Co-reporter:Hyungseob Lim, Jae Young Kim, Edward J. Evans, Amritesh Rai, Jun-Hyuk Kim, Bryan R. Wygant, and C. Buddie Mullins
ACS Applied Materials & Interfaces September 13, 2017 Volume 9(Issue 36) pp:30654-30654
Publication Date(Web):August 16, 2017
DOI:10.1021/acsami.7b08239
There has been debate on whether Ni(OH)2 is truly catalytically active for the photo/electrocatalytic oxygen evolution reaction. In this report, we synthesized a Ni(OH)2 cocatalyst on a hematite photoanode and showed that, as has been proposed in other studies, the current density varies as a function of scan rate, which arises due to a photoinduced capacitive charging effect. We discovered that this photoinduced charging of Ni2+/3+ can be overcome by mixing cerium nitrate into the Ni precursor solution. Under illumination, the NiCeOx cocatalyst on a hematite photoanode exhibited an approximately 200 mV cathodic shift in onset potential and a ∼53% enhancement in photocurrent at 1.23 V vs RHE. Material characterization by electrochemical impedance spectroscopy revealed that the Ni species create a p–n junction across the charge space region, which facilitates collection of the photogenerated holes by the cocatalyst layer, and core level X-ray photoelectron spectroscopy showed that Ce incorporated into the Ni-based cocatalyst layer may possibly induce the oxidation of the Ni species. In addition, we observed a reduction in binding energies of Ni after photoelectrochemical water splitting reactions, which suggests that the lattice oxygen of the NiCeOx is consumed in the catalytic cycle, forming oxygen vacancies. The NiCeOx cocatalyst, however, was incapable of passivating the surface recombination centers of the hematite photoanode, as indicated by the unaltered flat-band potential determined with Mott–Schottky analysis.Keywords: cerium; cocatalyst; hematite; nickel hydroxide; water splitting;
Co-reporter:Jie Lin, Jin-Myoung Lim, Duck Hyun Youn, Kenta Kawashima, Jun-Hyuk Kim, Yang Liu, Hang Guo, Graeme Henkelman, Adam Heller, and Charles Buddie Mullins
ACS Nano October 24, 2017 Volume 11(Issue 10) pp:10347-10347
Publication Date(Web):September 12, 2017
DOI:10.1021/acsnano.7b05294
Through a gelation–solvothermal method without heteroadditives, Cu–Sn–S composites self-assemble to form nanotubes, sub-nanotubes, and nanoparticles. The nanotubes with a Cu3–4SnS4 core and Cu2SnS3 shell can tolerate long cycles of expansion/contraction upon lithiation/delithiation, retaining a charge capacity of 774 mAh g–1 after 200 cycles with a high initial Coulombic efficiency of 82.5%. The importance of the Cu component for mitigation of the volume expansion and structural evolution upon lithiation is informed by density functional theory calculations. The self-generated template and calculated results can inspire the design of analogous Cu–M–S (M = metal) nanotubes for lithium batteries or other energy storage systems.Keywords: copper tin sulfide; core−shell; density functional theory; gelation−solvothermal; lithium battery; nanotube;
Co-reporter:Han Xiao, Joshua P. Pender, Mackenzie A. Meece-Rayle, J. Pedro de Souza, Kyle C. Klavetter, Heonjoo Ha, Jie Lin, Adam Heller, Christopher J. Ellison, and C. Buddie Mullins
ACS Applied Materials & Interfaces July 12, 2017 Volume 9(Issue 27) pp:22641-22641
Publication Date(Web):June 20, 2017
DOI:10.1021/acsami.7b07283
We report the synthesis and properties of a low-density (∼5 mg/cm3) and highly porous (99.6% void space) three-dimensional reduced graphene oxide (rGO)/poly(acrylic acid) (PAA) nanocomposite aerogel as the scaffold for cathode materials in lithium-ion batteries (LIBs). The rGO-PAA is both simple and starts from readily available graphite and PAA, thereby providing a scalable fabrication procedure. The scaffold can support as much as a 75 mg/cm2 loading of LiFePO4 (LFP) in a ∼430 μm thick layer, and the porosity of the aerogel is tunable by compression; the flexible aerogel can be compressed 30-fold (i.e., to as little as 3.3% of its initial volume) while retaining its mechanical integrity. Replacement of the Al foil by the rGO-PAA current collector of the slurry-cast LFP (1.45 ± 0.2 g/cm3 tap density) provides for exemplary mass loadings of 9 mgLFP/cm2 at 70 μm thickness and 1.4 g/cm3 density or 16 mgLFP/cm2 at 100 μm thickness and ∼1.6 g/cm3 density. When compared to Al foil, the distribution of LFP throughout the three-dimensional rGO-PAA framework doubles the effective LFP solution-contacted area at 9 mg/cm2 loading and increases it 2.5-fold at 16 mg/cm2 loading. Overall, the rGO-PAA current collector increases the volumetric capacity by increasing the effective electrode area without compromising the electrode density, which was compromised in past research where the effective electrode area has been increased by reducing the particle size.Keywords: 3D nanoarchitecture; aerogel; cathode; lithium-ion battery; nanocomposite; reduced graphene oxide;
Co-reporter:Gregory M. Mullen, Edward J. Evans Jr., Iliya Sabzevari, Brittany E. Long, Khalid Alhazmi, Bert D. Chandler, and C. Buddie Mullins
ACS Catalysis February 3, 2017 Volume 7(Issue 2) pp:1216-1216
Publication Date(Web):December 28, 2016
DOI:10.1021/acscatal.6b02960
In this study, we show that water influences the activity and selectivity of a ceria-supported gold catalyst for the oxidative dehydrogenation and esterification of ethanol in a fixed-bed flow reactor, resulting in the production of acetaldehyde and ethyl acetate, respectively. The rate of acetaldehyde formation was enhanced upon incorporation of water in the feed stream at low pressures; beyond this, the water had little effect on the kinetics of the primary oxidation reaction. However, water induced significant changes to ethyl acetate production, decreasing the reaction order in ethanol and slowing the reaction rate. Furthermore, a large kinetic isotope effect for esterification was observed upon incorporation of either D2O or d-ethanol in the system, suggesting that breaking an OH bond is associated with the rate-determining step for this reaction. Our observations are consistent with the esterification process occurring through secondary oxidation of acetaldehyde to acetate species on the ceria surface followed by the acid-catalyzed esterification of surface acetates with ethanol. These results highlight the importance of water in gold-catalyzed processes and show that water can direct reaction selectivity in addition to influencing activity (as many previous studies have shown for CO oxidation).Keywords: ceria; dehydrogenation; ester; ethanol; gold; isotope effect; water;
Co-reporter:Wen-Yueh Yu, Gregory M. Mullen, David W. Flaherty, and C. Buddie Mullins
Journal of the American Chemical Society August 6, 2014 Volume 136(Issue 31) pp:11070-11078
Publication Date(Web):July 14, 2014
DOI:10.1021/ja505192v
Pd–Au catalysts have shown exceptional performance for selective hydrogen production via HCOOH decomposition, a promising alternative to solve issues associated with hydrogen storage and distribution. In this study, we utilized temperature-programmed desorption (TPD) and reactive molecular beam scattering (RMBS) in an attempt to unravel the factors governing the catalytic properties of Pd–Au bimetallic surfaces for HCOOH decomposition. Our results show that Pd atoms at the Pd–Au surface are responsible for activating HCOOH molecules; however, the selectivity of the reaction is dictated by the identity of the surface metal atoms adjacent to the Pd atoms. Pd atoms that reside at Pd–Au interface sites tend to favor dehydrogenation of HCOOH, whereas Pd atoms in Pd(111)-like sites, which lack neighboring Au atoms, favor dehydration of HCOOH. These observations suggest that the reactivity and selectivity of HCOOH decomposition on Pd–Au catalysts can be tailored by controlling the arrangement of surface Pd and Au atoms. The findings in this study may prove informative for rational design of Pd–Au catalysts for associated reactions including selective HCOOH decomposition for hydrogen production and electro-oxidation of HCOOH in the direct formic acid fuel cell.
Co-reporter:Adrianna Brush;Shallaco McDonald;Robin Dupré;Shruti Kota;Gregory M. Mullen
Reaction Chemistry & Engineering (2016-Present) 2017 vol. 2(Issue 4) pp:512-520
Publication Date(Web):2017/08/01
DOI:10.1039/C7RE00038C
Transient techniques, such as Steady state isotopic transient kinetic analysis (SSITKA), are powerful methods for determining various mechanistic and kinetic insights into heterogeneously catalyzed gas-phase reactions. However, the reactor systems commonly used in these techniques underutilize the costly isotopically labeled reactants crucial to these experiments. In this manuscript, we describe a novel apparatus that allows more efficient utilization of isotopically labeled reactants. This pulse injection apparatus is relatively easy and inexpensive to install on new or existing reaction systems. Sample data and analysis of SSITKA experiments performed on this system are also included.
Co-reporter:E. J. Evans, Jr.;H. Li;Wen-Yueh Yu;G. M. Mullen;G. Henkelman
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 45) pp:30578-30589
Publication Date(Web):2017/11/22
DOI:10.1039/C7CP05097F
In this study, we have combined ultra-high vacuum (UHV) experiments and density functional theory (DFT) calculations to investigate ethanol (EtOH) dehydrogenation on Pd–Au model catalysts. Using EtOH reactive molecular beam scattering (RMBS), EtOH temperature-programmed desorption (TPD), and DFT calculations, we show how different Pd ensemble sizes on Au(111) can affect the mechanism for EtOH dehydrogenation and H2 production. The Au(111) surface with an initial coverage of 2 monolayers of Pd (2 ML Pd–Au) had the highest H2 yield. However, the 1 ML Pd–Au catalyst showed the highest selectivity and stability, yielding appreciable amounts of only H2 and acetaldehyde. Arrhenius plots of H2 production confirm that the mechanisms for EtOH dehydrogenation differed between 1 and 2 ML Pd–Au, supporting the perceived difference in selectivity between the two surfaces. DFT calculations support this difference in mechanism, showing a dependence of the initial dehydrogenation selectivity of EtOH on the size of Pd ensemble. DFT binding energies and EtOH TPD confirm that EtOH has increasing surface affinity with increasing Pd ensemble size and Pd coverage, indicating that surfaces with more Pd are more likely to induce an EtOH reaction instead of desorb. Our theoretical results show that the synergistic influence of atomic ensemble and electronic effects on Pd/Au(111) can lead to different H2 association energies and EtOH dehydrogenation capacities at different Pd ensembles. These results provide mechanistic insights into ethanol's dehydrogenation interactions with different sites on the Pd–Au surface and can potentially aid in bimetallic catalyst design for applications such as fuel cells.
Co-reporter:Sungmin Han;Edward J. Evans, Jr;Gregory M. Mullen
Chemical Communications 2017 vol. 53(Issue 28) pp:3990-3993
Publication Date(Web):2017/04/04
DOI:10.1039/C7CC01542A
Improved activation of adsorbed O2 by co-adsorbed H2O on the Pd–Au(111) surface has been observed. When co-adsorbed with H2O, O2 admolecules on the Pd–Au surface are more strongly bound via their interactions with H2O. This interaction leads to large enhancements in the dissociation of O2 as determined via the generation of CO2 upon exposure to CO.
Co-reporter:Duck Hyun Youn, Adam Heller, and C. Buddie Mullins
Chemistry of Materials 2016 Volume 28(Issue 5) pp:1343
Publication Date(Web):February 8, 2016
DOI:10.1021/acs.chemmater.5b04282
A composite of 3.5 nm Sn nanoparticles dispersed in nitrogen-doped carbon was prepared from low cost precursors, using simple equipment, by the simple process of hydrolyzing at 300 °C SnCl4 mixed with nitrilotriacetic acid and then pyrolyzing the complexed SnO2 at 650 °C. The affordable anode made with the composite retained at 0.2 A g–1 specific current a specific capacity of 660 mAh·g–1 at the 200th cycle and a 630 mAh·g–1 capacity at 400th cycle. At 1 A g–1 specific current the capacity was as 435 mAh·g–1.
Co-reporter:Bryan R. Wygant, Karalee A. Jarvis, William D. Chemelewski, Oluwaniyi Mabayoje, Hugo Celio, and C. Buddie Mullins
ACS Catalysis 2016 Volume 6(Issue 2) pp:1122
Publication Date(Web):January 4, 2016
DOI:10.1021/acscatal.5b02429
The poor kinetics of the oxygen evolution reaction (OER) are a considerable barrier to the development of water-derived hydrogen fuel. Previous work regarding theoretical calculations of the perovskite SrCoO3-δ (SCO) predicts a surface binding energy ideal for OER catalysis but could not be matched to experimental results due to the material’s propensity to form the incorrect trigonal crystal structure. By doping with iron and scandium, X-ray diffraction confirms that we have been able to synthesize a series of SCO catalysts of various crystal structures, culminating in cubic SCO. In doing so, we show that there is a limited correlation between the crystal structure and OER performance in alkaline media. Instead, the use of iron as a dopant is found to decrease the OER overpotential of the SCO by 40 mV in 0.1 M KOH and yield catalysts capable of performing water oxidation at an overpotential of 410 mV at 10 mA/cm2. The doped, cubic SCO catalysts are found to be more stable than the undoped material when tested for extended periods, showing only an approximate 3 mV increase in overpotential over a 2 h period at 10 mA/cm2. Our results show that proper doping of the B-site cation in SCO allows for tuning the structure, performance, and stability of the oxide as an OER electrocatalyst.Keywords: electrocatalyst; oxygen evolution reaction (OER); perovskite; strontium cobalt oxide; water oxidation
Co-reporter:Ding Tang, Alexander J. E. Rettie, Oluwaniyi Mabayoje, Bryan R. Wygant, Yanqing Lai, Yexiang Liu and C. Buddie Mullins
Journal of Materials Chemistry A 2016 vol. 4(Issue 8) pp:3034-3042
Publication Date(Web):02 Nov 2015
DOI:10.1039/C5TA07877F
Porous n-type Fe2V4O13 films on FTO substrates were prepared by a simplified successive ion layer adsorption and reaction method and characterized as photoelectrodes for photoelectrochemical (PEC) water oxidation. Synthesis parameters such as film thickness and annealing temperatures and durations were investigated to optimize the PEC performance. A band gap of ∼2.3 eV and a flat band potential of 0.5 V vs. RHE make Fe2V4O13 a promising photoanode material. Water oxidation was kinetically limited at the surface of Fe2V4O13 film as confirmed by tests in electrolyte with a hole scavenger (Na2SO3). Improved PEC performance was achieved by Mo and W doping because of enhanced carrier densities. The best performance was obtained by 2.5% W-doped Fe2V4O13 films (actual 0.8% W-doped), which efficiently oxidize water to O2via photogenerated holes as confirmed by oxygen evolution measurements. Moreover, the Fe2V4O13 photoanode displayed very stable photocurrent under illumination. Due to the suitable band gap and valence band position, Fe2V4O13 is a promising photoanode for solar water splitting. Co-catalyst loading and doping optimization are identified as routes to improve this material's performance further.
Co-reporter:Alexander J. E. Rettie, William D. Chemelewski, Bryan R. Wygant, Jeffrey Lindemuth, Jung-Fu Lin, David Eisenberg, Carolyn S. Brauer, Timothy J. Johnson, Toya N. Beiswenger, Richard D. Ash, Xiang Li, Jianshi Zhou and C. Buddie Mullins
Journal of Materials Chemistry A 2016 vol. 4(Issue 3) pp:559-567
Publication Date(Web):11 Dec 2015
DOI:10.1039/C5TC03368C
We report the synthesis of silicon-doped hematite (Si:α-Fe2O3) single crystals via chemical vapor transport, with Si incorporation on the order of 1019 cm−3. The conductivity, Seebeck and Hall effect were measured in the basal plane between 200 and 400 K. Distinct differences in electron transport were observed above and below the magnetic transition temperature of hematite at ∼265 K (the Morin transition, TM). Above 265 K, transport was found to agree with the adiabatic small-polaron model, the conductivity was characterized by an activation energy of ∼100 meV and the Hall effect was dominated by the weak ferromagnetism of the material. A room temperature electron drift mobility of ∼10−2 cm2 V−1 s−1 was estimated. Below TM, the activation energy increased to ∼160 meV and a conventional Hall coefficient could be determined. In this regime, the Hall coefficient was negative and the corresponding Hall mobility was temperature-independent with a value of ∼10−1 cm2 V−1 s−1. Seebeck coefficient measurements indicated that the silicon donors were fully ionized in the temperature range studied. Finally, we observed a broad infrared absorption upon doping and tentatively assign the feature at ∼0.8 eV to photon-assisted small-polaron hops. These results are discussed in the context of existing hematite transport studies.
Co-reporter:Duck Hyun Youn, Nicholas A. Patterson, Hunmin Park, Adam Heller, and C. Buddie Mullins
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 41) pp:27788
Publication Date(Web):September 26, 2016
DOI:10.1021/acsami.6b09857
The simple fabrication of composites of germanium nanoparticles dispersed on nitrogen-doped carbon nanospheres (Ge/NC) of varying nitrogen content and their performance in lithium ion battery anodes are reported. A heavily nitrogen-doped carbon gel was formed by condensing m-phenylenediamine with formaldehyde (PF-gel); a less heavily N-doped gel was formed by condensing resorcinol and m-phenylenediamine with formaldehyde (RPF-gel); and an undoped gel was formed by condensing resorcinol with formaldehyde (RF-gel). Pyrolises of the gels with GeCl4 at 750 °C produced nanocrystalline Ge composites with 7.5 atom % N-doped carbon, termed Ge/NC (PF), with 3.9% N-doped carbon, termed Ge/NC (RPF) and undoped carbon, termed Ge/C (RF). The heavily N-doped Ge/NC (PF) anode retained a reversible capacity of 684 mAhg–1 at a specific current of 0.2 Ag–1 after 200 cycles, versus 337 mAhg–1 retained by anode made with Ge/NC (RPF) and 278 mAhg–1 retained by anode made with undoped Ge/C (RF). At a specific current 2.0 Ag–1, the capacity of the Ge/NC (PF) anode was 472 mAhg–1, versus the 210 mAhg–1 of the Ge/NC (RPF) anode and 83 mAhg–1 of the Ge/C (RF) anode. The enhanced performance of the Ge/NC (PF) anode is attributed to the better electrical conductivity of Ge/NC (PF) and to the higher density of Li+ binding defects in its N-doped carbon.Keywords: anode; germanium; lithium ion battery; nitrogen-doped carbon
Co-reporter:Adrianne Brush, Edward J. Evans Jr., Gregory M. Mullen, Karalee Jarvis, C. Buddie Mullins
Fuel Processing Technology 2016 Volume 153() pp:111-120
Publication Date(Web):1 December 2016
DOI:10.1016/j.fuproc.2016.07.012
•A Ni-Mo-Carbide catalyst tested for the bi-reforming of methane reaction.•By varying the ratio of CO2:H2O, the resulting H2:CO ratio could be tuned.•Despite operating under favorable coking conditions no coking is observed.•The Ni/Mo2C catalyst deactivates via oxidation of Mo2C to MoO2.This study demonstrates the ability of Ni/Mo2C to catalyze the Methane Bireforming Reaction (combined Dry Methane Reforming Reaction, CH4 + CO2 ➔ 2H2 + 2CO, and Steam Methane Reforming Reaction, CH4 + H2O ➔ 3H2 + CO). By varying the ratio of CO2:H2O, the resulting H2:CO ratio could be tuned from 0.91 to 3.0, covering a wide range of Syn-gas (H2 + CO) ratios relevant to various hydrocarbon syntheses. We also document the unusual deactivation behavior of Ni/Mo2C in this system. The catalytic activity would change from very high (> 50% conversion) to very low (< 10% conversion) within 10 min. Despite running under conditions typically favorable for coking with a Ni catalyst (high temperature, 950 °C, and excess methane), XRD, TGA, TEM, SEM, and EDX results clearly show no evidence of coking during the reaction or after deactivation. In addition, the changes to the Ni/Mo2C catalyst seen after deactivation (oxidation of Mo2C to MoO2, Ni-phase changes, and catalyst morphology changes) could not be seen in the catalyst subjected to reaction conditions that were halted before deactivation could occur. This suggests a sudden, rapid deactivation “event” occurs in this catalytic system as opposed to gradual catalyst deactivation, a behavior more typically seen with catalysts.
