Co-reporter:Arun K. Ghosh, Margherita Brindisi, Prasanth R. Nyalapatla, Jun Takayama, Jean-Rene Ella-Menye, Sofiya Yashchuk, Johnson Agniswamy, Yuan-Fang Wang, Manabu Aoki, Masayuki Amano, Irene T. Weber, Hiroaki Mitsuya
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 19(Issue 19) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.bmc.2017.04.005
Based upon molecular insights from the X-ray structures of inhibitor-bound HIV-1 protease complexes, we have designed a series of isophthalamide-derived inhibitors incorporating substituted pyrrolidines, piperidines and thiazolidines as P2-P3 ligands for specific interactions in the S2-S3 extended site. Compound 4b has shown an enzyme Ki of 0.025 nM and antiviral IC50 of 69 nM. An X-ray crystal structure of inhibitor 4b-HIV-1 protease complex was determined at 1.33 Å resolution. We have also determined X-ray structure of 3b-bound HIV-1 protease at 1.27 Å resolution. These structures revealed important molecular insight into the inhibitor–HIV-1 protease interactions in the active site.Download high-res image (57KB)Download full-size image
Co-reporter:Arun K. Ghosh, Margherita Brindisi, Yu-Chen Yen, Emilio L. Cárdenas, Jean-Rene Ella-Menye, Nagaswamy Kumaragurubaran, Xiangping Huang, Jordan Tang, Andrew D. Mesecar
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 11(Issue 11) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.bmcl.2017.04.011
We report the design and synthesis of a series of BACE1 inhibitors incorporating mono- and bicyclic 6-substituted 2-oxopiperazines as novel P1′ and P2′ ligands and isophthalamide derivative as P2-P3 ligands. Among mono-substituted 2-oxopiperazines, inhibitor 5a with N-benzyl-2-oxopiperazine and isophthalamide showed potent BACE1 inhibitory activity (Ki = 2 nM). Inhibitor 5g, with N-benzyl-2-oxopiperazine and substituted indole-derived P2-ligand showed a reduction in potency. The X-ray crystal structure of 5g-bound BACE1 was determined and used to design a set of disubstituted 2-oxopiperazines and bicyclic derivatives that were subsequently investigated. Inhibitor 6j with an oxazolidinone derivative showed a BACE1 inhibitory activity of 23 nM and cellular EC50 of 80 nM.A series of BACE1 inhibitors incorporating mono- and bicyclic 2-oxopiperazines is described. Inhibitor 5a showed an enzyme Ki of 2 nM and a cellular EC50 value of 3.5 nM. An X-ray structural analysis of a related compound 5g-bound BACE1 provided insight into the ligand-binding site interactions.Download high-res image (83KB)Download full-size image
Co-reporter:Arun K. Ghosh, Anindya Sarkar
Tetrahedron Letters 2017 Volume 58, Issue 33(Issue 33) pp:
Publication Date(Web):16 August 2017
DOI:10.1016/j.tetlet.2017.07.010
•An enantioselective synthesis of hexahydro-4H-furo[2,3-b]pyran-4-ol is reported.•This ligand alcohol is a high-affinity ligand for potent HIV-1 protease inhibitors.•The key reaction is an efficient enzymatic desymmetrization of meso-diacetate.•Porcine pancreatic lipase mediated enzymatic reaction was done in 60 g scale.•The ligand alcohol was converted to a potent HIV-1 protease inhibitor.An enantioselective synthesis of (3aS,4S,7aR)-hexahydro-4H-furo[2,3-b]pyran-4-ol, a high-affinity nonpeptide ligand for a variety of potent HIV-1 protease inhibitors is described. The key steps involved a highly enantioselective enzymatic desymmetrization of meso-diacetate, an efficient transacetalization, and a highly diastereoselective reduction of a ketone. This route is amenable to large-scale synthesis using readily available starting materials.Download high-res image (53KB)Download full-size image
Co-reporter:Arun K. Ghosh, Emilio L. Cárdenas, Margherita Brindisi
Tetrahedron Letters 2017 Volume 58, Issue 43(Issue 43) pp:
Publication Date(Web):25 October 2017
DOI:10.1016/j.tetlet.2017.09.025
•Enantioselective syntheses of difluorinated aminoalkyl epoxides are reported.•The synthesis utilized Ti-enolate and boron-enolate asymmetric aldol reactions.•Aldol products were obtained with high diastereoselectivities and in good yields.•Curtius rearrangement of β-hydroxy acids installed the amine functionalities.•A fluorinated aminoalkyl oxirane was converted to a potent BACE1 inhibitor.Enantioselective syntheses of tert-butyl ((S)-2-(3,5-difluorophenyl)-1-((S)-oxiran-2-yl)ethyl)carbamate and ((S)-2-(3,5-difluorophenyl)-1-((R)-oxiran-2-yl)ethyl)carbamate are described. We utilized asymmetric syn- and anti-aldol reactions to set both stereogenic centers. We investigated ester-derived Ti-enolate aldol reactions as well as Evans’ diastereoselective syn-aldol reaction for these syntheses. We have converted optically active ((S)-2-(3,5-difluorophenyl)-1-((S)-oxiran-2-yl)ethyl)carbamate to a potent β-secretase inhibitor.Download high-res image (116KB)Download full-size image
Co-reporter:Arun K. Ghosh, W. Sean Fyvie, Margherita Brindisi, Melinda Steffey, Johnson Agniswamy, Yuan-Fang Wang, Manabu Aoki, Masayuki Amano, Irene T. Weber, Hiroaki Mitsuya
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 21(Issue 21) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.bmcl.2017.09.003
Design, synthesis, and evaluation of a new class of HIV-1 protease inhibitors containing diverse flexible macrocyclic P1′-P2′ tethers are reported. Inhibitor 5a with a pyrrolidinone-derived macrocycle exhibited favorable enzyme inhibitory and antiviral activity (Ki = 13.2 nM, IC50 = 22 nM). Further incorporation of heteroatoms in the macrocyclic skeleton provided macrocyclic inhibitors 5m and 5o. These compounds showed excellent HIV-1 protease inhibitory (Ki = 62 pM and 14 pM, respectively) and antiviral activity (IC50 = 5.3 nM and 2.0 nM, respectively). Inhibitor 5o also remained highly potent against a DRV-resistant HIV-1 variant.Download high-res image (81KB)Download full-size image
Co-reporter:Arun K. Ghosh, Bhavanam Sekhara Reddy, Yu-Chen Yen, Emilio L. Cárdenas, Kalapala Venkateswara Rao, Deborah Downs, Xiangping Huang, Jordan Tang and Andrew D. Mesecar
Chemical Science 2016 vol. 7(Issue 5) pp:3117-3122
Publication Date(Web):04 Feb 2016
DOI:10.1039/C5SC03718B
Design, synthesis and evaluation of very potent and selective β-secretase 2 (memapsin 1, BACE 2) inhibitors are described. The inhibitors were designed specifically to interact with the S2′-site of β-secretase 2 to provide >170000-fold selectivity over β-secretase (BACE 1) and >15000-fold selectivity over cathepsin D. BACE 2 is implicated in type 2 diabetes. The studies serve as an important guide to selective BACE 2 inhibitors.
Co-reporter:Arun K. Ghosh and Luke A. Kassekert
Organic Letters 2016 Volume 18(Issue 13) pp:3274-3277
Publication Date(Web):June 22, 2016
DOI:10.1021/acs.orglett.6b01523
Both epimers at C-21 in the proposed structure of (+)-callyspongiolide have been synthesized in a convergent and enantioselective manner. The 14-membered macrolide with a sensitive C2–C3 cis-olefin functionality was installed by a Yamaguchi macrolactonization of hydroxyl alkynoic acid followed by hydrogenation over Lindlar’s catalyst. The C5 methyl stereocenter was constructed by a ring-closing olefin metathesis followed by addition of methyl cuprate to an α,β-unsaturated δ-lactone. Other key reactions are chiral Corey–Bakshi–Shibata (CBS) reduction and Sonogashira coupling to conjoin the macrocyclic core and side chain.
Co-reporter:Arun K. Ghosh and Prasanth R. Nyalapatla
Organic Letters 2016 Volume 18(Issue 9) pp:2296-2299
Publication Date(Web):April 26, 2016
DOI:10.1021/acs.orglett.6b00942
An enantioselective total synthesis of (+)-amphirionin-4 has been accomplished in a convergent manner. The synthesis features an efficient enzymatic lipase resolution to access the tetrahydrofuranol core in optically active form. The functionalized tetrahydrofuran derivative was synthesized via an oxocarbenium ion-mediated highly diastereoselective syn-allylation reaction. The polyene side chain was synthesized using Stille coupling reactions. Nozaki–Hiyama–Kishi coupling was utilized to construct the C-8 stereocenter and complete the synthesis of (+)-amphirionin-4.
Co-reporter:Arun K. Ghosh, Anthony J. Tomaine, and Kelsey E. Cantwell
Organic Letters 2016 Volume 18(Issue 3) pp:396-399
Publication Date(Web):January 27, 2016
DOI:10.1021/acs.orglett.5b03411
Substituted oxocene derivatives have been synthesized by Lewis acid catalyzed reactions of ε-hydroxyalkene and substituted aromatic aldehydes. The Cu(OTf)2-bis-phosphine catalyzed reaction typically provides substituted dihydropyran derivatives through an olefin migration followed by a Prins cyclization. The corresponding reaction catalyzed by TMSOTf or BF3·OEt2 provided eight-membered cyclic ethers (oxocenes), selectively. This methodology provides convenient access to a variety of 2,4,8-trisubstituted oxocenes in good yields and excellent diastereoselectivities.
Co-reporter:Arun K. Ghosh; Heather L. Osswald;Gary Prato
Journal of Medicinal Chemistry 2016 Volume 59(Issue 11) pp:5172-5208
Publication Date(Web):January 22, 2016
DOI:10.1021/acs.jmedchem.5b01697
HIV-1 protease inhibitors continue to play an important role in the treatment of HIV/AIDS, transforming this deadly ailment into a more manageable chronic infection. Over the years, intensive research has led to a variety of approved protease inhibitors for the treatment of HIV/AIDS. In this review, we outline current drug design and medicinal chemistry efforts toward the development of next-generation protease inhibitors beyond the currently approved drugs.
Co-reporter:Arun K. Ghosh; Heather L. Osswald; Kristof Glauninger; Johnson Agniswamy; Yuan-Fang Wang; Hironori Hayashi; Manabu Aoki; Irene T. Weber;Hiroaki Mitsuya
Journal of Medicinal Chemistry 2016 Volume 59(Issue 14) pp:6826-6837
Publication Date(Web):July 7, 2016
DOI:10.1021/acs.jmedchem.6b00639
A series of potent HIV-1 protease inhibitors with a lipophilic adamantyl P1 ligand have been designed, synthesized, and evaluated. We have developed an enantioselective synthesis of adamantane-derived hydroxyethylamine isosteres utilizing Sharpless asymmetric epoxidation as the key step. Various inhibitors incorporating P1-adamantylmethyl in combination with P2 ligands such as 3-(R)-THF, 3-(S)-THF, bis-THF, and THF-THP were examined. The S1′ pocket was also probed with phenyl and phenylmethyl ligands. Inhibitor 15d, with an isobutyl P1′ ligand and a bis-THF P2 ligand, proved to be the most potent of the series. The cLogP value of inhibitor 15d is improved compared to inhibitor 2 with a phenylmethyl P1-ligand. X-ray structural studies of 15d, 15h, and 15i with HIV-1 protease complexes revealed molecular insight into the inhibitor–protein interaction.
Co-reporter:Arun K. Ghosh, Kai Lv, Nianchun Ma, Emilio L. Cárdenas, Kerstin A. Effenberger and Melissa S. Jurica
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 23) pp:5263-5271
Publication Date(Web):18 May 2016
DOI:10.1039/C6OB00725B
Herboxidiene is a potent inhibitor of spliceosomes. It exhibits excellent anticancer activity against multiple human cancer cell lines. Herein, we describe an enantioselective synthesis of a desmethyl derivative and the corresponding carba-derivatives of herboxidiene. The synthesis involved Suzuki coupling of a vinyl iodide with boronate as the key reaction. For the synthesis of carba-derivatives, the corresponding optically active cyclohexane-1,3-dicarbonyl derivatives were synthesized using an enantioselective desymmetrization of meso-anhydride. The biological properties of these derivatives were evaluated in an in vitro splicing assay.
Co-reporter:Arun K. Ghosh, Samuel Rodriguez
Tetrahedron Letters 2016 Volume 57(Issue 26) pp:2884-2887
Publication Date(Web):29 June 2016
DOI:10.1016/j.tetlet.2016.05.067
•Synthesis of the C3–C21 segment of the immunosuppressant FR252921 is achieved.•The C12 and C13 chiral centers were set by a diastereoselective non-aldol process.•Non-aldol route includes, alkylation, oxa-Michael reaction, and ring opening.•The C18 stereogenic center was installed using Braun’s acetate aldol reaction.•Chiral reduction of β-keto-ester with NaBH4 and tartaric acid was investigated.An enantioselective synthesis of the C3–C21 segment of the novel immunosuppressant FR252921 is described. The C12 and C13 stereogenic centers were constructed by a non-aldol process utilizing optically active 4-phenylbutyrolactone. The C18 stereogenic center was installed using Braun’s highly diastereoselective acetate aldol reaction. Other key steps involved Curtius rearrangement and Horner–Wadsworth–Emmons olefination reactions.
Co-reporter:Arun K. Ghosh; Xufen Yu; Heather L. Osswald; Johnson Agniswamy; Yuan-Fang Wang; Masayuki Amano; Irene T. Weber;Hiroaki Mitsuya
Journal of Medicinal Chemistry 2015 Volume 58(Issue 13) pp:5334-5343
Publication Date(Web):June 24, 2015
DOI:10.1021/acs.jmedchem.5b00676
We report the design, synthesis, X-ray structural studies, and biological evaluation of a novel series of HIV-1 protease inhibitors. We designed a variety of functionalized biphenyl derivatives to make enhanced van der Waals interactions in the S1 subsite of HIV-1 protease. These biphenyl derivatives were conveniently synthesized using a Suzuki–Miyaura cross-coupling reaction as the key step. We examined the potential of these functionalized biphenyl-derived P1 ligands in combination with 3-(S)-tetrahydrofuranyl urethane and bis-tetrahydrofuranyl urethane as the P2 ligands. Inhibitor 21e, with a 2-methoxy-1,1′-biphenyl derivative as P1 ligand and bis-THF as the P2 ligand, displayed the most potent enzyme inhibitory and antiviral activity. This inhibitor also exhibited potent activity against a panel of multidrug-resistant HIV-1 variants. A high resolution X-ray crystal structure of related Boc-derivative 17a-bound HIV-1 protease provided important molecular insight into the ligand-binding site interactions of the biphenyl core in the S1 subsite of HIV-1 protease.
Co-reporter:Arun K. Ghosh; Cuthbert D. Martyr; Heather L. Osswald; Venkat Reddy Sheri; Luke A. Kassekert; Shujing Chen; Johnson Agniswamy; Yuan-Fang Wang; Hironori Hayashi; Manabu Aoki; Irene T. Weber;Hiroaki Mitsuya
Journal of Medicinal Chemistry 2015 Volume 58(Issue 17) pp:6994-7006
Publication Date(Web):August 25, 2015
DOI:10.1021/acs.jmedchem.5b00900
Structure-based design, synthesis, and biological evaluation of a series of very potent HIV-1 protease inhibitors are described. In an effort to improve backbone ligand–binding site interactions, we have incorporated basic-amines at the C4 position of the bis-tetrahydrofuran (bis-THF) ring. We speculated that these substituents would make hydrogen bonding interactions in the flap region of HIV-1 protease. Synthesis of these inhibitors was performed diastereoselectively. A number of inhibitors displayed very potent enzyme inhibitory and antiviral activity. Inhibitors 25f, 25i, and 25j were evaluated against a number of highly-PI-resistant HIV-1 strains, and they exhibited improved antiviral activity over darunavir. Two high resolution X-ray structures of 25f- and 25g-bound HIV-1 protease revealed unique hydrogen bonding interactions with the backbone carbonyl group of Gly48 as well as with the backbone NH of Gly48 in the flap region of the enzyme active site. These ligand–binding site interactions are possibly responsible for their potent activity.
Co-reporter:Arun K. Ghosh;Margherita Brindisi
Journal of Medicinal Chemistry 2015 Volume 58(Issue 7) pp:2895-2940
Publication Date(Web):January 7, 2015
DOI:10.1021/jm501371s
The carbamate group is a key structural motif in many approved drugs and prodrugs. There is an increasing use of carbamates in medicinal chemistry and many derivatives are specifically designed to make drug–target interactions through their carbamate moiety. In this Perspective, we present properties and stabilities of carbamates, reagents and chemical methodologies for the synthesis of carbamates, and recent applications of carbamates in drug design and medicinal chemistry.
Co-reporter:Arun K. Ghosh, Cuthbert D. Martyr, Luke A. Kassekert, Prasanth R. Nyalapatla, Melinda Steffey, Johnson Agniswamy, Yuan-Fang Wang, Irene T. Weber, Masayuki Amano and Hiroaki Mitsuya
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 48) pp:11607-11621
Publication Date(Web):14 Oct 2015
DOI:10.1039/C5OB01930C
Design, synthesis, biological and X-ray crystallographic studies of a series of potent HIV-1 protease inhibitors are described. Various polar functionalities have been incorporated on the tetrahydropyranyl-tetrahydrofuran-derived P2 ligand to interact with the backbone atoms in the S2-subsite. The majority of the inhibitors showed very potent enzyme inhibitory and antiviral activity. Two high-resolution X-ray structures of 30b- and 30j-bound HIV-1 protease provide insight into ligand-binding site interactions. In particular, the polar functionalities on the P2-ligand appear to form unique hydrogen bonds with Gly48 amide NH and amide carbonyl groups in the flap region.
Co-reporter:Arun K. Ghosh, Chun-Xiao Xu, Heather L. Osswald
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:3314-3317
