Co-reporter:Jing Zhang, Jiye Huang, Zilan Song, Lin Guo, Wenxian Cai, Yun Wang, Xuechu Zhen, Ao Zhang
European Journal of Medicinal Chemistry 2014 Volume 85() pp:16-26
Publication Date(Web):6 October 2014
DOI:10.1016/j.ejmech.2014.07.059
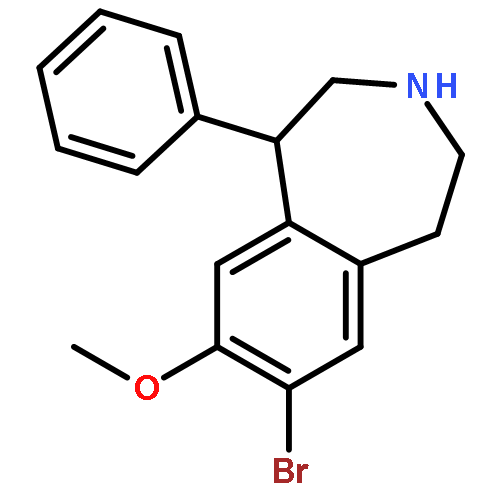
•Aryl-N3-benzazepine framework is the prototypical dopamine D1 receptor ligand.•Our study directly focused on modification of the metabolic site-catechol moiety.•Compounds 13b–d displayed Ki values of 270–370 nM at the D1 receptor.•Most compounds possess high affinity less than 10 nM at the 5-HT2A receptor.•13a was the most potent with a Ki value of 4.5 nM at the 5-HT2A receptor.A series of new benzazepines with modification on the catecholic fragment were designed. The 8-hydroxyl group, other than the 7-hydroxyl was confirmed crucial to the interaction with the dopamine D1 receptor. Subsequent replacement of the 7-hydroxyl with benzylamino groups was found tolerable. 7-(m-Chlorophenyl)methylamino- and 7-(m- or o-tolyl)methylamino-substituted benzazepines 13b–d displayed Ki values of 270–370 nM at the D1 receptor, which were slightly more potent than that of parent compound 1. In addition, 7-(arylmethyl)amino-benzazepines 13a–c were found possessing high binding affinities less than 10 nM at the 5-HT2A receptor. Among them, the non-substituted 7-benzylamino analogue 13a was the most potent showing a Ki values of 4.5 nM at the 5-HT2A receptor and a 5-HT2A/D1 selectivity of 147.
Co-reporter:Xiaolong Jiang;Zilan Song;Chang Xu;Qizheng Yao
European Journal of Organic Chemistry 2014 Volume 2014( Issue 2) pp:
Publication Date(Web):
DOI:10.1002/ejoc.201301295
Abstract
A tandem one-pot Friedel–Crafts alkylation/double cyclization process to conveniently assemble diaryl-fused 2,8-dioxabicyclo[3.3.1]nonanes with aryl substituents at the C-1 position was developed. 2-Hydroxychalcones and naphthol derivatives reacted in refluxing toluene with (D,L)-10-camphorsulfonic acid (CSA) as the catalyst. The transformation conveniently provided the corresponding 2,8-dioxabicyclo[3.3.1]nonanes fused with one phenyl and one naphthyl group in moderate to good yields, and various substitutions on both reaction partners were tolerated. More importantly, aryl- and aza-aryl-fused 2,8-dioxabicyclo[3.3.1]nonanes were prepared for the first time using this procedure.
Co-reporter:Pingyuan Wang, Shanshan Song, Zehong Miao, Guangfu Yang, and Ao Zhang
Organic Letters 2013 Volume 15(Issue 15) pp:3852-3855
Publication Date(Web):July 15, 2013
DOI:10.1021/ol401489x
An efficient one-pot synthesis of polyhydroxyalkyl-substituted pyrroles from 1,2-cyclopropa-3-pyranones with primary amines is reported. With 10% of InBr3 as the catalyst, both aryl- and alkylamines as well as various 1,2-cyclopropa-3-pyranones are well tolerated. This method is highly appealing because of its one-pot process, mild reaction conditions, substrate simplicity, and broad substrate scope.
Co-reporter:Na Ye ; Chuan-Huizi Chen ; TianTian Chen ; Zilan Song ; Jin-Xue He ; Xia-Juan Huan ; Shan-Shan Song ; Qiufeng Liu ; Yi Chen ; Jian Ding ; Yechun Xu ; Ze-Hong Miao
Journal of Medicinal Chemistry 2013 Volume 56(Issue 7) pp:2885-2903
Publication Date(Web):March 8, 2013
DOI:10.1021/jm301825t
A series of benzo[de][1,7]naphthyridin-7(8H)-ones possessing a functionalized long-chain appendage have been designed and evaluated as novel PARP1 inhibitors. The initial effort led to the first-generation PARP1 inhibitor 26 bearing a terminal phthalazin-1(2H)-one framework and showing remarkably high PARP1 inhibitory activity (0.31 nM) but only moderate potency in the cell. Further effort generated the second-generation lead 41, showing high potency against both the PARP1 enzyme and BRCA-deficient cells, especially for the BRCA1-deficient MDA-MB-436 cells (CC50 < 0.26 nM). Mechanistic studies revealed that the new PARP1 inhibitors significantly inhibited H2O2-triggered PARylation in SKOV3 cells, induced cellular accumulation of DNA double-strand breaks, and impaired cell-cycle progression in BRCA2-deficient cells. Significant potentiation on the cytotoxicity of Temozolomide was also observed. The unique structural character and exceptionally high potency of 41 made it stand out as a promising drug candidate worthy for further evaluation.
Co-reporter:Xuefeng Zhang, Xiaolong Jiang, Chunyong Ding, Qizheng Yao and Ao Zhang
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 8) pp:1383-1389
Publication Date(Web):12 Dec 2012
DOI:10.1039/C2OB27020J
An unexpected N-glycosidation reaction of anthracen-1-amine with glycals was identified, and its use in the synthesis of C1′ N-linked analogues of natural product marmycin A was explored. The structures of all these products were determined by 1D and 2D NMR, CD spectra, and X-ray crystal analysis. These products were then subjected to Friedel–Crafts acylation, Dess–Martin oxidation and nucleophilic addition leading to novel natural product analogues bearing a quaternary carbon center.
Co-reporter:Dongyu Wang ; Shanshan Song ; Ye Tian ; Youjun Xu ; Zehong Miao
Journal of Natural Products 2013 Volume 76(Issue 5) pp:974-978
Publication Date(Web):April 22, 2013
DOI:10.1021/np4001027
The first total synthesis of viequeamide A, a natural cyclic depsipeptide isolated from a marine button cyanobacterium, was achieved with the N-Me-Val–Thr peptide bond as the final macrocyclization site. The synthetic product gave nearly identical spectroscopic data to that reported for the natural product.
Co-reporter:Kui Wu;Meining Wang;Qizheng Yao
Chinese Journal of Chemistry 2013 Volume 31( Issue 1) pp:93-99
Publication Date(Web):
DOI:10.1002/cjoc.201201084
Abstract
Natural product mayamycin is the first example in the angucycline class featuring a C-glycoside linkage at the C5-position of the benz[a]anthracenone core with remarkable biological activities. We successfully synthesized the two retrosynthetic fragments, but found that the final C-glycosylation did not occur. Alternatively, an A-ring saturated aglycon was prepared, but the proposed C-glycosylation still did not proceed. Finally, a simplified substrate was used and the subsequent C-glycosylation went through smoothly, giving a two-ring less analogue of mayamycin.
Co-reporter:Dongyu Wang, Xian Jia and Ao Zhang
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 35) pp:7027-7030
Publication Date(Web):20 Jul 2012
DOI:10.1039/C2OB26002F
We have developed a practical method to assemble the proposed structure of natural product veraguamide A (1) by first preparing the three key fragments followed by optimization of the macrocyclization site. Although the synthetic product gave similar optical rotation to that reported for natural product, significant differences in the 1H and 13C NMR spectra were observed, especially the proton and carbon signals in the two N-MeVal moieties.
Co-reporter:Ziwen Wang, Meng Wu, Yi Wang, Zheng Li, Lei Wang, Guifang Han, Fazhong Chen, Yuxiu Liu, Kailiang Wang, Ao Zhang, Linghua Meng, Qingmin Wang
