
Publication Date(Web):
DOI:10.1002/anie.201304691
Co-reporter:Dr. K. C. Nicolaou;Dr. Christian Nilewski;Christopher R. H. Hale;Dr. Heraklidia A. Ioannidou;Dr. Abdelatif ElMarrouni;Lizanne G. Koch
Angewandte Chemie International Edition 2013 Volume 52( Issue 33) pp:
Publication Date(Web):
DOI:10.1002/anie.201305680
Co-reporter:Dr. K. C. Nicolaou;Dr. Christian Nilewski;Christopher R. H. Hale;Dr. Heraklidia A. Ioannidou;Dr. Abdelatif ElMarrouni;Lizanne G. Koch
Angewandte Chemie 2013 Volume 125( Issue 33) pp:8898-8904
Publication Date(Web):
DOI:10.1002/ange.201304691
Co-reporter:Dr. K. C. Nicolaou;Dr. Christian Nilewski;Christopher R. H. Hale;Dr. Heraklidia A. Ioannidou;Dr. Abdelatif ElMarrouni;Lizanne G. Koch
Angewandte Chemie 2013 Volume 125( Issue 33) pp:
Publication Date(Web):
DOI:10.1002/ange.201305680
Co-reporter:Om P. Mishra, Nicholas Simmons, Sonia Tyagi, Ralph Pietrofesa, Vladimir V. Shuvaev, Roman A. Valiulin, Philipp Heretsch, K.C. Nicolaou, Melpo Christofidou-Solomidou
Bioorganic & Medicinal Chemistry Letters 2013 23(19) pp: 5325-5328
Publication Date(Web):
DOI:10.1016/j.bmcl.2013.07.062
Co-reporter:K. C. Nicolaou; Quan Cai; Hongbao Sun; Bo Qin;Shugao Zhu
Journal of the American Chemical Society () pp:
Publication Date(Web):February 24, 2016
DOI:10.1021/jacs.5b12687
Trioxacarcins DC-45-A2, DC-45-A1, A, D, C7″-epi-C, and C have been synthesized through stereoselective strategies involving BF3·Et2O-catalyzed ketone–epoxide opening and gold-catalyzed glycosylation reactions, and the full structural assignment of trioxacacin C was deciphered via the syntheses of both of its C7″ epimers. The gathered knowledge sets the foundation for the design, synthesis, and biological evalution of analogues of these natural products as potential payloads for antibody–drug conjugates and other delivery systems for targeted and personalized cancer chemotherapy.
Co-reporter:Kyriacos C. Nicolaou and Roman A. Valiulin
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 25) pp:NaN4163-4163
Publication Date(Web):2013/05/17
DOI:10.1039/C3OB40654G
Reaction of 10-deacetylbaccatin III (III) and its 7-TES derivative (IV) with DAST under various conditions resulted in the formation of an array of new fluorinated and non-fluorinated 13-keto taxoid compounds (2a–4a) through a vinylogous pinacol–pinacolone rearrangement. Further fluorination of some of these products (2a, 3a) with NFSi or Selectfluor gave additional derivatives. Sodium borohydride reduction of the 13-keto group of these products (2a, 2b, 3a, 3b, 4a, 8, 9, 11–14) led to a series of 9α-hydroxy taxoid derivatives, which were esterified using the docetaxel side chain employing the corresponding protected β-lactam, followed by deprotection to furnish a library of docetaxel analogs and related compounds. A selected number of synthesized compounds (7, 10, 19a, 19b, 21a, 21b, 23, 27, 29, 34–36) were submitted to the National Cancer Institute (NCI) 60 cell line screening program and tested for cytotoxic properties. Taxoids 19a, 19b, 21a, 21b, 23, 27, 29, 34 and 35 were found to exhibit significant anticancer activity against various cancerous cell lines with 23, 27, and 29 being the most potent compounds, demonstrating GI50 values of ≤5 nM in several assays.
.jpg)
![[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetyloxy)pent-2-enoyl]amino}-3,6-dimethyltetrahydro-2H-pyran-2-yl]-3-methylpenta-1,3-dien-1-yl}-8-hydroxy-1,6-dioxaspiro[2.5]oct-5-yl]acetic acid](http://img.cochemist.com/ccimg/1427000/1426953-21-0.png)
![[(3R,5S,7R,8R)-7-{(1E,3E)-5-[(2S,3S,5R,6R)-5-{[(2Z,4S)-4-(acetyloxy)pent-2-enoyl]amino}-3,6-dimethyltetrahydro-2H-pyran-2-yl]-3-methylpenta-1,3-dien-1-yl}-8-hydroxy-1,6-dioxaspiro[2.5]oct-5-yl]acetic acid](http://img.cochemist.com/ccimg/1427000/1426953-21-0_b.png)