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2015.01.019
Synthesis of novel HIV-1 protease inhibitors incorporating dioxatriquinane-derived P2-ligands is described. The tricyclic ligand alcohol contains five contiguous chiral centers. The ligand alcohols were prepared in optically active form by an enzymatic asymmetrization of mesodiacetate, cascade radical cyclization, and Lewis acid catalyzed reduction as the key steps. Inhibitors with dioxatriquinane-derived P2-ligands exhibited low nanomolar HIV-1 protease activity.
Co-reporter:Arun K. Ghosh, Jun Takayama, Luke A. Kassekert, Jean-Rene Ella-Menye, Sofiya Yashchuk, Johnson Agniswamy, Yuan-Fang Wang, Manabu Aoki, Masayuki Amano, Irene T. Weber, Hiroaki Mitsuya
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 21) pp:4903-4909
Publication Date(Web):1 November 2015
DOI:10.1016/j.bmcl.2015.05.052
We describe the design, synthesis and biological evaluation of a series of novel HIV-1 protease inhibitors bearing isophthalamide derivatives as the P2–P3 ligands. We have investigated a range of acyclic and heterocyclic amides as the extended P2–P3 ligands. These inhibitors displayed good to excellent HIV-1 protease inhibitory activity. Also, a number of inhibitors showed very good antiviral activity in MT cells. Compound 5n has shown an enzyme Ki of 0.17 nM and antiviral IC50 of 14 nM. An X-ray crystal structure of inhibitor 5o-bound to HIV-1 protease was determined at 1.11 Å resolution. This structure revealed important molecular insight into the inhibitor–HIV-1 protease interactions in the active site.
Co-reporter:Dr. Arun K. Ghosh;Sofiya Yashchuk;Akira Mizuno;Nilanjana Chakraborty;Johnson Agniswamy;Yuan-Fang Wang;Manabu Aoki;Pedro Miguel Salcedo Gomez;Masayuki Amano; Irene T. Weber;Dr. Hiroaki Mitsuya
ChemMedChem 2015 Volume 10( Issue 1) pp:107-115
Publication Date(Web):
DOI:10.1002/cmdc.201402358
Abstract
The structure-based design, synthesis, biological evaluation, and X-ray structural studies of fluorine-containing HIV-1 protease inhibitors are described. The synthesis of both enantiomers of the gem-difluoro-bis-THF ligands was carried out in a stereoselective manner using a Reformatskii–Claisen reaction as the key step. Optically active ligands were converted into protease inhibitors. Two of these inhibitors, (3R,3aS,6aS)-4,4-difluorohexahydrofuro[2,3-b]furan-3-yl(2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl) carbamate (3) and (3R,3aS,6aS)-4,4-difluorohexahydrofuro[2,3-b]furan-3-yl(2S,3R)-3-hydroxy-4-((N-isobutyl-4-aminophenyl)sulfonamido)phenylbutan-2-yl) carbamate (4), exhibited HIV-1 protease inhibitory Ki values in the picomolar range. Both 3 and 4 showed very potent antiviral activity, with respective EC50 values of 0.8 and 3.1 nM against the laboratory strain HIV-1LAI. The two inhibitors exhibited better lipophilicity profiles than darunavir, and also showed much improved blood–brain barrier permeability in an in vitro model. A high-resolution X-ray structure of inhibitor 4 in complex with HIV-1 protease was determined, revealing that the fluorinated ligand makes extensive interactions with the S2 subsite of HIV-1 protease, including hydrogen bonding interactions with the protease backbone atoms. Moreover, both fluorine atoms on the bis-THF ligand formed strong interactions with the flap Gly 48 carbonyl oxygen atom.
Co-reporter:Dr. Arun K. Ghosh;Dr. Jordan Tang
ChemMedChem 2015 Volume 10( Issue 9) pp:1463-1466
Publication Date(Web):
DOI:10.1002/cmdc.201500216
Abstract
β-Secretase continues to be an attractive drug discovery target for the therapeutic intervention of Alzheimer’s disease (AD). This enzyme plays a critical role in the production of neurotoxic β-amyloid (Aβ) peptides in the brain. Over the years, extensive research efforts have led to the development of many promising classes of inhibitors against this protease. Many small-molecule, peptidomimetic, and nonpeptide β-secretase inhibitors have now overcome the key challenging development hurdles such as selectivity and brain penetration. A number of inhibitors have also shown further promise in reducing brain Aβ and rescuing cognitive decline in animal models. Recently, several β-secretase inhibitors have entered into preclinical and phase I studies, and at least one of these inhibitors has advanced to phase II/III human trials. The outlook on β-secretase inhibitor drugs for the treatment of AD patients is discussed herein.
Co-reporter:Arun K. Ghosh, Nianchun Ma, Kerstin A. Effenberger, and Melissa S. Jurica
Organic Letters 2014 Volume 16(Issue 11) pp:3154-3157
Publication Date(Web):May 28, 2014
DOI:10.1021/ol501345d
An enantioselective total synthesis of GEX1Q1 has been accomplished in a convergent manner. The C-5 asymmetric center has now been assigned through synthesis. GEX1Q1 displayed slightly better spliceosome inhibitory activity over its C-5 epimer. The salient features of this synthesis include an asymmetric hetero-Diels–Alder reaction to construct the tetrahydropyran ring and a Suzuki cross-coupling to assemble the key segments.
Co-reporter:Arun K. Ghosh, Anne M. Veitschegger, Venkata Reddy Sheri, Kerstin A. Effenberger, Beth E. Prichard, and Melissa S. Jurica
Organic Letters 2014 Volume 16(Issue 23) pp:6200-6203
Publication Date(Web):November 25, 2014
DOI:10.1021/ol503127r
An enantioselective total synthesis of spliceostatin E has been accomplished. The δ-lactone unit A was constructed from readily available (R)-glycidyl alcohol using a ring-closing olefin metathesis as the key reaction. A cross-metathesis of ring A containing δ-lactone and the functionalized tetrahydropyran B-ring provided spliceostatin E. Our biological evaluation of synthetic spliceostatin E revealed that it does not inhibit splicing in vitro and does not impact speckle morphology in cells. Spliceostatin E was reported to possess potent antitumor activity.
Co-reporter:Arun K. Ghosh, Gary E. Schiltz, Linah N. Rusere, Heather L. Osswald, D. Eric Walters, Masayuki Amano and Hiroaki Mitsuya
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 35) pp:6842-6854
Publication Date(Web):22 Jul 2014
DOI:10.1039/C4OB00738G
A series of potent macrocyclic HIV-1 protease inhibitors have been designed and synthesized. The compounds incorporated 16- to 19-membered macrocyclic rings between a nelfinavir-like P2 ligand and a tyrosine side chain containing a hydroxyethylamine sulfonamide isostere. All cyclic inhibitors are more potent than their corresponding acyclic counterparts. Saturated derivatives showed slight reduction of potency compared to the respective unsaturated derivatives. Compound 8a containing a 16-membered ring as the P1–P2 ligand showed the most potent enzyme inhibitory and antiviral activity.
Co-reporter:Arun K. Ghosh and Zhi-Hua Chen
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 22) pp:3567-3571
Publication Date(Web):10 Apr 2014
DOI:10.1039/C4OB00511B
Efficient intramolecular N/O-nucleophilic cyclization of 2-aryl indoles has been developed to afford the corresponding 2-aza-3-oxaindolines and 3-indolinones in 80–95% yield. The methods provided convenient access to fused imidazo[1,2-c]oxazolidinone, oxazolidine, or tetrahydro-1,3-oxazine cores under mild conditions.
Co-reporter:Arun K. Ghosh;Kai Lv
European Journal of Organic Chemistry 2014 Volume 2014( Issue 30) pp:6761-6768
Publication Date(Web):
DOI:10.1002/ejoc.201402812
Abstract
A convergent synthesis of carbocyclic sinefungin (2), its C-5 epimer 3a, and adenine-modified derivative 3b is described. The key features of our approach include the use of commercially available L-methionine and readily available (1R,4S)-4-hydroxy-2-cyclopentenyl acetate as starting materials, a cross-metathesis reaction, an enzymatic kinetic resolution, and a Staudinger reduction. The current synthesis is flexible and, therefore, provides convenient access to the synthesis of various carbocyclic sinefungin analogues for biological evaluation.
Co-reporter:Arun K. Ghosh, Chad Keyes, Anne M. Veitschegger
Tetrahedron Letters 2014 Volume 55(Issue 30) pp:4251-4254
Publication Date(Web):23 July 2014
DOI:10.1016/j.tetlet.2014.05.092
Catalytic FeCl3 in the presence of 4 Å molecular sieves has been shown to effect highly diastereoselective tandem Prins and Friedel–Crafts cyclization of substituted (E/Z)-6-phenylhex-3-en-1-ol and a variety of aldehydes to provide a range of polycyclic compounds in good to excellent yields. The reaction of an enantioenriched alcohol with an aldehyde provided the cyclization product without loss of optical activity. Furthermore, a Lewis acid catalyzed ring opening resulted in functionalized tetralin derivatives with multiple chiral centers.
Co-reporter:Arun K. Ghosh, Kalapala Venkateswara Rao, Siddhartha Akasapu
Tetrahedron Letters 2014 Volume 55(Issue 37) pp:5191-5194
Publication Date(Web):10 September 2014
DOI:10.1016/j.tetlet.2014.07.077
Aetheramides A and B are very potent anti-HIV agents. An enantioselective synthesis of a MEM-protected aetheramide A derivative is described. The synthesis was accomplished in a convergent and stereoselective manner. The key reactions involved asymmetric dihydroxylation, asymmetric allylation, asymmetric syn-aldol reactions, and asymmetric hydrogenation.
Co-reporter:Arun K. Ghosh, Zhi-Hua Chen, Kerstin A. Effenberger, and Melissa S. Jurica
The Journal of Organic Chemistry 2014 Volume 79(Issue 12) pp:5697-5709
Publication Date(Web):May 30, 2014
DOI:10.1021/jo500800k
FR901464 (1) and spliceostatin A (2) are potent inhibitors of spliceosomes. These compounds have shown remarkable anticancer activity against multiple human cancer cell lines. Herein, we describe efficient, enantioselective syntheses of FR901464, spliceostatin A, six corresponding diastereomers and an evaluation of their splicing activity. Syntheses of spliceostatin A and FR901464 were carried out in the longest linear sequence of 9 and 10 steps, respectively. To construct the highly functionalized tetrahydropyran A-ring, we utilized CBS reduction, Achmatowicz rearrangement, Michael addition, and reductive amination as key steps. The remarkable diastereoselectivity of the Michael addition was specifically demonstrated with different substrates under various reaction conditions. The side chain B was prepared from an optically active alcohol, followed by acetylation and hydrogenation over Lindlar’s catalyst. The other densely functionalized tetrahydropyran C-ring was derived from readily available (R)-isopropylidene glyceraldehyde through a route featuring 1,2-addition, cyclic ketalization, and regioselective epoxidation. These fragments were coupled together at a late stage through amidation and cross-metathesis in a convergent manner. Six key diastereomers were then synthesized to probe the importance of specific stereochemical features of FR901464 and spliceostatin A, with respect to their in vitro splicing activity.
Co-reporter:Arun K. Ghosh and Zhi-Hua Chen
Organic Letters 2013 Volume 15(Issue 19) pp:5088-5091
Publication Date(Web):September 19, 2013
DOI:10.1021/ol4024634
Enantioselective syntheses of FR901464 and spliceostatin A, potent spliceosome inhibitors, are described. The synthesis of FR901464 has been accomplished in a convergent manner in 10 linear steps (20 total steps). The A-tetrahydropyran ring was constructed from (R)-isopropylidene glyceraldehyde. The functionalized tetrahydropyran B-ring was synthesized utilizing a Corey–Bakshi–Shibata reduction, an Achmatowicz reaction, and a stereoselective Michael addition as the key steps. Coupling of A- and B-ring fragments was accomplished via cross-metathesis.
Co-reporter:Arun K. Ghosh ; Garth L. Parham ; Cuthbert D. Martyr ; Prasanth R. Nyalapatla ; Heather L. Osswald ; Johnson Agniswamy ; Yuan-Fang Wang ; Masayuki Amano ; Irene T. Weber ;Hiroaki Mitsuya
Journal of Medicinal Chemistry 2013 Volume 56(Issue 17) pp:6792-6802
Publication Date(Web):August 15, 2013
DOI:10.1021/jm400768f
The design, synthesis, and biological evaluation of a series of HIV-1 protease inhibitors incorporating stereochemically defined fused tricyclic P2 ligands are described. Various substituent effects were investigated to maximize the ligand-binding site interactions in the protease active site. Inhibitors 16a and 16f showed excellent enzyme inhibitory and antiviral activity, although the incorporation of sulfone functionality resulted in a decrease in potency. Both inhibitors 16a and 16f maintained activity against a panel of multidrug resistant HIV-1 variants. A high-resolution X-ray crystal structure of 16a-bound HIV-1 protease revealed important molecular insights into the ligand-binding site interactions, which may account for the inhibitor’s potent antiviral activity and excellent resistance profiles.
Co-reporter:Arun K. Ghosh, Khriesto A. Shurrush and Zachary L. Dawson
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 44) pp:7768-7777
Publication Date(Web):11 Oct 2013
DOI:10.1039/C3OB41541D
The asymmetric total synthesis of the anti-proliferative macrolide (+)-neopeltolide has been completed. The stereochemically defined trisubstituted tetrahydropyran ring was constructed via a catalytic hetero-Diels–Alder reaction creating two new chiral centers in a highly diastereoselective manner. The other key features of this synthesis included Brown's asymmetric allylation to install the requisite C-11 and C-13 stereocenters. The synthesis of the oxazole side chain consisted of a hydrozirconation of an alkynyl stannane to establish the Z stereochemistry, followed by a palladium catalyzed cross coupling to introduce the desired Z olefin in the oxazole side chain.
Co-reporter:Arun K. Ghosh, Bing Zhou
Tetrahedron Letters 2013 Volume 54(Issue 27) pp:3500-3502
Publication Date(Web):3 July 2013
DOI:10.1016/j.tetlet.2013.04.080
An enantioselective aza-Michael addition of indolines to α,β-unsaturated ketones was achieved using a bifunctional cinchona alkaloid-derived chiral squaramide derivative. Various β-indolinyl ketone derivatives were obtained in good to excellent yields and with high enantioselectivity. DDQ or MnO2 oxidation of indoline derivatives provided convenient access to various enantioenriched N-substituted indole derivatives.
Co-reporter:Arun K. Ghosh, Bing Zhou
Tetrahedron Letters 2013 Volume 54(Issue 19) pp:2311-2314
Publication Date(Web):8 May 2013
DOI:10.1016/j.tetlet.2013.02.030
Organocatalytic reactions of 3-olefinic oxindoles and pentane-1,5-dial were investigated to provide access to substituted spirocyclohexane oxindoles via Michael/aldol cascade reactions. Of particular interest, we have examined the stereochemical outcome of electron withdrawing and electron-donating groups on the oxindole ring nitrogen. Interestingly, we have observed that the N-protecting group on the oxindole has critical effect on aldol ring closure leading to ultimate stereochemical outcome of the hydroxyl center. The overall process is quite efficient and afforded products with multiple stereocenters in high yields and excellent enantioselectivities (>99% ee).
Co-reporter:Arun K. Ghosh and Jorden Kass
Organic Letters 2012 Volume 14(Issue 2) pp:510-512
Publication Date(Web):December 23, 2011
DOI:10.1021/ol203093g
A stereoselective synthesis of (−)-viridiofungin A is described. The convergent synthesis utilized a unique highly diastereoselective multicomponent reaction between optically active phenyldihydrofuran and an α-ketoester to provide two chiral centers including a quarternary carbon center in a single step. Other key steps include an acyloxycarbonium ion-mediated tetrahydrofuran ring-opening reaction and a Julia–Kocienski olefination.
Co-reporter:Arun K. Ghosh, Cuthbert D. Martyr, and Chun-Xiao Xu
Organic Letters 2012 Volume 14(Issue 8) pp:2002-2005
Publication Date(Web):April 4, 2012
DOI:10.1021/ol300494q
A new and convenient synthesis of benzo-fused 8-oxabicyclo[3.2.1]octane and 9-oxabicyclo[4.2.1]nonane derivatives are described. The reaction involved a TiCl4-mediated tandem carbonyl or imine addition followed by a Friedel–Crafts cyclization to provide these functionalized derivatives in good to excellent yields and high diastereoselectivity.
Co-reporter:Arun K. Ghosh and David D. Anderson
Organic Letters 2012 Volume 14(Issue 18) pp:4730-4733
Publication Date(Web):September 6, 2012
DOI:10.1021/ol301886g
An enantioselective and convergent total synthesis of pladienolide B (1) is described. Pladienolide B binds to the SF3b complex of a spliceosome and inhibits mRNA splicing activity. The synthesis features an epoxide opening reaction, an asymmetric reduction of a β-keto ester, and a cross metathesis strategy for the side chain synthesis.
Co-reporter:Arun K. Ghosh, Xu Cheng, and Bing Zhou
Organic Letters 2012 Volume 14(Issue 19) pp:5046-5049
Publication Date(Web):September 14, 2012
DOI:10.1021/ol302273r
An enantioselective synthesis of (+)-lithospermic acid, a potent anti-HIV agent, has been accomplished in a convergent manner in nine steps. The synthesis features an enantioselective intramolecular oxa-Michael addition catalyzed by a quinidine derivative, a hypervalent iodine-mediated rearrangement of chromanone to dihydrobenzofuran, an enantioselective α-oxyamination, and an intermolecular C–H olefination.