European Journal of Medicinal Chemistry 2012 Volume 51() pp:250-258
Publication Date(Web):May 2012
DOI:10.1016/j.ejmech.2012.02.048
A series of phenanthroindolizidine and phenanthroquinolizidine alkaloids and their 14-amino-derivatives (1–44) were prepared and systematically evaluated for their anti-tumor activities against A549 and HL60 cell lines. The bioassay results showed that most of these alkaloids possess good anti-tumor activities. Especially, compounds 15, 22, 28, 33–36, 40 and 42 displayed low nanomolar or subnanomolar levels of anti-tumor activity. The configuration of (13aS,14S)-14-hydroxyphenanthroindolizidines and (14aR,15R)-15-hydroxyphenanthroquinolizidines was confirmed to be optimal. 14-Amino-phenanthroindolizidines with increased polarity possess good anti-tumor activity, especially for compounds 26 and 28. Most of the phenanthroquinolizidine alkaloids exhibited higher anti-tumor activity than that of phenanthroindolizidine alkaloids. Our present study provides fundamental support for development and optimization of phenanthroindolizidine and phenanthroquinolizidine alkaloids as potential anti-tumor drugs.A series of phenanthroindolizidine and phenanthroquinolizidine alkaloids and their 14-amino-derivatives (1–44) were prepared and systematically evaluated for their anti-tumor activities against A549 and HL60 cell lines.Highlights► A series of phenanthroindolizidine and phenanthroquinolizidine alkaloids and their 14-amino-derivatives were prepared. ► We systematically evaluated their anti-tumor activities against A549 and HL60 cell lines. ► Most of these alkaloids possess good anti-tumor activities. ► Compounds 15, 22, 28, 33–36, 40 and 42 display low nanomolar or subnanomolar levels of anti-tumor activity.
Co-reporter:Kui Wu, Jing Ai, Qiufeng Liu, TianTian Chen, Ailing Zhao, Xia Peng, Yuanxiang Wang, Yinchun Ji, Qizheng Yao, Yechun Xu, Meiyu Geng, Ao Zhang
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 20) pp:6368-6372
Publication Date(Web):15 October 2012
DOI:10.1016/j.bmcl.2012.08.075
Two series of new analogues were designed by replacing the quinoline scaffold of our earlier lead 2 (zgwatinib) with quinoxaline and pyrido[2,3-d]pyrimidine frameworks. Moderate c-Met inhibitory activity was observed in the quinoxaline series. Among the pyrido[2,3-d]pyrimidine series, compounds 13a–c possessing an O-linkage were inactive, whilst the N-linked analogues 15a–c retained c-Met inhibitory potency. Highest activity was observed in the 3-nitrobenzyl analog 15b that showed an IC50 value of 6.5 nM. Further structural modifications based on this compound were undergoing.
Co-reporter:Yuanxiang Wang, Jing Ai, Jinfeng Yue, Xia Peng, Yinchun Ji, Ailing Zhao, Xin Gao, Ying Wang, Yi Chen, Gang Liu, Zhaobing Gao, Meiyu Geng and Ao Zhang
MedChemComm 2012 vol. 3(Issue 11) pp:1423-1427
Publication Date(Web):31 Aug 2012
DOI:10.1039/C2MD20192E
A preclinical study on our previously discovered highly potent c-Met inhibitor 1 (zgwatinib) demonstrated its significant toxicity, and a SAR campaign was conducted to finely tune down the hERG inhibition without affecting the c-Met potency. Compounds 11, 12 and 39 stood out as new c-Met inhibitors with IC50 values of <3.0 nM and being nearly inactive against hERG channels.
Co-reporter:Na Ye, Xiangtao Xu, Fuying Li, Mengmeng Ning, Zhiqing Liu, Yuqing Cao, Ying Leng, Ao Zhang
Tetrahedron Letters 2012 Volume 53(Issue 35) pp:4738-4742
Publication Date(Web):29 August 2012
DOI:10.1016/j.tetlet.2012.06.111
RuCl3/NaIO4-initiated oxidation of oxiranemethanols was investigated, and two products, oxirane-2-carboxylic acid and (5′-oxotetrahydrofuran-3-yl)acetic acid, were obtained in variant ratios. The product structures were determined and a tentative mechanism involving oxidation-rearrangement-oxidation process was proposed. Glucokinase enzymatic assay revealed that oxiranecarboxamides 4a–c retained moderate GK activation potency with amide 4a showing an EC50 value of 584 nM and a high activation fold of 3.14. However, (5′-oxotetrahydrofuran-3-yl)acetamide 11a is inactive. This study not only provided an alternative protocol to access (5′-oxotetrahydrofuran-3-yl)acetic acid analogs, but also yielded nanomolar GA activators (esp. 4a) for further structure–activity relationship study.
Co-reporter:Weiwei Mao, Mengmeng Ning, Zhiqing Liu, Qingzhang Zhu, Ying Leng, Ao Zhang
Bioorganic & Medicinal Chemistry 2012 20(9) pp: 2982-2991
Publication Date(Web):
DOI:10.1016/j.bmc.2012.03.008
Co-reporter:Yuanxiang Wang ; Jing Ai ; Ying Wang ; Yi Chen ; Lu Wang ; Gang Liu ; Meiyu Geng
Journal of Medicinal Chemistry 2011 Volume 54(Issue 7) pp:2127-2142
Publication Date(Web):March 15, 2011
DOI:10.1021/jm101340q
By use of an improved synthetic strategy, a series of 3,5-disubstituted and 3,5,7-trisubstituted quinolines were readily prepared. 3,5,7-Trisubstituted quinolines 21a−c, 21l, and 27a−c were identified as the most potent c-Met inhibitors with IC50 of less than 1.0 nM. Compound 21b showed the most promising overall PK profile and has high potency and extraordinary selectivity to c-Met against c-Met family member Ron and 12 other tyrosine kinases. It produced constitutive inhibition of c-Met phosphorylation in c-Met dependent cell lines. At doses of 100 mg/kg, compound 21b showed statistically significant tumor growth inhibition (68−69%) in both NIH-3T3-TPR-Met and U-87 MG human gliobastoma xenograft models. These results clearly indicated that compound 21b is a potent and highly selective c-Met inhibitor. Its favorable in vitro and in vivo profiles warrant further investigation.
Co-reporter:Hai Zhang ; Na Ye ; Shanglin Zhou ; Lin Guo ; Longtai Zheng ; Zhili Liu ; Bo Gao ; Xuechu Zhen
Journal of Medicinal Chemistry 2011 Volume 54(Issue 13) pp:4324-4338
Publication Date(Web):May 18, 2011
DOI:10.1021/jm200347t
A series of new aporphine analogues (aporlogues) were synthesized bearing a C-, N-, or O-linkage at the C11 position. Lipoic ester (−)-15 was identified as a full agonist at the dopamine D2 and serotonin 5-HT1A receptors with Ki values of 174 and 66 nM, respectively. It elicited antiparkinsonian action on Parkinsin’s disease (PD) rats with minor dyskinesia. Chronic use of (−)-15 reduced l-DOPA-induced dyskinesia (LID) without attenuating the antiparkinsonian effect. These results suggest that 5-HT1A and D2 dual-receptor agonist (−)-15 may present a novel candidate drug in the treatment of PD and LID.
Co-reporter:Yuanxiang Wang, Jing Ai, Gang Liu, Meiyu Geng and Ao Zhang
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 17) pp:5930-5933
Publication Date(Web):01 Jul 2011
DOI:10.1039/C1OB05830D
An effective one-pot synthesis of quinolines bearing diverse C3-piperazinyl functions was developed by using a modified Friedländer's protocol. The method not only enables the synthesis of our early reported c-Met inhibitor on a large scale, but also provides a way to generate novel multi-substituted quinolines for further structure–activity relationship (SAR) study.