Co-reporter:William D. Chemelewski, Oluwaniyi Mabayoje, Ding Tang, Alexander J. E. Rettie and C. Buddie Mullins
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 3) pp:1644-1648
Publication Date(Web):07 Dec 2015
DOI:10.1039/C5CP05154A
Hematite is a promising material for photoelectrochemical (PEC) water splitting. While it has a low bandgap of ∼2.1 eV it is still larger than the optimal value of ∼1.8 eV. Previous work on epitaxial films has shown that Cr-doping leads to a shift of the bandgap as measured optically, but more importantly, also as measured by photoconductivity – to a value as low as 1.6 eV. We extend this work to polycrystalline films and attempt to use Cr-doping to lower the photon energy for which photocurrent can be generated. Our polycrystalline films show strong agreement with epitaxial films with regards to optical measurements of the direct and indirect bandgap. Furthermore, we find that Cr-doped polycrystalline films show photoconductivity at notably lower photon energies than undoped films, consistent with epitaxial results. However, when using Cr-doped films for photoelectrochemistry we find little to no shift of the photocurrent onset. We outline a number of proposals for why this could be the case, with a focus on the possibility of the existence of separate O 2p and Cr 3d states that would impact PEC but not PC behaviour.
Co-reporter:Alexander J. E. Rettie; William D. Chemelewski; David Emin
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 3) pp:471-479
Publication Date(Web):January 13, 2016
DOI:10.1021/acs.jpclett.5b02143
Transition-metal oxides are a promising class of semiconductors for the oxidation of water, a process that underpins both photoelectrochemical water splitting and carbon dioxide reduction. However, these materials are limited by very slow charge transport. This is because, unlike conventional semiconductors, material aspects of metal oxides favor the formation of slow-moving, self-trapped charge carriers: small polarons. In this Perspective, we seek to highlight the salient features of small-polaron transport in metal oxides, offer guidelines for their experimental characterization, and examine recent transport studies of two prototypical oxide photoanodes: tungsten-doped monoclinic bismuth vanadate (W:BiVO4) and titanium-doped hematite (Ti:α-Fe2O3). Analysis shows that conduction in both materials is well-described by the adiabatic small-polaron model, with electron drift mobility (distinct from the Hall mobility) values on the order of 10–4 and 10–2 cm2 V–1 s–1, respectively. Future directions to build a full picture of charge transport in this family of materials are discussed.
Co-reporter:Duck Hyun Youn, Shannon K. Stauffer, Penghao Xiao, Hunmin Park, Yejin Nam, Andrei Dolocan, Graeme HenkelmanAdam Heller, C. Buddie Mullins
ACS Nano 2016 Volume 10(Issue 12) pp:
Publication Date(Web):November 22, 2016
DOI:10.1021/acsnano.6b04214
Composites of nitrogen-doped reduced graphene oxide (NRGO) and nanocrystalline tin sulfides were synthesized, and their performance as lithium ion battery anodes was evaluated. Following the first cycle the composite consisted of Li2S/LixSn/NRGO. The conductive NRGO cushions the stress associated with the expansion of lithiation of Sn, and the noncycling Li2S increases the residual Coulombic capacity of the cycled anode because (a) Sn domains in the composite formed of unsupported SnS2 expand only by 63% while those in the composite formed of unsupported SnS expand by 91% and (b) Li percolates rapidly at the boundary between the Li2S and LixSn nanodomains. The best cycling SnS2/NRGO-derived composite retained a specific capacity of 562 mAh g–1 at the 200th cycle at 0.2 A g–1 rate.Keywords: lithium ion battery anode; reduced graphene oxide; SnS; SnS2;
Co-reporter:Oluwaniyi Mabayoje, Ahmed Shoola, Bryan R. Wygant, and C. Buddie Mullins
ACS Energy Letters 2016 Volume 1(Issue 1) pp:195
Publication Date(Web):May 31, 2016
DOI:10.1021/acsenergylett.6b00084
Oxygen evolution catalysts composed of a metal (Ni, Co, or Fe) and a pnictide or chalcogenide (P, S, or Se) counterion are a promising class of electrocatalysts for the oxygen evolution reaction (OER), an important reaction for the photoelectrochemical splitting of water. We synthesized a nickel-based oxygen evolution catalyst derived from pulse-electrodeposited nickel sulfide. This catalyst was found to produce current densities of 10 mA/cm2 at the relatively low overpotential of 320 mV in alkaline electrolyte (1 M KOH). Importantly, we found that the sulfur anion in the nickel sulfide is depleted in the active form of the electrocatalyst and that the NiS is converted into an amorphous nickel oxide in the potential range where water is oxidized to oxygen. The superior catalytic activity of this nickel sulfide is thus unrelated to the sulfur anions in the active catalyst but is instead related to the metal sulfide’s ability to act as a precursor to a highly active nickel oxide OER electrocatalyst. The nickel oxide derived from nickel sulfide was found to be amorphous with a relatively high surface area, two factors that have been previously shown to be important in oxygen evolution electrocatalysis.
Co-reporter:Sean M. Wood, Codey H. Pham, Rodrigo Rodriguez, Sindhu S. Nathan, Andrei D. Dolocan, Hugo Celio, J. Pedro de Souza, Kyle C. Klavetter, Adam Heller, and C. Buddie Mullins
ACS Energy Letters 2016 Volume 1(Issue 2) pp:414
Publication Date(Web):July 26, 2016
DOI:10.1021/acsenergylett.6b00259
Adding 10 mM KPF6 to the 1 M LiPF6 in ethylene carbonate/dimethyl carbonate electrolyte of symmetrical Li | Li cells eliminated the growth of dendrites at 0.5 mA cm–2 current density and massively reduced, but did not eliminate, the growth of dendrites at 2.5 mA cm–2. The added KPF6 increased the fraction of inorganic salts in the solid electrolyte interface, making it thinner and more Li+ conductive. It overcame the growth of dendrites resulting from inadequate nucleation density but not dendrite growth into the depletion layer, which scales with the layer’s thickness, i.e., the current density.
Co-reporter:Emily J. Powell, Sean M. Wood, Hugo Celio, Adam Heller and C. Buddie Mullins
Journal of Materials Chemistry A 2015 vol. 3(Issue 46) pp:23442-23447
Publication Date(Web):19 Oct 2015
DOI:10.1039/C5TA04941E
Mainstream rechargeable lithium battery materials research of the past 20 years has focused on nano-particulate materials, where Li+-diffusion lengths exceeded at designated cycling rates the particle radii, and where the particles slipped rather than broke upon their expansion and shrinkage in lithiation/delithiation cycles. Here we show that in intrinsically rapidly Li+-transporting macrocrystalline germanium and even more so in a dispersion of non-cycling Li2Te in macrocrystalline germanium it is unnecessary to use nanocrystalline materials and that Li2Te increases the retained capacity at 1C rate after 500 cycles. A dispersion of 17.65 atom% of crystalline GeTe in 82.35 atom% crystalline Ge was synthesized by quenching from the melt followed by high energy ball milling to 1 μm–5 μm particle size. The particles, as well as similarly made and similarly sized pure Ge particles were incorporated in electrodes, which were galvanostatically lithiated/delithiated. In the initial cycle, GeTe is reduced to LixGe alloys and Li2Te. In 500 1C cycles of LixGe delithiation/Ge lithiation the capacity of the pure Ge faded more rapidly than that of the Ge electrodes containing Li2Te, which retained 96% of their initial capacity after 500 cycles at 1C rate.
Co-reporter:Hoang X. Dang, Kyle C. Klavetter, Melissa L. Meyerson, Adam Heller and C. Buddie Mullins
Journal of Materials Chemistry A 2015 vol. 3(Issue 25) pp:13500-13506
Publication Date(Web):27 May 2015
DOI:10.1039/C5TA02131F
In a 100 cycle test at 0.5 C-rate a negative electrode formed of micro-sized Sn0.9Se0.1 particles retains a specific capacity of 500 mA h g−1 with a coulombic efficiency of 99.6%. In contrast, a control electrode made with pure Sn retains only a 200 mA h g−1 capacity with a 98.7% efficiency. The improvement in electrochemical performance of the Sn/Se alloy is attributed to the reduced inactive Se-phase preventing agglomeration of Sn to a size susceptible to particle fracture. The Sn/Se alloy particles are manufacturable, being made by melting the 9:1 atomic ratio mixture of Sn and Se, quenching and jet-milling.
Co-reporter:Kyle C. Klavetter, J. Pedro de Souza, Adam Heller and C. Buddie Mullins
Journal of Materials Chemistry A 2015 vol. 3(Issue 11) pp:5829-5834
Publication Date(Web):09 Feb 2015
DOI:10.1039/C5TA00319A
Slurry cast electrodes with μm-sized Ge0.9Se0.1 particles cycle stably at ∼800 mA h g−1 with ∼99.9% efficiency for 900 1C-rate cycles while electrodes with μm-size pure Ge particles lose 1/3rd of their capacity after five C/5 cycles. The difference is attributed to an inactive glassy Li–Se–Ge phase forming in the Ge active material of the Ge0.9Se0.1 particle.
Co-reporter:Wen-Yueh Yu, Liang Zhang, Gregory M. Mullen, Edward J. Evans, Graeme Henkelman and C. Buddie Mullins
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 32) pp:20588-20596
Publication Date(Web):15 Jul 2015
DOI:10.1039/C5CP03515E
It has been reported that Pd–Au bimetallic catalysts display improved catalytic performance after adequate calcination. In this study, a model catalyst study was conducted to investigate the effects of annealing in oxygen on the surface structures of Pd–Au alloys by comparing the physicochemical properties of Pd/Au(111) surfaces that were annealed in ultrahigh vacuum (UHV) versus in an oxygen ambient. Auger electron spectroscopy (AES) and Basin hopping simulations reveal that the presence of oxygen can inhibit the diffusion of surface Pd atoms into the subsurface of the Au(111) sample. Reflection–absorption infrared spectroscopy using CO as a probe molecule (CO-RAIRS) and King–Wells measurements of O2 uptake suggest that surfaces annealed in an oxygen ambient possess more contiguous Pd sites than surfaces annealed under UHV conditions. The oxygen-annealed Pd/Au(111) surface exhibited a higher activity for CO oxidation in reactive molecular beam scattering (RMBS) experiments. This enhanced activity likely results from the higher oxygen uptake and relatively facile dissociation of oxygen admolecules due to stronger adsorbate–surface interactions as suggested by temperature-programmed desorption (TPD) measurements. These observations provide fundamental insights into the surface phenomena of Pd–Au alloys, which may prove beneficial in the design of future Pd–Au catalysts.
Co-reporter:Gregory M. Mullen, Liang Zhang, Edward J. Evans, Ting Yan, Graeme Henkelman and C. Buddie Mullins
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 6) pp:4730-4738
Publication Date(Web):07 Jan 2015
DOI:10.1039/C4CP04739G
Gold catalysts display high activity and good selectivity for partial oxidation of a number of alcohol species. In this work, we discuss the effects of oxygen adatoms and surface hydroxyls on the selectivity for oxidation of allylic alcohols (allyl alcohol and crotyl alcohol) on gold surfaces. Utilizing temperature programmed desorption (TPD), reactive molecular beam scattering (RMBS), and density functional theory (DFT) techniques, we provide evidence to suggest that the selectivity displayed towards partial oxidation versus combustion pathways is dependent on the type of oxidant species present on the gold surface. TPD and RMBS results suggest that surface hydroxyls promote partial oxidation of allylic alcohols to their corresponding aldehydes with very high selectivity, while oxygen adatoms promote both partial oxidation and combustion pathways. DFT calculations indicate that oxygen adatoms can react with acrolein to promote the formation of a bidentate surface intermediate, similar to structures that have been shown to decompose to generate combustion products over other transition metal surfaces. Surface hydroxyls do not readily promote such a process. Our results help explain phenomena observed in previous studies and may prove useful in the design of future catalysts for partial oxidation of alcohols.
Co-reporter:Hoang X. Dang, Melissa L. Meyerson, Adam Heller and C. Buddie Mullins
RSC Advances 2015 vol. 5(Issue 100) pp:82012-82017
Publication Date(Web):15 Sep 2015
DOI:10.1039/C5RA18046E
Tin forms a series of sodium alloys, some with a large change in volume, sufficient to fracture the sodiated/de-sodiated tin electrodes. In a series of Sn/C and Sn/Se/C electrodes, made similarly by ball milling the elements, the sodiation/de-sodiation cycling stability in the 0.01–1.00 V vs. Na+/Na voltage window, in which the initially formed Na2Se is electrochemically inactive, is best at an Sn:Se atomic ratio of 9:2. At this ratio the retained capacity is ∼300 mA h g−1 after 150 cycles at 0.17 A g−1 rate versus only 70 mA h g−1 in the absence of Na2Se. The improvement is attributed to prevention of crystallization of the Na–Sn alloys by the Na2Se.
Co-reporter:Wen-Yueh Yu
The Journal of Physical Chemistry C 2015 Volume 119(Issue 21) pp:11754-11762
Publication Date(Web):April 29, 2015
DOI:10.1021/acs.jpcc.5b02970
Co-reporter:Hoang X. Dang
The Journal of Physical Chemistry C 2015 Volume 119(Issue 26) pp:14524-14531
Publication Date(Web):November 7, 2014
DOI:10.1021/jp508349g
NiV2O6 films were successfully fabricated and characterized as photoanodes for photoelectrochemical (PEC) water oxidation. The films were synthesized by vacuum codeposition of Ni and V followed by annealing in air. The resulting triclinic NiV2O6 films were n-type semiconductors with optical transitions at ∼2.1 eV (indirect) and 2.4 eV (direct). Photoelectrochemical testing in 1 M KOH showed a photoelectrochemical band gap of ∼2.4 eV and that ∼45% of the photocurrent came from light with λ > 420 nm. NiV2O6 electrodes showed quite stable photocurrent under bias in basic electrolyte, indicative of chemical stability against photocorrosion despite its relatively small band gap. The flat band potential of NiV2O6 was ∼0.6 V vs RHE, thus allowing photogenerated holes to oxidize water thermodynamically, and confirmed by oxygen evolution measurements (Faradaic efficiency ∼80%). Suitable oxygen evolution catalysts and n-type doping are suggested to improve the PEC performance of this material further. Regarding the search for inexpensive photoanodes capable of harvesting photons in the visible region of the solar spectrum, the availability of Ni and V makes NiV2O6 a promising anode material for photoelectrochemical use.
Co-reporter:William D. Chemelewski
The Journal of Physical Chemistry C 2015 Volume 119(Issue 48) pp:26803-26808
Publication Date(Web):November 10, 2015
DOI:10.1021/acs.jpcc.5b06658
α-Ag3VO4 has shown promise as a photocatalyst for decomposition of organics and H2O in particle dispersion studies, but no thin film studies of Ag3VO4 have looked at its photoelectrochemical (PEC) properties. Addressing this deficiency, we grow films via successive ionic layer adsorption and reaction (SILAR) and characterize the material using standard physical and PEC techniques. We confirm a low bandgap of 2.2 eV and report the first results on chemical and electrochemical stability, intrinsic doping behavior, flat-band potential, and potential dependence of photocurrent. While our results are not initially promising with respect to the applicability of Ag3VO4 to solar water splitting, they highlight the most important property changes necessary to make Ag3VO4 competitive with better known photocatalysts and the salience of thin-film studies for PEC material characterization.
Co-reporter:Wenlong Guo
The Journal of Physical Chemistry C 2015 Volume 119(Issue 49) pp:27220-27227
Publication Date(Web):November 18, 2015
DOI:10.1021/acs.jpcc.5b07219
Thin films of two copper-based metal vanadates (CuV2O6 and Cu2V2O7) were synthesized by a facile drop-casting method. The primary photoelectrochemical (PEC) and physical properties of these two materials including photocurrent response, band gap, flat band potential, incident photon to current conversion efficiency, chemical stability, and oxygen evolution faradaic efficiency were researched. The photocurrent density of CuV2O6 and Cu2V2O7 films at 1.23 V vs RHE in 0.1 M sodium borate buffer solution was about 25 and 35 μA/cm2, respectively. At 1.58 V vs RHE, however, the photocurrent density reached approximately 220 and 120 μA/cm2, respectively. Although the photocurrents observed for these two materials at 1.23 V vs RHE were relatively low, the photocurrents were much higher when tested with sodium sulfite as a hole scavenger. Suitable oxygen evolution catalysts are therefore expected to improve the PEC performance of these materials.
Co-reporter:Ming Pan, Jinlong Gong, Guangbin Dong, and C. Buddie Mullins
Accounts of Chemical Research 2014 Volume 47(Issue 3) pp:750
Publication Date(Web):December 10, 2013
DOI:10.1021/ar400172u
Historically, scientists have considered gold an inert catalyst constituent. However, in recent decades, chemists have discovered that nanoscale gold shows exceptional activity for many chemical reactions. They have investigated model gold surfaces in order to obtain fundamental understanding of catalytic properties. In this Account, we present our current understanding of oxidation and hydrogenation reactions on the Au(111) single crystal as a planar representative of gold catalysts, revealing the interesting surface chemistry of gold.We begin by comparing two inverse reactions, alcohol oxidation and aldehyde hydrogenation, on a Au(111) surface. Beyond the expected different chemistry, we observe intriguing similarities since the same surface is employed. First, both molecular oxygen and hydrogen have high barriers to dissociation on Au(111), and frequently chemists study reactions here by using atomic O and H to populate the surfaces. Recombinative desorption features of oxygen and hydrogen are apparent at ∼500 and ∼110 K, lower than other transition metals. These results indicate that oxygen and hydrogen have low desorption activation energies and weakly chemisorb on the surface, likely leading to selective reactions. On the oxygen-precovered Au(111) surface, alcohols are selectively oxidized to aldehydes. Similarly, weakly bound hydrogen atoms on Au(111) also show chemoselective reactivity for hydrogenation of propionaldehyde and acetone.The second similarity is that the gold surface activates self-coupling of alcohol or aldehyde with oxygen or hydrogen, resulting in the formation of esters and ethers, respectively, in alcohol oxidation and aldehyde hydrogenation. During these two reactions, both alkoxy groups and alcohol-like species show up as intermediates, which likely play a key role in the formation of coupling products. In addition, the cross coupling reaction between alcohol and aldehyde occurs on both O- and H-modified surfaces, yielding the production of esters and ethers, respectively. Thus, we can tune the molecular structure of both esters and ethers by selecting the corresponding aldehyde and alcohol for the coupling reaction.These studies indicate that gold is a versatile active catalyst for various reactions, including oxidation and hydrogenation transformations. Despite the very different chemistry for these two reactions, we can establish an intrinsic relationship due to the distinct catalytic properties of gold. It can show activity for selective reactions on both O- and H-covered Au(111) and further induce the coupling reaction between surface reactants and adsorbed O/H to produce esters and ethers. This comparison demonstrates the unique surface chemistry of gold and enhances understanding of its catalytic properties.
Co-reporter:Sean P. Berglund ; Huichao He ; William D. Chemelewski ; Hugo Celio ; Andrei Dolocan
Journal of the American Chemical Society 2014 Volume 136(Issue 4) pp:1535-1544
Publication Date(Web):January 6, 2014
DOI:10.1021/ja411604k
p-Si/W2C photocathodes are synthesized by evaporating tungsten metal in an ambient of ethylene gas to form tungsten semicarbide (W2C) thin films on top of p-type silicon (p-Si) substrates. As deposited the thin films contain crystalline W2C with a bulk W:C atomic ratio of approximately 2:1. The W2C films demonstrate catalytic activity for the hydrogen evolution reaction (HER), and p-Si/W2C photocathodes produce cathodic photocurrent at potentials more positive than 0.0 V vs RHE while bare p-Si photocathodes do not. The W2C films are an effective support for Pt nanoparticles allowing for a considerable reduction in Pt loading. p-Si/W2C/Pt photocathodes with Pt nanoparticles achieve photocurrent onset potentials and limiting photocurrent densities that are comparable to p-Si/Pt photocathodes with Pt loading nine times higher. This makes W2C an earth abundant alternative to pure Pt for use as an electrocatalyst on photocathodes for the HER.