Co-reporter:Arun K. Ghosh ; Kalapala Venkateswara Rao ; Navnath D. Yadav ; David D. Anderson ; Navnath Gavande ; Xiangping Huang ; Simon Terzyan ;Jordan Tang
Journal of Medicinal Chemistry 2012 Volume 55(Issue 21) pp:9195-9207
Publication Date(Web):September 6, 2012
DOI:10.1021/jm3008823
The structure-based design, synthesis, and X-ray structure of protein–ligand complexes of exceptionally potent and selective β-secretase inhibitors are described. The inhibitors are designed specifically to interact with S1′ active site residues to provide selectivity over memapsin 1 and cathepsin D. Inhibitor 5 has exhibited exceedingly potent inhibitory activity (Ki = 17 pM) and high selectivity over BACE 2 (>7000-fold) and cathepsin D (>250000-fold). A protein–ligand crystal structure revealed important molecular insight into these selectivities. These interactions may serve as an important guide to design selectivity over the physiologically important aspartic acid proteases.
Co-reporter:Arun K. Ghosh;Xu Cheng;Ruoli Bai;Ernest Hamel
European Journal of Organic Chemistry 2012 Volume 2012( Issue 22) pp:4130-4139
Publication Date(Web):
DOI:10.1002/ejoc.201200286
Abstract
A detailed account of the enantioselective total synthesis of (–)-zampanolide, a macrolide marine natural product with high anticancer activity, is described. For the synthesis of the 4-methylenetetrahydropyran unit of (–)-zampanolide, we initially relied upon an oxidative C–H activation of an alkenyl ether and intramolecular cyclization to provide the substituted tetrahydropyran ring. However, this strategy was unsuccessful. Subsequently, we found that a cinnamyl ether is critical for the successful oxidative intramolecular cyclization reaction. The synthesis also features a cross-metathesis reaction for the construction of a trisubstituted olefin, a ring-closing metathesis to form a highly functionalized macrolactone, and a chiral phosphoric acid promoted formation of an N-acyl aminal to furnish (–)-zampanolide stereoselectively and in good yield. The synthetic (–)-zampanolide had effects on cultured cells and on tubulin assembly consistent with the properties reported for the natural product.
Co-reporter:Arun K. Ghosh, Satyendra Pandey, Sudhakar Gangarajula, Sarang Kulkarni, Xiaoming Xu, Kalapala Venkateswara Rao, Xiangping Huang, Jordan Tang
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 17) pp:5460-5465
Publication Date(Web):1 September 2012
DOI:10.1016/j.bmcl.2012.07.043
Structure-based design, synthesis, and biological evaluation of a series of dihydroquinazoline-derived β-secretase inhibitors incorporating thiazole and pyrazole-derived P2-ligands are described. We have identified inhibitor 4f which has shown potent enzyme inhibitory (Ki = 13 nM) and cellular (IC50 = 21 nM in neuroblastoma cells) assays. A model of 4f was created based upon the X-ray structure of 3a-bound β-secretase. The model suggested possible interactions in the active site.β-Secretase inhibitor 4f showed an enzyme Ki of 13 nM and a cellular IC50 value of 21 nM. A molecular model of 4f provided insight into the ligand-binding site interactions.
Co-reporter:Arun K. Ghosh, Bruno D. Chapsal, Melinda Steffey, Johnson Agniswamy, Yuan-Fang Wang, Masayuki Amano, Irene T. Weber, Hiroaki Mitsuya
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 6) pp:2308-2311
Publication Date(Web):15 March 2012
DOI:10.1016/j.bmcl.2012.01.061
The design, synthesis, and biological evaluation of novel C3-substituted cyclopentyltetrahydrofuranyl (Cp-THF)-derived HIV-1 protease inhibitors are described. Various C3-functional groups on the Cp-THF ligand were investigated in order to maximize the ligand-binding site interactions in the flap region of the protease. Inhibitors 3c and 3d have displayed the most potent enzyme inhibitory and antiviral activity. Both inhibitors have maintained impressive activity against a panel of multidrug resistant HIV-1 variants. A high-resolution X-ray crystal structure of 3c-bound HIV-1 protease revealed a number of important molecular insights into the ligand-binding site interactions.
Co-reporter:Arun K. Ghosh, Xu Cheng
Tetrahedron Letters 2012 Volume 53(Issue 20) pp:2568-2570
Publication Date(Web):16 May 2012
DOI:10.1016/j.tetlet.2012.03.041
Oxidative activation of benzyl or cinnamyl ether bearing allylsilane derivatives using a catalytic amount of DDQ and 2 equiv of CAN in the presence of PPTS provided functionalized 4-methylenetetrahydropyrans in good yields and excellent diastereoselectivity. The reaction could be applied to the synthesis of a variety of substituted tetrahydropyran derivatives.
Co-reporter:Arun K. Ghosh, Daniel R. Nicponski, Jorden Kass
Tetrahedron Letters 2012 Volume 53(Issue 29) pp:3699-3702
Publication Date(Web):18 July 2012
DOI:10.1016/j.tetlet.2012.04.006
In situ-generated (bis-DPPMB)–Cu(OTf)2 complex has been examined to catalyze a tandem olefin migration and Prins cyclization of an alkenol with various aldehydes. The reaction proceeded with electron-rich aromatic aldehydes at room temperature and provided functionalized tetrahydropyrans in good yields. An efficient synthesis of the bis-DPPMB ligand has also been described.
Co-reporter:Arun K. Ghosh and Guo-Bao Ren
The Journal of Organic Chemistry 2012 Volume 77(Issue 5) pp:2559-2565
Publication Date(Web):February 10, 2012
DOI:10.1021/jo202631e
Stereoselective syntheses of both functionalized tetrahydropyran subunits of (−)-lasonolide A are described. These tetrahydropyran rings were constructed using catalytic asymmetric hetero Diels–Alder reactions as the key steps. The C22 quaternary stereocenter present in the upper tetrahydropyran ring was constructed by a stereoselective alkylation, and the C9 hydroxy stereochemistry of the bottom tetrahydropyran was constructed by a stereoselective epoxidation followed by a regioselective epoxide opening reaction.
Co-reporter:Dr. Arun K. Ghosh;David D. Anderson;Dr. Irene T. Weber;Dr. Hiroaki Mitsuya
Angewandte Chemie International Edition 2012 Volume 51( Issue 8) pp:1778-1802
Publication Date(Web):
DOI:10.1002/anie.201102762
Abstract
The evolution of drug resistance is one of the most fundamental problems in medicine. In HIV/AIDS, the rapid emergence of drug-resistant HIV-1 variants is a major obstacle to current treatments. HIV-1 protease inhibitors are essential components of present antiretroviral therapies. However, with these protease inhibitors, resistance occurs through viral mutations that alter inhibitor binding, resulting in a loss of efficacy. This loss of potency has raised serious questions with regard to effective long-term antiretroviral therapy for HIV/AIDS. In this context, our research has focused on designing inhibitors that form extensive hydrogen-bonding interactions with the enzyme’s backbone in the active site. In doing so, we limit the protease’s ability to acquire drug resistance as the geometry of the catalytic site must be conserved to maintain functionality. In this Review, we examine the underlying principles of enzyme structure that support our backbone-binding concept as an effective means to combat drug resistance and highlight their application in our recent work on antiviral HIV-1 protease inhibitors.
Co-reporter:Dr. Arun K. Ghosh;David D. Anderson;Dr. Irene T. Weber;Dr. Hiroaki Mitsuya
Angewandte Chemie 2012 Volume 124( Issue 8) pp:1812-1838
Publication Date(Web):
DOI:10.1002/ange.201102762
Abstract
Die Entstehung von Arzneimittelresistenzen ist eines der grundlegenden Probleme der Medizin. Dass sich bei HIV/AIDS so schnell resistente HIV-1-Varianten einstellen, ist ein großes Hindernis für die modernen Therapien. Wichtige Zielmoleküle der derzeitigen antiretroviralen Therapien sind Inhibitoren der HIV-1-Protease. Das Virus entwickelt Resistenzen, wenn Mutationen die Inhibitorbindung des Enzyms verändern und somit die Effizienz mindern. Um diese Resistenzbildung zu vermeiden, haben wir Inhibitoren erforscht, die am aktiven Zentrum der Protease Wasserstoffbrücken zum Proteinrückgrat bilden. Weil sich die Geometrie am katalytischen Zentrum ohne Funktionsverlust nicht ändern kann, schränken solche Wechselwirkungen die Möglichkeiten der Protease zur Resistenzbildung erheblich ein. Hier diskutieren wir das enzymatische Strukturprinzip, auf dem unser Konzept der Rückgratbindung beruht. Besonderes Augenmerk richten wir auf die Anwendung des Konzepts in unseren jüngeren Arbeiten zu antiviralen Inhibitoren der HIV-1-Protease.
Co-reporter:Arun K. Ghosh and Daniel R. Nicponski
Organic Letters 2011 Volume 13(Issue 16) pp:4328-4331
Publication Date(Web):July 28, 2011
DOI:10.1021/ol2016675
Metal–ligand complexes of Cu(OTf)2 with an appropriate bisphosphine ligand have been shown to effectively catalyze the formation of substituted tetrahydropyrans via a sequential olefin migration and Prins-type cyclization. This methodology provides convenient access to a variety of functionalized tetrahydropyrans in excellent diastereoselectivities and good to excellent yields.
Co-reporter:Arun K. Ghosh and Jianfeng Li
Organic Letters 2011 Volume 13(Issue 1) pp:66-69
Publication Date(Web):December 2, 2010
DOI:10.1021/ol102549a
A stereoselective synthesis of (+)-herboxidiene is described. The convergent synthesis utilized a Suzuki cross-coupling reaction to assemble the key segments. The synthesis of the functionalized tetrahydropyran ring utilized an Achmatowicz reaction as the key step. The synthesis of the C10−C19 segment was accomplished using Brown’s crotylboration, asymmetric alkylation, and a stereoselective allylic chlorination reactions.
Co-reporter:Arun K. Ghosh ; Bruno D. Chapsal ; Abigail Baldridge ; Melinda P. Steffey ; D. Eric Walters ; Yasuhiro Koh ; Masayuki Amano ;Hiroaki Mitsuya
Journal of Medicinal Chemistry 2011 Volume 54(Issue 2) pp:622-634
Publication Date(Web):December 31, 2010
DOI:10.1021/jm1012787
The design, synthesis, and evaluation of a new series of hexahydrofuropyranol-derived HIV-1 protease inhibitors are described. We have designed a stereochemically defined hexahydrofuropyranol-derived urethane as the P2-ligand. The current ligand is designed based upon the X-ray structure of 1a-bound HIV-1 protease. The synthesis of (3aS,4S,7aR)-hexahydro-2H-furo[2,3-b]pyran-4-ol, (−)-7, was carried out in optically active form. Incorporation of this ligand provided inhibitor 35a, which has shown excellent enzyme inhibitory activity and antiviral potency. Our structure−activity studies have indicated that the stereochemistry and the position of oxygens in the ligand are important to the observed potency of the inhibitor. Inhibitor 35a has maintained excellent potency against multidrug-resistant HIV-1 variants. An active site model of 35a was created based upon the X-ray structure of 1b-bound HIV-1 protease. The model offers molecular insights regarding ligand-binding site interactions of the hexahydrofuropyranol-derived novel P2-ligand.
Co-reporter:Arun K. Ghosh ; Bruno D. Chapsal ; Garth L. Parham ; Melinda Steffey ; Johnson Agniswamy ; Yuan-Fang Wang ; Masayuki Amano ; Irene T. Weber ;Hiroaki Mitsuya
Journal of Medicinal Chemistry 2011 Volume 54(Issue 16) pp:5890-5901
Publication Date(Web):July 29, 2011
DOI:10.1021/jm200649p
We report the design, synthesis, biological evaluation, and the X-ray crystal structure of a novel inhibitor bound to the HIV-1 protease. Various C3-functionalized cyclopentanyltetrahydrofurans (Cp-THF) were designed to interact with the flap Gly48 carbonyl or amide NH in the S2-subsite of the HIV-1 protease. We investigated the potential of those functionalized ligands in combination with hydroxyethylsulfonamide isosteres. Inhibitor 26 containing a 3-(R)-hydroxyl group on the Cp-THF core displayed the most potent enzyme inhibitory and antiviral activity. Our studies revealed a preference for the 3-(R)-configuration over the corresponding 3-(S)-derivative. Inhibitor 26 exhibited potent activity against a panel of multidrug-resistant HIV-1 variants. A high resolution X-ray structure of 26-bound HIV-1 protease revealed important molecular insight into the ligand-binding site interactions.
Co-reporter:Arun K. Ghosh, Cuthbert D. Martyr, Melinda Steffey, Yuan-Fang Wang, Johnson Agniswamy, Masayuki Amano, Irene T. Weber, and Hiroaki Mitsuya
ACS Medicinal Chemistry Letters 2011 Volume 2(Issue 4) pp:298
Publication Date(Web):January 27, 2011
DOI:10.1021/ml100289m
We investigated substituted bis-THF-derived HIV-1 protease inhibitors in order to enhance ligand-binding site interactions in the HIV-1 protease active site. In this context, we have carried out convenient syntheses of optically active bis-THF and C4-substituted bis-THF ligands using a [2,3]-sigmatropic rearrangement as the key step. The synthesis provided convenient access to a number of substituted bis-THF derivatives. Incorporation of these ligands led to a series of potent HIV-1 protease inhibitors. Inhibitor 23c turned out to be the most potent (Ki = 2.9 pM; IC50 = 2.4 nM) among the inhibitors. An X-ray structure of 23c-bound HIV-1 protease showed extensive interactions of the inhibitor with the protease active site, including a unique water-mediated hydrogen bond to the Gly-48 amide NH in the S2 site.Keywords (keywords): bis-THF; Darunavir; design; drug-resistant; HIV-1 protease inhibitors; synthesis; X-ray structure
Co-reporter:Arun K. Ghosh and Jorden Kass
Chemical Communications 2010 vol. 46(Issue 8) pp:1218-1220
Publication Date(Web):25 Jan 2010
DOI:10.1039/B924807B
We have developed a practical synthesis of unique nucleoside derivatives via TiCl4 promoted multicomponent reaction of optically active dihydrofuran, ethyl pyruvate/glyoxylate, and a TMS protected nucleobase in a single-pot operation.
Co-reporter:Arun K. Ghosh and Hao Yuan
Organic Letters 2010 Volume 12(Issue 14) pp:3120-3123
Publication Date(Web):June 18, 2010
DOI:10.1021/ol101105v
Enantioselective total syntheses of the proposed structures of macrolide cytotoxic agents iriomoteolide-1a and -1b have been accomplished. The synthesis was carried out in a convergent and stereoselective manner. However, the present work suggests that the reported structures have been assigned incorrectly. The synthesis features Julia−Kocienski olefination, Sharpless asymmetric epoxidation, Brown asymmetric crotylboration, a Sakurai reaction, an aldol reaction, and enzymatic resolution as the key steps.
Co-reporter:Arun K. Ghosh ; Jun Takayama ; Kalapala Venkateswara Rao ; Kiira Ratia ; Rima Chaudhuri ; Debbie C. Mulhearn ; Hyun Lee ; Daniel B. Nichols ; Surendranath Baliji ; Susan C. Baker ; Michael E. Johnson ;Andrew D. Mesecar
Journal of Medicinal Chemistry 2010 Volume 53(Issue 13) pp:4968-4979
Publication Date(Web):June 9, 2010
DOI:10.1021/jm1004489
The design, synthesis, X-ray crystal structure, molecular modeling, and biological evaluation of a series of new generation SARS-CoV PLpro inhibitors are described. A new lead compound 3 (6577871) was identified via high-throughput screening of a diverse chemical library. Subsequently, we carried out lead optimization and structure−activity studies to provide a series of improved inhibitors that show potent PLpro inhibition and antiviral activity against SARS-CoV infected Vero E6 cells. Interestingly, the (S)-Me inhibitor 15h (enzyme IC50 = 0.56 μM; antiviral EC50 = 9.1 μM) and the corresponding (R)-Me 15g (IC50 = 0.32 μM; antiviral EC50 = 9.1 μM) are the most potent compounds in this series, with nearly equivalent enzymatic inhibition and antiviral activity. A protein−ligand X-ray structure of 15g-bound SARS-CoV PLpro and a corresponding model of 15h docked to PLpro provide intriguing molecular insight into the ligand-binding site interactions.
Co-reporter:Arun K. Ghosh, Zachary L. Dawson, Deuk Kyu Moon, Ruoli Bai, Ernest Hamel
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 17) pp:5104-5107
Publication Date(Web):1 September 2010
DOI:10.1016/j.bmcl.2010.07.023
Synthesis and biological evaluation of jasplakinolide analogs are described. The synthesis of analogs utilized a diastereoselective syn-aldol reaction and an orthoester Claisen rearrangement as key steps. All synthetic analogs were evaluated for their ability to disrupt the actin cytoskeleton. Compounds 2, 3, and 4 essentially displayed similar activity to jasplakinolide.Eight new jasplakinolide derivatives were synthesized and evaluated for antitumor activity by measuring their ability to interfere with the actin cytoskeleton. Simplified derivative 4 is essentially as active as natural jasplakinolide.
Co-reporter:A. Jonathan Singh, Chun-Xiao Xu, Xiaoming Xu, Lyndon M. West, Anja Wilmes, Ariane Chan, Ernest Hamel, John H. Miller, Peter T. Northcote and Arun K. Ghosh
The Journal of Organic Chemistry 2010 Volume 75(Issue 1) pp:2-10
Publication Date(Web):December 3, 2009
DOI:10.1021/jo9021265