Co-reporter:Na Ye, QianQian Wu, Liyuan Zhu, Longtai Zheng, Bo Gao, Xuechu Zhen, Ao Zhang
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 6) pp:1999-2008
Publication Date(Web):15 March 2011
DOI:10.1016/j.bmc.2011.01.053
A series of new aporphine analogues (aporlogues) were prepared from appropriate aporphine precursors and arylpiperazines using the Click reaction protocol. These compounds displayed good to high affinity at the D3 receptor, low or no affinity at the D1 and D2 receptors. Compounds 7f and 11c stood out as the most potent at the D3 receptor among our newly synthesized aporlogues with Ki values of 2.67 and 1.14 nM, respectively. Further assay at the 5-HT1A receptor revealed that aporlogues 7f and 11c also showed high affinity at this receptor with Ki values of 9.68 and 7.59 nM, respectively. They were 3.6- and 6.6-fold more potent at the D3 over 5-HT1A receptors. Such D3/5-HT1A dual property of these compounds may be useful in the treatment of several brain disorders.A series of new aporphine analogues (aporlogues) were prepared from appropriate aporphine precursors and arylpiperazines using the Click reaction protocol. Compounds 7f and 11c stood out as the most potent at the D3 receptor among our newly synthesized aporlogues with Ki values of 2.67 and 1.14 nM, respectively. Further assay at the 5-HT1A receptor revealed that aporlogues 7f and 11c also showed high affinity at this receptor.
Co-reporter:Ailing Zhao, Xin Gao, Yuanxiang Wang, Jing Ai, Ying Wang, Yi Chen, Meiyu Geng, Ao Zhang
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 13) pp:3906-3918
Publication Date(Web):1 July 2011
DOI:10.1016/j.bmc.2011.05.038
A series of thieno[2,3-d]pyrimidines and furo[2,3-d]pyrimidines were synthesized and evaluated for the c-Met inhibition. Thieno[2,3-d]pyrimidine 6b stood out as the most potent showing an IC50 of 35.7 nM. This compound displayed high inhibitory effect on cell proliferation in BaF3-TPR-Met cells and showed high selectivity for c-Met family against other 14 tested kinases. However, compound 6b was found ineffective in the c-Met-dependent U-87MG human gliobastoma xenograft model that may be relevant to its poor PK profile.A series of thieno[2,3-d]pyrimidines and furo[2,3-d]pyrimidines were synthesized and evaluated for the c-Met inhibition. Thieno[2,3-d]pyrimidine 6b stood out as the most potent showing an IC50 of 35.7 nM. This compound displayed high inhibitory effect on cell proliferation in BaF3-TPR-Met cells and showed high selectivity for c-Met family against other 14 tested kinases. However, compound 6b was found ineffective in the c-Met-dependent U-87MG human gliobastoma xenograft model that may be relevant to its poor PK profile.
Co-reporter:Jun Sun, Xuefeng Zhang, Fulong Li, Chunyong Ding, Wenjuan Wu, Ao Zhang
Tetrahedron Letters 2011 Volume 52(Issue 43) pp:5693-5696
Publication Date(Web):26 October 2011
DOI:10.1016/j.tetlet.2011.08.106
Reaction of methyl 4,6-O-benzylidene-3(2)-deoxy-hexopyranosid-2(3)-ulose (1) with various arylamines under Bargellini reaction conditions was investigated. A series of unique enaminoketones 3–12 was obtained unexpectedly under basic conditions in 52–72% yield.
Co-reporter:Zhiqing Liu, Qingzhang Zhu, Fuying Li, Lina Zhang, Ying Leng and Ao Zhang
MedChemComm 2011 vol. 2(Issue 6) pp:531-535
Publication Date(Web):14 Apr 2011
DOI:10.1039/C1MD00002K
A series of novel arylacetamides were designed to further explore the GK binding property at the aminothiazole C5 position. The C5-amide substituted aminothiazoles 7a–f generally displayed decreased potency, whereas most of the C5-triazole substituted aminothiazoles retained good GK potency. Triazole 15 with a hydroxyethyl side chain was the most potent among the current series possessing an EC50 value of 0.18 μM. Its R-enantiomerR-15 showed similar potency (0.22 μM) that deserves for further evaluation.
Co-reporter:Fulong Li, Chunyong Ding, Meining Wang, Qizheng Yao, and Ao Zhang
The Journal of Organic Chemistry 2011 Volume 76(Issue 8) pp:2820-2827
Publication Date(Web):March 9, 2011
DOI:10.1021/jo200243d
To turn side products into major products, a novel strategy to access biologically active 4-aminocyclopent-2-enones was developed. These compounds were originally identified as side products but became major products when 3,5-dimethylpyran-3,4-diol 7a was used as the substrate and 30% InBr3 as the catalyst. Aryl- or heteroarylamines as well as variously substituted glycals can be used in this reaction, and the corresponding 4-aminocyclopent-2-enones were obtained in moderate to good yields. These compounds can be further used to prepare 4-aminocarbocyclic nucleosides.
Co-reporter:Chengtang Du, Fulong Li, Xuefeng Zhang, Wenxiang Hu, Qizheng Yao, and Ao Zhang
The Journal of Organic Chemistry 2011 Volume 76(Issue 21) pp:8833-8839
Publication Date(Web):October 3, 2011
DOI:10.1021/jo2015483
The cyclization reactions of arylamines with 2-deoxy-d-ribose or glycals were reinvestigated in the current report. In the montmorillonite KSF- or InCl3-initiated reactions of 2-deoxy-d-ribose with arylamines, a pair of diastereomeric tetrahydro-2H-pyran-fused tetrahydroquinolines was obtained in a nearly 1:1 ratio where the structure of one diastereomer was incorrectly assigned in the literature. Meanwhile, the diastereoselectivity in InBr3-catalyzed cyclization of glycals with arylamines was also incorrectly reported previously. It was found that high diastereomeric selectivity was achieved only when a C5-substituted glycal was used; otherwise, a pair of diastereomers was obtained in moderate yield with 1:1 diastereomeric ratio. Furthermore, tetrahydrofuran-fused tetrahydroquinolines 5b and 5b′ were also prepared successfully by using TBDPS-protected ribose as the glycal precursor and montmorillonite KSF as the activator.
Co-reporter:Jing Zhang, Kui Wu, Meining Wang, Jianqin Jiang, Ao Zhang
Tetrahedron 2011 67(5) pp: 842-848
Publication Date(Web):
DOI:10.1016/j.tet.2010.12.038
Co-reporter:Shanghui Tu;Chunyong Ding;Wenxiang Hu;Fulong Li;Qizheng Yao
Molecular Diversity 2011 Volume 15( Issue 1) pp:91-99
Publication Date(Web):2011 February
DOI:10.1007/s11030-010-9241-x
An efficient three-step approach was developed to assemble indole- or benzofuran-fused benzocarbazole-1,4-diones in 42–53% overall yield. This approach includes AgOAc-promoted oxidative cyclization of 2,6-di-bromocyclohexene-1,4-dione with indol-3-ylpropanoid acid, condensation of the resulting bromocarbazole intermediates with phenols or anilines, followed by Pd(OAc)-catalyzed cyclization. Such convenient synthetic protocol and the novelty of the corresponding products will largely assist our drug design and development program.
Co-reporter:Yi Zhang, Zhaobin Han, Fuying Li, Kuiling Ding and Ao Zhang
Chemical Communications 2010 vol. 46(Issue 1) pp:156-158