Co-reporter:William D. Chemelewski ; Heung-Chan Lee ; Jung-Fu Lin ; Allen J. Bard
Journal of the American Chemical Society 2014 Volume 136(Issue 7) pp:2843-2850
Publication Date(Web):January 29, 2014
DOI:10.1021/ja411835a
Reaching the goal of economical photoelectrochemical (PEC) water splitting will likely require the combination of efficient solar absorbers with high activity electrocatalysts for the hydrogen and oxygen evolution reactions (HER and OER). Toward this goal, we synthesized an amorphous FeOOH (a-FeOOH) phase that has not previously been studied as an OER catalyst. The a-FeOOH films show activity comparable to that of another OER cocatalyst, Co-borate (Co–Bi), in 1 M Na2CO3, reaching 10 mA/cm2 at an overpotential of ∼550 mV for 10 nm thick films. Additionally, the a-FeOOH thin films absorb less than 3% of the solar photons (AM1.5G) with energy greater than 1.9 eV, are homogeneous over large areas, and act as a protective layer separating the solution from the solar absorber. The utility of a-FeOOH in a realistic system is tested by depositing on amorphous Si triple junction solar cells with a photovoltaic efficiency of 6.8%. The resulting a-FeOOH/a-Si devices achieve a total water splitting efficiency of 4.3% at 0 V vs RHE in a three-electrode configuration and show no decrease in efficiency over the course of 4 h.
Co-reporter:Gregory M. Mullen ; Liang Zhang ; Edward J. Evans ; Jr.; Ting Yan ; Graeme Henkelman
Journal of the American Chemical Society 2014 Volume 136(Issue 17) pp:6489-6498
Publication Date(Web):April 6, 2014
DOI:10.1021/ja502347d
Partial oxidation of alcohols is a topic of great interest in the field of gold catalysis. In this work, we provide evidence that the partial oxidation of allyl alcohol to its corresponding aldehyde, acrolein, over oxygen-precovered gold surfaces occurs via multiple reaction pathways. Utilizing temperature-programmed desorption (TPD) with isotopically labeled water and oxygen species, reactive molecular beam scattering, and density functional theory (DFT) calculations, we demonstrate that the reaction mechanism for allyl alcohol oxidation is influenced by the relative proportions of atomic oxygen and hydroxyl species on the gold surface. Both atomic oxygen and hydroxyl species are shown to be active for allyl alcohol oxidation, but each displays a different pathway of oxidation, as indicated by TPD measurements and DFT calculations. The hydroxyl hydrogen of allyl alcohol is readily abstracted by either oxygen adatoms or adsorbed hydroxyl species on the gold surface to generate a surface-bound allyloxide intermediate, which then undergoes α-dehydrogenation via interaction with an oxygen adatom or surface hydroxyl species to generate acrolein. Mediation of a second allyloxide with the hydroxyl species lowers the activation barrier for the α-dehydrogenation process. A third pathway exists in which two hydroxyl species recombine to generate water and an oxygen adatom, which subsequently dehydrogenates allyloxide. This work may aid in the understanding of oxidative catalysis over gold and the effect of water therein.
Co-reporter:Stephen E. Fosdick, Sean P. Berglund, C. Buddie Mullins, and Richard M. Crooks
ACS Catalysis 2014 Volume 4(Issue 5) pp:1332
Publication Date(Web):March 31, 2014
DOI:10.1021/cs500168t
Here, we report the development of a parallel electrocatalyst screening platform for the hydrogen evolution reaction (HER) using bipolar electrodes (BPEs). Electrocatalyst candidates are subjected to screening in a N2-purged bipolar electrochemical cell where a pair of driving electrodes produce an electric field in the electrolyte solution. The HER occurring at the BPE cathodes is electrically coupled to the electrodissolution of an array of Cr microbands present at the BPE anodes. The readout of this device is simple, where the species that dissolve the most Cr microbands are identified as the most promising electrocatalyst candidates for further evaluation. We demonstrate the utility of this technique by comparing several bi- and trimetallic systems involving Co, Fe, Ni, Mo, and W, which are compared directly with pure Pt. Of all the compositions tested, Ni8–Mo2 is demonstrated to be the most active for the HER in a neutral electrolyte solution.Keywords: electrocatalysis; electrochemistry; high-throughput screening
Co-reporter:Alexander J. E. Rettie, Kyle C. Klavetter, Jung-Fu Lin, Andrei Dolocan, Hugo Celio, Ashioma Ishiekwene, Heather L. Bolton, Kristen N. Pearson, Nathan T. Hahn, and C. Buddie Mullins
Chemistry of Materials 2014 Volume 26(Issue 4) pp:1670
Publication Date(Web):February 3, 2014
DOI:10.1021/cm403969r
We report the incorporation of sulfur or iodine into monoclinic tungsten trioxide (S:WO3 or I:WO3 respectively), with the aim to improve its visible light-harvesting ability. Films were synthesized by spray pyrolysis with either ammonium sulfide or iodide added to the aqueous WO3 precursor solutions. Red shifts of the absorption spectra were observed with S and I incorporation (from ∼2.7 to 2.6 and 2.1 eV respectively), likely due to the formation of intragap impurity bands. S:WO3 samples exhibited enhanced photoelectrochemical (PEC) performance at low S concentrations, but this quickly deteriorated with increasing S content. Incident photon conversion efficiency (IPCE) data showed that this initial improvement was driven by improved collection efficiency at longer wavelengths. Conversely, photocurrent decreased at all levels of I addition. IPCE measurements for these films showed only a marginal increase in efficiency at longer wavelengths, indicating that the extra absorbed photons did not contribute significantly to the photocurrent. Time of flight-secondary ion mass spectrometry (ToF-SIMS) depth profiling revealed a uniform distribution of S throughout the S:WO3 films, but showed surface segregation of I in the I:WO3 samples. Raman and X-ray photoelectron spectrometry (XPS) showed that S and I substituted for oxygen, but in the case of S, other pathways such as interstitial incorporation and cation substitution could not be ruled out. The complexities of intentionally adding nonmetal impurities to metal oxide systems are highlighted in the context of the existing body of literature.
Co-reporter:Paul R. Abel, Kyle C. Klavetter, Karalee Jarvis, Adam Heller and C. Buddie Mullins
Journal of Materials Chemistry A 2014 vol. 2(Issue 44) pp:19011-19018
Publication Date(Web):01 Oct 2014
DOI:10.1039/C4TA04496G
Nanocolumnar, sub-stoichiometric germanium sulfide thin-films with compositions of Ge0.9S0.1 and Ge0.95S0.05, deposited by glancing angle deposition, were investigated as lithium storage materials. The materials are amorphous and homogeneous as deposited, but lithiation induces phase separation leading to the formation of poorly-crystallized lithium sulfide inclusions during the first cycle. The presence of these inclusions raises the lithium diffusion coefficient above that of pure germanium and provides superior capacity retention at high rates. While the lithium sulfide is non-cycling, the low weight percentage of sulfur necessary for enhanced lithiation/de-lithiation does not significantly reduce the specific lithium storage capacity of the films relative to that of germanium. In addition to high capacity and superior lithium transport, the sub-stoichiometric germanium sulfide thin-films show excellent cycling stability at high rates, retaining 88% of their initial capacity after 500 cycles at a rate of 20 C.
Co-reporter:William D. Chemelewski, Jacob R. Rosenstock and C. Buddie Mullins
Journal of Materials Chemistry A 2014 vol. 2(Issue 36) pp:14957-14962
Publication Date(Web):16 Jul 2014
DOI:10.1039/C4TA03078H
The oxygen evolution reaction (OER) is one important bottleneck in the development of economical photoelectrochemical (PEC) water splitting materials. To help address this we report the electrodeposition of Ni-doped FeOOH (Ni:FeOOH) as an OER electrocatalyst. The deposition method is applicable to a wide range of photoanodes and catalytic films as thin as a few nanometers can be easily grown. The Ni:FeOOH films with 5–20% Ni content reach 10 mA cm−2 in 0.1 M NaOH at an overpotential ranging from 420–460 mV initially, and improve with anodization at 10 mA cm−2 to below 400 mV. Deposition on triple junction solar cells results in a full PEC system with higher performance and a more cathodic peak power potential compared to undoped FeOOH electrocatalysts.
Co-reporter:Kyle C. Klavetter, Jonathan L. Snider, J. Pedro de Souza, Han Tu, Trevor H. Cell, Joon Hee Cho, Chistopher J. Ellison, Adam Heller and C. Buddie Mullins
Journal of Materials Chemistry A 2014 vol. 2(Issue 35) pp:14459-14467
Publication Date(Web):16 Jul 2014
DOI:10.1039/C4TA03201B
A free-standing electrode film composed of high tap density SnO2 particles and carboxymethyl cellulose binder with Super-P Li (SP-Li) conductive carbon was formed from an aqueous slurry cast by doctor-blading. Upon air-drying, the free-standing film spontaneously evolved via delamination from the substrate as the slurry solvent evaporated. The electrodes cut from the free-standing film were ∼5 μm thick with a SnO2 loading of ∼0.5 mg cm−2. The films were found to be easily handled, flexed and folded. For evaluation of the durability of the free-standing films, the tensile strength and elongation at break were measured: 13 MPa and 1.7%. The robustness of the electrically conductive network was measured with a four-point probe: the initial electrical resistivity of the film (0.6 Ω cm) was observed to increase by 6% after folding, applying pressure to the crease and unfolding. When tested in a coin cell, the electrode cycled stably with near 100% coulombic efficiency at up to 2 C and without capacity fade for 100 cycles at 1 C. To adjust the areal capacity of the cell, multiple free-standing films could be stacked. An electrode formed from several stacked films with an active material mass loading of greater than 4 mg cm−2 was found to cycle stably at 2.6 mA h cm−2 tested at 0.33 mA cm−2 current density. For evaluating cycling performance of the electrode while flexed, an electrode was placed in a once-folded pouch cell for testing at 1 C and cycled stably for 20 cycles before slight capacity fade was observed. For free-standing electrodes, 1D or 2D carbons such as carbon nanotubes (CNT) or graphene are commonly used to provide both mechanical strength and electrical conductivity. Here, CNTs were substituted for the SP-Li and similar free-standing films were made and compared. With CNT, the electrode strength at break as well as the electronic conductivity increased, but, despite this, the cycling performance of the electrodes made using the low-cost SP-Li carbon exceeds that of the electrodes made with orders-of-magnitude more expensive 1D carbon.
Co-reporter:Kyle. C. Klavetter, Stephany Garcia, Naween Dahal, Jonathan L. Snider, J. Pedro de Souza, Trevor H. Cell, Mark A. Cassara, Adam Heller, Simon M. Humphrey and C. Buddie Mullins
Journal of Materials Chemistry A 2014 vol. 2(Issue 34) pp:14209-14221
Publication Date(Web):10 Jul 2014
DOI:10.1039/C4TA02684E
High surface area (367 m2 g−1) meso-porous Co3O4 was investigated as the precursor of the anode material for lithium and also sodium ion batteries. Co3O4 is considered a potential anode material due to its theoretical capacity of 890 mA h g−1, over twice that of graphite. This comparatively higher capacity can be safely charged at rapid rates owing to a relatively high Li-insertion potentials, but, consequently, the discharged energy is yielded at an average potential near 2 V vs. Li/Li+, with full Li-extraction achieved over a continuum of potentials up to 3 V. The products of the lithium reduction of Co3O4 cycle stably from 0.01–3.0 V vs. Li/Li+ with 600–900 mA h g−1 capacity retention at C rates from 1–5; the products of its sodium reduction cycle stably from 0.01–3.0 V vs. Na/Na+ at C-rates up to 1 C with a lower 150–400 mA h g−1 capacity retention owing to greater ionic impedance. TEM, SAED and XRD were used to examine the cycled material and the stable performance is attributed to finding that the mesoporous structure is retained. Evaluation of five electrolyte formulations testing EC, FEC and Cl-EC showed that the stable meso-porous structure was best cycled with 5% FEC in EC:DEC at high charge/discharge rates, retaining 77% of its initial capacity at 5 C in a rate test. Comparison of the AC impedance spectra and of the XPS of the SEIs formed in the presence and in the absence of 5 vol% FEC shows that the SEI formed in the presence of FEC contains lithium fluoride and its carbonate layer is thinner than that formed in its absence, resulting in lesser impedance to Li migration through the SEI and facile ion de-solvation, improving the cycling performance. In cycling stability tests with EC:DEC, irregular cycling behaviour attributable to abrupt rises in cell resistance was regularly observed after testing over a few hundred cycles. Long-term cycling irregularities are inhibited by halogenated solvents and completely eliminated by adding fluoroethylene carbonate (FEC).
Co-reporter:Huichao He, Sean P. Berglund, Alexander J. E. Rettie, William D. Chemelewski, Peng Xiao, Yunhuai Zhang and C. Buddie Mullins
Journal of Materials Chemistry A 2014 vol. 2(Issue 24) pp:9371-9379
Publication Date(Web):24 Apr 2014
DOI:10.1039/C4TA00895B
Because of the potential for application in photoelectrochemical cells for water splitting, the synthesis of nanostructured BiVO4 is receiving increasing attention. Here we report a simple new drop-casting method for the first time to synthesize un-doped and doped bismuth vanadate (BiVO4) nanoflake array films. Synthesis parameters such as the amount of polyethylene glycol 600 (PEG-600) and the precursor solution drying time are investigated to optimize the films for photoelectrochemical water oxidation. The BiVO4 films consisting of nanoflakes with an average thickness of 20 nm and length of 2 μm were synthesized from a precursor solution containing Bi3+, V3+ and PEG-600 with a Bi:V: PEG-600 volume ratio of 2:2:1, dried at 135 °C for 55 min. Photoelectrochemical measurements show that the BiVO4 nanoflake array films have higher photoelectrochemical activity than the BiVO4 nanoparticle films. Additionally, the nanoflake arrays were tested after incorporating W and Mo to enhance the photoelectrochemical activity. The 2% W, 6% Mo co-doped BiVO4 nanoflake array films demonstrate the best photoelectrochemical activity with photocurrent densities about 2 times higher than the un-doped BiVO4 nanoflake films and greater than the photocurrents of individually Mo doped or W doped BiVO4 films. The origin of enhanced photoelectrochemical activity for the co-doped film may be due to the improved conductivity through the BiVO4 or slightly enhanced water oxidation kinetics.
Co-reporter:Sean M. Wood, Kyle C. Klavetter, Adam Heller and C. Buddie Mullins
Journal of Materials Chemistry A 2014 vol. 2(Issue 20) pp:7238-7243
Publication Date(Web):26 Mar 2014
DOI:10.1039/C4TA01167H
The reversible charging of a lead chalcogenide, PbTe, was studied for use as the anode material in a Li-ion cell and compared to PbO. A similar series of Li–Pb alloys were formed but with Li2Te present instead of Li2O. In the presence of Li2Te, rapid Li–Pb alloying and dealloying were observed in the potential range of 0.01–0.7 V. In the potential range of 0.8–2.5 V, Li2Te formed and decomposed reversibly. Electrodes were cycled stably for 100 cycles at a C/5 rate in both potential domains. The electrodes were also cycled stably at rates up to 10C. The presence of Li2Te reduced the overpotential required at higher charge and discharge rates by acting as a superionic conductor to improve lithium ion diffusion. These results recommend this material for potential use in low-power applications such as cell phones.
Co-reporter:Yiqing Sun, William D. Chemelewski, Sean P. Berglund, Chun Li, Huichao He, Gaoquan Shi, and C. Buddie Mullins
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 8) pp:5494
Publication Date(Web):March 26, 2014
DOI:10.1021/am405628r
We report the growth of well-defined antimony-doped tin oxide (ATO) nanorods as a conductive scaffold to improve hematite’s photoelectrochemical water oxidation performance. The hematite grown on ATO exhibits greatly improved performance for photoelectrochemical water oxidation compared to hematite grown on flat fluorine-doped tin oxide (FTO). The optimized photocurrent density of hematite on ATO is 0.67 mA/cm2 (0.6 V vs Ag/AgCl), which is much larger than the photocurrent density of hematite on flat FTO (0.03 mA/cm2). Using H2O2 as a hole scavenger, it is shown that the ATO nanorods indeed act as a useful scaffold and enhanced the bulk charge separation efficiency of hematite from 2.5% to 18% at 0.4 V vs Ag/AgCl.Keywords: Fe2O3; nanorod scaffold; PEC; photoelectrochemistry; water oxidation; water splitting;
Co-reporter:Paul R. Abel, Meredith G. Fields, Adam Heller, and C. Buddie Mullins
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 18) pp:15860
Publication Date(Web):August 26, 2014
DOI:10.1021/am503365k
The sodium electrochemistry of evaporatively deposited tin, germanium, and alloys of the two elements is reported. Limiting the sodium stripping voltage window to 0.75 V versus Na/Na+ improves the stability of the tin and tin-rich compositions on repeated sodiation/desodiation cycles, whereas the germanium and germanium-rich alloys were stable up to 1.5 V. The stability of the electrodes could be correlated to the surface mobility of the alloy species during deposition suggesting that tin must be effectively immobilized in order to be successfully utilized as a stable electrode. While the stability of the alloys is greatly increased by the presence of germanium, the specific Coulombic capacity of the alloy decreases with increasing germanium content due to the lower Coulombic capacity of germanium. Additionally, the presence of germanium in the alloy suppresses the formation of intermediate phases present in the electrochemical sodiation of tin. Four-point probe resistivity measurements of the different compositions show that electrical resistivity increases with germanium content. Pure germanium is the most resistive yet exhibited the best electrochemical performance at high current densities which indicates that electrical resistivity is not rate limiting for any of the tested compositions.Keywords: alloy; germanium; sodium-ion battery; tin
Co-reporter:Wen-Yueh Yu, Gregory M. Mullen, and C. Buddie Mullins
The Journal of Physical Chemistry C 2014 Volume 118(Issue 4) pp:2129-2137
Publication Date(Web):January 8, 2014
DOI:10.1021/jp411299e
Pd–Au bimetallic catalysts have shown potential applications in numerous heterogeneous reactions in which hydrogen and CO act as reactants, intermediates, or products. A fundamental understanding of the interplay between coadsorbed H and CO on the Pd–Au surface is necessary for improving the understanding of catalytic performance. In this study, the interactions of hydrogen and CO with Pd/Au(111) model surfaces were investigated by temperature-programmed desorption (TPD) and molecular beam scattering (MBS) experiments, carried out under ultra-high-vacuum conditions. Our results reveal that CO adsorbs competitively on the hydrogen-precovered Pd–Au surface, causing surface H adatoms to diffuse away from stronger-binding sites (e.g., Pd(111)-like islands) to weaker-binding sites (e.g., Pd–Au alloy sites and subsurface), as evidenced by a shift of the H2 desorption feature to lower temperatures in TPD measurements. Additionally, evolution of H2 was observed when a CO molecular beam was impinged onto the H-precovered Pd–Au surface, providing direct evidence that CO induces recombinative desorption of H adatoms. The presence of H adatoms on the Pd–Au surface was found to decrease the initial sticking probability of CO during MBS experiments but had little influence on CO desorption during subsequent TPD measurements.
Co-reporter:Hoang X. Dang;Yong-Mao Lin;Kyle C. Klavetter;Trevor H. Cell; Adam Heller; C. Buddie Mullins
ChemElectroChem 2014 Volume 1( Issue 1) pp:158-164
Publication Date(Web):
DOI:10.1002/celc.201300139
Abstract
Lithium uptake and release by pure, morphology-controlled Ta2O5 films is studied. Porous nanocolumnar films, reactively e-beam deposited at 70° versus normal, are rapidly lithiated and delithiated, withstanding volume-change-associated stresses. Their reversible gravimetric Coulombic capacities at 1 C, 2 C, 5 C, and 10 C rates are, respectively, 252, 225, 192, 168 mAh g−1. After 100 cycles at 1 C rate the films retain 96 % of their Coulombic capacity and 99 % of their Coulombic efficiency. The rapid lithiation/delithiation is attributed to formation of amorphous, Li+ permeable, nanoporous LiTaO3.
Co-reporter:Paul R. Abel ; Kyle C. Klavetter ; Adam Heller
The Journal of Physical Chemistry C 2014 Volume 118(Issue 31) pp:17407-17412
Publication Date(Web):July 8, 2014
DOI:10.1021/jp504327e
Thin films of amorphous nanocolumnar germanium subselenide Ge0.9Se0.1, with a lithiation/delithiation capacity of 1.2 Ahr g–1, retain a capacity of 0.8 Ahr g–1 when lithiated in 1.2 min and 0.5 Ahr g–1 when lithiated in 36 s. After 1000 cycles of 72 s lithiation/72 s delithiation, the films retain a capacity of 0.8 Ahr g–1. For delithiation in 3.3 s, the capacity is 0.9 Ahr g–1, and the specific delithiation current is 1.34 kA g–1 (kiloampere per gram).