![2-METHYL-2-PROPANYL [(3R,6S)-6-(2,3-DIFLUOROPHENYL)-2-OXO-3-AZEPANYL]CARBAMATE](http://img.cochemist.com/ccimg/1313100/1313028-09-9.png)
![2-METHYL-2-PROPANYL [(3R,6S)-6-(2,3-DIFLUOROPHENYL)-2-OXO-3-AZEPANYL]CARBAMATE](http://img.cochemist.com/ccimg/1313100/1313028-09-9_b.png)













![BICYCLO[1.1.1]PENTANE-1-CARBOXYLIC ACID, 3-AMINO-, METHYL ESTER (9CI)](http://img.cochemist.com/ccimg/758700/758684-88-7.png)
![BICYCLO[1.1.1]PENTANE-1-CARBOXYLIC ACID, 3-AMINO-, METHYL ESTER (9CI)](http://img.cochemist.com/ccimg/758700/758684-88-7_b.png)
![OCTANAL, 3-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-, (3S)-](http://img.cochemist.com/ccimg/676500/676456-67-0.png)
![OCTANAL, 3-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-, (3S)-](http://img.cochemist.com/ccimg/676500/676456-67-0_b.png)
![Methyl 3-(Boc-amino)-bicyclo[1.1.1]pentane-1-carboxylate](http://img.cochemist.com/ccimg/676400/676371-64-5.png)
![Methyl 3-(Boc-amino)-bicyclo[1.1.1]pentane-1-carboxylate](http://img.cochemist.com/ccimg/676400/676371-64-5_b.png)
![1-OCTANOL, 3-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-, (3S)-](http://img.cochemist.com/ccimg/676300/676236-08-1.png)
![1-OCTANOL, 3-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-, (3S)-](http://img.cochemist.com/ccimg/676300/676236-08-1_b.png)







![Benzenamine, 2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-6-methyl-](http://img.cochemist.com/ccimg/347200/347196-63-8.png)
![Benzenamine, 2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-6-methyl-](http://img.cochemist.com/ccimg/347200/347196-63-8_b.png)
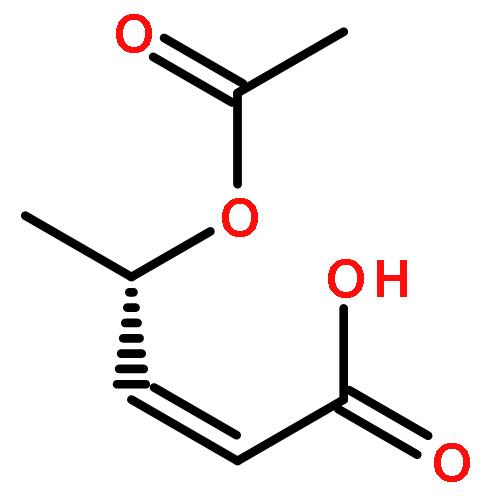
![Bicyclo[1.1.1]pentane-1-carboxylic acid,3-[[(1,1-dimethylethoxy)carbonyl]amino]-](http://img.cochemist.com/ccimg/303800/303752-38-7.png)
![Bicyclo[1.1.1]pentane-1-carboxylic acid,3-[[(1,1-dimethylethoxy)carbonyl]amino]-](http://img.cochemist.com/ccimg/303800/303752-38-7_b.png)






![2-Cyclohexen-1-ol, 4,5-bis[[(1,1-dimethylethyl)dimethylsilyl]oxy]-,(1S,4R,5S)-](http://img.cochemist.com/ccimg/184200/184110-47-2.png)
![2-Cyclohexen-1-ol, 4,5-bis[[(1,1-dimethylethyl)dimethylsilyl]oxy]-,(1S,4R,5S)-](http://img.cochemist.com/ccimg/184200/184110-47-2_b.png)
![Bicyclo[1.1.1]pentane-1-carboxylic acid, 3-formyl-, methyl ester (9CI)](http://img.cochemist.com/ccimg/180500/180464-92-0.png)
![Bicyclo[1.1.1]pentane-1-carboxylic acid, 3-formyl-, methyl ester (9CI)](http://img.cochemist.com/ccimg/180500/180464-92-0_b.png)
![Bicyclo[1.1.1]pentane-1-carboxylicacid, 3-(hydroxymethyl)-, methyl ester](http://img.cochemist.com/ccimg/180500/180464-87-3.png)
![Bicyclo[1.1.1]pentane-1-carboxylicacid, 3-(hydroxymethyl)-, methyl ester](http://img.cochemist.com/ccimg/180500/180464-87-3_b.png)