Publication Date(Web):24 Nov 2009
DOI:10.1039/B919902K
The enantioselective hydrogenation of a series of challenging substrates, α-aryl-β-substituted acrylic acids, was realized with high efficiency and enantioselectivity (up to 96%) under the catalysis of Ir(I) complex of Spiro-based P,N ligand, SpinPHOX.
Co-reporter:Chunyong Ding;Shanghui Tu;Qizheng Yao;Fulong Li;Yuanxiang Wang;Wenxiang Hu
Advanced Synthesis & Catalysis 2010 Volume 352( Issue 5) pp:847-853
Publication Date(Web):
DOI:10.1002/adsc.200900789
Abstract
A three-step, one-pot tandem reaction including radical nucleophilic alkylation/cyclization/aromatization was developed using 0.3 equivalents of silver(I) acetate (AgOAc) as the catalyst and 2 equivalents of ammonium persulfate [(NH4)2S2O8] as the oxidant. This strategy is highly efficient for the assembly of pentacyclic complex carbazoles from aryl-fused bromobenzoquinones and indol-3-ylpropanoic acid acids in 52–72% overall yields (three steps). This new approach provides a significant improvement over the previously reported methods and would greatly facilitate analog library construction of pentacyclic complex carbazoles and benefit further biological evaluation of these compounds.
Co-reporter:Zhili Liu ; Hai Zhang ; Na Ye ; Jing Zhang ; QianQian Wu ; Peihua Sun ; Linyong Li ; Xuechu Zhen
Journal of Medicinal Chemistry 2010 Volume 53(Issue 3) pp:1319-1328
Publication Date(Web):December 30, 2009
DOI:10.1021/jm9015763
A series of new aporphine analogues were synthesized and pharmacologically evaluated. 11-Allyloxy-(17), 11-propargyloxy-(20), and dihydrofuro-(19) aporphines displayed the highest affinity at the 5-HT1A receptor with Ki values of 12.0, 14.0, and 6.7 nM, respectively. The high binding potential of the diastereomeric mixture of aporphine 19 was found residing in the cis-diastereomer (cis-19). [35S]GTPγS function assays on 5-HT1A receptor indicated that aporphines 17 and 20 were partial agonists, while trans-19 behaved as a high efficacy full antagonist and cis-19 was a full agonist. The agonistic property of cis-19 at the 5-HT1A receptor was further confirmed in vitro and in vivo. This compound may be useful as a potential treatment for anxiety.
Co-reporter:Fuying Li, Qingzhang Zhu, Yi Zhang, Ying Feng, Ying Leng, Ao Zhang
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 11) pp:3875-3884
Publication Date(Web):1 June 2010
DOI:10.1016/j.bmc.2010.04.038
A series of N-thiazole substituted arylacetamides were designed on the basis of metabolic mechanism of the aminothiazole fragment as glucokinase (GK) activators for the treatment of type 2 diabetes. Instead of introducing a substituent to block the metabolic sensitive C-5 position on the thiazole core directly, a wide variety of C-4 or both C-4 and C-5 substitutions were explored. Compound R-9k bearing an iso-propyl group as the C-4 substituent was found possessing the highest GK activation potency with an EC50 of 0.026 μM. This compound significantly increased both glucose uptake and glycogen synthesis in rat primary cultured hepatocytes. Moreover, single oral administration of compound R-9k exerted significant reduction of blood glucose levels in both ICR and ob/ob mice. These promising results indicated that compound R-9k is a potent orally active GK activator, and is warranted for further investigation as a new anti-diabetic treatment.A series of N-thiazole substituted arylacetamides were designed as metabolic stable glucokinase (GK) activators for the treatment of type 2 diabetes. Compound R-9k, with an EC50 of 0.026 μM significantly increased both glucose uptake and glycogen synthesis in rat primary cultured hepatocytes. Single oral administration of compound R-9k exerted significant reduction of blood glucose levels in both ICR and ob/ob mice.
Co-reporter:Fuying Li;Chenlei Yin;Jie Chen;Jinggen Liu;Xin Xie
Chemical Biology & Drug Design 2009 Volume 74( Issue 4) pp:335-342
Publication Date(Web):
DOI:10.1111/j.1747-0285.2009.00849.x
Mono- and bis-indolomorphinans were synthesized through a multi-step synthetic approach from the alkaloid, thebaine, to further explore the C-ring SAR (structure-activity relationship) of morphinan scaffold. Both mono-indoles displayed good binding affinity and selectivity for the δ receptor, with compound 6b possessed the highest Ki value of 1.45 nm at this receptor. Bisindolomorphinans 7a,b did not have appreciable affinity for both δ and κ receptors, but moderate binding at the μ receptor was observed. Functional assays indicated that the newly synthesized mono-indole 6b was δ-agonist, opposite to the δ-antagonist profile of naltrindole. Bisindoles 7a,b were μ-agonists. This work further confirms that the phenol component in opioids is essential for higher binding to the opioid receptors. The different binding ability, receptor selectivity, and the functional activity profiles of naltrindole 2, monoindole 6b, and bisindole 7b clearly indicated that they interact with the opioid receptors in different modes.
Co-reporter:Jing Zhang, Hai Zhang, Wenxian Cai, Leiping Yu, Xuechu Zhen, Ao Zhang
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 14) pp:4873-4880
Publication Date(Web):15 July 2009
DOI:10.1016/j.bmc.2009.06.019
A series of new 1-aryl-3-benzazepine derivatives containing an arylpiperazinyl function as the N3 substituent were synthesized by combining a D1 receptor agonistic pharmacophore and a 5-HT1A receptor pharmacophore through Click reaction. Interestingly, these compounds generally do not have good binding affinity at the D1 receptor, but most compounds are potent at both D2 and 5-HT1A receptors. Compound 8h, containing 1-m-tolyl-benzazepine scaffold and 2-methoxyphenylpiperazine core, displayed good affinity at all tested receptors, with Ki values of 144, 80, and 133 nM, for the D1, D2, and 5-HT1A receptors, respectively. Compound 13 with the triazole moiety formed differently from that in 8h showed the highest affinity at the D2 receptor with Ki value of 19 nM. This compound also showed moderate affinity at the 5-HT1A (Ki, 105 nM), and D1 (Ki, 551 nM) receptors. Functional assays indicated that both compounds 13 and 8h are antagonists at D1 and D2 receptors, whereas full agonistic activity at the 5-HT1A receptor was observed. In agreement with the binding affinity, compound 13 is a high efficacy D2 antagonist and 5-HT1A agonist.
Co-reporter:Fuying Li, Linghuan Gaob, Chenlei Yin, Jie Chen, Jinggen Liu, Xin Xie, Ao Zhang
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 16) pp:4603-4606
Publication Date(Web):15 August 2009
DOI:10.1016/j.bmcl.2009.06.093