Co-reporter:Alexander J. E. Rettie ; Shirin Mozaffari ; Martin D. McDaniel ; Kristen N. Pearson ; John G. Ekerdt ; John T. Markert
The Journal of Physical Chemistry C 2014 Volume 118(Issue 46) pp:26543-26550
Publication Date(Web):October 29, 2014
DOI:10.1021/jp5082824
We report pulsed laser deposition (PLD) synthesis of epitaxial and polycrystalline monoclinic bismuth vanadate (BiVO4, BVO) thin films. X-ray diffraction (XRD), atomic force microscopy, X-ray photoelectron spectroscopy, and scanning electron microscopy were used to characterize the samples. Epitaxial, c-axis oriented growth was achieved using single crystal yttria-stabilized zirconia (100), a substrate temperature of 575–600 °C, and an oxygen pressure of 7.8 mTorr. The volatility of Bi necessitated a large excess (Bi:V = ∼6:1) of this element in the ceramic targets to obtain stoichiometric films. XRD confirmed a BVO (001)||YSZ (001) and BVO [100]||YSZ [100] epitaxial relationship. Film growth was 3-D, and the morphology was discontinuous, consisting of irregular, smooth grains. Additionally, dense, continuous polycrystalline films were deposited on fluorine-doped tin oxide (FTO) on glass substrates at room temperature with stoichiometric targets and postdeposition annealing in air. Evaluation of these samples as photoanodes yielded photocurrents of ∼0.15 and ∼0.05 mA cm–2 at 1.23 V vs RHE under backside AM1.5G illumination with and without a hole scavenger (Na2SO3), respectively. We argue that the photocurrents are due to the high oxygen content inherent in the PLD process and suggest that these continuous films may be well-suited to investigating oxygen-related defects in BVO.
Co-reporter:Ming Pan, Adrian J. Brush, Zachary D. Pozun, Hyung Chul Ham, Wen-Yueh Yu, Graeme Henkelman, Gyeong S. Hwang and C. Buddie Mullins
Chemical Society Reviews 2013 vol. 42(Issue 12) pp:5002-5013
Publication Date(Web):27 Feb 2013
DOI:10.1039/C3CS35523C
Supported gold nanoparticles have recently been shown to possess intriguing catalytic activity for hydrogenation reactions, particularly for selective hydrogenation reactions. However, fundamental studies that can provide insight into the reaction mechanisms responsible for this activity have been largely lacking. In this tutorial review, we highlight several recent model experiments and theoretical calculations on a well-structured gold surface that provide some insights. In addition to the behavior of hydrogen on a model gold surface, we review the reactivity of hydrogen on a model gold surface in regards to NO2 reduction, chemoselective CO bond hydrogenation, ether formation, and O–H bond dissociation in water and alcohols. Those studies indicate that atomic hydrogen has a weak interaction with gold surfaces which likely plays a key role in the unique hydrogenative chemistry of classical gold catalysts.
Co-reporter:Alexander J. E. Rettie ; Heung Chan Lee ; Luke G. Marshall ; Jung-Fu Lin ; Cigdem Capan ; Jeffrey Lindemuth @; John S. McCloy ; Jianshi Zhou ; Allen J. Bard
Journal of the American Chemical Society 2013 Volume 135(Issue 30) pp:11389-11396
Publication Date(Web):July 8, 2013
DOI:10.1021/ja405550k
Bismuth vanadate (BiVO4) is a promising photoelectrode material for the oxidation of water, but fundamental studies of this material are lacking. To address this, we report electrical and photoelectrochemical (PEC) properties of BiVO4 single crystals (undoped, 0.6% Mo, and 0.3% W:BiVO4) grown using the floating zone technique. We demonstrate that a small polaron hopping conduction mechanism dominates from 250 to 400 K, undergoing a transition to a variable-range hopping mechanism at lower temperatures. An anisotropy ratio of ∼3 was observed along the c axis, attributed to the layered structure of BiVO4. Measurements of the ac field Hall effect yielded an electron mobility of ∼0.2 cm2 V–1 s–1 for Mo and W:BiVO4 at 300 K. By application of the Gärtner model, a hole diffusion length of ∼100 nm was estimated. As a result of low carrier mobility, attempts to measure the dc Hall effect were unsuccessful. Analyses of the Raman spectra showed that Mo and W substituted for V and acted as donor impurities. Mott–Schottky analysis of electrodes with the (001) face exposed yielded a flat band potential of 0.03–0.08 V versus the reversible H2 electrode, while incident photon conversion efficiency tests showed that the dark coloration of the doped single crystals did not result in additional photocurrent. Comparison of these intrinsic properties to those of other metal oxides for PEC applications gives valuable insight into this material as a photoanode.
Co-reporter:Huichao He, Sean P. Berglund, Peng Xiao, William D. Chemelewski, Yunhuai Zhang and C. Buddie Mullins
Journal of Materials Chemistry A 2013 vol. 1(Issue 41) pp:12826-12834
Publication Date(Web):06 Sep 2013
DOI:10.1039/C3TA13239K
To improve the photoelectrochemical activity of WO3, Bi2S3/WO3 heterojunction films were designed by coupling WO3 films with varying amounts of urchin-like Bi2S3 nanospheres. The WO3 films were composed of WO3 nanoprism arrays, which were synthesized using a solvothermal method. After coating a single layer of Bi2S3 on top of the WO3 film, the resulting Bi2S3/WO3 heterojunction film showed enhanced photoelectrochemical activity. At 1.2 V vs. Ag/AgCl, the initial photocurrent density of the Bi2S3/WO3 heterojunction film with one layer of Bi2S3 was 1.33 mA cm−2 in 0.1 M Na2SO4 and 1.19 mA cm−2 in a 0.2 M NaCl mixed water–ethanol solution, which was 40% and 32% higher than the bare WO3 film under the same conditions, respectively. The optimal number of Bi2S3 layers for coupling with the WO3 film was found to be 3 layers, which had the highest photocurrent density and IPCE values. The photoelectrochemical activity of Bi2S3/WO3 heterojunction film was not stable for water oxidation due to photocorrosion in aqueous electrolyte, but it was stable in the NaCl mixed water–ethanol solution and a non-aqueous solution containing iodide/triiodide as a redox couple. The origin of enhanced photoelectrochemical activity of the Bi2S3/WO3 heterojunction film was primarily ascribed to the band potential matching between WO3 and Bi2S3, which is advantageous for charge separation.
Co-reporter:Son Hoang, Sean P. Berglund, Raymond R. Fullon, Ryan L. Minter and C. Buddie Mullins
Journal of Materials Chemistry A 2013 vol. 1(Issue 13) pp:4307-4315
Publication Date(Web):07 Feb 2013
DOI:10.1039/C3TA01384G
We report a facile, scalable, and low cost chemical bath deposition of vertically aligned TiO2 nanoplatelet arrays on various substrates including fluorine-doped tin oxide coated glass substrates and their applications for photoelectrochemical (PEC) water splitting and dye sensitized solar cells. The TiO2 arrays consisting of single crystal rutile nanoplatelets with heights (film thicknesses) of up to 1 μm, lengths of up to 130 nm, and widths of ∼5 nm were grown via controlling oxidation and hydrolysis of TiCl3 at low pH (0.71–0.85) and low TiCl3 concentration (8–40 mM). As a photoanode for water oxidation in a PEC water splitting cell, the TiO2 nanoplatelets show excellent charge separation characteristics with a saturated photocurrent in 1 M KOH electrolyte under AM 1.5 G illumination of ∼0.4 mA cm−2 reached at an exceptionally low bias of −0.6 V vs. Ag/AgCl (0.4 V vs. reversible hydrogen electrode). Dye sensitized solar cells assembled using N719 dye sensitized-TiO2 nanoplatelet arrays also show promising performance with photoconversion efficiencies of 1.28% for as-synthesized (no thermal post-treatment) and 3.7% for annealed TiO2 nanoplatelets.
Co-reporter:Yong-Mao Lin, Paul R. Abel, Asha Gupta, John B. Goodenough, Adam Heller, and C. Buddie Mullins
ACS Applied Materials & Interfaces 2013 Volume 5(Issue 17) pp:8273
Publication Date(Web):August 19, 2013
DOI:10.1021/am4023994
Sn0.9Cu0.1 nanoparticles were synthesized via a surfactant-assisted wet chemistry method, which were then investigated as an anode material for ambient temperature rechargeable sodium ion batteries. The Sn0.9Cu0.1 nanoparticle-based electrodes exhibited a stable capacity of greater than 420 mA h g–1 at 0.2 C rate, retaining 97% of their maximum observed capacity after 100 cycles of sodium insertion/deinsertion. Their performance is considerably superior to electrodes made with either Sn nanoparticles or Sn microparticles.Keywords: anode; Na-ion battery; nanoparticles; tin copper alloy;
Co-reporter:Kyle C. Klavetter, Sean M. Wood, Yong-Mao Lin, Jonathan L. Snider, Nicholas C. Davy, Aaron M. Chockla, Dwight K. Romanovicz, Brian A. Korgel, Joo-Woon Lee, Adam Heller, C. Buddie Mullins
Journal of Power Sources 2013 Volume 238() pp:123-136
Publication Date(Web):15 September 2013
DOI:10.1016/j.jpowsour.2013.02.091
We report stable, high capacity cycling performance over 2500 deep cycles at variable C-rates (1C, 5C and 10C) for slurry-cast Li-ion battery anodes made using commercially-available germanium nanopowder. The determining factor in cycling performance was the use of fluoroethylene carbonate (FEC) rather than ethylene carbonate (EC) as a co-solvent in the electrolyte. Cycling tests for the FEC-based electrode showed stable performance close to 700 mAh g−1 through 500 cycles at 10C with near 100% Coulombic efficiency. These results show that a Ge-based slurry-cast electrode using active material structured only as a simple particle can be used to create an electrode system which is a candidate for optimization and scale-up. These cycling improvements obtained using the FEC-based electrolyte complements recent progress in Ge-based electrode research which has focused on improving performance through tailored structural and chemical modifications to the active material structure. The effect of the electrolyte on Li-ion transport, electrode stability toward oxidation, and electrode and SEI structural stability was studied using electrochemical impedance spectroscopy, differential capacity profiles, SEM and cross-sectional TEM imaging where we characterize the evolution of the electrode structure cycled with the FEC-based electrolyte considering the type and extent of SEI growth, particle agglomeration and fracturing.Highlights► Ge-based slurry cast Li-ion anode performs stably at high C-rates for 2500 cycles. ► Fluoroethylene carbonate (FEC) based electrolyte critical to anode performance. ► Stable performance and near 100% Coulombic efficiency at 10C for 500 cycles. ► dq/dV show thermodynamic reversibility of FEC- but not EC-based electrode. ► Cross-sectional TEM with SEM show structure of cycled anode and SEI.
Co-reporter:Sean P. Berglund, Heung Chan Lee, Paul D. Núñez, Allen J. Bard and C. Buddie Mullins
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 13) pp:4554-4565
Publication Date(Web):11 Feb 2013
DOI:10.1039/C3CP50540E
A new dispenser and scanner system is used to create and screen Bi–M–Cu oxide arrays for cathodic photoactivity, where M represents 1 of 22 different transition and post-transition metals. Over 3000 unique Bi:M:Cu atomic ratios are screened. Of the 22 metals tested, 10 show a M–Cu oxide with higher photoactivity than CuO and 10 show a Bi–M–Cu oxide with higher photoactivity than CuBi2O4. Cd, Zn, Sn, and Co produce the most photoactive M–Cu oxides, all showing a 200–300% improvement in photocurrent over CuO. Ag, Cd, and Zn produce the highest photoactivity Bi–M–Cu oxides with a 200–400% improvement over CuBi2O4. Most notable is a Bi–Ag–Cu oxide (Bi:Ag:Cu atomic ratio of 22:3:11) which shows 4 times higher photocurrent than CuBi2O4. This material is capable of evolving hydrogen under illumination in neutral electrolyte solutions at 0.6 V vs. RHE when Pt is added to the surface as an electrocatalyst.
Co-reporter:Son Hoang;Thong Q. Ngo;Sean P. Berglund;Raymond R. Fullon; John G. Ekerdt ; C. Buddie Mullins
ChemPhysChem 2013 Volume 14( Issue 10) pp:2270-2276
Publication Date(Web):
DOI:10.1002/cphc.201201092
Abstract
Niobium-modified TiO2 hierarchical spherical micrometer-size particles, which consist of many nanowires, are synthesized by solvothermal synthesis and studied as photoelectrodes for water photo-oxidation and dye-sensitized solar cell (DSSC) applications. Incorporation of Nb leads to a rutile-to-anatase TiO2 phase transition in the TiO2 hierarchical spheres (HSs), with the anatase percentage increasing from 0 % for the pristine TiO2 HSs to 47.6 % for the 1.82 at. % Nb-incorporated TiO2 sample. Incorporation of Nb leads to significant improvements in water photo-oxidation with the photocurrents reaching 70.5 μA cm−2 at 1.23 V versus the reversible hydrogen electrode, compared with 28.3 μA cm−2 for the pristine TiO2 sample. The photoconversion efficiency of Nb:TiO2 HS-based DSSCs reaches 6.09±0.15 % at 0.25 at. % Nb, significantly higher than that for the pristine TiO2 HS cells (3.99±0.02 %). In addition, the incident-photon-to-current efficiency spectra for DSSCs show that employing TiO2 and Nb:TiO2 HSs provides better light harvesting, especially of long-wavelength photons, than anatase TiO2 nanoparticle-based DSSCs.
Co-reporter:Yong-Mao Lin, Kyle C. Klavetter, Adam Heller, and C. Buddie Mullins
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 6) pp:999-1004
Publication Date(Web):March 8, 2013
DOI:10.1021/jz4003058
Amorphous GeO2 nanoparticles were prepared via a surfactant-assisted hydrothermal process. The effect of the reaction temperature and the surfactant concentration on the morphology of GeO2 particles were investigated. Particles of less than 300 nm were obtained when using 1,2-diaminopropane surfactant in a synthesis carried out at 180◦C. The synthesized germanium oxide nanoparticles were evaluated for their utility as the active anode material in Li-ion batteries. The electrode prepared with this material exhibited a stable capacity ∼600 mAh g–1 at 0.2 C rate for up to 150 cycles in a conventional electrolyte containing ethylene carbonate (EC). The cyclability of the GeO2 nanoparticle electrode was further improved by using a fluorinated ethylene carbonate (FEC) based electrolyte, which showed capacities greater than 600 mAh g–1 and retained more than 96% of their capacity after 500 cycles at 0.2 C rate. The effect of different electrolyte systems was studied by using electrochemical impedance spectroscopy and electron microscopy.Keywords: anode; fluoroethylene carbonate; germanium dioxide; lithium ion battery; nanoparticles;
Co-reporter:Gregory M. Mullen;Jinlong Gong;Ting Yan;Ming Pan
Topics in Catalysis 2013 Volume 56( Issue 15-17) pp:1499-1511
Publication Date(Web):2013 November
DOI:10.1007/s11244-013-0143-x
Water has important effects on several reactions occurring over gold catalysts. In this work, we review studies demonstrating the interactions of water in surface chemistry and catalysis over gold, with specific emphasis on the ability of water to enhance activity for the CO oxidation reaction and the role water plays in the mechanism of the water–gas shift reaction. Water significantly influences these and other catalytic reactions over gold catalysts.
Co-reporter:Paul R. Abel, Aaron M. Chockla, Yong-Mao Lin, Vincent C. Holmberg, Justin T. Harris, Brian A. Korgel, Adam Heller, and C. Buddie Mullins
ACS Nano 2013 Volume 7(Issue 3) pp:2249
Publication Date(Web):February 22, 2013
DOI:10.1021/nn3053632
Both silicon and germanium are leading candidates to replace the carbon anode of lithium ions batteries. Silicon is attractive because of its high lithium storage capacity while germanium, a superior electronic and ionic conductor, can support much higher charge/discharge rates. Here we investigate the electronic, electrochemical and optical properties of Si(1-x)Gex thin films with x = 0, 0.25, 0.5, 0.75, and 1. Glancing angle deposition provided amorphous films of reproducible nanostructure and porosity. The film’s composition and physical properties were investigated by X-ray photoelectron spectroscopy, four-point probe conductivity, Raman, and UV–vis absorption spectroscopy. The films were assembled into coin cells to test their electrochemical properties as a lithium-ion battery anode material. The cells were cycled at various C-rates to determine the upper limits for high rate performance. Adjusting the composition in the Si(1-x)Gex system demonstrates a trade-off between rate capability and specific capacity. We show that high-capacity silicon anodes and high-rate germanium anodes are merely the two extremes; the composition of Si(1-x)Gex alloys provides a new parameter to use in electrode optimization.Keywords: germanium; glancing angle deposition; lithium-ion battery; SiGe; silicon
Co-reporter:Sean P. Berglund, Son Hoang, Ryan L. Minter, Raymond R. Fullon, and C. Buddie Mullins
The Journal of Physical Chemistry C 2013 Volume 117(Issue 48) pp:25248-25258
Publication Date(Web):November 18, 2013
DOI:10.1021/jp4073747
A total of 35 elements were investigated as single component metal oxides and as dopants for TiO2 for use in dye-sensitized solar cells (DSCs). An array dispenser and scanner system was utilized for high-throughput testing of the single component metal oxides and hundreds of doped TiO2 compositions. The optimal dopant concentrations were identified and summarized according to their effect on short circuit current (ISC) and open circuit voltage (VOC). New dopant candidates were discovered, including several post-transition metals, which showed improvements in DSC performance when incorporated at compositions of 6% In TiO2, 6% Sn TiO2, and 6% Sb TiO2 (metals basis). These compositions were used to synthesize scaled-up DSC devices for detailed characterization. Incorporation of 6% In into TiO2 enhanced VOC substantially and increased ISC slightly, 6% Sn improved ISC while preserving VOC, and 6% Sb increased ISC significantly but decreased VOC. When coincorporated, Cr and Sb showed a complementary interaction, which reduced resistance to charge transport in the DSC device. The 2% Cr, 6% Sb TiO2 showed higher ISC and VOC values than 6% Sb TiO2 and considerably higher ISC than 2% Cr TiO2. The fill-factor for 2% Cr, 6% Sb TiO2 was relatively low, but the maximum power was higher than the other compositions due to the higher currents. The VOC values for DSC devices with different compositions correlated linearly with the measured flat-band potentials (EFB).
Co-reporter:Wen-Yueh Yu, Gregory M. Mullen, and C. Buddie Mullins
The Journal of Physical Chemistry C 2013 Volume 117(Issue 38) pp:19535-19543
Publication Date(Web):August 26, 2013
DOI:10.1021/jp406736b
Pd–Au bimetallic catalysts have shown promising performance in numerous reactions that involve hydrogen. Fundamental studies of hydrogen interactions with Pd–Au surfaces could provide useful insights into the reaction mechanisms over Pd–Au catalysts, which may, in turn, guide future catalyst design. In this study, the interactions of hydrogen (i.e., adsorption, absorption, diffusion, and desorption) with Pd/Au(111) model surfaces were studied using temperature-programmed desorption (TPD) under ultrahigh-vacuum conditions. Our experimental results reveal Pd–Au bimetallic surfaces readily dissociate H2 and yet also weakly bind H adatoms, properties that could be beneficial for catalytic reactions involving hydrogen. The presence of contiguous Pd sites, characterized by reflection–absorption infrared spectroscopy using CO as a probe molecule (CO-RAIRS), was found to be vital for the dissociative adsorption of H2 at 77 K. The H adatom binds to Pd–Au alloy sites more strongly than to Au(111) but more weakly than to Pd(111) as indicated by its desorption temperature (∼200 K). With hydrogen exposure at slightly higher temperatures (i.e., 100–150 K), extension of a low-temperature desorption feature was observed, suggesting the formation of subsurface H atoms (or H absorption). Experiments using deuterium indicate that H–D exchange over the Pd–Au bimetallic surface obeys Langmuir–Hinshelwood kinetics and that H/D adatoms are mobile on the surface at low temperatures.
Co-reporter:Paul R. Abel, Yong-Mao Lin, Tania de Souza, Chia-Yun Chou, Asha Gupta, John B. Goodenough, Gyeong S. Hwang, Adam Heller, and C. Buddie Mullins
The Journal of Physical Chemistry C 2013 Volume 117(Issue 37) pp:18885-18890
Publication Date(Web):August 21, 2013
DOI:10.1021/jp407322k
Both nanocolumnar and dense germanium thin films, synthesized by evaporative deposition, were tested as a potential anode material for sodium-ion batteries. The reversible capacity of the nanocolumnar films was found to be 430 mAh/g, which is higher than the theoretical capacity of 369 mAh/g. The nanocolumnar films retained 88% of their initial capacity after 100 cycles at C/5, whereas the dense films began to deteriorate after ∼15 cycles. Additionally, the nanocolumnar films were stable at charge/discharge rates up to 27C (10 A/g). The diffusion coefficient for sodium in germanium was estimated, from impedance analysis of the dense films, to be ∼10–13 cm2 s–1. Modeling of diffusion in the sodium- germanium system predicts that sodium diffusion in the near-surface layers of the material is significantly faster than in the bulk. These results show that small feature sizes are critical for rapid, reversible electrochemical sodiation of germanium.