![Carbamic acid, [(1R)-1-(cyanomethyl)-2-methylpropyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/172700/172695-23-7.png)
![Carbamic acid, [(1R)-1-(cyanomethyl)-2-methylpropyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/172700/172695-23-7_b.png)


![Benzene, 1-[(4-iodobutoxy)methyl]-4-methoxy-](http://img.cochemist.com/ccimg/165000/164933-38-4.png)
![Benzene, 1-[(4-iodobutoxy)methyl]-4-methoxy-](http://img.cochemist.com/ccimg/165000/164933-38-4_b.png)








![Pyridine, 2-bromo-6-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/150100/150058-63-2.png)
![Pyridine, 2-bromo-6-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/150100/150058-63-2_b.png)
![Silane, trimethyl[(3Z)-6-[tris(1-methylethyl)silyl]-3-hexene-1,5-diynyl]-](http://img.cochemist.com/ccimg/149800/149776-79-4.png)
![Silane, trimethyl[(3Z)-6-[tris(1-methylethyl)silyl]-3-hexene-1,5-diynyl]-](http://img.cochemist.com/ccimg/149800/149776-79-4_b.png)










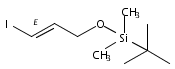
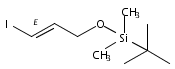


![Propanal, 3-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/112900/112897-03-7.png)
![Propanal, 3-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/112900/112897-03-7_b.png)


























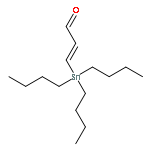



![Spiro[2,13-methano-10H-anthra[1,2-b]-1,3-dioxolo[4,5-d]pyran-14,2'-oxiran]-10-one, 13-[(4-C-acetyl-2,6-dideoxy-α-L-xylo-hexopyranosyl)oxy]-7-[(4-O-](/data/chemimg/1335600/81552-36-5.png)
![Spiro[2,13-methano-10H-anthra[1,2-b]-1,3-dioxolo[4,5-d]pyran-14,2'-oxiran]-10-one, 13-[(4-C-acetyl-2,6-dideoxy-α-L-xylo-hexopyranosyl)oxy]-7-[(4-O-](/data/chemimg/1335600/81552-36-5_b.png)
![2-(dimethoxymethyl)-8,10,12,13a-tetrahydroxy-7-methoxy-5-methyl-3a,4,8,9,10,13a-hexahydro-11H-spiro[2,4-epoxyfuro[3,2-b]naphtho[2,3-h]chromene-1,2'-oxiran]-11-one](http://img.cochemist.com/ccimg/81600/81552-35-4.png)
![2-(dimethoxymethyl)-8,10,12,13a-tetrahydroxy-7-methoxy-5-methyl-3a,4,8,9,10,13a-hexahydro-11H-spiro[2,4-epoxyfuro[3,2-b]naphtho[2,3-h]chromene-1,2'-oxiran]-11-one](http://img.cochemist.com/ccimg/81600/81552-35-4_b.png)
![Silane, trimethyl[[1-(phenylmethoxy)ethenyl]oxy]-](http://img.cochemist.com/ccimg/81200/81171-44-0.png)
![Silane, trimethyl[[1-(phenylmethoxy)ethenyl]oxy]-](http://img.cochemist.com/ccimg/81200/81171-44-0_b.png)






![1-[4-(METHYLSULFONYL)PHENYL]-1-PROPANAMINE](http://img.cochemist.com/ccimg/74300/74272-73-4.png)
![1-[4-(METHYLSULFONYL)PHENYL]-1-PROPANAMINE](http://img.cochemist.com/ccimg/74300/74272-73-4_b.png)








![Benzenamine, 2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/69600/69589-21-5.png)
![Benzenamine, 2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/69600/69589-21-5_b.png)













![9H-Pyrido[3,4-b]indol-6-ol](http://img.cochemist.com/ccimg/59000/58982-28-8.png)
![9H-Pyrido[3,4-b]indol-6-ol](http://img.cochemist.com/ccimg/59000/58982-28-8_b.png)








![Silane, [(1-methoxyethenyl)oxy]trimethyl-](http://img.cochemist.com/ccimg/36900/36850-80-3.png)
![Silane, [(1-methoxyethenyl)oxy]trimethyl-](http://img.cochemist.com/ccimg/36900/36850-80-3_b.png)