A series of skeletal rearranged indolomorphinans 7a–d were obtained by N-demethylation of 3-methoxy-N-methyl-14-hydroxymorphinan-6-one 12 followed by N-realkylation, reduction and Fischer indole cyclization. The structure of the novel skeleton was confirmed by X-ray analysis. These new indoles displayed moderate binding affinity and selectivity at the μ receptor, with compound 7b showing the highest affinity at this receptor with a Ki value of 40 nM, and 6- and 25-fold selectivity against δ and κ receptors, respectively. Function assays showed that indolopropellanes 7b and 7c possessed full agonistic activity at all the opioid receptors indicating a different interaction model existed.A series of skeletal rearranged indolomorphinans 7a–d was synthesized, and their binding affinity and functional activity for opioid receptors were evaluated.
Co-reporter:Jing Zhang Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 42) pp:11119-11122
Publication Date(Web):
DOI:10.1002/chem.200901197
Co-reporter:Yuanxiang Wang, Ao Zhang
Tetrahedron 2009 65(34) pp: 6986-6990
Publication Date(Web):
DOI:10.1016/j.tet.2009.06.049
Co-reporter:Fuying Li;Chenlei Yin;Jie Chen;Jinggen Liu ;Xin Xie
ChemMedChem 2009 Volume 4( Issue 12) pp:2103-2110
Publication Date(Web):
DOI:10.1002/cmdc.200900308
Abstract
A series of new 14-hydroxymorphinan analogues with a thiazole or imidazo[2,1-b]thiazole fragment as the heterocyclic function fused to ring C were designed and synthesized. These compounds can be viewed as the result of a direct modification at ring C of the 14-hydroxymorphinan scaffold. Among these compounds, three were identified as having potent binding affinity (∼1 nM) at both κ and μ receptors, and acting as agonists at κ and partial agonists or antagonists at μ receptors. In view of the promising results from studies on compounds with mixed κ and μ receptor activities, these new compounds warrant further investigation.
Co-reporter:Zhili Liu, Xuetao Chen, Peihua Sun, Leiping Yu, Xuechu Zhen, Ao Zhang
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 18) pp:8335-8338
Publication Date(Web):15 September 2008
DOI:10.1016/j.bmc.2008.08.056
A small series of N-propylnoraporphin-11-O-yl carboxylic esters with variant ester lengths were synthesized and their binding potencies at dopamine receptors (D1, D2) and serotonin receptors (5-HT1A, 5-HT2A) were evaluated. Monoesters 3a–f showed binding potency of 100 nM or less for the D2 receptor, and potency of 10–30 nM for the 5-HT1A receptor. Butyryl ester 3d was found to be the best compound possessing the highest potency for both receptors, with Ki values of 55 and 12 nM for D2 and 5-HT1A receptors, respectively. There is no correlation between the binding potency and the length of the monoesters, but the diesters 9 and 10 were inactive for the D2 receptor. The dual binding profile of these monoesters for the D2 and 5-HT1A receptors may be useful for the treatment of neuropsychiatric disorders.A small series of N-propylnoraporphin-11-O-yl carboxylic esters with variant ester lengths were synthesized. Although the diesters 9 and 10 were inactive for the D2 receptor, all of the aporphine monoesters displayed high dual binding profile for the D2 and 5-HT1A receptors, with butyryl ester 3d as the best possessing the highest potency for both receptors, with Ki values of 55 and 12 nM, respectively.
Co-reporter:Zhili Liu, Xuetao Chen, Leiping Yu, Xuechu Zhen, Ao Zhang
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 14) pp:6675-6681
Publication Date(Web):15 July 2008
DOI:10.1016/j.bmc.2008.05.077
A series of apomorphine ((−)-1, APO)-derived analogues ((±)-3, (−)-4-(−)-6) were designed and synthesized by hybridizing APO with a privileged 2-aminothiazole functionality which was lent from the orally available anti-parkinsonian drug, pramipexole (2). Among these hybridized compounds, catecholic aporphine (−)-6 shows good affinity at the D2 receptor with Ki of 328 nM, slightly less potent (3-fold), but more selective against the D1 receptor than that of the parent compound, APO. Although possessing reduced affinity at the D2 receptor, aporphines 15 and 18 show significant potency at both the D1 and 5-HT1A receptors. The former compound is equipotent at both receptors (Ki: 116 and 151 nM, respectively), while the latter is 8-fold more potent at the D1 (Ki: 78 nM) than at the 5-HT1A receptors (Ki: 640 nM). These results indicate that the catechol fragment is critical for the D2 receptor binding of the anti-parkinsonian drug, APO ((−)-1), but not necessary for binding at the D1 and 5-HT1A receptors.A series of 2-aminothiazole-privileged aporphine analogues were designed and synthesized. These compounds displayed variant binding affinity at dopamine D1, D2 and serotonin 5-HT1A receptors.
Co-reporter:Ao Zhang, Chunyong Ding, Chen Cheng and Qizheng Yao
ACS Combinatorial Science 2007 Volume 9(Issue 6) pp:916
Publication Date(Web):October 10, 2007
DOI:10.1021/cc700135h
Co-reporter:Yuanxiang Wang, Jing Ai, Gang Liu, Meiyu Geng and Ao Zhang
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 17) pp:NaN5933-5933
Publication Date(Web):2011/07/01
DOI:10.1039/C1OB05830D
An effective one-pot synthesis of quinolines bearing diverse C3-piperazinyl functions was developed by using a modified Friedländer's protocol. The method not only enables the synthesis of our early reported c-Met inhibitor on a large scale, but also provides a way to generate novel multi-substituted quinolines for further structure–activity relationship (SAR) study.
Co-reporter:Dongyu Wang, Xian Jia and Ao Zhang
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 35) pp:NaN7030-7030
Publication Date(Web):2012/07/20
DOI:10.1039/C2OB26002F
We have developed a practical method to assemble the proposed structure of natural product veraguamide A (1) by first preparing the three key fragments followed by optimization of the macrocyclization site. Although the synthetic product gave similar optical rotation to that reported for natural product, significant differences in the 1H and 13C NMR spectra were observed, especially the proton and carbon signals in the two N-MeVal moieties.
Co-reporter:Xuefeng Zhang, Xiaolong Jiang, Chunyong Ding, Qizheng Yao and Ao Zhang
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 8) pp:NaN1389-1389
Publication Date(Web):2012/12/12
DOI:10.1039/C2OB27020J
An unexpected N-glycosidation reaction of anthracen-1-amine with glycals was identified, and its use in the synthesis of C1′ N-linked analogues of natural product marmycin A was explored. The structures of all these products were determined by 1D and 2D NMR, CD spectra, and X-ray crystal analysis. These products were then subjected to Friedel–Crafts acylation, Dess–Martin oxidation and nucleophilic addition leading to novel natural product analogues bearing a quaternary carbon center.
Co-reporter:Yi Zhang, Zhaobin Han, Fuying Li, Kuiling Ding and Ao Zhang
Chemical Communications 2010 - vol. 46(Issue 1) pp:NaN158-158
Publication Date(Web):2009/11/24
DOI:10.1039/B919902K
The enantioselective hydrogenation of a series of challenging substrates, α-aryl-β-substituted acrylic acids, was realized with high efficiency and enantioselectivity (up to 96%) under the catalysis of Ir(I) complex of Spiro-based P,N ligand, SpinPHOX.