Co-reporter:Son Hoang, Siwei Guo, Nathan T. Hahn, Allen J. Bard, and C. Buddie Mullins
Nano Letters 2012 Volume 12(Issue 1) pp:26-32
Publication Date(Web):November 23, 2011
DOI:10.1021/nl2028188
We report hydrothermal synthesis of single crystalline TiO2 nanowire arrays with unprecedented small feature sizes of ∼5 nm and lengths up to 4.4 μm on fluorine-doped tin oxide substrates. A substantial amount of nitrogen (up to 1.08 atomic %) can be incorporated into the TiO2 lattice via nitridation in NH3 flow at a relatively low temperature (500 °C) because of the small cross-section of the nanowires. The low-energy threshold of the incident photon to current efficiency (IPCE) spectra of N-modified TiO2 samples is at ∼520 nm, corresponding to 2.4 eV. We also report a simple cobalt treatment for improving the photoelectrochemical (PEC) performance of our N-modified TiO2 nanowire arrays. With the cobalt treatment, the IPCE of N-modified TiO2 samples in the ultraviolet region is restored to equal or higher values than those of the unmodified TiO2 samples, and it remains as high as ∼18% at 450 nm. We propose that the cobalt treatment enhances PEC performance via two mechanisms: passivating surface states on the N-modified TiO2 surface and acting as a water oxidation cocatalyst.
Co-reporter:Ming Pan ; Hyung Chul Ham ; Wen-Yueh Yu ; Gyeong S. Hwang
Journal of the American Chemical Society 2012 Volume 135(Issue 1) pp:436-442
Publication Date(Web):December 11, 2012
DOI:10.1021/ja3096575
We have discovered that NO2 is reduced to NO at 77 K by hydrogen precovered gold in vacuum. Here, we investigate the partial reduction of NO2 to NO on an atomic-hydrogen populated model gold catalyst for a more fundamental understanding of the surface chemistry of hydrogenation. Gold-based catalysts have been found to be active for many hydrogenation reactions, but few related fundamental studies have been conducted. Our experimental results reveal a high catalytic activity for gold: indeed, NO2 is reduced to NO with 100% conversion and 100% selectivity at temperatures lower than 120 K. Density functional theory calculations and reflection–absorption infrared spectroscopy measurements indicate that HNO2 and N2O3 are intermediates which are highly dependent on surface hydrogen concentrations; subsequent hydrogenation of HNO2 and dissociation of N2O3 upon annealing induces the production of NO and H2O.
Co-reporter:Son Hoang ; Sean P. Berglund ; Nathan T. Hahn ; Allen J. Bard
Journal of the American Chemical Society 2012 Volume 134(Issue 8) pp:3659-3662
Publication Date(Web):February 8, 2012
DOI:10.1021/ja211369s
We report a synergistic effect involving hydrogenation and nitridation cotreatment of TiO2 nanowire (NW) arrays that improves the water photo-oxidation performance under visible light illumination. The visible light (>420 nm) photocurrent of the cotreated TiO2 is 0.16 mA/cm2 and accounts for 41% of the total photocurrent under simulated AM 1.5 G illumination. Electron paramagnetic resonance (EPR) spectroscopy reveals that the concentration of Ti3+ species in the bulk of the TiO2 following hydrogenation and nitridation cotreatment is significantly higher than that of the sample treated solely with ammonia. It is believed that the interaction between the N-dopant and Ti3+ is the key to the extension of the active spectrum and the superior visible light water photo-oxidation activity of the hydrogenation and nitridation cotreated TiO2 NW arrays.
Co-reporter:Yong-Mao Lin, Rajaram K. Nagarale, Kyle C. Klavetter, Adam Heller and C. Buddie Mullins
Journal of Materials Chemistry A 2012 vol. 22(Issue 22) pp:11134-11139
Publication Date(Web):30 Apr 2012
DOI:10.1039/C2JM16328D
Li-ion battery anodes made of SnO2 nanoparticles and a TiO2-supported SnO2 nanocomposite formed of equimolar amounts of the Sn and Ti oxides were investigated, respectively. By limiting the voltage window of the charge/discharge cycles to the range 50 mV–1.0 V, both the SnO2-based anode and the SnO2/TiO2-based anode show improved cycling stability. Compared to the SnO2 nanoparticle based anodes, the TiO2-support-SnO2 nanocomposite anodes exhibit better cyclability and higher Coulombic efficiency. During the first lithiation process, Li+ conducting LixTiO2 is formed in the SnO2/TiO2 composite, which structurally/mechanically supports the electrode. The anode made of amorphous TiO2-cassiterite SnO2 retained a reversible capacity of ∼500 mAh g−1 (based on the weight of SnO2) or ∼320 mAh g−1 (based on the weight of SnO2/TiO2) at 0.2 C after 100 cycles and at a rate as fast as 5 C retained a stable reversible capacity of ∼340 mAh g−1 (based on the weight of SnO2) or ∼220 mAh g−1 (based on the weight of SnO2/TiO2).
Co-reporter:Yong-Mao Lin, Kyle C. Klavetter, Paul R. Abel, Nicholas C. Davy, Jonathan L. Snider, Adam Heller and C. Buddie Mullins
Chemical Communications 2012 vol. 48(Issue 58) pp:7268-7270
Publication Date(Web):30 May 2012
DOI:10.1039/C2CC31712E
Electrodes composed of silicon nanoparticles (SiNP) were prepared by slurry casting and then electrochemically tested in a fluoroethylene carbonate (FEC)-based electrolyte. The capacity retention after cycling was significantly improved compared to electrodes cycled in a traditional ethylene carbonate (EC)-based electrolyte.
Co-reporter:Sean P. Berglund, Alexander J. E. Rettie, Son Hoang and C. Buddie Mullins
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 19) pp:7065-7075
Publication Date(Web):23 Mar 2012
DOI:10.1039/C2CP40807D
Porous, nanostructured BiVO4 films are incorporated with Mo and W by simultaneous evaporation of Bi, V, Mo, and W in vacuum followed by oxidation in air. Synthesis parameters such as the Bi:V:Mo:W atomic ratio and deposition angle are adjusted to optimize the films for photoelectrochemical (PEC) water oxidation. Films synthesized with a Bi:V:Mo:W atomic ratio of 46:46:6:2 (6% Mo, 2% W) demonstrate the best PEC performance with photocurrent densities 10 times higher than for pure BiVO4 and greater than previously reported for Mo and W containing BiVO4. The films consist of a directional, nanocolumnar layer beneath an irregular surface structure. Backside illumination utilizes light scattering off the irregular surface structure resulting in 30–45% higher photocurrent densities than for frontside illumination. To improve the kinetics for water oxidation Pt is photo-deposited onto the surface of the 6% Mo, 2% W BiVO4 films as an electrocatalyst. These films achieve quantum efficiencies of 37% at 1.1 V vs. RHE and 50% at 1.6 V vs. RHE for 450 nm light.
Co-reporter:Ming Pan;Dr. Zachary D. Pozun;Adrian J. Brush; Graeme Henkelman; C. Buddie Mullins
ChemCatChem 2012 Volume 4( Issue 9) pp:1241-1244
Publication Date(Web):
DOI:10.1002/cctc.201200311
Co-reporter:William D. Chemelewski ; Nathan T. Hahn
The Journal of Physical Chemistry C 2012 Volume 116(Issue 8) pp:5255-5261
Publication Date(Web):February 2, 2012
DOI:10.1021/jp210877u
The photoelectrochemical water oxidation performance under simulated solar irradiation of hematite (α-Fe2O3) films synthesized by coevaporation of pure Si and Fe in an oxygen ambient, a process known as reactive ballistic deposition, is studied as a function of Si doping level and film porosity, ranging from dense films to nanocolumnar films. It is found that Si segregates to the hematite surface, does not improve the bulk conductivity, and lowers the optical absorption coefficient. Nevertheless, the photoelectrochemical performance of Si-doped, porous films is significantly improved relative to undoped, porous films. However, the improvement relative to dense, undoped films is marginal. It is concluded that Si acts to passivate the hematite surface and aids charge transfer to the solution. Additionally, from incident photon conversion efficiency measurements it is found that Si doping and porosity have little effect on the normalized spectral response of 100 nm thick hematite films.
Co-reporter:Son Hoang ; Siwei Guo
The Journal of Physical Chemistry C 2012 Volume 116(Issue 44) pp:23283-23290
Publication Date(Web):October 15, 2012
DOI:10.1021/jp309743u
We report a synthesis of N- and Ta-coincorporated TiO2 (N,Ta:TiO2) and Ta-incorporated TiO2 (Ta:TiO2) nanowire (NW) arrays and their application as photoanodes for water photooxidation. Tantalum is incorporated into TiO2 NWs with concentrations ranging from 0.11 to 3.47 atomic % by a simple solvothermal synthesis. N,Ta:TiO2 nanowires are prepared via nitridation of Ta:TiO2 nanowires in NH3 flow at a relatively low temperature (500 °C). N,Ta:TiO2 NWs with the optimum Ta concentration of 0.29 atomic % also demonstrate significant enhancement in photoelectrochemical performance with the photocurrent reaching 0.52 and 0.18 mA/cm2 under AM 1.5 G and visible light (>420 nm) illumination, compared with 0.26 and 0.13 mA/cm2 for that of N:TiO2 NWs, although the active spectrum of the N,Ta:TiO2 NW sample only extends to ∼520 nm (2.38 eV), compared to ∼540 nm (2.30 eV) for N:TiO2 NWs. We believe that the enhancement shown by the N,Ta-coincorporated sample is due to fewer recombination centers from charge compensation effects and suppression of the formation of an amorphous layer on the nanowires during the nitridation process.
Co-reporter:Nathan T. Hahn ; Alexander J. E. Rettie ; Susanna K. Beal ; Raymond R. Fullon
The Journal of Physical Chemistry C 2012 Volume 116(Issue 47) pp:24878-24886
Publication Date(Web):October 31, 2012
DOI:10.1021/jp3088397
BiSI (indirect band gap = 1.57 eV) is a recently discovered photoelectrode material possessing promising optical properties for use in alternative thin film solar cells. In this work, we study the effects of selenium doping on BiSI film properties and also demonstrate the incorporation of BiS1–xSexI films into both electrochemical and solid state solar cells. Tuning the band gap of BiS1–xSexI by substituting selenium for sulfur was accomplished by substituting various amounts of SeO2 for thiourea in the BiSI spray pyrolysis precursor solutions. This strategy was employed to reduce the direct band gap of BiS1–xSexI films from 1.63 eV to as low as 1.48 eV, as measured by UV–vis–NIR diffuse reflectance spectroscopy for x = 0.4. Both electrochemical and solid state solar cell devices utilizing n-BiSI as the light absorbing material demonstrated open circuit voltages of nearly 0.4 V. The electrochemical devices showed much higher short circuit currents and power conversion efficiencies than the solid state devices. Power conversion efficiencies of up to 0.25 and 0.012% were measured for electrochemical and solid state devices, respectively, under AM1.5G illumination.
Co-reporter:Paul R. Abel, Yong-Mao Lin, Hugo Celio, Adam Heller, and C. Buddie Mullins
ACS Nano 2012 Volume 6(Issue 3) pp:2506
Publication Date(Web):February 28, 2012
DOI:10.1021/nn204896n
Silicon and partially oxidized silicon thin films with nanocolumnar morphology were synthesized by evaporative deposition at a glancing angle, and their performance as lithium-ion battery anodes was evaluated. The incorporated oxygen concentration was controlled by varying the partial pressure of water during the deposition and monitored by quartz crystal microbalance, X-ray photoelectron spectroscopy. In addition to bulk oxygen content, surface oxidation and annealing at low temperature affected the cycling stability and lithium-storage capacity of the films. By simultaneously optimizing all three, films of ∼2200 mAh/g capacity were synthesized. Coin cells made with the optimized films were reversibly cycled for ∼120 cycles with virtually no capacity fade. After 300 cycles, 80% of the initial reversible capacity was retained.Keywords: lithium-ion battery; nanostructured; reactive ballistic deposition; silicon; SiOx
Co-reporter:Hoang X. Dang, Nathan T. Hahn, Hyun S. Park, Allen J. Bard, and C. Buddie Mullins
The Journal of Physical Chemistry C 2012 Volume 116(Issue 36) pp:19225-19232
Publication Date(Web):August 15, 2012
DOI:10.1021/jp307369z
Nanostructured Ta3N5 photoanodes (band gap of ∼2.0 eV) were synthesized via a two-step process: first, nanocolumnar Ta2O5 films were deposited by evaporation of tantalum metal in a vacuum chamber in a low pressure oxygen ambient followed by heating in an ammonia gas flow to convert Ta2O5 into orthorhombic Ta3N5. Under Xe lamp irradiation (∼73 mW/cm2), a 100 nm nanoporous Ta3N5 electrode achieved an anodic photocurrent of ∼1.4 mA/cm2 at +0.5 V versus Ag/AgCl in 1 M KOH solution. By comparison, a dense film achieved ∼0.4 mA/cm2 clearly illustrating the importance of nanostructuring for improving the performance of Ta3N5 photoanodes. However, Ta3N5 films suffered from inherent self-oxidation under light illumination, and application of a cobalt cocatalyst layer was found to improve the stability as well as photocatalytic activity of the Ta3N5 films.
Co-reporter:Nathan T. Hahn, Son Hoang, Jeffrey L. Self, and C. Buddie Mullins
ACS Nano 2012 Volume 6(Issue 9) pp:7712
Publication Date(Web):August 14, 2012
DOI:10.1021/nn3031063
Bismuth oxy-iodide is a potentially interesting visible-light-active photocatalyst; yet there is little research regarding its photoelectrochemical properties. Herein we report the synthesis of BiOI nanoplatelet photoelectrodes by spray pyrolysis on fluorine-doped tin oxide substrates at various temperatures. The films exhibited n-type conductivity, most likely due to the presence of anion vacancies, and optimized films possessed incident photon conversion efficiencies of over 20% in the visible range for the oxidation of I– to I3– at 0.4 V vs Ag/AgCl in acetonitrile. Visible-light photons (λ > 420 nm) contributed approximately 75% of the overall photocurrent under AM1.5G illumination, illustrating their usefulness under solar light illumination. A deposition temperature of 260 °C was found to result in the best performance due to the balance of morphology, crystallinity, impurity levels, and optical absorption, leading to photocurrents of roughly 0.9 mA/cm2 at 0.4 V vs Ag/AgCl. Although the films performed stably in acetonitrile, their performance decreased significantly upon extended exposure to water, which was apparently caused by a loss of surface iodine and subsequent formation of an insulating bismuth hydroxide layer.Keywords: layered semiconductor; solar energy; visible-light photocatalyst; water splitting
Co-reporter:Ming Pan, Zachary D. Pozun, Wen-Yueh Yu, Graeme Henkelman, and C. Buddie Mullins
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 14) pp:1894-1899
Publication Date(Web):July 3, 2012
DOI:10.1021/jz3007707
A fundamental understanding of the interactions between coadsorbed water and hydrogen on metallic surfaces is critical to many chemical processes including catalysis and electrochemistry. Here, we report on the strong and intricate interactions between coadsorbed H/D and water on the close-packed (111) surface of gold. Deuterium isotopic labeling shows H/D exchange in H–D2O and D–H2O systems, indicating water dissociation and suggesting a nonrandom scrambling process by revealing the origin of hydrogen evolution (from surface H atoms or from water molecules) during annealing. In this reaction, the protonation of the H-bonding ice network (i.e., the formation of (H2O)nH+) is energetically favorable and is responsible for water dissociation. Density functional theory (DFT) modeling suggests that the thermodynamics and structure of the protonated clusters are predominant factors for yielding the traceable H2 desorption features from the surface interaction with H atoms, providing insights into reaction mechanisms.Keywords: density functional theory; gold catalysis; gold surface chemistry; interaction of water and hydrogen; isotope effect; protonated water clusters; temperature-programmed desorption;
Co-reporter:Nathan T. Hahn, Jeffrey L. Self, and C. Buddie Mullins
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 11) pp:1571-1576
Publication Date(Web):May 25, 2012
DOI:10.1021/jz300515p
The development of improved solar energy conversion materials is critical to the growth of a sustainable energy infrastructure in the coming years. We report the deposition of polycrystalline BiSI thin films exhibiting promising photoelectrochemical properties on both metal foils and fluorine-doped tin-oxide-coated glass slides using a single-source chemical spray pyrolysis technique. Their strong light absorption in the visible range and well-crystallized layered structure give rise to their excellent photoelectrochemical performance through improved electron–hole generation and separation. The structure and surface composition of the films are dependent on deposition temperature, resulting in dramatic differences in performance over the temperature range studied. These results reveal the potential of n-BiSI as an alternative thin film solar energy conversion material and may stimulate further investigation into V–VI–VII compounds for these applications.Keywords: metal sulfides; photoelectrochemical; photovoltaics; semiconductors; solar cells; spray pyrolysis;
Co-reporter:Ming Pan, Adrian J. Brush, Guangbin Dong, and C. Buddie Mullins
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 17) pp:2512-2516
Publication Date(Web):August 22, 2012
DOI:10.1021/jz301105e
Ethers are an important group of organic compounds that are primarily prepared via homogeneous catalysis, which can lead to operational and environmental issues. Here we demonstrate the production of ethers via heterogeneous catalysis over H adatom-covered gold at temperatures lower than 250 K. Symmetrical ethers can be formed via a self-coupling reaction of corresponding aldehydes; for example, homocoupling of acetaldehyde and propionaldehyde yields diethyl ether and di-n-propyl ether, respectively. In addition, coupling reactions between alcohols and aldehydes, with different carbon chain lengths, are observed via the production of the corresponding unsymmetrical ethers. A reaction mechanism is proposed, suggesting that an alcohol-like intermediate via partial hydrogenation of aldehydes on the surface plays a key role in these reactions. These surface chemical reactions suggest possible heterogeneous routes to low-temperature production of ethers.Keywords: alcohol; aldehyde; ether synthesis; gold; hydrogen;
Co-reporter:Adrian J. Brush, Ming Pan, and C. Buddie Mullins
The Journal of Physical Chemistry C 2012 Volume 116(Issue 39) pp:20982-20989
Publication Date(Web):September 6, 2012
DOI:10.1021/jp308099y
Gold has been shown to exhibit promising catalytic activity, and understanding the fundamental interactions of reactants and hydrogen atoms on a gold surface is key to gaining insight into hydrogenation reaction mechanisms. In this paper, we report that the adsorption of methanol onto a H-precovered Au(111) surface induces an adsorbate structure, or set of structures, on the surface involving both methanol and hydrogen adatoms with a wide range of stability on the surface. Coadsorption of H/MeOD or D/MeOH indicates H/D exchange between the two surface species, providing evidence that the H-precovered gold surface can dissociate the methanol O–H bond at low temperature (<120 K). These isotopic experiments also demonstrate that hydrogen/deuterium atoms released from a methanol molecule desorb at higher temperatures than hydrogen/deuterium atoms originating from the surface, providing insight into the adsorbate structure(s) present. Additionally, the presence of MeOH on the surface is shown to inhibit the ability of adsorbed MeOD to undergo hydrogen exchange, providing additional clues regarding the exchange reaction mechanism. These phenomena are also shown to exist for ethanol on H-precovered Au(111), suggesting that this behavior may be common to alcohols or species with an O–H functional group in general. These observations give insight into the behavior of the O–H group on a gold surface, which can aid in determining reaction mechanisms and directing future catalytic research.