![Bicyclo[2.2.1]hept-5-en-2-one, (1S,4S)-](http://img.cochemist.com/ccimg/16700/16620-79-4.png)
![Bicyclo[2.2.1]hept-5-en-2-one, (1S,4S)-](http://img.cochemist.com/ccimg/16700/16620-79-4_b.png)













![1H-BENZO[F][2]BENZOFURAN-3-ONE](http://img.cochemist.com/ccimg/4800/4711-50-6.png)
![1H-BENZO[F][2]BENZOFURAN-3-ONE](http://img.cochemist.com/ccimg/4800/4711-50-6_b.png)


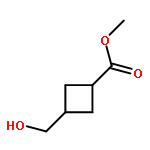





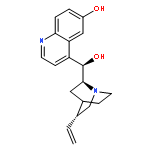
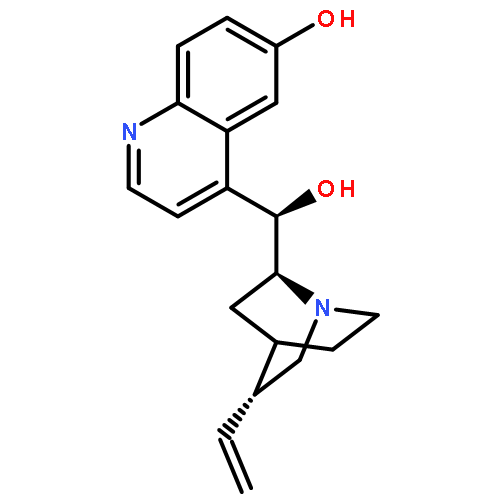




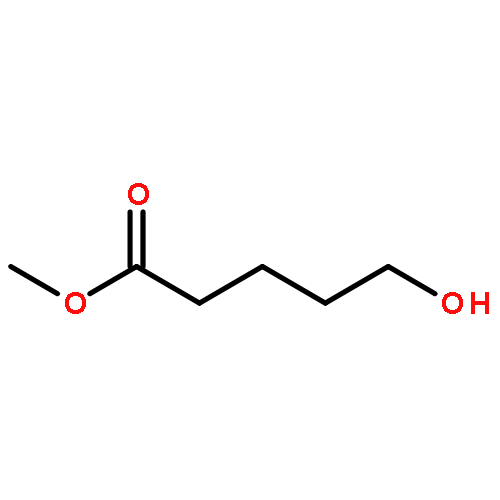
![2-Cyclopenten-1-one, 3-[2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]ethyl]-](http://img.cochemist.com/ccimg/150900/150892-92-5.png)
![2-Cyclopenten-1-one, 3-[2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]ethyl]-](http://img.cochemist.com/ccimg/150900/150892-92-5_b.png)


![Bicyclo[1.1.1]pentane-1,3-dicarboxylic acid, monomethyl ester](http://img.cochemist.com/ccimg/83300/83249-10-9.png)
![Bicyclo[1.1.1]pentane-1,3-dicarboxylic acid, monomethyl ester](http://img.cochemist.com/ccimg/83300/83249-10-9_b.png)
![2,2-DIFLUORO-5H-[1,3]DIOXOLO[4,5-F]INDOLE-6,7-DIONE](http://img.cochemist.com/ccimg/74200/74141-12-1.png)
![2,2-DIFLUORO-5H-[1,3]DIOXOLO[4,5-F]INDOLE-6,7-DIONE](http://img.cochemist.com/ccimg/74200/74141-12-1_b.png)
![Pentacyclo[4.2.0.02,5.03,8.04,7]octane-1,4-dicarboxylicacid, 1-methyl ester](http://img.cochemist.com/ccimg/24600/24539-28-4.png)
![Pentacyclo[4.2.0.02,5.03,8.04,7]octane-1,4-dicarboxylicacid, 1-methyl ester](http://img.cochemist.com/ccimg/24600/24539-28-4_b.png)


![1,5-Methano-1H,7H-furo[3,4-d]xanthene-7,13-dione, 3,3a,4,5-tetrahydro-3,3-dimethyl-1-(3-methyl-2-buten-1-yl)-, (1R,3aS,5S,12aS)-](/data/chemimg/177500/1033873-84-5.png)
![1,5-Methano-1H,7H-furo[3,4-d]xanthene-7,13-dione, 3,3a,4,5-tetrahydro-3,3-dimethyl-1-(3-methyl-2-buten-1-yl)-, (1R,3aS,5S,12aS)-](/data/chemimg/177500/1033873-84-5_b.png)