![9-ethyl-8-iodo-6,6-dimethyl-11-oxo-6,11-dihydro-5h-benzo[b]carbaz Ole-3-carbonitrile](http://img.cochemist.com/ccimg/1256600/1256584-80-1.png)
![9-ethyl-8-iodo-6,6-dimethyl-11-oxo-6,11-dihydro-5h-benzo[b]carbaz Ole-3-carbonitrile](http://img.cochemist.com/ccimg/1256600/1256584-80-1_b.png)
![(2S)-2-[(4-METHOXYPHENYL)SULFANYLCARBONYLAMINO]PENTANEDIOIC ACID](http://img.cochemist.com/ccimg/1256600/1256584-75-4.png)
![(2S)-2-[(4-METHOXYPHENYL)SULFANYLCARBONYLAMINO]PENTANEDIOIC ACID](http://img.cochemist.com/ccimg/1256600/1256584-75-4_b.png)

























![3-[4-[2-[4-(2,3-dichlorophenyl)piperazin-1-yl]ethyl]cyclohexyl]-1,1-dimethylurea](http://img.cochemist.com/ccimg/839800/839712-12-8.png)
![3-[4-[2-[4-(2,3-dichlorophenyl)piperazin-1-yl]ethyl]cyclohexyl]-1,1-dimethylurea](http://img.cochemist.com/ccimg/839800/839712-12-8_b.png)