Peloruside B (2), a natural congener of peloruside A (1), was isolated in sub-milligram quantities from the New Zealand marine sponge Mycale hentscheli. Peloruside B promotes microtubule polymerization and arrests cells in the G2/M phase of mitosis similar to paclitaxel, and its bioactivity was comparable to that of peloruside A. NMR-directed isolation, structure elucidation, structure confirmation by total synthesis, and bioactivity of peloruside B are described in this article. The synthesis features Sharpless dihydroxylation, Brown’s asymmetric allylboration reaction, reductive aldol coupling, Yamaguchi macrolactonization, and selective methylation.
Co-reporter:Arun K. Ghosh
The Journal of Organic Chemistry 2010 Volume 75(Issue 23) pp:7967-7989
Publication Date(Web):October 11, 2010
DOI:10.1021/jo101606g
In this Perspective, I outline my group’s research involving the chemical syntheses of medicinally important natural products, exploration of their bioactivity, and the development of new asymmetric carbon−carbon bond-forming reactions. This paper also highlights our approach to molecular design and synthesis of conceptually novel inhibitors against target proteins involved in the pathogenesis of human diseases, including AIDS and Alzheimer’s disease.
Co-reporter:Dr. Arun K. Ghosh;Dr. Chun-Xiao Xu;Dr. Kalapala Venkateswara Rao;Abigail Baldridge;Johnson Agniswamy;Yuan-Fang Wang;Dr. Irene T. Weber;Manabu Aoki;Salcedo Gomez Pedro Miguel;Masayuki Amano;Dr. Hiroaki Mitsuya
ChemMedChem 2010 Volume 5( Issue 11) pp:1850-1854
Publication Date(Web):
DOI:10.1002/cmdc.201000318
Co-reporter:Dr. Arun K. Ghosh;Dr. Chun-Xiao Xu;Dr. Kalapala Venkateswara Rao;Abigail Baldridge;Johnson Agniswamy;Yuan-Fang Wang;Dr. Irene T. Weber;Manabu Aoki;Salcedo Gomez Pedro Miguel;Masayuki Amano;Dr. Hiroaki Mitsuya
ChemMedChem 2010 Volume 5( Issue 11) pp:
Publication Date(Web):
DOI:10.1002/cmdc.201090049
Co-reporter:Arun K. Ghosh and Jianfeng Li
Organic Letters 2009 Volume 11(Issue 18) pp:4164-4167
Publication Date(Web):August 20, 2009
DOI:10.1021/ol901691d
An enantioselective synthesis of marine alkaloid brevisamide was accomplished in a convergent manner. The synthesis utilized an enantioselective hetero-Diels−Alder reaction which sets three chiral centers in compound 11. The synthesis also features a modified Wolff−Kishner reduction, Rubottom oxidation, and Suzuki−Miyaura coupling to furnish brevisamide.
Co-reporter:Arun K. Ghosh and Chun-Xiao Xu
Organic Letters 2009 Volume 11(Issue 9) pp:1963-1966
Publication Date(Web):April 8, 2009
DOI:10.1021/ol900412u
The total synthesis of the proposed structure of anticancer agent stereocalpin A is described. The synthesis features a diastereoselective synthesis of a 5-hydroxy-2,4-dimethyl-3-oxooctanoic acid unit with asymmetric anti- and syn-aldol reactions as the key steps. Initial cycloamidation led to complete epimerization at the C-11 stereocenter due to unique steric constraints in the 12-membered depsipeptide ring. A late-stage methylation strategy led to the synthesis of the proposed structure of stereocalpin A.
Co-reporter:Arun K. Ghosh ; Sofiya Leshchenko-Yashchuk ; David D. Anderson ; Abigail Baldridge ; Marcus Noetzel ; Heather B. Miller ; Yunfeng Tie ; Yuan-Fang Wang ; Yasuhiro Koh ; Irene T. Weber ;Hiroaki Mitsuya
Journal of Medicinal Chemistry 2009 Volume 52(Issue 13) pp:3902-3914
Publication Date(Web):May 27, 2009
DOI:10.1021/jm900303m
Structure-based design, synthesis, and biological evaluation of a series of novel HIV-1 protease inhibitors are described. In an effort to enhance interactions with protease backbone atoms, we have incorporated stereochemically defined methyl-2-pyrrolidinone and methyl oxazolidinone as the P1′-ligands. These ligands are designed to interact with Gly-27′ carbonyl and Arg-8 side chain in the S1′-subsite of the HIV protease. We have investigated the potential of these ligands in combination with our previously developed bis-tetrahydrofuran (bis-THF) and cyclopentanyltetrahydrofuran (Cp-THF) as the P2-ligands. Inhibitor 19b with a (R)-aminomethyl-2-pyrrolidinone and a Cp-THF was shown to be the most potent compound. This inhibitor maintained near full potency against multi-PI-resistant clinical HIV-1 variants. A high resolution protein−ligand X-ray crystal structure of 19b-bound HIV-1 protease revealed that the P1′-pyrrolidinone heterocycle and the P2-Cp-ligand are involved in several critical interactions with the backbone atoms in the S1′ and S2 subsites of HIV-1 protease.
Co-reporter:Arun K. Ghosh
Journal of Medicinal Chemistry 2009 Volume 52(Issue 8) pp:2163-2176
Publication Date(Web):March 26, 2009
DOI:10.1021/jm900064c
Co-reporter:Arun K. Ghosh ; Jun Takayama ; Yoann Aubin ; Kiira Ratia ; Rima Chaudhuri ; Yahira Baez ; Katrina Sleeman ; Melissa Coughlin ; Daniel B. Nichols ; Debbie C. Mulhearn ; Bellur S. Prabhakar ; Susan C. Baker ; Michael E. Johnson ;Andrew D. Mesecar
Journal of Medicinal Chemistry 2009 Volume 52(Issue 16) pp:5228-5240
Publication Date(Web):July 31, 2009
DOI:10.1021/jm900611t
We describe here the design, synthesis, molecular modeling, and biological evaluation of a series of small molecule, nonpeptide inhibitors of SARS-CoV PLpro. Our initial lead compound was identified via high-throughput screening of a diverse chemical library. We subsequently carried out structure−activity relationship studies and optimized the lead structure to potent inhibitors that have shown antiviral activity against SARS-CoV infected Vero E6 cells. Upon the basis of the X-ray crystal structure of inhibitor 24-bound to SARS-CoV PLpro, a drug design template was created. Our structure-based modification led to the design of a more potent inhibitor, 2 (enzyme IC50 = 0.46 μM; antiviral EC50 = 6 μM). Interestingly, its methylamine derivative, 49, displayed good enzyme inhibitory potency (IC50 = 1.3 μM) and the most potent SARS antiviral activity (EC50 = 5.2 μM) in the series. We have carried out computational docking studies and generated a predictive 3D-QSAR model for SARS-CoV PLpro inhibitors.
Co-reporter:Arun K. Ghosh ; Sarang Kulkarni ; David D. Anderson ; Lin Hong ; Abigail Baldridge ; Yuan-Fang Wang ; Alexander A. Chumanevich ; Andrey Y. Kovalevsky ; Yasushi Tojo ; Masayuki Amano ; Yasuhiro Koh ; Jordan Tang ; Irene T. Weber ;Hiroaki Mitsuya ▽
Journal of Medicinal Chemistry 2009 Volume 52(Issue 23) pp:7689-7705
Publication Date(Web):September 11, 2009
DOI:10.1021/jm900695w
The structure-based design, synthesis, and biological evaluation of a series of nonpeptidic macrocyclic HIV protease inhibitors are described. The inhibitors are designed to effectively fill in the hydrophobic pocket in the S1′−S2′ subsites and retain all major hydrogen bonding interactions with the protein backbone similar to darunavir (1) or inhibitor 2. The ring size, the effect of methyl substitution, and unsaturation within the macrocyclic ring structure were assessed. In general, cyclic inhibitors were significantly more potent than their acyclic homologues, saturated rings were less active than their unsaturated analogues and a preference for 10- and 13-membered macrocylic rings was revealed. The addition of methyl substituents resulted in a reduction of potency. Both inhibitors 14b and 14c exhibited marked enzyme inhibitory and antiviral activity, and they exerted potent activity against multidrug-resistant HIV-1 variants. Protein−ligand X-ray structures of inhibitors 2 and 14c provided critical molecular insights into the ligand-binding site interactions.
Co-reporter:Arun K. Ghosh, Hao Yuan
Tetrahedron Letters 2009 50(13) pp: 1416-1418
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.01.043
Co-reporter:ArunK. Ghosh Dr. ;Kai Xi
Angewandte Chemie 2009 Volume 121( Issue 29) pp:5476-5479
Publication Date(Web):
DOI:10.1002/ange.200902338
Co-reporter:Arun K. Ghosh and Kai Xi
The Journal of Organic Chemistry 2009 Volume 74(Issue 3) pp:1163-1170
Publication Date(Web):January 5, 2009
DOI:10.1021/jo802261f
An enantioselective synthesis of platensimycin, a novel antibiotic natural product that inhibits bacterial β-ketoacyl-(acyl-carrier-protein) synthase (FabF), is described. Our synthetic strategy for the construction of the oxatetracyclic core involved an intramolecular Diels−Alder reaction. Our preliminary studies provided a complex tetracyclic product by first undergoing an interesting 1,5-hydride shift followed by a Diels−Alder reaction. Further optimization of the diene’s electronic properties, by incorporation of a methoxy group, led to the oxatetracyclic core of platensimycin. The three required chiral centers, including two all-carbon quaternary chiral centers, were built in the intramolecular Diels−Alder step. The synthesis utilized natural (+)-carvone as the key chiral starting material, which determined the stereochemistry of the final product. The synthesis also featured an efficient Petasis olefination, a hydroboration sequence, a Gais’s asymmetric Horner−Wadsworth−Emmons reaction, and a mercury salt catalyzed enol ether isomerization.
Co-reporter:ArunK. Ghosh Dr. ;Kai Xi
Angewandte Chemie International Edition 2009 Volume 48( Issue 29) pp:5372-5375
Publication Date(Web):
DOI:10.1002/anie.200902338
Co-reporter:Arun K. Ghosh, Bruno D. Chapsal, Irene T. Weber and Hiroaki Mitsuya
Accounts of Chemical Research 2008 Volume 41(Issue 1) pp:78
Publication Date(Web):August 28, 2007
DOI:10.1021/ar7001232
The discovery of human immunodeficiency virus (HIV) protease inhibitors (PIs) and their utilization in highly active antiretroviral therapy (HAART) have been a major turning point in the management of HIV/acquired immune-deficiency syndrome (AIDS). However, despite the successes in disease management and the decrease of HIV/AIDS-related mortality, several drawbacks continue to hamper first-generation protease inhibitor therapies. The rapid emergence of drug resistance has become the most urgent concern because it renders current treatments ineffective and therefore compels the scientific community to continue efforts in the design of inhibitors that can efficiently combat drug resistance. The present line of research focuses on the presumption that an inhibitor that can maximize interactions in the HIV-1 protease active site, particularly with the enzyme backbone atoms, will likely retain these interactions with mutant enzymes. Our structure-based design of HIV PIs specifically targeting the protein backbone has led to exceedingly potent inhibitors with superb resistance profiles. We initially introduced new structural templates, particulary nonpeptidic conformationally constrained P2 ligands that would efficiently mimic peptide binding in the S2 subsite of the protease and provide enhanced bioavailability to the inhibitor. Cyclic ether derived ligands appeared as privileged structural features and allowed us to obtain a series of potent PIs. Following our structure-based design approach, we developed a high-affinity 3(R),3a(R),6a(R)-bis-tetrahydrofuranylurethane (bis-THF) ligand that maximizes hydrogen bonding and hyrophobic interactions in the protease S2 subsite. Combination of this ligand with a range of different isosteres led to a series of exceedingly potent inhibitors. Darunavir, initially TMC-114, which combines the bis-THF ligand with a sulfonamide isostere, directly resulted from this line of research. This inhibitor displayed unprecedented enzyme inhibitory potency (Ki = 16 pM) and antiviral activity (IC90 = 4.1 nM). Most importantly, it consistently retained is potency against highly drug-resistant HIV strains. Darunavirʼs IC50 remained in the low nanomolar range against highly mutated HIV strains that displayed resistance to most available PIs. Our detailed crystal structure analyses of darunavir-bound protease complexes clearly demonstrated extensive hydrogen bonding between the inhibitor and the protease backbone. Most strikingly, these analyses provided ample evidence of the unique contribution of the bis-THF as a P2-ligand. With numerous hydrogen bonds, bis-THF was shown to closely and tightly bind to the backbone atoms of the S2 subsite of the protease. Such tight interactions were consistently observed with mutant proteases and might therefore account for the unusually high resistance profile of darunavir. Optimization attempts of the backbone binding in other subsites of the enzyme, through rational modifications of the isostere or tailor made P2 ligands, led to equally impressive inhibitors with excellent resistance profiles. The concept of targeting the protein backbone in current structure-based drug design may offer a reliable strategy for combating drug resistance.
Co-reporter:Arun K. Ghosh ; Sandra Gemma ; Abigail Baldridge ; Yuan-Fang Wang ; Andrey Yu. Kovalevsky ; Yashiro Koh ; Irene T. Weber ;Hiroaki Mitsuya
Journal of Medicinal Chemistry 2008 Volume 51(Issue 19) pp:6021-6033
Publication Date(Web):September 11, 2008
DOI:10.1021/jm8004543
We report the design, synthesis, and biological evaluation of a series of novel HIV-1 protease inhibitors. The inhibitors incorporate stereochemically defined flexible cyclic ethers/polyethers as high affinity P2-ligands. Inhibitors containing small ring 1,3-dioxacycloalkanes have shown potent enzyme inhibitory and antiviral activity. Inhibitors 3d and 3h are the most active inhibitors. Inhibitor 3d maintains excellent potency against a variety of multi-PI-resistant clinical strains. Our structure−activity studies indicate that the ring size, stereochemistry, and position of oxygens are important for the observed activity. Optically active synthesis of 1,3-dioxepan-5-ol along with the syntheses of various cyclic ether and polyether ligands have been described. A protein−ligand X-ray crystal structure of 3d-bound HIV-1 protease was determined. The structure revealed that the P2-ligand makes extensive interactions including hydrogen bonding with the protease backbone in the S2-site. In addition, the P2-ligand in 3d forms a unique water-mediated interaction with the NH of Gly-48.
Co-reporter:Arun K. Ghosh, Sandra Gemma, Jun Takayama, Abigail Baldridge, Sofiya Leshchenko-Yashchuk, Heather B. Miller, Yuan-Fang Wang, Andrey Y. Kovalevsky, Yashiro Koh, Irene T. Weber and Hiroaki Mitsuya
Organic & Biomolecular Chemistry 2008 vol. 6(Issue 20) pp:3703-3713
Publication Date(Web):11 Aug 2008
DOI:10.1039/B809178A
Recently, we designed a series of novel HIV-1 protease inhibitors incorporating a stereochemically defined bicyclic fused cyclopentyl (Cp-THF) urethane as the high affinity P2-ligand. Inhibitor 1 with this P2-ligand has shown very impressive potency against multi-drug-resistant clinical isolates. Based upon the 1-bound HIV-1 protease X-ray structure, we have now designed and synthesized a number of meso-bicyclic ligands which can conceivably interact similarly to the Cp-THF ligand. The design of meso-ligands is quite attractive as they do not contain any stereocenters. Inhibitors incorporating urethanes of bicyclic-1,3-dioxolane and bicyclic-1,4-dioxane have shown potent enzyme inhibitory and antiviral activities. Inhibitor 2 (Ki = 0.11 nM; IC50 = 3.8 nM) displayed very potent antiviral activity in this series. While inhibitor 3 showed comparable enzyme inhibitory activity (Ki = 0.18 nM) its antiviral activity (IC50 = 170 nM) was significantly weaker than inhibitor 2. Inhibitor 2 maintained an antiviral potency against a series of multi-drug resistant clinical isolates comparable to amprenavir. A protein–ligand X-ray structure of 3-bound HIV-1 protease revealed a number of key hydrogen bonding interactions at the S2-subsite. We have created an active model of inhibitor 2 based upon this X-ray structure.
Co-reporter:Arun K. Ghosh, Gangli Gong, Valerie Grum-Tokars, Debbie C. Mulhearn, Susan C. Baker, Melissa Coughlin, Bellur S. Prabhakar, Katrina Sleeman, Michael E. Johnson, Andrew D. Mesecar
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 20) pp:5684-5688
Publication Date(Web):15 October 2008
DOI:10.1016/j.bmcl.2008.08.082
Design, synthesis and biological evaluation of a series of 5-chloropyridine ester-derived severe acute respiratory syndrome-coronavirus chymotrypsin-like protease inhibitors is described. Position of the carboxylate functionality is critical to potency. Inhibitor 10 with a 5-chloropyridinyl ester at position 4 of the indole ring is the most potent inhibitor with a SARS-CoV 3CLpro IC50 value of 30 nM and an antiviral EC50 value of 6.9 μM. Molecular docking studies have provided possible binding modes of these inhibitors.Design, synthesis and biological evaluation of a series of chloropyridyl ester-derived SARS-CoV 3CLpro Inhibitors are described Inhibitors 10 has shown potent activity in both enzyme inhibitory and antiviral assays
Co-reporter:Arun K. Ghosh, Sarang S. Kulkarni, Chun-Xiao Xu, Khriesto Shurrush
Tetrahedron: Asymmetry 2008 Volume 19(Issue 8) pp:1020-1026
Publication Date(Web):1 May 2008
DOI:10.1016/j.tetasy.2008.04.005