Co-reporter:Jing Wu, Nellymar Membreno, Wen-Yueh Yu, Jaclyn D. Wiggins-Camacho, David W. Flaherty, C. Buddie Mullins, and Keith J. Stevenson
The Journal of Physical Chemistry C 2012 Volume 116(Issue 40) pp:21208-21215
Publication Date(Web):September 12, 2012
DOI:10.1021/jp305937b
Vanadium oxide (V2O5) is a multifaceted material possessing desirable redox properties, including accessibility to multiple valence states, which make it attractive as a cathode for lithium ion batteries and microbatteries. Studies show that performance of this electrode material is dependent on the electrolyte employed and that solid electrolyte interphase (SEI) layer formation is responsible for the fade in capacity with multiple cycling. Nanostructured V2O5 thin films synthesized through reactive ballistic deposition (RBD) were studied with electrochemical methods, ex situ Raman and ex situ XPS in two widely used electrolytes: LiClO4/propylene carbonate (PC) and LiPF6/diethyl carbonate (DEC) + ethylene carbonate (EC). Films cycled in LiPF6/DEC+EC experienced a 32% greater capacity fade between the first and second lithiathion/delithiation cycles than those cycled in LiClO4/PC, due to a redox-induced change in the surface morphology and composition and an irreversible transformation into an amorphous state as monitored by ex situ Raman. From X-ray photoelectron spectroscopy (XPS), it was shown that V2O5 cycled in LiPF6/DEC+EC contained a high atomic concentration percentage of fluoride (16.18%) in comparison with V2O5 electrodes cycled in LiClO4/PC (3.94%). No significant amounts of carbonates, oxalates, or oxyfluorophosphates typically associated with SEI formation were found when V2O5 was cycled in either electrolyte. The results obtained suggest instead that HF, formed upon water contamination of the electrolyte, reacts with V2O5 through a self-catalyzed process both at open circuit and under applied potential. The formation of vanadium oxyfluorides causes active mass loss and severe capacity fade upon discharging/charging.
Co-reporter:Yong-Mao Lin, Paul R. Abel, David W. Flaherty, Jing Wu, Keith J. Stevenson, Adam Heller, and C. Buddie Mullins
The Journal of Physical Chemistry C 2011 Volume 115(Issue 5) pp:2585-2591
Publication Date(Web):December 28, 2010
DOI:10.1021/jp110474y
Amorphous TiO2 film electrodes of controllable and reproducible nanostructure and porosity were grown via evaporation of titanium in an oxygen ambient (i.e., reactive ballistic deposition (RBD)). The cyclability, rate capability, and Coulombic capacity of the electrodes depended on their morphology and porosity, which varied with the angle of incidence of the evaporated titanium. When films are deposited via evaporation at a glancing angle of 80° with respect to surface normal, nanocolumnar arrays with high internal porosity, high surface area, and optimal pore size and connectivity can be prepared. The optimized films deposited at 80° exhibit a reversible lithium capacity of ∼285 mA h g−1 at a low cycling rate (0.2 C) and maintain a reversible capacity near 200 mA h g−1 at rates as high as 5 C. About 70% of the theoretical capacity (235 mA h g−1) was retained with indiscernible capacity decay after 100 cycles at 1 C. The total charge stored in the TiO2 RBD films involves both surface capacitive and diffusional processes.
Co-reporter:Yong-Mao Lin, Paul R. Abel, Adam Heller, and C. Buddie Mullins
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 22) pp:2885-2891
Publication Date(Web):October 28, 2011
DOI:10.1021/jz201363j
Hydrothermally synthesized single-crystalline hematite (α-Fe2O3) nanorods were investigated as an anode material for Li-ion batteries. Electrodes prepared with this material exhibited initial reversible capacities of 908 mAh g–1 at 0.2 C rate and 837 mAh g–1 at 0.5 C rate, and these capacities were completely retained after numerous cycles. The α-Fe2O3 nanorods average ∼40 nm in diameter and ∼400 nm in length providing a short path for lithium-ion diffusion and effective accommodation of the strain generated from volume expansion during the lithiation/delithiation process.Keywords: anode; hematite; lithium ion battery; nanorods; α-Fe2O3;
Co-reporter:Ming Pan, David W. Flaherty, and C. Buddie Mullins
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 12) pp:1363-1367
Publication Date(Web):May 19, 2011
DOI:10.1021/jz200577n
Gold-based classical high surface area catalysts have been widely investigated for hydrogenation reactions, but fundamental studies on model catalysts are lacking. We present experimental measurements of the reaction of hydrogen adatoms and adsorbed acetaldehyde on the Au(111) surface employing temperature-programmed desorption. Here, we show that chemisorbed hydrogen adatoms bind weakly with desorption peaks at ∼110 K, indicating an activation energy for recombinative desorption of ∼28 kJ/mol. We further demonstrate that acetaldehyde (CH3CHO) can be hydrogenated to ethanol (CH3CH2OH) on the H-atom-precovered Au(111) surface at cryogenic temperatures. Isotopic experiments employing D atoms indicate a lower hydrogenation reactivity.Keywords: acetaldehyde hydrogenation; Au(111) single crystal; deuterium; hydrogen; isotopic effect; temperature-programmed desorption; ultrahigh vacuum;
Co-reporter:Ting Yan ; Jinlong Gong ; David W. Flaherty
The Journal of Physical Chemistry C 2011 Volume 115(Issue 5) pp:2057-2065
Publication Date(Web):December 20, 2010
DOI:10.1021/jp109295u
The effect of moisture on CO oxidation on Au/TiO2(110) model catalysts is investigated using temperature-programmed desorption and molecular beam reactive scattering under ultrahigh vacuum (UHV) conditions. Oxygen exchange is observed between adsorbed atomic oxygen and isotopically labeled water. Coadsorbed water (H218O) takes part in CO oxidation on Oa precovered Au/TiO2(110) model catalysts, leading to the formation of C16O18O and C16O16O. The amount of C16O18O produced increases with increasing water coverages; however, the total amount of CO2 produced decreases. Although coadsorbed Oa and H2O have a minimal influence on the initial adsorption probability of CO, the total uptake of CO decreases as H2O coverages increase. Interestingly, the adsorption of water induces desorption of predeposited molecularly chemisorbed O2. Thus, adsorbed water slightly inhibits CO oxidation on atomic oxygen precovered Au/TiO2(110) model catalysts under UHV conditions.
Co-reporter:Sean P. Berglund ; David W. Flaherty ; Nathan T. Hahn ; Allen J. Bard
The Journal of Physical Chemistry C 2011 Volume 115(Issue 9) pp:3794-3802
Publication Date(Web):February 16, 2011
DOI:10.1021/jp1109459
Nanostructured BiVO4 films were synthesized by coevaporation of bismuth and vanadium in an oxygen ambient, a process referred to as reactive ballistic deposition (RBD). The films were tested in various electrolyte solutions to assess their activity for photoelectrochemical water oxidation. Deposition parameters, including the V/Bi atomic flux ratio and the incident angle of deposition, were adjusted. Films deposited with excess vanadium (V/Bi = 2) and incident angles of deposition at 65° showed the highest initial photocurrents with IPCE values above 21% for light wavelengths of 340−460 nm (in 0.5 M Na2SO4 at 1.0 V vs Ag/AgCl). With continued illumination the excess vanadium in these films dissolved into the electrolyte and the photocurrents dropped by 60−75% before reaching steady state. The steady-state photocurrent and IPCE values (above 14% for 340−460 nm light) were higher than the initial values for films synthesized with stoichiometric amounts of vanadium and bismuth (V/Bi = 1) and incident angles of deposition at 65°. Stoichiometric BiVO4 films remained stable under illumination but their photocurrents were limited by surface reaction kinetics. The addition of cobalt as an electrocatalyst to the surface of these films increased their photocurrent by a factor of 3.
Co-reporter:David W. Flaherty, R. Alan May, Sean P. Berglund, Keith J. Stevenson and C. Buddie Mullins
Chemistry of Materials 2010 Volume 22(Issue 2) pp:319
Publication Date(Web):December 30, 2009
DOI:10.1021/cm902184m
High surface area, porous titanium carbide films have been synthesized at room temperature via reactive ballistic deposition (RBD). X-ray diffraction and X-ray photoelectron spectroscopy show that evaporative deposition of titanium in an ethylene ambient environment allows for low temperature (35 °C) synthesis of nanocrystalline titanium carbide, a material which typically requires high processing temperatures to produce. Angle-dependent RBD allows for the controlled tuning of TiC nanostructure and porosity where changing the deposition angle from near normal incidence (13°) to more glancing angles (50−85°) changes the film morphology from relatively nonporous, dense TiC to a continuous, reticulated TiC and finally to discrete, nanocolumnar TiC. The influence of the deposition angle on TiC optical constants, porosity, specific surface area, and the pore size distribution has been investigated using hybrid quartz crystal microbalance and ellipsometric porosimetry. Notably, TiC films deposited at 35 °C at an angle of 70° have a specific surface area of 710 m2·g−1 and a mean Kelvin radius of 1.8 nm, making them attractive materials for application in catalysis, energy conversion, and storage.
Co-reporter:Nathan T. Hahn, Heechang Ye, David W. Flaherty, Allen J. Bard and C. Buddie Mullins
ACS Nano 2010 Volume 4(Issue 4) pp:1977
Publication Date(Web):April 2, 2010
DOI:10.1021/nn100032y
We report the preparation of α-Fe2O3 electrodes using a technique known as reactive ballistic deposition in which iron metal is evaporatively deposited in an oxygen ambient for photoelectrochemical (PEC) water oxidation. By manipulating synthesis parameters such as deposition angle, film thickness, and annealing temperature, we find that it is possible to optimize the structural and morphological properties of such films in order to improve their PEC efficiency. Incident photon to current conversion efficiencies (IPCE) are used to calculate an AM1.5 photocurrent of 0.55 mA/cm2 for optimized films with an IPCE reaching 10% at 420 nm in 1 M KOH at +0.5 V versus Ag/AgCl. We also note that the commonly observed low photoactivity of extremely thin hematite films on fluorine-doped tin oxide substrates may be improved by modification of annealing conditions in some cases.Keywords: hematite; nanostructured; photocatalysis; solar; water splitting
Co-reporter:Jinlong Gong and C. Buddie Mullins
Accounts of Chemical Research 2009 Volume 42(Issue 8) pp:1063
Publication Date(Web):July 9, 2009
DOI:10.1021/ar8002706
Because of gold’s resistance to oxidation and corrosion, historically chemists have considered this metal inert. However, decades ago, researchers discovered that highly dispersed gold particles on metal oxides are highly chemically active, particularly in low-temperature CO oxidations. These seminal findings spurred considerable interest in investigations and applications of gold-based materials. Since the discovery of gold’s chemical activity at the nanoscale, researchers found that bulk gold also has interesting catalytic properties. Thus, it is important to understand and contrast the intrinsic chemical properties of bulk gold with those of nanoparticle Au. Despite numerous studies, the structure and active site of supported Au nanoclusters and the active oxygen species remain elusive, and model studies under well-controlled conditions could help identify these species. The {111} facet has the lowest surface energy and is the most stable and prevalent configuration of most supported gold nanoparticles. Therefore, a molecular-level understanding of the physical properties and surface chemistry of Au(111) could provide mechanistic details regarding the nature of Au-based catalysts and lead to improved catalytic processes. This Account focuses on our current understanding of oxidative chemistry on well-defined gold single crystals, predominantly from recent investigations on Au(111) that we have performed using modern surface science techniques. Our model system strategy allows us to control reaction conditions, which assists in the identification of reaction intermediates, the determination of the elementary reaction steps, and the evaluation of reaction energetics for rate-limiting steps. We have employed temperature-programmed desorption (TPD), molecular beam reactive scattering (MBRS), and Auger electron spectroscopy (AES) to evaluate surface oxidative chemistry. In some cases, we have combined these results with density functional theory (DFT) calculations. By controlling the reaction parameters that determine product selectivity, we have examined the chemical properties of bulk gold. Based on our investigations, the surface-bound oxygen atoms are metastable at low temperature. We also demonstrate that the oxygen atoms and formed hydroxyls are responsible for some of the distinct chemical behavior of gold and participate in surface reactions either as a Brønsted base or a nucleophilic base. We observe similar reaction patterns on gold surfaces to those on copper and silver surfaces, suggesting that the acid−base reactions that have been observed on copper and silver may also occur on gold. Our model chemical studies on gold surfaces have provided intrinsic fundamental insights into high surface area gold-based catalysts and the origin of the reactive oxygen species.
Co-reporter:Ting Yan, Jinlong Gong and C. Buddie Mullins
Journal of the American Chemical Society 2009 Volume 131(Issue 44) pp:16189-16194
Publication Date(Web):October 21, 2009
DOI:10.1021/ja9062986
Direct evidence for C−O bond cleavage in the partial oxidation of 2-butanol on oxygen precovered Au(111) is provided using temperature programmed desorption (TPD) and molecular beam reactive scattering (MBRS) under ultrahigh vacuum (UHV) conditions. The oxygen precovered Au(111) surface can promote the partial oxidation of 2-butanol into 2-butanone with near 100% selectivity at low oxygen coverages, while 2-butanol adsorbs and desorbs molecularly on the clean Au(111) surface. Both C2H5C16OCH3 and C2H5C18OCH3 are observed in TPD after 2-butanol (C2H5CH16OHCH3) was dosed onto Au(111) precovered with 18Oa. This oxygen exchange phenomenon serves as strong evidence for the C−O bond cleavage in 2-butanol partial oxidation to 2-butanone. Two surface intermediates are proposed for the selective oxidation of 2-butanol: 2-butoxide and η2-aldehyde. As oxygen coverage increases, full oxidation is activated in addition to selective partial oxidation.
Co-reporter:Ming Pan, Son Hoang, Jinlong Gong and C. Buddie Mullins
Chemical Communications 2009 (Issue 47) pp:7300-7302
Publication Date(Web):07 Oct 2009
DOI:10.1039/B914308D
Although CO does not dissociate on the clean Ir(111) surface, the addition of atomic oxygen induces COdissociation at low temperatures (lower than 400 K); similarly, COdissociation has also been observed on water co-adsorbed Ir(111) or water and oxygen co-adsorbed Ir(111).
Co-reporter:Jinlong Gong, Ting Yan and C. Buddie Mullins
Chemical Communications 2009 (Issue 7) pp:761-763
Publication Date(Web):12 Jan 2009
DOI:10.1039/B821050K
Oxidative dehydrogenation of amines using heterogeneous gold catalysts has unanticipated potential; chemisorbed atomic oxygen is used to activate propylamine, producing propionitrile and/or propionaldehyde on a single-crystal Au(111) surface.
Co-reporter:Rotimi A. Ojifinni, Jinlong Gong, David W. Flaherty, Tae S. Kim and C. Buddie Mullins
The Journal of Physical Chemistry C 2009 Volume 113(Issue 22) pp:9820-9825
Publication Date(Web):May 7, 2009
DOI:10.1021/jp9022019
We present results of an investigation into the effect of annealing on the reactivity of atomic oxygen adsorbed on Au(111) employing reactive molecular beam scattering (RMBS) and temperature-programmed desorption (TPD) techniques. Isotopically labeled water (e.g., H218O and D216O), carbon monoxide (CO), and oxygen-labeled carbon dioxide (C18O2) were used as probe molecules to investigate the reactivity of adsorbed oxygen. Our results show that the reactivity of atomic oxygen-precovered Au(111) is significantly altered by annealing. The annealed surfaces were prepared by depositing atomic oxygen (16O or 18O) at 77 K followed by annealing to temperatures ranging from 100−420 K before dosing probe molecules (H218O, CO, or C18O2) at 77 K. Without exception, annealing dramatically diminishes the reactivity of oxygen for all three probe reactions. In the case of the oxygen−water interactions, TPD indicates that annealing decreases the amount of oxygen isotope scrambling between oxygen and water. Additionally, the activity of the oxygen-precovered Au(111) surface for the CO oxidation reaction decreases monotonically as the surface is incrementally annealed to increasing temperatures. The decrease in activity is indicated both by diminishing CO2 production during reactive molecular beam scattering conducted at 77 K and by subsequent O2 TPD following the CO RMBS experiments. A similar loss of activity due to annealing is observed for the formation and decomposition of surface carbonate on Au(111) as detected by oxygen isotope exchange between adsorbed atomic oxygen (16Oa) and C18O2. These observations are attributed to the thermally induced stabilization of metastable oxygen species, suggesting that the metastable oxygen species are responsible for greater reactivity on the unannealed surface. A plausible explanation is that, at lower temperatures, the adsorbed atomic oxygen species reside in a metastable state from which the kinetic barrier to reaction is lower than when adsorbed or annealed at higher temperatures.
Co-reporter:Son Hoang, Ming Pan and C. Buddie Mullins
The Journal of Physical Chemistry C 2009 Volume 113(Issue 52) pp:21745-21754
Publication Date(Web):December 3, 2009
DOI:10.1021/jp907324n
The decomposition and oxidation of 2-propanol on clean and O-precovered Ir(111) were studied using temperature-programmed desorption and molecular beam reactive scattering. On the clean surface, 2-propanol reacts via a 2-propoxide intermediate, followed by β-hydride elimination (the reaction-limiting step), producing acetone and adsorbed hydrogen at 225 K. Reactive scattering measurements show that the acetone production from the reaction of 2-propanol reduces with increasing surface temperature from 333 to 600 K. At 600 K, 2-propanol reacts mainly via nonselective decomposition, producing CO, H2, and adsorbed C. No acetone production was observed when a beam of 2-propanol was impinged on the sample at 600 K. We speculate that at high surface temperature (600 K) the formation of acetone in the η1 (O) binding configuration is inhibited, leaving only η2 (C, O) acetone species, which tend to nonselectively decompose to CO, H2, and Cad. On the O-covered Ir(111) surface, the thermal-induced reaction of 2-propanol also occurs via a 2-propoxide intermediate with β-C−H bond activation. The reaction mechanism changes with surface concentration of oxygen, causing changes in the kinetics of acetone and water evolution. Interestingly, acetone is formed upon impinging a 2-propanol beam on O−Ir(111) at 600 K. We propose that the role of surface oxygen includes (1) acting as a Brønsted base, (2) acting as a precursor of a nucleophilic attacker in the β-C−H cleavage step, and (3) altering the surface interaction with the produced acetone.
Co-reporter:David W. Flaherty, Nathan T. Hahn, Domingo Ferrer, Todd R. Engstrom, Paul L. Tanaka and C. Buddie Mullins
The Journal of Physical Chemistry C 2009 Volume 113(Issue 29) pp:12742-12752
Publication Date(Web):June 11, 2009
DOI:10.1021/jp904236v
High surface area, porous titanium carbide (TiC) films have been synthesized employing physical vapor deposition of titanium at glancing angles under high vacuum within an ethylene ambient. The composition, surface area, and morphology of the TiC films were studied as a function of deposition conditions including ethylene pressure, titanium deposition angle, substrate temperature during growth, and postdeposition annealing temperature. At high or glancing deposition angles (∼80−85°) synthesis produces films composed of arrays of porous nanocolumns of TiC, while deposition at more moderate angles, less than 70°, results in continuous, reticulated films. The maximum specific surface area (840 m2/g) is obtained by growth with an incident titanium deposition angle of 65°, an ethylene pressure of 1.5 × 10−7 Torr, and a substrate growth temperature of ∼350 K. This result is in contrast to previous investigations using related physical vapor deposition techniques which have generally shown that films with the greatest porosity and surface area are grown by deposition at cryogenic temperatures (T ≤ 77 K). The fact that the surface area is maximized at this uncharacteristically high growth temperature implies that thermally induced decomposition of ethylene and the subsequent desorption of reaction byproducts are important steps for the synthesis of these materials. Not only does deposition of TiC at 350 K result in high specific surface areas, but electron diffraction measurements indicate that these films are polycrystalline. Titanium carbide films created in this study are thermally robust and resistant to sintering, retaining greater than 70% of their initial surface area after annealing to 1000 K. The ability to deposit TiC near room temperature should allow these films to be deposited onto a wide variety of substrates.
Co-reporter:Jinlong Gong and C. Buddie Mullins
The Journal of Physical Chemistry C 2008 Volume 112(Issue 45) pp:17631-17634
Publication Date(Web):2017-2-22
DOI:10.1021/jp805871j
We report the enhanced formation and decomposition of carbonate (CO3 ↔ CO2 + Oa) from the reaction of C18O2 preadsorbed on Au(111) with an 16O atomic beam. The amount of formed carbonate increases significantly (by a factor of ∼4) compared to thermally accommodated C18O2 and 16Oa coadsorbed on the Au(111) surface. The results suggest that the reaction occurs prior to the accommodation of the incident atomic oxygen via a precursor-mediated mechanism.