![5-Chloro-N2-[2-methoxy-4-[4-(4-methyl-1-piperazinyl)-1-piperidinyl]phenyl]-N4-[2-[(1-methylethyl)sulfonyl]phenyl]-2,4-pyrimidinediamine](http://img.cochemist.com/ccimg/761500/761439-42-3.png)
![5-Chloro-N2-[2-methoxy-4-[4-(4-methyl-1-piperazinyl)-1-piperidinyl]phenyl]-N4-[2-[(1-methylethyl)sulfonyl]phenyl]-2,4-pyrimidinediamine](http://img.cochemist.com/ccimg/761500/761439-42-3_b.png)








![Benzoic acid, 4-[(2,2-dimethyl-1-oxopropyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/299500/299418-33-0.png)
![Benzoic acid, 4-[(2,2-dimethyl-1-oxopropyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/299500/299418-33-0_b.png)





![Ethanone, 1-(3-phenyltricyclo[3.3.1.13,7]dec-1-yl)-](http://img.cochemist.com/ccimg/209400/209336-43-6.png)
![Ethanone, 1-(3-phenyltricyclo[3.3.1.13,7]dec-1-yl)-](http://img.cochemist.com/ccimg/209400/209336-43-6_b.png)




![(3aR,6aS)-2-Methyloctahydropyrrolo[3,4-c]pyrrole](http://img.cochemist.com/ccimg/172800/172739-03-6.png)
![(3aR,6aS)-2-Methyloctahydropyrrolo[3,4-c]pyrrole](http://img.cochemist.com/ccimg/172800/172739-03-6_b.png)