Asymmetric multicomponent reactions of optically active phenyl dihydrofuran, keto ester or N-tosyl imino ester, and allylsilane provided functionalized phenyl tetrahydrofurans with multiple stereogenic centers diastereoselectively. Cleavage of the resulting substituted tetrahydrofurans readily provided acyclic derivatives with three contiguous asymmetric centers via an acyloxycarbenium ion intermediate. Ring closing olefin metathesis, using Grubbs catalyst, afforded functionalized cyclopentene derivatives in optically active form. A one-pot tandem tetrahydrofuran ring cleavage followed by ring closing olefin metathesis also provided functionalized cyclopentenes in good yield..(R)-2-Phenyl-2,3-dihydrofuranC10H10O4[α]D23=-66(c1.05,CHCl3)Absolute configuration: (R)(R)-Ethyl2-((2R,3R,5R)-2-allyl-5-phenyltetrahydrofuran-3-yl)-2-hydroxypropanoateC18H24O4[α]D23=+15.7(c1.3,CHCl3)Absolute configuration: (2R,3R,5R)(S)-Ethyl2-((2R,3S,5R)-2-allyl-5-phenyltetrahydrofuran-3-yl)-2-hydroxy-4-methoxybutanoateC20H28O5[α]D25=-15.3(c1.2,CHCl3)Absolute configuration: (S,2R,3S,5R)(3R,4S,5R)-3-(Ethoxycarbonyl)-1-methoxy-4-styryloct-7-ene-3,5-diyl diacetateC10H10O[α]D23=+44.9(c1.0,CHCl3)Absolute configuration: (3R,4S,5R)(R)-Ethyl 2-acetoxy-2-((1S,5R)-5-acetoxycyclopent-2-enyl)propanoateC14H20O6[α]D23=-3.5(c1.0,CHCl3)Absolute configuration: (R,1S,5R)(R)-Ethyl 2-acetoxy-2-((1S,5R)-5-acetoxycyclopent-2-enyl)-4-methoxybutanoateC16H24O7[α]D23=-3.2(c1.0,CHCl3)Absolute configuration: (R,1S,5R)(S)-Ethyl 2-acetoxy-2-((1S,5R)-5-acetoxycyclopent-2-enyl)-4-methoxybutanoateC10H10O[α]D23=-5.5(c1.0,CHCl3)Absolute configuration: (S,1S,5R)(R)-Ethyl 2-((2R,3R,5R)-2-allyl-5-phenyltetrahydrofuran-3-yl)-2-(4-methylphenylsulfonamido)acetateC24H29NO5S[α]D23=-11.8(c0.9,CHCl3)Absolute configuration: (R,2R,3R,5R)(S)-4-((2R,3R,5R)-2-Allyl-5-phenyltetrahydrofuran-3-yl)-3-tosyloxazolidin-2-oneC23H25NO5S[α]D23=-32.5(c1.6,CHCl3)Absolute configuration: (S,2R,3R,5R)(3R,4R)-3-(2-Oxo-3-tosyloxazolidin-4-yl)-1-phenylhepta-1,6-diene-4-yl acetateC25H27NO6S[α]D23=+41.4(c1.05,CHCl3)Absolute configuration: (3R,4R)(1R,2R)-2-((R)-2-Oxo-3-tosyloxazolidin-4-yl)cyclopent-3-enyl acetateC17H19NO6S[α]D23=-87.4(c0.85,CHCl3)Absolute configuration: (1R,2R,R)
Co-reporter:ArunK. Ghosh Dr. ;Gangli Gong Dr.
Chemistry – An Asian Journal 2008 Volume 3( Issue 10) pp:1811-1823
Publication Date(Web):
DOI:10.1002/asia.200800164
Abstract
A detailed account of the enantioselective total synthesis of (−)-lasonolide A is described. Our initial synthetic route to the top tetrahydropyran ring involved Evans asymmetric alkylation as the key step. Initially, we relied on the diastereoselective alkylation of an α-alkoxyacetimide derivative containing an α′ stereogenic center and investigated such an asymmetric alkylation reaction. Although alkylation proceeded in good yield, the lack of diastereoselectivity prompted us to explore alternative routes. Our subsequent successful synthetic strategies involved highly diastereoselective cycloaddition routes to both tetrahydropyran rings of lasonolide A. The top tetrahydropyran ring was constructed stereoselectively by an intramolecular 1,3-dipolar cycloaddition reaction. The overall process constructed a bicyclic isoxazoline, which was later unravelled to a functionalized tetrahydropyran ring as well as a quaternary stereocenter present in the molecule. The lower tetrahydropyran ring was assembled by a Jacobsen catalytic asymmetric hetero-Diels–Alder reaction as the key step. The synthesis also features a Lewis acid catalyzed epoxide opening to form a substituted ether stereoselectively.
Co-reporter:Surendranath Baliji;Scott Pegan;Kiira Ratia;Melissa Coughlin;Jun Takayama;Katrina Sleeman;Rima Chaudhuri;Wentao Fu;Bellur S. Prabhakar;Michael E. Johnson;Susan C. Baker;Andrew D. Mesecar
PNAS 2008 Volume 105 (Issue 42 ) pp:16119-16124
Publication Date(Web):2008-10-21
DOI:10.1073/pnas.0805240105
We report the discovery and optimization of a potent inhibitor against the papain-like protease (PLpro) from the coronavirus
that causes severe acute respiratory syndrome (SARS-CoV). This unique protease is not only responsible for processing the
viral polyprotein into its functional units but is also capable of cleaving ubiquitin and ISG15 conjugates and plays a significant
role in helping SARS-CoV evade the human immune system. We screened a structurally diverse library of 50,080 compounds for
inhibitors of PLpro and discovered a noncovalent lead inhibitor with an IC50 value of 20 μM, which was improved to 600 nM via synthetic optimization. The resulting compound, GRL0617, inhibited SARS-CoV
viral replication in Vero E6 cells with an EC50 of 15 μM and had no associated cytotoxicity. The X-ray structure of PLpro in complex with GRL0617 indicates that the compound
has a unique mode of inhibition whereby it binds within the S4-S3 subsites of the enzyme and induces a loop closure that shuts
down catalysis at the active site. These findings provide proof-of-principle that PLpro is a viable target for development
of antivirals directed against SARS-CoV, and that potent noncovalent cysteine protease inhibitors can be developed with specificity
directed toward pathogenic deubiquitinating enzymes without inhibiting host DUBs.
Co-reporter:Arun K. Ghosh, Zachary L. Dawson, Hiroaki Mitsuya
Bioorganic & Medicinal Chemistry 2007 Volume 15(Issue 24) pp:7576-7580
Publication Date(Web):15 December 2007
DOI:10.1016/j.bmc.2007.09.010
Our structure-based design strategies which specifically target the HIV-1 protease backbone, resulted in a number of exceedingly potent nonpeptidyl inhibitors. One of these inhibitors, darunavir (TMC114), contains a privileged, structure-based designed high-affinity P2 ligand, 3(R)(R),3a(S)(S),6a(R)(R)-bis-tetrahydrofuranylurethane (bis-THF). Darunavir has recently been approved for the treatment of HIV/AIDS patients harboring multidrug-resistant HIV-1 variants that do not respond to previously existing HAART regimens.This perspective article describes our structure-based design efforts targeting the protein-backbone of HIV-1 protease to combat drug- resistance. Darunavir has been recently approved for the treatment of drug- resistant HIV.
Co-reporter:Arun K. Ghosh, Kai Xi, Valerie Grum-Tokars, Xiaoming Xu, Kiira Ratia, Wentao Fu, Katherine V. Houser, Susan C. Baker, Michael E. Johnson, Andrew D. Mesecar
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 21) pp:5876-5880
Publication Date(Web):1 November 2007
DOI:10.1016/j.bmcl.2007.08.031
Structure-based design, synthesis, and biological evaluation of a series of peptidomimetic severe acute respiratory syndrome-coronavirus chymotrypsin-like protease inhibitors are described. These inhibitors were designed and synthesized based upon our X-ray crystal structure of inhibitor 1 bound to SARS-CoV 3CLpro. Incorporation of Boc-Ser as the P4-ligand resulted in enhanced SARS-CoV 3CLpro inhibitory activity. Structural analysis of the inhibitor-bound X-ray structure revealed high binding affinity toward the enzyme.Structure-based design, synthesis, and biological evaluation of a series of peptidomimetic SARS-CoV 3CLpro inhibitors are described. Inhibitor 3-bound SAR-3CLpro X-ray crystal structure provided molecular insight into the ligand-binding site interactions.
Co-reporter:Arun K. Ghosh, Gary Schiltz, Ramu Sridhar Perali, Sofiya Leshchenko, Stephanie Kay, D. Eric Walters, Yasuhiro Koh, Kenji Maeda, Hiroaki Mitsuya
Bioorganic & Medicinal Chemistry Letters 2006 Volume 16(Issue 7) pp:1869-1873
Publication Date(Web):1 April 2006
DOI:10.1016/j.bmcl.2006.01.011
A series of novel oxyindole-derived HIV-1 protease inhibitors were designed and synthesized based upon our X-ray crystal structure of inhibitor 2 (TMC-114) bound to HIV-1 protease. The effects of substituents, spirocyclic rings, and ring sizes have been investigated. A number of inhibitors exhibited low nanomolar inhibitory potencies against HIV protease.A series of novel oxyindole-derived HIV-1 protease inhibitors were designed and synthesized. A number of inhibitors exhibited low nanomolar inhibitory potencies against HIV protease.
Co-reporter:Arun K. Ghosh ;Perali Ramu Sridhar Dr.;Nagaswamy Kumaragurubaran Dr.;Yasuhiro Koh Dr.;Irene T. Weber ;Hiroaki Mitsuya
ChemMedChem 2006 Volume 1(Issue 9) pp:
Publication Date(Web):22 AUG 2006
DOI:10.1002/cmdc.200600103
Two inhibitors that incorporate bis-THF as an effective high-affinity P2 ligand for the HIV-1 protease substrate binding site maintain impressive potency against mutant strains resistant to currently approved protease inhibitors. Crystallographic structures of protein–ligand complexes help to explain the superior antiviral property of these inhibitors and their potency against a wide spectrum of HIV-1 strains.
Co-reporter:Arun K. Ghosh, Prasanth R. Nyalapatla
Tetrahedron (6 April 2017) Volume 73(Issue 14) pp:
Publication Date(Web):6 April 2017
DOI:10.1016/j.tet.2017.02.031
The total syntheses of (−)-amphirionin-4 and (+)-amphirionin-4 have been achieved in a convergent and enantioselective manner. The tetrahydrofuranol cores of amphirionin-4 were constructed in optically active form by enzymatic resolution of racemic cis-3-hydroxy-5-methyldihydrofuran-2(3H)-one. The polyene side chain was efficiently synthesized using Stille coupling. The remote C8-stereocenter was constructed using the Nozaki-Hiyama-Kishi coupling reaction. Detailed 1H NMR studies of Mosher esters of (−)-amphirionin-4 and (+)-amphirionin-4 were carried out to support the assignment of the absolute configurations of the C-4 and C-8 asymmetric centers of amphirionin-4.
Co-reporter:Arun K. Ghosh, Sandra Gemma, Jun Takayama, Abigail Baldridge, Sofiya Leshchenko-Yashchuk, Heather B. Miller, Yuan-Fang Wang, Andrey Y. Kovalevsky, Yashiro Koh, Irene T. Weber and Hiroaki Mitsuya
Organic & Biomolecular Chemistry 2008 - vol. 6(Issue 20) pp:NaN3713-3713
Publication Date(Web):2008/08/11
DOI:10.1039/B809178A
Recently, we designed a series of novel HIV-1 protease inhibitors incorporating a stereochemically defined bicyclic fused cyclopentyl (Cp-THF) urethane as the high affinity P2-ligand. Inhibitor 1 with this P2-ligand has shown very impressive potency against multi-drug-resistant clinical isolates. Based upon the 1-bound HIV-1 protease X-ray structure, we have now designed and synthesized a number of meso-bicyclic ligands which can conceivably interact similarly to the Cp-THF ligand. The design of meso-ligands is quite attractive as they do not contain any stereocenters. Inhibitors incorporating urethanes of bicyclic-1,3-dioxolane and bicyclic-1,4-dioxane have shown potent enzyme inhibitory and antiviral activities. Inhibitor 2 (Ki = 0.11 nM; IC50 = 3.8 nM) displayed very potent antiviral activity in this series. While inhibitor 3 showed comparable enzyme inhibitory activity (Ki = 0.18 nM) its antiviral activity (IC50 = 170 nM) was significantly weaker than inhibitor 2. Inhibitor 2 maintained an antiviral potency against a series of multi-drug resistant clinical isolates comparable to amprenavir. A protein–ligand X-ray structure of 3-bound HIV-1 protease revealed a number of key hydrogen bonding interactions at the S2-subsite. We have created an active model of inhibitor 2 based upon this X-ray structure.
Co-reporter:Arun K. Ghosh and Zhi-Hua Chen
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 22) pp:NaN3571-3571
Publication Date(Web):2014/04/10
DOI:10.1039/C4OB00511B
Efficient intramolecular N/O-nucleophilic cyclization of 2-aryl indoles has been developed to afford the corresponding 2-aza-3-oxaindolines and 3-indolinones in 80–95% yield. The methods provided convenient access to fused imidazo[1,2-c]oxazolidinone, oxazolidine, or tetrahydro-1,3-oxazine cores under mild conditions.
Co-reporter:Arun K. Ghosh, Cuthbert D. Martyr, Luke A. Kassekert, Prasanth R. Nyalapatla, Melinda Steffey, Johnson Agniswamy, Yuan-Fang Wang, Irene T. Weber, Masayuki Amano and Hiroaki Mitsuya
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 48) pp:NaN11621-11621
Publication Date(Web):2015/10/14
DOI:10.1039/C5OB01930C
Design, synthesis, biological and X-ray crystallographic studies of a series of potent HIV-1 protease inhibitors are described. Various polar functionalities have been incorporated on the tetrahydropyranyl-tetrahydrofuran-derived P2 ligand to interact with the backbone atoms in the S2-subsite. The majority of the inhibitors showed very potent enzyme inhibitory and antiviral activity. Two high-resolution X-ray structures of 30b- and 30j-bound HIV-1 protease provide insight into ligand-binding site interactions. In particular, the polar functionalities on the P2-ligand appear to form unique hydrogen bonds with Gly48 amide NH and amide carbonyl groups in the flap region.
Co-reporter:Arun K. Ghosh, Khriesto A. Shurrush and Zachary L. Dawson
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 44) pp:NaN7777-7777
Publication Date(Web):2013/10/11
DOI:10.1039/C3OB41541D
The asymmetric total synthesis of the anti-proliferative macrolide (+)-neopeltolide has been completed. The stereochemically defined trisubstituted tetrahydropyran ring was constructed via a catalytic hetero-Diels–Alder reaction creating two new chiral centers in a highly diastereoselective manner. The other key features of this synthesis included Brown's asymmetric allylation to install the requisite C-11 and C-13 stereocenters. The synthesis of the oxazole side chain consisted of a hydrozirconation of an alkynyl stannane to establish the Z stereochemistry, followed by a palladium catalyzed cross coupling to introduce the desired Z olefin in the oxazole side chain.
Co-reporter:Arun K. Ghosh, Gary E. Schiltz, Linah N. Rusere, Heather L. Osswald, D. Eric Walters, Masayuki Amano and Hiroaki Mitsuya
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 35) pp:NaN6854-6854
Publication Date(Web):2014/07/22
DOI:10.1039/C4OB00738G
A series of potent macrocyclic HIV-1 protease inhibitors have been designed and synthesized. The compounds incorporated 16- to 19-membered macrocyclic rings between a nelfinavir-like P2 ligand and a tyrosine side chain containing a hydroxyethylamine sulfonamide isostere. All cyclic inhibitors are more potent than their corresponding acyclic counterparts. Saturated derivatives showed slight reduction of potency compared to the respective unsaturated derivatives. Compound 8a containing a 16-membered ring as the P1–P2 ligand showed the most potent enzyme inhibitory and antiviral activity.
Co-reporter:Arun K. Ghosh, Kai Lv, Nianchun Ma, Emilio L. Cárdenas, Kerstin A. Effenberger and Melissa S. Jurica
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 23) pp:NaN5271-5271
Publication Date(Web):2016/05/18
DOI:10.1039/C6OB00725B
Herboxidiene is a potent inhibitor of spliceosomes. It exhibits excellent anticancer activity against multiple human cancer cell lines. Herein, we describe an enantioselective synthesis of a desmethyl derivative and the corresponding carba-derivatives of herboxidiene. The synthesis involved Suzuki coupling of a vinyl iodide with boronate as the key reaction. For the synthesis of carba-derivatives, the corresponding optically active cyclohexane-1,3-dicarbonyl derivatives were synthesized using an enantioselective desymmetrization of meso-anhydride. The biological properties of these derivatives were evaluated in an in vitro splicing assay.
Co-reporter:Arun K. Ghosh, Luke A. Kassekert and Joseph D. Bungard
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 48) pp:NaN11370-11370
Publication Date(Web):2016/10/20
DOI:10.1039/C6OB02051H
We have elucidated the complete absolute configuration of callyspongiolide and unambiguously assigned its stereochemistry at the C-21 center through synthesis. Four stereoisomers of callyspongiolide were synthesized in a convergent and enantioselective manner. A late-stage Sonogashira coupling forges the diene-ynic side chain. Other notable reactions are Yonemitsu's variation of Yamaguchi macrolactonization to cyclize an alkynic seco acid, highly trans-selective Julia–Kocienski olefination, CBS reduction to set the C-21 stereocenter, and methyl cuprate addition to an unsaturated pyranone to install the C-5 methyl center.
Co-reporter:Arun K. Ghosh and Jorden Kass
Chemical Communications 2010 - vol. 46(Issue 8) pp:NaN1220-1220
Publication Date(Web):2010/01/25
DOI:10.1039/B924807B
We have developed a practical synthesis of unique nucleoside derivatives via TiCl4 promoted multicomponent reaction of optically active dihydrofuran, ethyl pyruvate/glyoxylate, and a TMS protected nucleobase in a single-pot operation.
Co-reporter:Arun K. Ghosh, Bhavanam Sekhara Reddy, Yu-Chen Yen, Emilio L. Cárdenas, Kalapala Venkateswara Rao, Deborah Downs, Xiangping Huang, Jordan Tang and Andrew D. Mesecar
Chemical Science (2010-Present) 2016 - vol. 7(Issue 5) pp:NaN3122-3122
Publication Date(Web):2016/02/04
DOI:10.1039/C5SC03718B
Design, synthesis and evaluation of very potent and selective β-secretase 2 (memapsin 1, BACE 2) inhibitors are described. The inhibitors were designed specifically to interact with the S2′-site of β-secretase 2 to provide >170000-fold selectivity over β-secretase (BACE 1) and >15000-fold selectivity over cathepsin D. BACE 2 is implicated in type 2 diabetes. The studies serve as an important guide to selective BACE 2 inhibitors.