Co-reporter:Jinlong Gong;David W. Flaherty;Ting Yan
ChemPhysChem 2008 Volume 9( Issue 17) pp:2461-2466
Publication Date(Web):
DOI:10.1002/cphc.200800680
Co-reporter:Jinlong Gong;Rotimi A. Ojifinni;Tae S. Kim;James D. Stiehl
Topics in Catalysis 2007 Volume 44( Issue 1-2) pp:57-63
Publication Date(Web):2007 June
DOI:10.1007/s11244-007-0278-8
This paper presents results of an investigation of low-temperature CO oxidation and the role of moisture on an atomic oxygen covered Au(111) surface by employing molecular beam scattering techniques under ultrahigh vacuum (UHV) conditions. The effect of atomic oxygen precoverage on CO oxidation was examined at sample temperatures as low as 77 K. Prompt CO2 production was observed when the CO beam impinges on the sample followed by a rapid decay of CO2 production in all cases. At oxygen precoverages above 0.5 ML, CO2 production decreases with increasing oxygen precoverage primarily due to the decrease in CO uptake. CO oxidation at 77 K goes through a precursor mediated reaction mechanism, where CO is in a precursor or trapped state and oxygen atoms are in a chemisorbed state. The role of adsorbed water was studied by using isotopically labeled water [H218O] to distinguish the oxygen species from that used in oxygen atom exposures [16O]. Evidence is presented that shows activated water or OH groups formed from water can directly participate in oxidizing CO on an atomic oxygen covered Au(111) surface.
Co-reporter:B.A. Ferguson, C.T. Reeves, C.B. Mullins
Surface Science 1999 Volume 437(1–2) pp:L748-L754
Publication Date(Web):20 August 1999
DOI:10.1016/S0039-6028(99)00716-5
The initial trapping probability of disilane on bare and monohydride-terminated Si(100)-2×1 surfaces was measured using molecular beam techniques. On the bare surface, trapping is shown to be very efficient, with near unit trapping probability at incident kinetic energies up to 0.2 eV, and nearly 50% trapping at 1 eV kinetic energy. The monohydride-terminated surface displays significantly less trapping efficiency than the bare surface. Interestingly, trapping probabilities decrease as the angle of incidence is increased, with the decrease becoming more pronounced at higher kinetic energies; this effect is discussed in terms of parallel momentum accommodation dynamics.
Co-reporter:Ting Yan, Daniel W. Redman, Wen-Yueh Yu, David W. Flaherty, José A. Rodriguez, C. Buddie Mullins
Journal of Catalysis (October 2012) Volume 294() pp:216-222
Publication Date(Web):1 October 2012
DOI:10.1016/j.jcat.2012.07.024
CO oxidation is studied at pressures between 4 and 100 Torr and temperatures from 400 K to 670 K on inverse model catalysts made of Fe2O3 nanoclusters grown on a Au(1 1 1) single crystal surface. The addition of Fe2O3 nanoclusters transformed the inert Au(1 1 1) single crystal into an active catalyst for CO oxidation. The catalytic activity increases with iron oxide coverage initially and then decreases when the iron oxide coverage is greater than 0.5 monolayers. Additionally, when the iron oxide particles form a continuous film on Au(1 1 1), there is no catalytic activity. These experimental results strongly suggest that the active sites for CO oxidation are located at the iron oxide/gold perimeter. Kinetic measurements suggest that CO oxidation by chemisorbed oxygen at the Fe2O3/Au perimeter is likely to be the rate-limiting step. Carbon deposition observed via a post-reaction Auger electron spectra suggests that multiple reaction pathways are involved in CO oxidation over Fe2O3/Au(1 1 1).Graphical abstractFe2O3 clusters supported on Au(1 1 1) is an active catalyst for CO oxidation. The perimeter between iron oxide and gold is the active center. The rate-limiting step is CO oxidation by chemisorbed molecular oxygen at the perimeter.Download high-res image (43KB)Download full-size imageHighlights► Fe2O3 particles supported on the Au(1 1 1) single crystal were studied as CO oxidation catalysts. ► Active centers are on the Fe2O3/Au(1 1 1) perimeter. ► CO oxidation by chemisorbed O2 at the perimeter is the kinetically relevant step.
Co-reporter:David W. Flaherty, Wen-Yueh Yu, Zachary D. Pozun, Graeme Henkelman, C. Buddie Mullins
Journal of Catalysis (1 September 2011) Volume 282(Issue 2) pp:278-288
Publication Date(Web):1 September 2011
DOI:10.1016/j.jcat.2011.06.024
The behavior of monofunctional platinum, Pt(1 1 1), for the water–gas shift reaction has been investigated using experimental and theoretical methods. Kinetic and isotopic measurements performed from 525 to 675 K are consistent with an associative mechanism for the water–gas shift reaction in which carbon monoxide is oxidized by a hydroxyl group. The kinetically-relevant step consists of the unimolecular decomposition of an adsorbed carboxylate intermediate. The turnover frequency of Pt(1 1 1) is five times greater than that observed on Cu(1 1 1) under identical conditions (612 K, 26 Torr CO, 10 Torr H2O); however, Pt(1 1 1) loses activity over time due to the formation of carbonaceous deposits, a process not observed in similar studies of Cu(1 1 1). Our experimental and theoretical results suggest that CO dissociates via two pathways: the Boudouard reaction and through a COH intermediate. Nucleation of carbon at step-edges and subsequent oligomerization deactivate the catalyst. These results provide insight into the synergistic roles of noble metal clusters and active supports for the water–gas shift reaction.Graphical abstractKinetic and isotopic measurements suggest that the water-gas shift reaction proceeds by an associative mechanism on Pt(1 1 1). The kinetically relevant step consists of unimolecular decomposition of adsorbed carboxylate intermediates. The surface loses activity over time due to the formation of carbonaceous deposits which nucleate at step-edges and subsequently oligomerize.Download high-res image (93KB)Download full-size imageHighlights► Monofunctional Pt(1 1 1) as water–gas shift catalyst. ► Data are consistent with associative mechanism. ► Unimolecular decomposition of carboxylate intermediate kinetically-relevant step. ► Activity loss due to carbonaceous deposits.
Co-reporter:David W. Flaherty, Sean P. Berglund, C. Buddie Mullins
Journal of Catalysis (1 January 2010) Volume 269(Issue 1) pp:33-43
Publication Date(Web):1 January 2010
DOI:10.1016/j.jcat.2009.10.012
Selective decomposition of formic acid is important as a prototype to study selective bond cleavage of oxygenates. We demonstrate that carbon-modified Mo(1 1 0), C–Mo(1 1 0), is up to 15 times more selective for the dehydrogenation of formic acid than Mo(1 1 0). Reflection absorption infrared spectroscopy (RAIRS) indicates that carbidic carbon blocks active sites for C–O bond cleavage, decreasing the rate of dehydration. Steady-state reactive molecular beam scattering (RMBS) shows that dehydration is the dominant reaction pathway on clean Mo(1 1 0), while C–Mo(1 1 0) selectively promotes dehydrogenation. Kinetic analysis of RMBS data reveals that formic acid dehydrogenation on Mo(1 1 0) has an activation energy of 34.4 ± 3.3 kJ mol−1 while the C–Mo(1 1 0) surface promotes distinct pathways for dehydrogenation with an activation energy of only 12.8 ± 1.0 kJ mol−1. RAIRS spectra suggest the new pathways include the formation of monodentate formate, and at temperatures of 500 K and greater, direct activation of the C–H bond to form carboxyl, both of which decompose via a CO2δ- intermediate to evolve CO2 and H2.Mo2C is up to 15 times more selective towards formic acid dehydrogenation than Mo. Suppressed C–O bond dissociation leading to the formation of monodentate formate and carboxyl is responsible.Download high-res image (60KB)Download full-size image
Co-reporter:Wen-Yueh Yu ; Gregory M. Mullen ; David W. Flaherty
Journal of the American Chemical Society () pp:
Publication Date(Web):July 14, 2014
DOI:10.1021/ja505192v
Pd–Au catalysts have shown exceptional performance for selective hydrogen production via HCOOH decomposition, a promising alternative to solve issues associated with hydrogen storage and distribution. In this study, we utilized temperature-programmed desorption (TPD) and reactive molecular beam scattering (RMBS) in an attempt to unravel the factors governing the catalytic properties of Pd–Au bimetallic surfaces for HCOOH decomposition. Our results show that Pd atoms at the Pd–Au surface are responsible for activating HCOOH molecules; however, the selectivity of the reaction is dictated by the identity of the surface metal atoms adjacent to the Pd atoms. Pd atoms that reside at Pd–Au interface sites tend to favor dehydrogenation of HCOOH, whereas Pd atoms in Pd(111)-like sites, which lack neighboring Au atoms, favor dehydration of HCOOH. These observations suggest that the reactivity and selectivity of HCOOH decomposition on Pd–Au catalysts can be tailored by controlling the arrangement of surface Pd and Au atoms. The findings in this study may prove informative for rational design of Pd–Au catalysts for associated reactions including selective HCOOH decomposition for hydrogen production and electro-oxidation of HCOOH in the direct formic acid fuel cell.
Co-reporter:Sungmin Han, Edward J. Evans, Gregory M. Mullen and C. Buddie Mullins
Chemical Communications 2017 - vol. 53(Issue 28) pp:NaN3993-3993
Publication Date(Web):2017/03/16
DOI:10.1039/C7CC01542A
Improved activation of adsorbed O2 by co-adsorbed H2O on the Pd–Au(111) surface has been observed. When co-adsorbed with H2O, O2 admolecules on the Pd–Au surface are more strongly bound via their interactions with H2O. This interaction leads to large enhancements in the dissociation of O2 as determined via the generation of CO2 upon exposure to CO.
Co-reporter:Jinlong Gong, Ting Yan and C. Buddie Mullins
Chemical Communications 2009(Issue 7) pp:NaN763-763
Publication Date(Web):2009/01/12
DOI:10.1039/B821050K
Oxidative dehydrogenation of amines using heterogeneous gold catalysts has unanticipated potential; chemisorbed atomic oxygen is used to activate propylamine, producing propionitrile and/or propionaldehyde on a single-crystal Au(111) surface.
Co-reporter:Son Hoang, Sean P. Berglund, Raymond R. Fullon, Ryan L. Minter and C. Buddie Mullins
Journal of Materials Chemistry A 2013 - vol. 1(Issue 13) pp:NaN4315-4315
Publication Date(Web):2013/02/07
DOI:10.1039/C3TA01384G
We report a facile, scalable, and low cost chemical bath deposition of vertically aligned TiO2 nanoplatelet arrays on various substrates including fluorine-doped tin oxide coated glass substrates and their applications for photoelectrochemical (PEC) water splitting and dye sensitized solar cells. The TiO2 arrays consisting of single crystal rutile nanoplatelets with heights (film thicknesses) of up to 1 μm, lengths of up to 130 nm, and widths of ∼5 nm were grown via controlling oxidation and hydrolysis of TiCl3 at low pH (0.71–0.85) and low TiCl3 concentration (8–40 mM). As a photoanode for water oxidation in a PEC water splitting cell, the TiO2 nanoplatelets show excellent charge separation characteristics with a saturated photocurrent in 1 M KOH electrolyte under AM 1.5 G illumination of ∼0.4 mA cm−2 reached at an exceptionally low bias of −0.6 V vs. Ag/AgCl (0.4 V vs. reversible hydrogen electrode). Dye sensitized solar cells assembled using N719 dye sensitized-TiO2 nanoplatelet arrays also show promising performance with photoconversion efficiencies of 1.28% for as-synthesized (no thermal post-treatment) and 3.7% for annealed TiO2 nanoplatelets.
Co-reporter:Gregory M. Mullen, Liang Zhang, Edward J. Evans, Ting Yan, Graeme Henkelman and C. Buddie Mullins
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 6) pp:NaN4738-4738
Publication Date(Web):2015/01/07
DOI:10.1039/C4CP04739G
Gold catalysts display high activity and good selectivity for partial oxidation of a number of alcohol species. In this work, we discuss the effects of oxygen adatoms and surface hydroxyls on the selectivity for oxidation of allylic alcohols (allyl alcohol and crotyl alcohol) on gold surfaces. Utilizing temperature programmed desorption (TPD), reactive molecular beam scattering (RMBS), and density functional theory (DFT) techniques, we provide evidence to suggest that the selectivity displayed towards partial oxidation versus combustion pathways is dependent on the type of oxidant species present on the gold surface. TPD and RMBS results suggest that surface hydroxyls promote partial oxidation of allylic alcohols to their corresponding aldehydes with very high selectivity, while oxygen adatoms promote both partial oxidation and combustion pathways. DFT calculations indicate that oxygen adatoms can react with acrolein to promote the formation of a bidentate surface intermediate, similar to structures that have been shown to decompose to generate combustion products over other transition metal surfaces. Surface hydroxyls do not readily promote such a process. Our results help explain phenomena observed in previous studies and may prove useful in the design of future catalysts for partial oxidation of alcohols.
Co-reporter:Sean P. Berglund, Heung Chan Lee, Paul D. Núñez, Allen J. Bard and C. Buddie Mullins
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 13) pp:NaN4565-4565
Publication Date(Web):2013/02/11
DOI:10.1039/C3CP50540E
A new dispenser and scanner system is used to create and screen Bi–M–Cu oxide arrays for cathodic photoactivity, where M represents 1 of 22 different transition and post-transition metals. Over 3000 unique Bi:M:Cu atomic ratios are screened. Of the 22 metals tested, 10 show a M–Cu oxide with higher photoactivity than CuO and 10 show a Bi–M–Cu oxide with higher photoactivity than CuBi2O4. Cd, Zn, Sn, and Co produce the most photoactive M–Cu oxides, all showing a 200–300% improvement in photocurrent over CuO. Ag, Cd, and Zn produce the highest photoactivity Bi–M–Cu oxides with a 200–400% improvement over CuBi2O4. Most notable is a Bi–Ag–Cu oxide (Bi:Ag:Cu atomic ratio of 22:3:11) which shows 4 times higher photocurrent than CuBi2O4. This material is capable of evolving hydrogen under illumination in neutral electrolyte solutions at 0.6 V vs. RHE when Pt is added to the surface as an electrocatalyst.
Co-reporter:Sean P. Berglund, Alexander J. E. Rettie, Son Hoang and C. Buddie Mullins
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 19) pp:NaN7075-7075
Publication Date(Web):2012/03/23
DOI:10.1039/C2CP40807D
Porous, nanostructured BiVO4 films are incorporated with Mo and W by simultaneous evaporation of Bi, V, Mo, and W in vacuum followed by oxidation in air. Synthesis parameters such as the Bi:V:Mo:W atomic ratio and deposition angle are adjusted to optimize the films for photoelectrochemical (PEC) water oxidation. Films synthesized with a Bi:V:Mo:W atomic ratio of 46:46:6:2 (6% Mo, 2% W) demonstrate the best PEC performance with photocurrent densities 10 times higher than for pure BiVO4 and greater than previously reported for Mo and W containing BiVO4. The films consist of a directional, nanocolumnar layer beneath an irregular surface structure. Backside illumination utilizes light scattering off the irregular surface structure resulting in 30–45% higher photocurrent densities than for frontside illumination. To improve the kinetics for water oxidation Pt is photo-deposited onto the surface of the 6% Mo, 2% W BiVO4 films as an electrocatalyst. These films achieve quantum efficiencies of 37% at 1.1 V vs. RHE and 50% at 1.6 V vs. RHE for 450 nm light.
Co-reporter:Wen-Yueh Yu, Liang Zhang, Gregory M. Mullen, Edward J. Evans, Graeme Henkelman and C. Buddie Mullins
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 32) pp:NaN20596-20596
Publication Date(Web):2015/07/15
DOI:10.1039/C5CP03515E
It has been reported that Pd–Au bimetallic catalysts display improved catalytic performance after adequate calcination. In this study, a model catalyst study was conducted to investigate the effects of annealing in oxygen on the surface structures of Pd–Au alloys by comparing the physicochemical properties of Pd/Au(111) surfaces that were annealed in ultrahigh vacuum (UHV) versus in an oxygen ambient. Auger electron spectroscopy (AES) and Basin hopping simulations reveal that the presence of oxygen can inhibit the diffusion of surface Pd atoms into the subsurface of the Au(111) sample. Reflection–absorption infrared spectroscopy using CO as a probe molecule (CO-RAIRS) and King–Wells measurements of O2 uptake suggest that surfaces annealed in an oxygen ambient possess more contiguous Pd sites than surfaces annealed under UHV conditions. The oxygen-annealed Pd/Au(111) surface exhibited a higher activity for CO oxidation in reactive molecular beam scattering (RMBS) experiments. This enhanced activity likely results from the higher oxygen uptake and relatively facile dissociation of oxygen admolecules due to stronger adsorbate–surface interactions as suggested by temperature-programmed desorption (TPD) measurements. These observations provide fundamental insights into the surface phenomena of Pd–Au alloys, which may prove beneficial in the design of future Pd–Au catalysts.
Co-reporter:William D. Chemelewski, Oluwaniyi Mabayoje, Ding Tang, Alexander J. E. Rettie and C. Buddie Mullins
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 3) pp:NaN1648-1648
Publication Date(Web):2015/12/07
DOI:10.1039/C5CP05154A
Hematite is a promising material for photoelectrochemical (PEC) water splitting. While it has a low bandgap of ∼2.1 eV it is still larger than the optimal value of ∼1.8 eV. Previous work on epitaxial films has shown that Cr-doping leads to a shift of the bandgap as measured optically, but more importantly, also as measured by photoconductivity – to a value as low as 1.6 eV. We extend this work to polycrystalline films and attempt to use Cr-doping to lower the photon energy for which photocurrent can be generated. Our polycrystalline films show strong agreement with epitaxial films with regards to optical measurements of the direct and indirect bandgap. Furthermore, we find that Cr-doped polycrystalline films show photoconductivity at notably lower photon energies than undoped films, consistent with epitaxial results. However, when using Cr-doped films for photoelectrochemistry we find little to no shift of the photocurrent onset. We outline a number of proposals for why this could be the case, with a focus on the possibility of the existence of separate O 2p and Cr 3d states that would impact PEC but not PC behaviour.
Co-reporter:Yong-Mao Lin, Rajaram K. Nagarale, Kyle C. Klavetter, Adam Heller and C. Buddie Mullins
Journal of Materials Chemistry A 2012 - vol. 22(Issue 22) pp:NaN11139-11139
Publication Date(Web):2012/04/30
DOI:10.1039/C2JM16328D
Li-ion battery anodes made of SnO2 nanoparticles and a TiO2-supported SnO2 nanocomposite formed of equimolar amounts of the Sn and Ti oxides were investigated, respectively. By limiting the voltage window of the charge/discharge cycles to the range 50 mV–1.0 V, both the SnO2-based anode and the SnO2/TiO2-based anode show improved cycling stability. Compared to the SnO2 nanoparticle based anodes, the TiO2-support-SnO2 nanocomposite anodes exhibit better cyclability and higher Coulombic efficiency. During the first lithiation process, Li+ conducting LixTiO2 is formed in the SnO2/TiO2 composite, which structurally/mechanically supports the electrode. The anode made of amorphous TiO2-cassiterite SnO2 retained a reversible capacity of ∼500 mAh g−1 (based on the weight of SnO2) or ∼320 mAh g−1 (based on the weight of SnO2/TiO2) at 0.2 C after 100 cycles and at a rate as fast as 5 C retained a stable reversible capacity of ∼340 mAh g−1 (based on the weight of SnO2) or ∼220 mAh g−1 (based on the weight of SnO2/TiO2).
Co-reporter:Alexander J. E. Rettie, William D. Chemelewski, Bryan R. Wygant, Jeffrey Lindemuth, Jung-Fu Lin, David Eisenberg, Carolyn S. Brauer, Timothy J. Johnson, Toya N. Beiswenger, Richard D. Ash, Xiang Li, Jianshi Zhou and C. Buddie Mullins
Journal of Materials Chemistry A 2016 - vol. 4(Issue 3) pp:NaN567-567
Publication Date(Web):2015/12/11
DOI:10.1039/C5TC03368C
We report the synthesis of silicon-doped hematite (Si:α-Fe2O3) single crystals via chemical vapor transport, with Si incorporation on the order of 1019 cm−3. The conductivity, Seebeck and Hall effect were measured in the basal plane between 200 and 400 K. Distinct differences in electron transport were observed above and below the magnetic transition temperature of hematite at ∼265 K (the Morin transition, TM). Above 265 K, transport was found to agree with the adiabatic small-polaron model, the conductivity was characterized by an activation energy of ∼100 meV and the Hall effect was dominated by the weak ferromagnetism of the material. A room temperature electron drift mobility of ∼10−2 cm2 V−1 s−1 was estimated. Below TM, the activation energy increased to ∼160 meV and a conventional Hall coefficient could be determined. In this regime, the Hall coefficient was negative and the corresponding Hall mobility was temperature-independent with a value of ∼10−1 cm2 V−1 s−1. Seebeck coefficient measurements indicated that the silicon donors were fully ionized in the temperature range studied. Finally, we observed a broad infrared absorption upon doping and tentatively assign the feature at ∼0.8 eV to photon-assisted small-polaron hops. These results are discussed in the context of existing hematite transport studies.