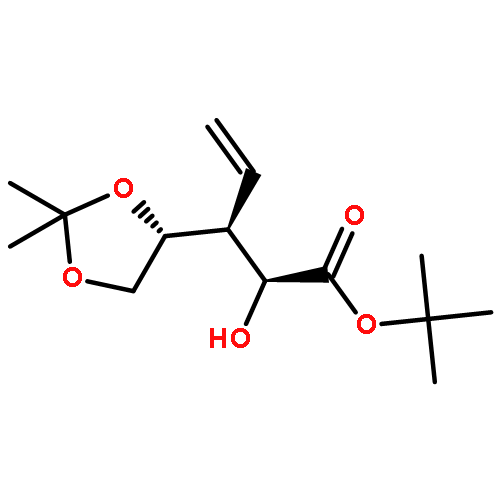

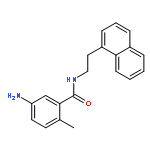
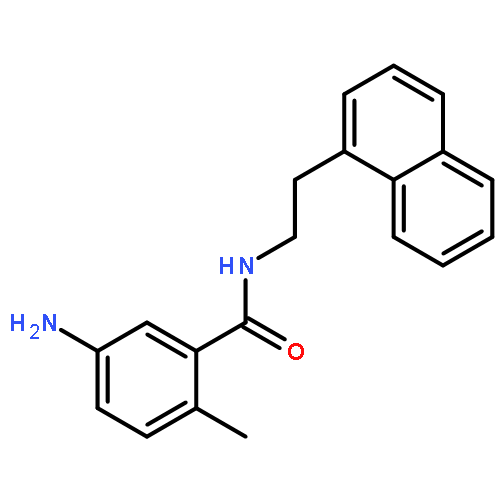



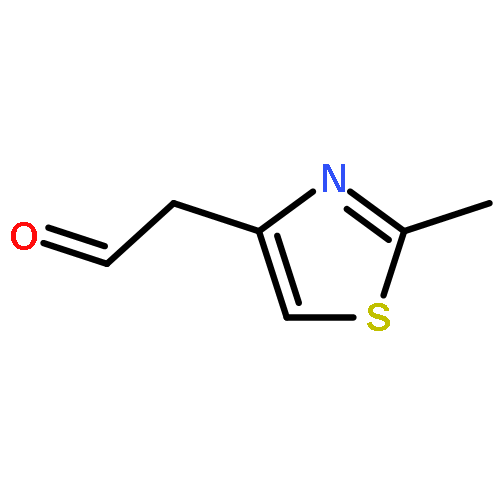


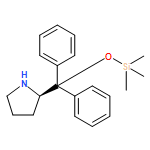



![N-{(2S,3R)-3-Hydroxy-4-[(3-methoxybenzyl)amino]-1-phenyl-2-butany l}-5-[methyl(methylsulfonyl)amino]-N'-[(1R)-1-phenylethyl]isophth alamide](http://img.cochemist.com/ccimg/913100/913071-81-5.png)
![N-{(2S,3R)-3-Hydroxy-4-[(3-methoxybenzyl)amino]-1-phenyl-2-butany l}-5-[methyl(methylsulfonyl)amino]-N'-[(1R)-1-phenylethyl]isophth alamide](http://img.cochemist.com/ccimg/913100/913071-81-5_b.png)
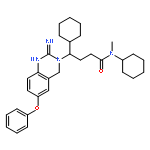
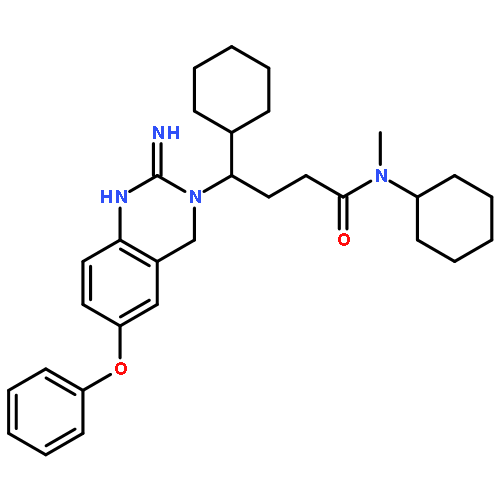
![Carbamic acid, N-[(1S)-1-cyclohexyl-4-(cyclohexylmethylamino)-4-oxobutyl]-, 1,1-dimethylethyl ester](/data/chemimg/1375500/876761-26-1.png)
![Carbamic acid, N-[(1S)-1-cyclohexyl-4-(cyclohexylmethylamino)-4-oxobutyl]-, 1,1-dimethylethyl ester](/data/chemimg/1375500/876761-26-1_b.png)
![D-Tryptophan, N-[(1,1-dimethylethoxy)carbonyl]-L-alanyl-N-methyl-](http://img.cochemist.com/ccimg/873400/873326-27-3.png)
![D-Tryptophan, N-[(1,1-dimethylethoxy)carbonyl]-L-alanyl-N-methyl-](http://img.cochemist.com/ccimg/873400/873326-27-3_b.png)


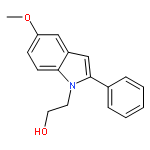

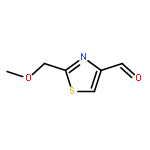





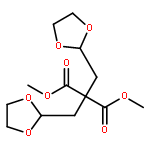
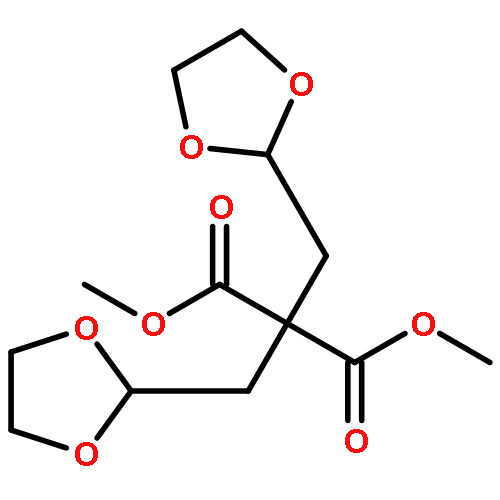
![(3aR,6aR)-3a,4,5,6a-tetrahydrofuro[2,3-b]furan-3(2H)-one](http://img.cochemist.com/ccimg/809300/809286-93-9.png)
![(3aR,6aR)-3a,4,5,6a-tetrahydrofuro[2,3-b]furan-3(2H)-one](http://img.cochemist.com/ccimg/809300/809286-93-9_b.png)
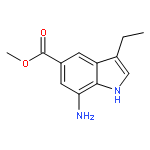
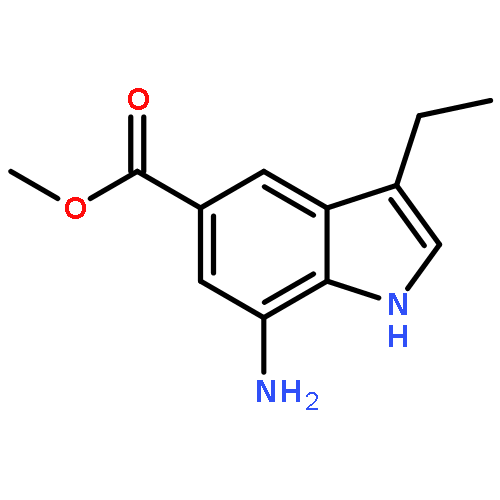
![1H-Tetrazole, 5-[[7-(2-heptyl-1,3-dioxolan-2-yl)heptyl]sulfonyl]-1-phenyl-](http://img.cochemist.com/ccimg/777900/777891-05-1.png)
![1H-Tetrazole, 5-[[7-(2-heptyl-1,3-dioxolan-2-yl)heptyl]sulfonyl]-1-phenyl-](http://img.cochemist.com/ccimg/777900/777891-05-1_b.png)

![Benzoic acid,3-[methyl(methylsulfonyl)amino]-5-[[[(1R)-1-phenylethyl]amino]carbonyl]-](http://img.cochemist.com/ccimg/695300/695215-92-0.png)
![Benzoic acid,3-[methyl(methylsulfonyl)amino]-5-[[[(1R)-1-phenylethyl]amino]carbonyl]-](http://img.cochemist.com/ccimg/695300/695215-92-0_b.png)


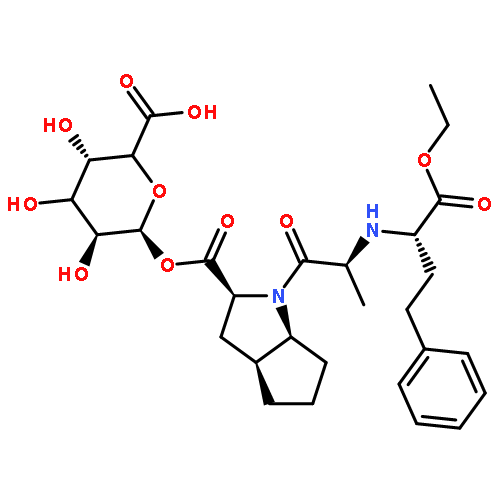


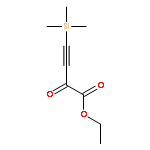
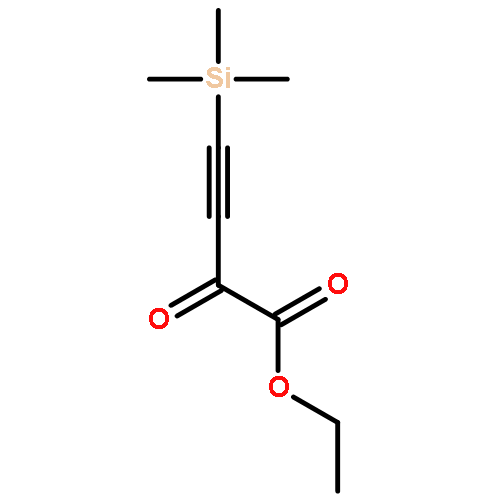


![Carbamic acid,N-[(1S,2R)-3-[(1,3-benzodioxol-5-ylsulfonyl)(2-methylpropyl)amino]-2-hydroxy-1-[[4-[(2-methyl-4-thiazolyl)methoxy]phenyl]methyl]propyl]-,(3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester](http://img.cochemist.com/ccimg/313700/313682-08-5.png)
![Carbamic acid,N-[(1S,2R)-3-[(1,3-benzodioxol-5-ylsulfonyl)(2-methylpropyl)amino]-2-hydroxy-1-[[4-[(2-methyl-4-thiazolyl)methoxy]phenyl]methyl]propyl]-,(3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester](http://img.cochemist.com/ccimg/313700/313682-08-5_b.png)
![4-Pentenamide, N-[(1R,2R)-2-hydroxy-1-methyl-2-phenylethyl]-N,2-dimethyl-, (2S)-](/data/chemimg/120300/277297-37-7.png)
![4-Pentenamide, N-[(1R,2R)-2-hydroxy-1-methyl-2-phenylethyl]-N,2-dimethyl-, (2S)-](/data/chemimg/120300/277297-37-7_b.png)
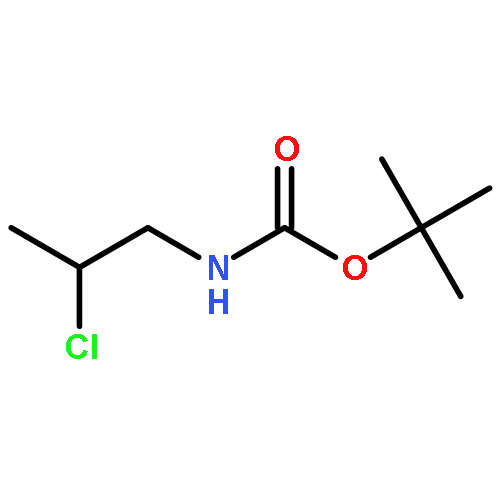
![4-Thiazolidinecarboxamide, 5,5-dimethyl-N-[(2-methylphenyl)methyl]-, (4R)-](/data/chemimg/1798300/226998-04-5.png)
![4-Thiazolidinecarboxamide, 5,5-dimethyl-N-[(2-methylphenyl)methyl]-, (4R)-](/data/chemimg/1798300/226998-04-5_b.png)
![2-Oxazolidinone, 3-[(2S,3S)-3-hydroxy-2,4-dimethyl-1-oxo-4-penten-1-yl]-4-(phenylmethyl)-, (4S)-](/data/chemimg/1798100/226703-78-2.png)
![2-Oxazolidinone, 3-[(2S,3S)-3-hydroxy-2,4-dimethyl-1-oxo-4-penten-1-yl]-4-(phenylmethyl)-, (4S)-](/data/chemimg/1798100/226703-78-2_b.png)
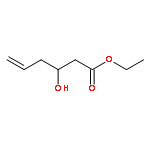
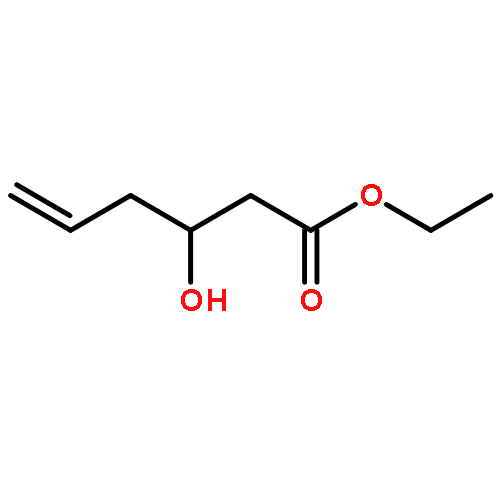
![Propanamide, N-[(1R,2R)-2-hydroxy-1-methyl-2-phenylethyl]-N-methyl-](http://img.cochemist.com/ccimg/192100/192060-67-6.png)
![Propanamide, N-[(1R,2R)-2-hydroxy-1-methyl-2-phenylethyl]-N-methyl-](http://img.cochemist.com/ccimg/192100/192060-67-6_b.png)