Co-reporter:Huichao He, Sean P. Berglund, Peng Xiao, William D. Chemelewski, Yunhuai Zhang and C. Buddie Mullins
Journal of Materials Chemistry A 2013 - vol. 1(Issue 41) pp:NaN12834-12834
Publication Date(Web):2013/09/06
DOI:10.1039/C3TA13239K
To improve the photoelectrochemical activity of WO3, Bi2S3/WO3 heterojunction films were designed by coupling WO3 films with varying amounts of urchin-like Bi2S3 nanospheres. The WO3 films were composed of WO3 nanoprism arrays, which were synthesized using a solvothermal method. After coating a single layer of Bi2S3 on top of the WO3 film, the resulting Bi2S3/WO3 heterojunction film showed enhanced photoelectrochemical activity. At 1.2 V vs. Ag/AgCl, the initial photocurrent density of the Bi2S3/WO3 heterojunction film with one layer of Bi2S3 was 1.33 mA cm−2 in 0.1 M Na2SO4 and 1.19 mA cm−2 in a 0.2 M NaCl mixed water–ethanol solution, which was 40% and 32% higher than the bare WO3 film under the same conditions, respectively. The optimal number of Bi2S3 layers for coupling with the WO3 film was found to be 3 layers, which had the highest photocurrent density and IPCE values. The photoelectrochemical activity of Bi2S3/WO3 heterojunction film was not stable for water oxidation due to photocorrosion in aqueous electrolyte, but it was stable in the NaCl mixed water–ethanol solution and a non-aqueous solution containing iodide/triiodide as a redox couple. The origin of enhanced photoelectrochemical activity of the Bi2S3/WO3 heterojunction film was primarily ascribed to the band potential matching between WO3 and Bi2S3, which is advantageous for charge separation.
Co-reporter:Paul R. Abel, Kyle C. Klavetter, Karalee Jarvis, Adam Heller and C. Buddie Mullins
Journal of Materials Chemistry A 2014 - vol. 2(Issue 44) pp:NaN19018-19018
Publication Date(Web):2014/10/01
DOI:10.1039/C4TA04496G
Nanocolumnar, sub-stoichiometric germanium sulfide thin-films with compositions of Ge0.9S0.1 and Ge0.95S0.05, deposited by glancing angle deposition, were investigated as lithium storage materials. The materials are amorphous and homogeneous as deposited, but lithiation induces phase separation leading to the formation of poorly-crystallized lithium sulfide inclusions during the first cycle. The presence of these inclusions raises the lithium diffusion coefficient above that of pure germanium and provides superior capacity retention at high rates. While the lithium sulfide is non-cycling, the low weight percentage of sulfur necessary for enhanced lithiation/de-lithiation does not significantly reduce the specific lithium storage capacity of the films relative to that of germanium. In addition to high capacity and superior lithium transport, the sub-stoichiometric germanium sulfide thin-films show excellent cycling stability at high rates, retaining 88% of their initial capacity after 500 cycles at a rate of 20 C.
Co-reporter:Huichao He, Sean P. Berglund, Alexander J. E. Rettie, William D. Chemelewski, Peng Xiao, Yunhuai Zhang and C. Buddie Mullins
Journal of Materials Chemistry A 2014 - vol. 2(Issue 24) pp:NaN9379-9379
Publication Date(Web):2014/04/24
DOI:10.1039/C4TA00895B
Because of the potential for application in photoelectrochemical cells for water splitting, the synthesis of nanostructured BiVO4 is receiving increasing attention. Here we report a simple new drop-casting method for the first time to synthesize un-doped and doped bismuth vanadate (BiVO4) nanoflake array films. Synthesis parameters such as the amount of polyethylene glycol 600 (PEG-600) and the precursor solution drying time are investigated to optimize the films for photoelectrochemical water oxidation. The BiVO4 films consisting of nanoflakes with an average thickness of 20 nm and length of 2 μm were synthesized from a precursor solution containing Bi3+, V3+ and PEG-600 with a Bi:V: PEG-600 volume ratio of 2:2:1, dried at 135 °C for 55 min. Photoelectrochemical measurements show that the BiVO4 nanoflake array films have higher photoelectrochemical activity than the BiVO4 nanoparticle films. Additionally, the nanoflake arrays were tested after incorporating W and Mo to enhance the photoelectrochemical activity. The 2% W, 6% Mo co-doped BiVO4 nanoflake array films demonstrate the best photoelectrochemical activity with photocurrent densities about 2 times higher than the un-doped BiVO4 nanoflake films and greater than the photocurrents of individually Mo doped or W doped BiVO4 films. The origin of enhanced photoelectrochemical activity for the co-doped film may be due to the improved conductivity through the BiVO4 or slightly enhanced water oxidation kinetics.
Co-reporter:Sean M. Wood, Kyle C. Klavetter, Adam Heller and C. Buddie Mullins
Journal of Materials Chemistry A 2014 - vol. 2(Issue 20) pp:NaN7243-7243
Publication Date(Web):2014/03/26
DOI:10.1039/C4TA01167H
The reversible charging of a lead chalcogenide, PbTe, was studied for use as the anode material in a Li-ion cell and compared to PbO. A similar series of Li–Pb alloys were formed but with Li2Te present instead of Li2O. In the presence of Li2Te, rapid Li–Pb alloying and dealloying were observed in the potential range of 0.01–0.7 V. In the potential range of 0.8–2.5 V, Li2Te formed and decomposed reversibly. Electrodes were cycled stably for 100 cycles at a C/5 rate in both potential domains. The electrodes were also cycled stably at rates up to 10C. The presence of Li2Te reduced the overpotential required at higher charge and discharge rates by acting as a superionic conductor to improve lithium ion diffusion. These results recommend this material for potential use in low-power applications such as cell phones.
Co-reporter:William D. Chemelewski, Jacob R. Rosenstock and C. Buddie Mullins
Journal of Materials Chemistry A 2014 - vol. 2(Issue 36) pp:NaN14962-14962
Publication Date(Web):2014/07/16
DOI:10.1039/C4TA03078H
The oxygen evolution reaction (OER) is one important bottleneck in the development of economical photoelectrochemical (PEC) water splitting materials. To help address this we report the electrodeposition of Ni-doped FeOOH (Ni:FeOOH) as an OER electrocatalyst. The deposition method is applicable to a wide range of photoanodes and catalytic films as thin as a few nanometers can be easily grown. The Ni:FeOOH films with 5–20% Ni content reach 10 mA cm−2 in 0.1 M NaOH at an overpotential ranging from 420–460 mV initially, and improve with anodization at 10 mA cm−2 to below 400 mV. Deposition on triple junction solar cells results in a full PEC system with higher performance and a more cathodic peak power potential compared to undoped FeOOH electrocatalysts.
Co-reporter:Kyle C. Klavetter, Jonathan L. Snider, J. Pedro de Souza, Han Tu, Trevor H. Cell, Joon Hee Cho, Chistopher J. Ellison, Adam Heller and C. Buddie Mullins
Journal of Materials Chemistry A 2014 - vol. 2(Issue 35) pp:NaN14467-14467
Publication Date(Web):2014/07/16
DOI:10.1039/C4TA03201B
A free-standing electrode film composed of high tap density SnO2 particles and carboxymethyl cellulose binder with Super-P Li (SP-Li) conductive carbon was formed from an aqueous slurry cast by doctor-blading. Upon air-drying, the free-standing film spontaneously evolved via delamination from the substrate as the slurry solvent evaporated. The electrodes cut from the free-standing film were ∼5 μm thick with a SnO2 loading of ∼0.5 mg cm−2. The films were found to be easily handled, flexed and folded. For evaluation of the durability of the free-standing films, the tensile strength and elongation at break were measured: 13 MPa and 1.7%. The robustness of the electrically conductive network was measured with a four-point probe: the initial electrical resistivity of the film (0.6 Ω cm) was observed to increase by 6% after folding, applying pressure to the crease and unfolding. When tested in a coin cell, the electrode cycled stably with near 100% coulombic efficiency at up to 2 C and without capacity fade for 100 cycles at 1 C. To adjust the areal capacity of the cell, multiple free-standing films could be stacked. An electrode formed from several stacked films with an active material mass loading of greater than 4 mg cm−2 was found to cycle stably at 2.6 mA h cm−2 tested at 0.33 mA cm−2 current density. For evaluating cycling performance of the electrode while flexed, an electrode was placed in a once-folded pouch cell for testing at 1 C and cycled stably for 20 cycles before slight capacity fade was observed. For free-standing electrodes, 1D or 2D carbons such as carbon nanotubes (CNT) or graphene are commonly used to provide both mechanical strength and electrical conductivity. Here, CNTs were substituted for the SP-Li and similar free-standing films were made and compared. With CNT, the electrode strength at break as well as the electronic conductivity increased, but, despite this, the cycling performance of the electrodes made using the low-cost SP-Li carbon exceeds that of the electrodes made with orders-of-magnitude more expensive 1D carbon.
Co-reporter:Kyle. C. Klavetter, Stephany Garcia, Naween Dahal, Jonathan L. Snider, J. Pedro de Souza, Trevor H. Cell, Mark A. Cassara, Adam Heller, Simon M. Humphrey and C. Buddie Mullins
Journal of Materials Chemistry A 2014 - vol. 2(Issue 34) pp:NaN14221-14221
Publication Date(Web):2014/07/10
DOI:10.1039/C4TA02684E
High surface area (367 m2 g−1) meso-porous Co3O4 was investigated as the precursor of the anode material for lithium and also sodium ion batteries. Co3O4 is considered a potential anode material due to its theoretical capacity of 890 mA h g−1, over twice that of graphite. This comparatively higher capacity can be safely charged at rapid rates owing to a relatively high Li-insertion potentials, but, consequently, the discharged energy is yielded at an average potential near 2 V vs. Li/Li+, with full Li-extraction achieved over a continuum of potentials up to 3 V. The products of the lithium reduction of Co3O4 cycle stably from 0.01–3.0 V vs. Li/Li+ with 600–900 mA h g−1 capacity retention at C rates from 1–5; the products of its sodium reduction cycle stably from 0.01–3.0 V vs. Na/Na+ at C-rates up to 1 C with a lower 150–400 mA h g−1 capacity retention owing to greater ionic impedance. TEM, SAED and XRD were used to examine the cycled material and the stable performance is attributed to finding that the mesoporous structure is retained. Evaluation of five electrolyte formulations testing EC, FEC and Cl-EC showed that the stable meso-porous structure was best cycled with 5% FEC in EC:DEC at high charge/discharge rates, retaining 77% of its initial capacity at 5 C in a rate test. Comparison of the AC impedance spectra and of the XPS of the SEIs formed in the presence and in the absence of 5 vol% FEC shows that the SEI formed in the presence of FEC contains lithium fluoride and its carbonate layer is thinner than that formed in its absence, resulting in lesser impedance to Li migration through the SEI and facile ion de-solvation, improving the cycling performance. In cycling stability tests with EC:DEC, irregular cycling behaviour attributable to abrupt rises in cell resistance was regularly observed after testing over a few hundred cycles. Long-term cycling irregularities are inhibited by halogenated solvents and completely eliminated by adding fluoroethylene carbonate (FEC).
Co-reporter:Emily J. Powell, Sean M. Wood, Hugo Celio, Adam Heller and C. Buddie Mullins
Journal of Materials Chemistry A 2015 - vol. 3(Issue 46) pp:NaN23447-23447
Publication Date(Web):2015/10/19
DOI:10.1039/C5TA04941E
Mainstream rechargeable lithium battery materials research of the past 20 years has focused on nano-particulate materials, where Li+-diffusion lengths exceeded at designated cycling rates the particle radii, and where the particles slipped rather than broke upon their expansion and shrinkage in lithiation/delithiation cycles. Here we show that in intrinsically rapidly Li+-transporting macrocrystalline germanium and even more so in a dispersion of non-cycling Li2Te in macrocrystalline germanium it is unnecessary to use nanocrystalline materials and that Li2Te increases the retained capacity at 1C rate after 500 cycles. A dispersion of 17.65 atom% of crystalline GeTe in 82.35 atom% crystalline Ge was synthesized by quenching from the melt followed by high energy ball milling to 1 μm–5 μm particle size. The particles, as well as similarly made and similarly sized pure Ge particles were incorporated in electrodes, which were galvanostatically lithiated/delithiated. In the initial cycle, GeTe is reduced to LixGe alloys and Li2Te. In 500 1C cycles of LixGe delithiation/Ge lithiation the capacity of the pure Ge faded more rapidly than that of the Ge electrodes containing Li2Te, which retained 96% of their initial capacity after 500 cycles at 1C rate.
Co-reporter:Hoang X. Dang, Kyle C. Klavetter, Melissa L. Meyerson, Adam Heller and C. Buddie Mullins
Journal of Materials Chemistry A 2015 - vol. 3(Issue 25) pp:NaN13506-13506
Publication Date(Web):2015/05/27
DOI:10.1039/C5TA02131F
In a 100 cycle test at 0.5 C-rate a negative electrode formed of micro-sized Sn0.9Se0.1 particles retains a specific capacity of 500 mA h g−1 with a coulombic efficiency of 99.6%. In contrast, a control electrode made with pure Sn retains only a 200 mA h g−1 capacity with a 98.7% efficiency. The improvement in electrochemical performance of the Sn/Se alloy is attributed to the reduced inactive Se-phase preventing agglomeration of Sn to a size susceptible to particle fracture. The Sn/Se alloy particles are manufacturable, being made by melting the 9:1 atomic ratio mixture of Sn and Se, quenching and jet-milling.
Co-reporter:Kyle C. Klavetter, J. Pedro de Souza, Adam Heller and C. Buddie Mullins
Journal of Materials Chemistry A 2015 - vol. 3(Issue 11) pp:NaN5834-5834
Publication Date(Web):2015/02/09
DOI:10.1039/C5TA00319A
Slurry cast electrodes with μm-sized Ge0.9Se0.1 particles cycle stably at ∼800 mA h g−1 with ∼99.9% efficiency for 900 1C-rate cycles while electrodes with μm-size pure Ge particles lose 1/3rd of their capacity after five C/5 cycles. The difference is attributed to an inactive glassy Li–Se–Ge phase forming in the Ge active material of the Ge0.9Se0.1 particle.
Co-reporter:Ding Tang, Alexander J. E. Rettie, Oluwaniyi Mabayoje, Bryan R. Wygant, Yanqing Lai, Yexiang Liu and C. Buddie Mullins
Journal of Materials Chemistry A 2016 - vol. 4(Issue 8) pp:NaN3042-3042
Publication Date(Web):2015/11/02
DOI:10.1039/C5TA07877F
Porous n-type Fe2V4O13 films on FTO substrates were prepared by a simplified successive ion layer adsorption and reaction method and characterized as photoelectrodes for photoelectrochemical (PEC) water oxidation. Synthesis parameters such as film thickness and annealing temperatures and durations were investigated to optimize the PEC performance. A band gap of ∼2.3 eV and a flat band potential of 0.5 V vs. RHE make Fe2V4O13 a promising photoanode material. Water oxidation was kinetically limited at the surface of Fe2V4O13 film as confirmed by tests in electrolyte with a hole scavenger (Na2SO3). Improved PEC performance was achieved by Mo and W doping because of enhanced carrier densities. The best performance was obtained by 2.5% W-doped Fe2V4O13 films (actual 0.8% W-doped), which efficiently oxidize water to O2via photogenerated holes as confirmed by oxygen evolution measurements. Moreover, the Fe2V4O13 photoanode displayed very stable photocurrent under illumination. Due to the suitable band gap and valence band position, Fe2V4O13 is a promising photoanode for solar water splitting. Co-catalyst loading and doping optimization are identified as routes to improve this material's performance further.
Co-reporter:Ming Pan, Adrian J. Brush, Zachary D. Pozun, Hyung Chul Ham, Wen-Yueh Yu, Graeme Henkelman, Gyeong S. Hwang and C. Buddie Mullins
Chemical Society Reviews 2013 - vol. 42(Issue 12) pp:NaN5013-5013
Publication Date(Web):2013/02/27
DOI:10.1039/C3CS35523C
Supported gold nanoparticles have recently been shown to possess intriguing catalytic activity for hydrogenation reactions, particularly for selective hydrogenation reactions. However, fundamental studies that can provide insight into the reaction mechanisms responsible for this activity have been largely lacking. In this tutorial review, we highlight several recent model experiments and theoretical calculations on a well-structured gold surface that provide some insights. In addition to the behavior of hydrogen on a model gold surface, we review the reactivity of hydrogen on a model gold surface in regards to NO2 reduction, chemoselective CO bond hydrogenation, ether formation, and O–H bond dissociation in water and alcohols. Those studies indicate that atomic hydrogen has a weak interaction with gold surfaces which likely plays a key role in the unique hydrogenative chemistry of classical gold catalysts.
Co-reporter:Ming Pan, Son Hoang, Jinlong Gong and C. Buddie Mullins
Chemical Communications 2009(Issue 47) pp:NaN7302-7302
Publication Date(Web):2009/10/07
DOI:10.1039/B914308D
Although CO does not dissociate on the clean Ir(111) surface, the addition of atomic oxygen induces COdissociation at low temperatures (lower than 400 K); similarly, COdissociation has also been observed on water co-adsorbed Ir(111) or water and oxygen co-adsorbed Ir(111).
Co-reporter:Yong-Mao Lin, Kyle C. Klavetter, Paul R. Abel, Nicholas C. Davy, Jonathan L. Snider, Adam Heller and C. Buddie Mullins
Chemical Communications 2012 - vol. 48(Issue 58) pp:NaN7270-7270
Publication Date(Web):2012/05/30
DOI:10.1039/C2CC31712E
Electrodes composed of silicon nanoparticles (SiNP) were prepared by slurry casting and then electrochemically tested in a fluoroethylene carbonate (FEC)-based electrolyte. The capacity retention after cycling was significantly improved compared to electrodes cycled in a traditional ethylene carbonate (EC)-based electrolyte.
.jpg)















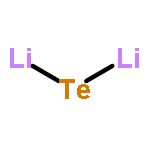
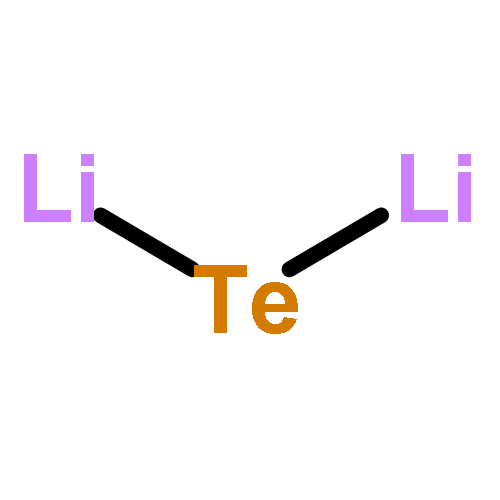





















![2-[4-(aminoiminomethyl)phenyl]-1H-Indole-6-carboximidamide](http://img.cochemist.com/ccimg/47200/47165-04-8.png)
![2-[4-(aminoiminomethyl)phenyl]-1H-Indole-6-carboximidamide](http://img.cochemist.com/ccimg/47200/47165-04-8_b.png)
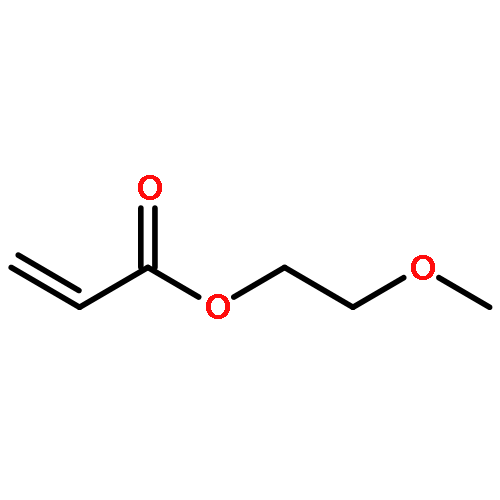


![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)