![(4R)-3-{(2S,3S)-2-hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4-phenylbutanoyl}-5,5-dimethyl-N-(2-methylbenzyl)-1,3-thiazolidine-4-carboxamide](http://img.cochemist.com/ccimg/186600/186538-00-1.png)
![(4R)-3-{(2S,3S)-2-hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4-phenylbutanoyl}-5,5-dimethyl-N-(2-methylbenzyl)-1,3-thiazolidine-4-carboxamide](http://img.cochemist.com/ccimg/186600/186538-00-1_b.png)
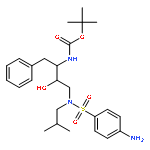


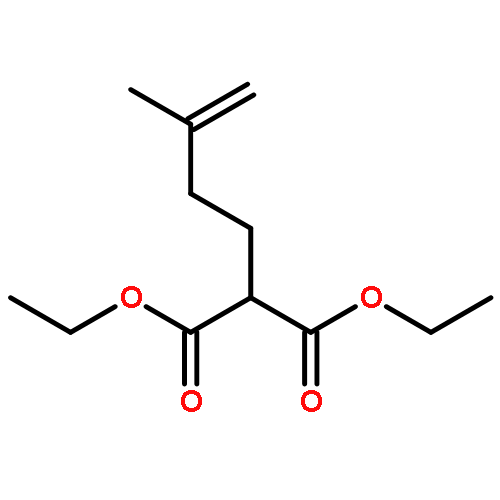
![2-Pyridinesulfonamide,N-[3-[(1S)-1-[(6R)-5,6-dihydro-4-hydroxy-2-oxo-6-(2-phenylethyl)-6-propyl-2H-pyran-3-yl]propyl]phenyl]-5-(trifluoromethyl)-](http://img.cochemist.com/ccimg/174600/174590-27-3.png)
![2-Pyridinesulfonamide,N-[3-[(1S)-1-[(6R)-5,6-dihydro-4-hydroxy-2-oxo-6-(2-phenylethyl)-6-propyl-2H-pyran-3-yl]propyl]phenyl]-5-(trifluoromethyl)-](http://img.cochemist.com/ccimg/174600/174590-27-3_b.png)
![Phosphine oxide, [2-(dibromomethyl)phenyl]diphenyl-](/data/chemimg/2215600/163272-09-1.png)
![Phosphine oxide, [2-(dibromomethyl)phenyl]diphenyl-](/data/chemimg/2215600/163272-09-1_b.png)


![Furo[2,3-b]furan-3-ol, hexahydro-, (3S,3aR,6aS)-](http://img.cochemist.com/ccimg/157000/156928-10-8.png)
![Furo[2,3-b]furan-3-ol, hexahydro-, (3S,3aR,6aS)-](http://img.cochemist.com/ccimg/157000/156928-10-8_b.png)


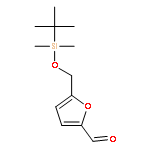
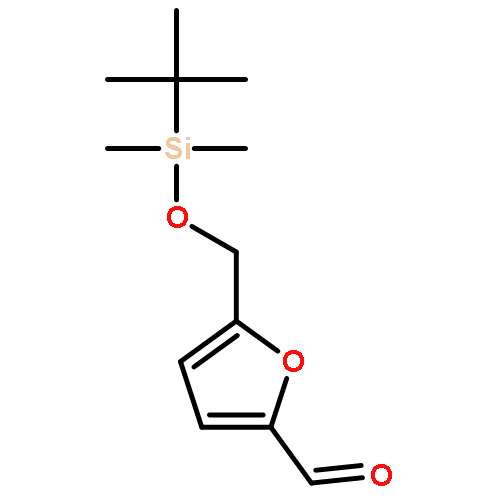
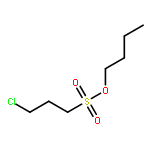




![Oxirane, [[(4-methoxyphenyl)methoxy]methyl]-, (2S)-](http://img.cochemist.com/ccimg/144100/144069-33-0.png)
![Oxirane, [[(4-methoxyphenyl)methoxy]methyl]-, (2S)-](http://img.cochemist.com/ccimg/144100/144069-33-0_b.png)








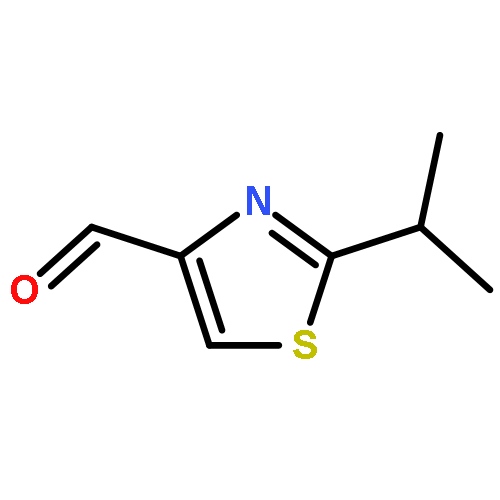
![(2,R,5S)-5-(tert-butoxycarbonylamino)-3-methyl-2-[(2-bromo-1H-indol-3-yl)methyl]-4-oxo-3-azahexanoic acid](http://img.cochemist.com/ccimg/131800/131791-79-2.png)
![(2,R,5S)-5-(tert-butoxycarbonylamino)-3-methyl-2-[(2-bromo-1H-indol-3-yl)methyl]-4-oxo-3-azahexanoic acid](http://img.cochemist.com/ccimg/131800/131791-79-2_b.png)
![2-Propanone, 1-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]-](/data/chemimg/120000/120342-04-3.png)
![2-Propanone, 1-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]-](/data/chemimg/120000/120342-04-3_b.png)
![4-Nonenoic acid, 8-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,4,6-trimethyl-, (2S,4E,6R,8S)-](/data/chemimg/1677800/117289-84-6.png)
![4-Nonenoic acid, 8-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,4,6-trimethyl-, (2S,4E,6R,8S)-](/data/chemimg/1677800/117289-84-6_b.png)
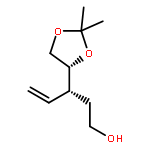
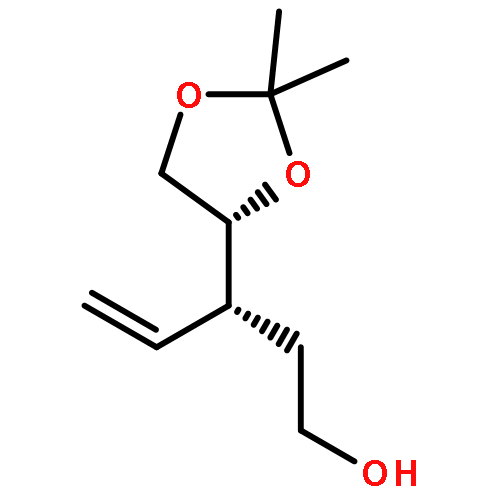
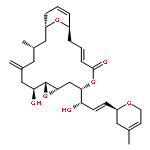
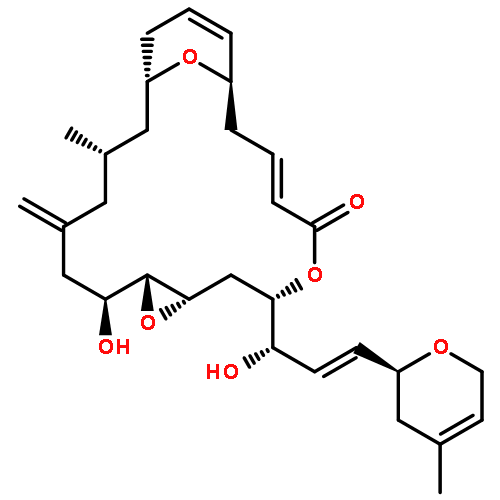
![2-Cyclopenten-1-ol, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, acetate,(1R,4S)-](http://img.cochemist.com/ccimg/115100/115074-49-2.png)
![2-Cyclopenten-1-ol, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, acetate,(1R,4S)-](http://img.cochemist.com/ccimg/115100/115074-49-2_b.png)
![Propanal, 3-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/112900/112897-03-7.png)
![Propanal, 3-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/112900/112897-03-7_b.png)


![2-Oxiranemethanol, 2-[(phenylmethoxy)methyl]-, (2R)-](/data/chemimg/108800/109240-65-5.png)
![2-Oxiranemethanol, 2-[(phenylmethoxy)methyl]-, (2R)-](/data/chemimg/108800/109240-65-5_b.png)
![Silane, (1,1-dimethylethyl)[(2S)-oxiranylmethoxy]diphenyl-](http://img.cochemist.com/ccimg/107100/107033-46-5.png)
![Silane, (1,1-dimethylethyl)[(2S)-oxiranylmethoxy]diphenyl-](http://img.cochemist.com/ccimg/107100/107033-46-5_b.png)




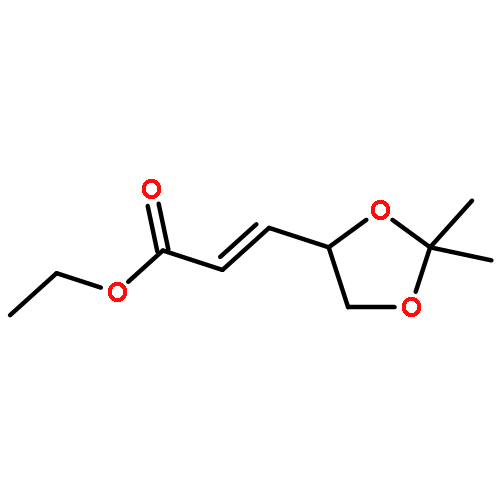


![2(3H)-Furanone,5-[[[(1,1-dimethylethyl)diphenylsilyl]oxy]methyl]dihydro-, (S)-](http://img.cochemist.com/ccimg/102800/102717-29-3.png)
![2(3H)-Furanone,5-[[[(1,1-dimethylethyl)diphenylsilyl]oxy]methyl]dihydro-, (S)-](http://img.cochemist.com/ccimg/102800/102717-29-3_b.png)
![(1S)-1-[(2R)-OXIRAN-2-YL]PROP-2-EN-1-OL](http://img.cochemist.com/ccimg/100100/100017-22-9.png)
![(1S)-1-[(2R)-OXIRAN-2-YL]PROP-2-EN-1-OL](http://img.cochemist.com/ccimg/100100/100017-22-9_b.png)
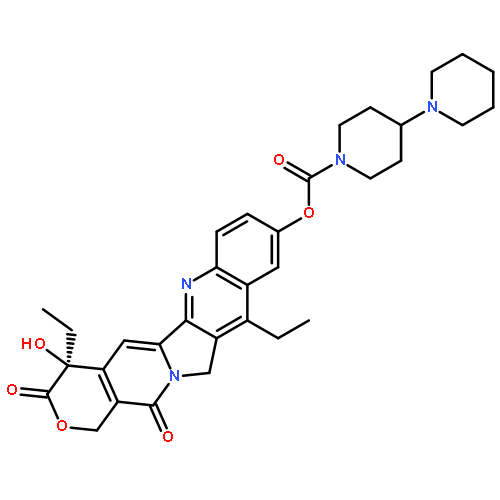
![2-Propenoic acid,3-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]-, ethyl ester, (2Z)-](http://img.cochemist.com/ccimg/92000/91926-90-8.png)
![2-Propenoic acid,3-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]-, ethyl ester, (2Z)-](http://img.cochemist.com/ccimg/92000/91926-90-8_b.png)

![Acetic acid, [[(4-methylphenyl)sulfonyl]imino]-, ethyl ester](http://img.cochemist.com/ccimg/83700/83670-53-5.png)
![Acetic acid, [[(4-methylphenyl)sulfonyl]imino]-, ethyl ester](http://img.cochemist.com/ccimg/83700/83670-53-5_b.png)
![2-PROPEN-1-OL, 3-[(4S)-2,2-DIMETHYL-1,3-DIOXOLAN-4-YL]-, (2Z)-](http://img.cochemist.com/ccimg/80600/80532-35-0.png)
![2-PROPEN-1-OL, 3-[(4S)-2,2-DIMETHYL-1,3-DIOXOLAN-4-YL]-, (2Z)-](http://img.cochemist.com/ccimg/80600/80532-35-0_b.png)
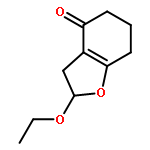

![(S)-2-[(benzyloxycarbonyl)amino]-4-(methylsulfinyl)butanoic acid methyl ester](http://img.cochemist.com/ccimg/75300/75266-39-6.png)
![(S)-2-[(benzyloxycarbonyl)amino]-4-(methylsulfinyl)butanoic acid methyl ester](http://img.cochemist.com/ccimg/75300/75266-39-6_b.png)



![Phosphine oxide, [2-(bromomethyl)phenyl]diphenyl-](/data/chemimg/1143600/68718-85-4.png)
![Phosphine oxide, [2-(bromomethyl)phenyl]diphenyl-](/data/chemimg/1143600/68718-85-4_b.png)


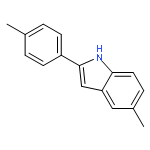






![Butanoic acid, 2-[[(1,1-dimethylethoxy)carbonyl]amino]-4-(methylsulfinyl)-, methyl ester, (2S)-](/data/chemimg/1902000/63701-09-7.png)
![Butanoic acid, 2-[[(1,1-dimethylethoxy)carbonyl]amino]-4-(methylsulfinyl)-, methyl ester, (2S)-](/data/chemimg/1902000/63701-09-7_b.png)

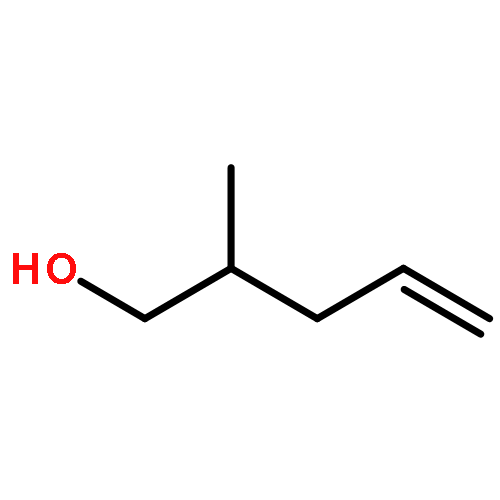


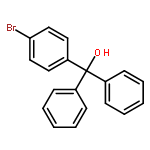
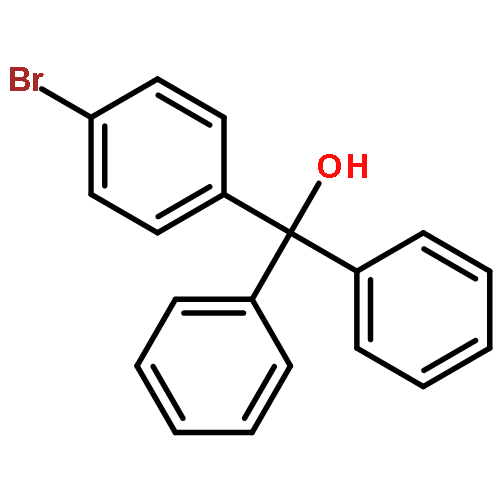
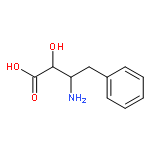
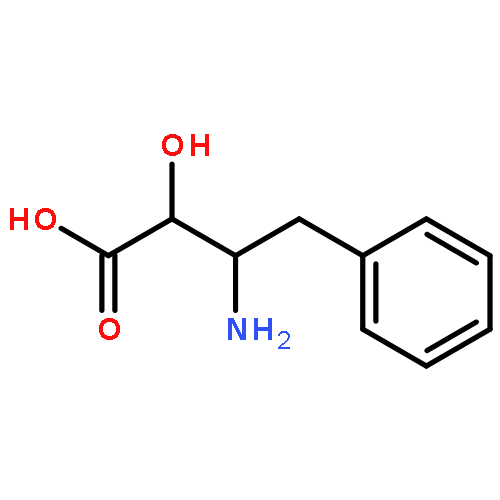

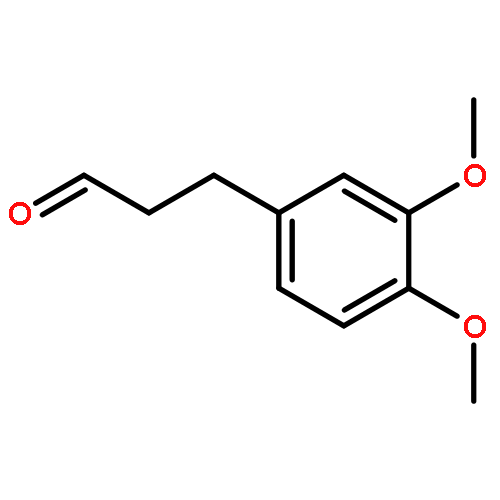
![2-CYCLOPENTEN-1-OL, 4-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-, (1S,4S)-](http://img.cochemist.com/ccimg/61800/61740-31-6.png)
![2-CYCLOPENTEN-1-OL, 4-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-, (1S,4S)-](http://img.cochemist.com/ccimg/61800/61740-31-6_b.png)
![2-Propen-1-one, 1-phenyl-3-[4-(trifluoromethyl)phenyl]-](/data/chemimg/1516700/61637-11-4.png)
![2-Propen-1-one, 1-phenyl-3-[4-(trifluoromethyl)phenyl]-](/data/chemimg/1516700/61637-11-4_b.png)





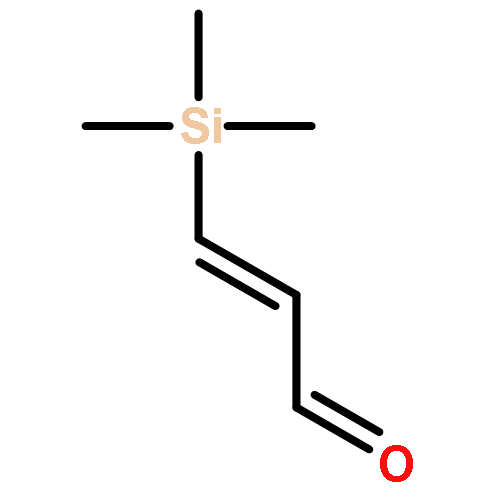
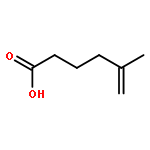
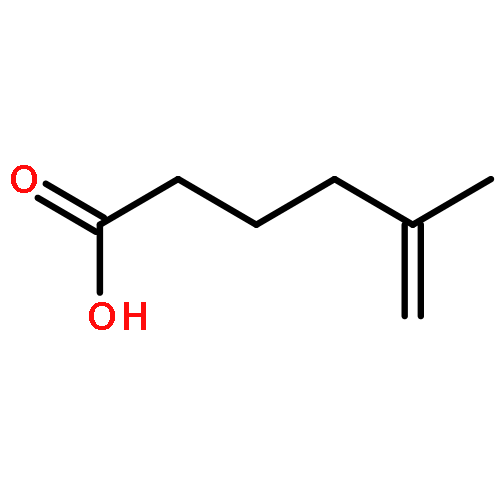


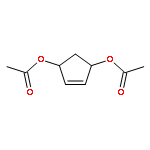

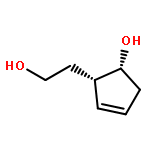

![2H-Cyclopenta[b]furan-2-one,3,3a,6,6a-tetrahydro-, (3aS,6aR)-](http://img.cochemist.com/ccimg/54500/54483-22-6.png)
![2H-Cyclopenta[b]furan-2-one,3,3a,6,6a-tetrahydro-, (3aS,6aR)-](http://img.cochemist.com/ccimg/54500/54483-22-6_b.png)
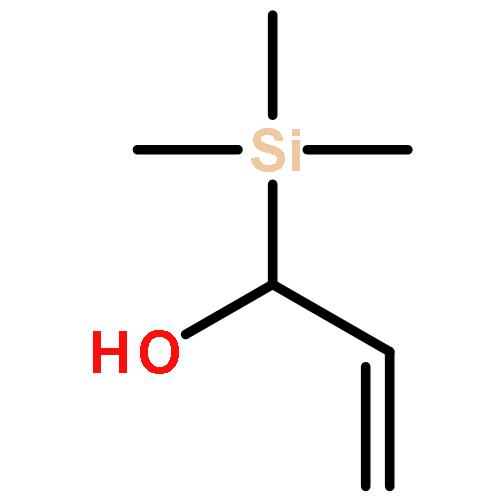

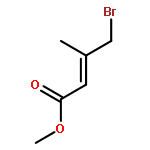
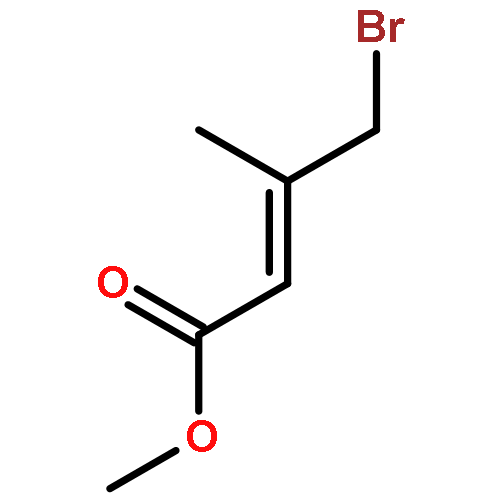

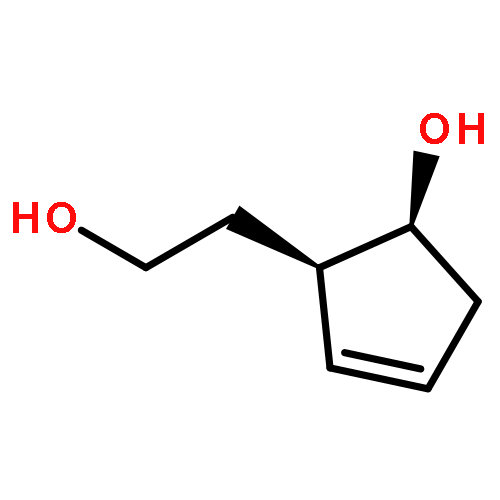
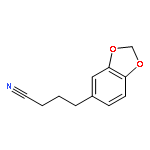
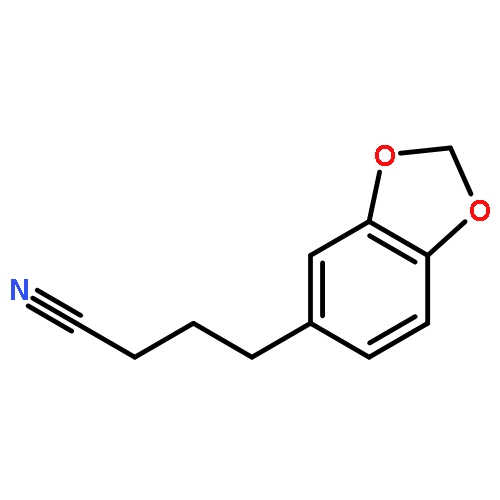



















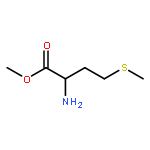





![3-BENZO[1,3]DIOXOL-5-YL-PROPAN-1-OL](http://img.cochemist.com/ccimg/7100/7031-03-0.png)
![3-BENZO[1,3]DIOXOL-5-YL-PROPAN-1-OL](http://img.cochemist.com/ccimg/7100/7031-03-0_b.png)











![1H-Purin-6-amine, N-[(3-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/4300/4217-23-6.png)
![1H-Purin-6-amine, N-[(3-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/4300/4217-23-6_b.png)
















![Phosphine, [2-[(diphenylphosphino)methyl]phenyl]diphenyl-](/data/chemimg/3720900/1384422-33-6.png)
![Phosphine, [2-[(diphenylphosphino)methyl]phenyl]diphenyl-](/data/chemimg/3720900/1384422-33-6_b.png)
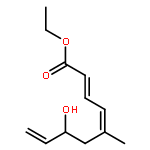
![6,21-Dioxabicyclo[15.3.1]heneicosa-2,8,10,14-tetraene-5-carboxaldehyde, 3,11-dimethyl-19-methylene-7,13-dioxo-, (1S,2E,5S,8E,10Z,14E,17S)-](/data/chemimg/125700/596812-41-8.png)
![6,21-Dioxabicyclo[15.3.1]heneicosa-2,8,10,14-tetraene-5-carboxaldehyde, 3,11-dimethyl-19-methylene-7,13-dioxo-, (1S,2E,5S,8E,10Z,14E,17S)-](/data/chemimg/125700/596812-41-8_b.png)
![2-Pentenamide, 4-(acetyloxy)-N-[(2R,3R,5S,6S)-tetrahydro-6-[(2E,4E)-5-[(3R,4R,5R,7S)-4-hydroxy-7-methoxy-7-methyl-1,6-dioxaspiro[2.5]oct-5-yl]-3-methyl-2,4-pentadien-1-yl]-2,5-dimethyl-2H-pyran-3-yl]-, (2Z,4S)-](/data/chemimg/3738700/391611-36-2.png)
![2-Pentenamide, 4-(acetyloxy)-N-[(2R,3R,5S,6S)-tetrahydro-6-[(2E,4E)-5-[(3R,4R,5R,7S)-4-hydroxy-7-methoxy-7-methyl-1,6-dioxaspiro[2.5]oct-5-yl]-3-methyl-2,4-pentadien-1-yl]-2,5-dimethyl-2H-pyran-3-yl]-, (2Z,4S)-](/data/chemimg/3738700/391611-36-2_b.png)




![2-Pentenamide,4-(acetyloxy)-N-[(2R,3R,5S,6S)-6-[(2E,4E)-5-[(3R,4R,5R,7S)-4,7-dihydroxy-7-methyl-1,6-dioxaspiro[2.5]oct-5-yl]-3-methyl-2,4-pentadien-1-yl]tetrahydro-2,5-dimethyl-2H-pyran-3-yl]-,(2Z,4S)-](http://img.cochemist.com/ccimg/146500/146478-72-0.png)
![2-Pentenamide,4-(acetyloxy)-N-[(2R,3R,5S,6S)-6-[(2E,4E)-5-[(3R,4R,5R,7S)-4,7-dihydroxy-7-methyl-1,6-dioxaspiro[2.5]oct-5-yl]-3-methyl-2,4-pentadien-1-yl]tetrahydro-2,5-dimethyl-2H-pyran-3-yl]-,(2Z,4S)-](http://img.cochemist.com/ccimg/146500/146478-72-0_b.png)
![2-Cyclopenten-1-ol, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (1R,4S)-](http://img.cochemist.com/ccimg/120600/120520-91-4.png)
![2-Cyclopenten-1-ol, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (1R,4S)-](http://img.cochemist.com/ccimg/120600/120520-91-4_b.png)
![2-Pentenal, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2E)-](http://img.cochemist.com/ccimg/118700/118635-66-8.png)
![2-Pentenal, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2E)-](http://img.cochemist.com/ccimg/118700/118635-66-8_b.png)
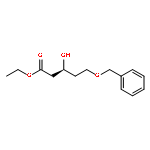
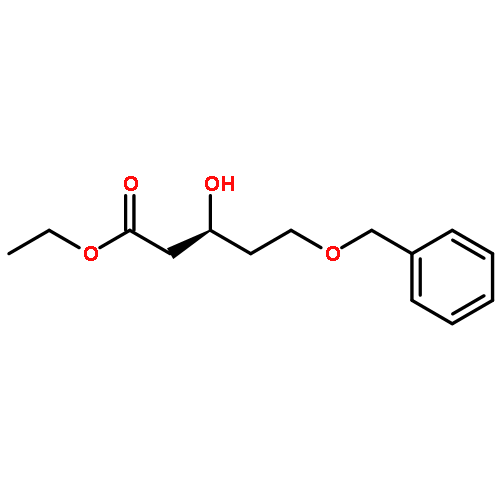


![(2s,3s)-4-[(e)-3-[(1r)-1-carboxy-2-(3,4-dihydroxyphenyl)ethoxy]-3-oxoprop-1-enyl]-2-(3,4-dihydroxyphenyl)-7-hydroxy-2,3-dihydro-1-benzofuran-3-carboxylic Acid](http://img.cochemist.com/ccimg/28900/28831-65-4.png)
![(2s,3s)-4-[(e)-3-[(1r)-1-carboxy-2-(3,4-dihydroxyphenyl)ethoxy]-3-oxoprop-1-enyl]-2-(3,4-dihydroxyphenyl)-7-hydroxy-2,3-dihydro-1-benzofuran-3-carboxylic Acid](http://img.cochemist.com/ccimg/28900/28831-65-4_b.png)

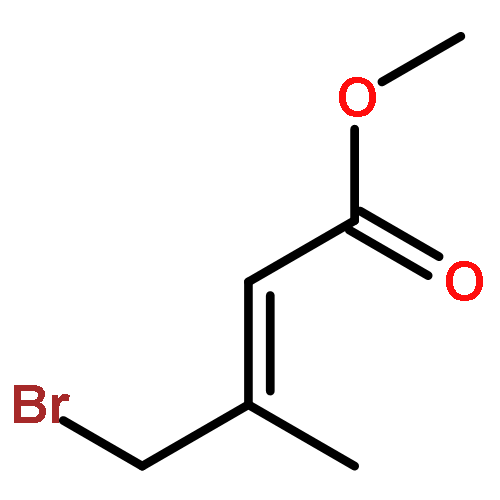
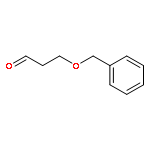
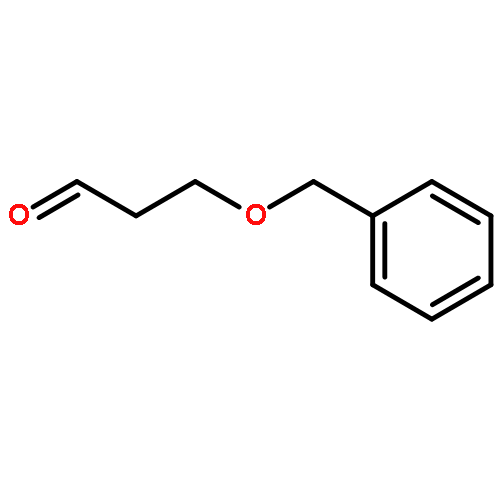
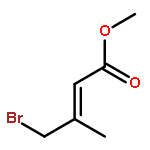
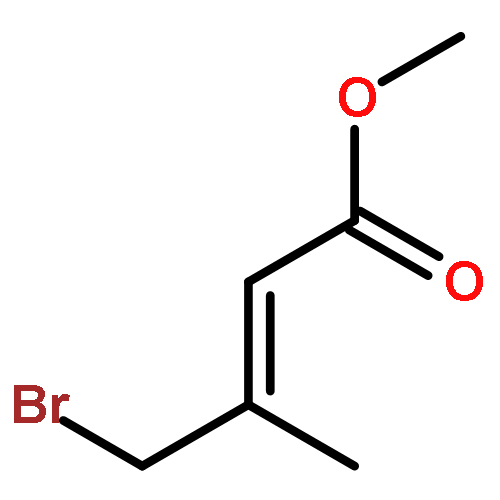
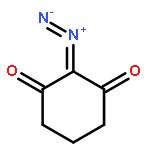

![N-[1(S)-Benzyl-2(R)-hydroxy-3-[N-isobutyl-N-(4-methoxyphenylsulfonyl)amino]propyl]carbamic acid (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester](http://img.cochemist.com/ccimg/206400/206362-00-7.png)
![N-[1(S)-Benzyl-2(R)-hydroxy-3-[N-isobutyl-N-(4-methoxyphenylsulfonyl)amino]propyl]carbamic acid (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester](http://img.cochemist.com/ccimg/206400/206362-00-7_b.png)
![Acetaldehyde, 2-[[(4-methylphenyl)sulfonyl]oxy]-](/data/chemimg/110700/172328-35-7.png)
![Acetaldehyde, 2-[[(4-methylphenyl)sulfonyl]oxy]-](/data/chemimg/110700/172328-35-7_b.png)
amino]-1-(phenylmethyl)propyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/159100/159006-03-8.png)
amino]-1-(phenylmethyl)propyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/159100/159006-03-8_b.png)
![L-glycero-L-gluco-Heptitol,5,6-anhydro-6-C-[(2S,3E,5E)-6-[(2S,3S,6R)-6-(carboxymethyl)tetrahydro-3-methyl-2H-pyran-2-yl]-2-methyl-3,5-heptadien-1-yl]-1,4,7-trideoxy-4-methyl-3-O-methyl-](http://img.cochemist.com/ccimg/142900/142861-00-5.png)
![L-glycero-L-gluco-Heptitol,5,6-anhydro-6-C-[(2S,3E,5E)-6-[(2S,3S,6R)-6-(carboxymethyl)tetrahydro-3-methyl-2H-pyran-2-yl]-2-methyl-3,5-heptadien-1-yl]-1,4,7-trideoxy-4-methyl-3-O-methyl-](http://img.cochemist.com/ccimg/142900/142861-00-5_b.png)