![Propanamide, N-[1,1'-biphenyl]-4-yl-2,2-dimethyl-](http://img.cochemist.com/ccimg/150800/150787-00-1.png)
![Propanamide, N-[1,1'-biphenyl]-4-yl-2,2-dimethyl-](http://img.cochemist.com/ccimg/150800/150787-00-1_b.png)




![2,7-diazaspiro[3.5]nonane](http://img.cochemist.com/ccimg/136100/136098-14-1.png)
![2,7-diazaspiro[3.5]nonane](http://img.cochemist.com/ccimg/136100/136098-14-1_b.png)
![BUTANEDIOIC ACID;PIPERIDIN-1-YL-[3-[[4-(2-PROPAN-2-YLOXYPHENYL)PIPERAZIN-1-YL]METHYL]PHENYL]METHANONE](http://img.cochemist.com/data/images/134208-18-7.png)
![BUTANEDIOIC ACID;PIPERIDIN-1-YL-[3-[[4-(2-PROPAN-2-YLOXYPHENYL)PIPERAZIN-1-YL]METHYL]PHENYL]METHANONE](http://img.cochemist.com/data/images/134208-18-7_b.png)


![5-Boc-hexahydropyrrolo[3,4-b]pyrrole](http://img.cochemist.com/ccimg/132500/132414-81-4.png)
![5-Boc-hexahydropyrrolo[3,4-b]pyrrole](http://img.cochemist.com/ccimg/132500/132414-81-4_b.png)












![4,8:11,15-Dimethano-20H-bisbenzofuro[2,3-a:3',2'-i]dipyrido[4,3-b:3',4'-h]carbazole-1,8a,10a,18-tetrol,7,12-bis(cyclopropylmethyl)-5,6,7,8,9,10,11,12,13,14,19a,20b-dodecahydro-,(4bS,8R,8aS,10aS,11R,14aS,19aR,20bR)-](http://img.cochemist.com/ccimg/105700/105618-26-6.png)
![4,8:11,15-Dimethano-20H-bisbenzofuro[2,3-a:3',2'-i]dipyrido[4,3-b:3',4'-h]carbazole-1,8a,10a,18-tetrol,7,12-bis(cyclopropylmethyl)-5,6,7,8,9,10,11,12,13,14,19a,20b-dodecahydro-,(4bS,8R,8aS,10aS,11R,14aS,19aR,20bR)-](http://img.cochemist.com/ccimg/105700/105618-26-6_b.png)

![Methyl 4-[(z)-c-chloro-n-hydroxycarbonimidoyl]benzoate](http://img.cochemist.com/ccimg/101100/101023-70-5.png)
![Methyl 4-[(z)-c-chloro-n-hydroxycarbonimidoyl]benzoate](http://img.cochemist.com/ccimg/101100/101023-70-5_b.png)
![3,12-Epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin-10(3H)-one,octahydro-3,6-dimethyl-9-methylene-, (3R,5aS,6R,8aS,12S,12aR)-](http://img.cochemist.com/ccimg/101100/101020-89-7.png)
![3,12-Epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin-10(3H)-one,octahydro-3,6-dimethyl-9-methylene-, (3R,5aS,6R,8aS,12S,12aR)-](http://img.cochemist.com/ccimg/101100/101020-89-7_b.png)


![4,5,6,7-Tetrahydrothiazolo[5,4-c]pyridin-2-amine](http://img.cochemist.com/ccimg/97900/97817-23-7.png)
![4,5,6,7-Tetrahydrothiazolo[5,4-c]pyridin-2-amine](http://img.cochemist.com/ccimg/97900/97817-23-7_b.png)
![3-[2-(Trifluoromethyl)phenyl]propanoic acid](http://img.cochemist.com/ccimg/94100/94022-99-8.png)
![3-[2-(Trifluoromethyl)phenyl]propanoic acid](http://img.cochemist.com/ccimg/94100/94022-99-8_b.png)









![2-Methyl-octahydro-pyrrolo[3,4-c]pyrrole](http://img.cochemist.com/ccimg/86800/86732-28-7.png)
![2-Methyl-octahydro-pyrrolo[3,4-c]pyrrole](http://img.cochemist.com/ccimg/86800/86732-28-7_b.png)




![2-(3,4-dichlorophenyl)-N-methyl-N-[(1R,2R)-2-(pyrrolidin-1-yl)cyclohexyl]acetamide methanesulfonate (1:1)](http://img.cochemist.com/ccimg/84000/83913-06-8.png)
![2-(3,4-dichlorophenyl)-N-methyl-N-[(1R,2R)-2-(pyrrolidin-1-yl)cyclohexyl]acetamide methanesulfonate (1:1)](http://img.cochemist.com/ccimg/84000/83913-06-8_b.png)






![Phenanthro[1,2-b]furan-10,11-dione, 1-(hydroxymethyl)-6-methyl-](/data/chemimg/2084200/76829-01-1.png)
![Phenanthro[1,2-b]furan-10,11-dione, 1-(hydroxymethyl)-6-methyl-](/data/chemimg/2084200/76829-01-1_b.png)





![10aH-9,10b-Epoxypyrano[4,3,2-jk][2]benzoxepin-2(3H)-one,octahydro-3,6,9-trimethyl-, (3R,3aS,6R,6aS,9S,10aS,10bR)-](http://img.cochemist.com/ccimg/72900/72826-63-2.png)
![10aH-9,10b-Epoxypyrano[4,3,2-jk][2]benzoxepin-2(3H)-one,octahydro-3,6,9-trimethyl-, (3R,3aS,6R,6aS,9S,10aS,10bR)-](http://img.cochemist.com/ccimg/72900/72826-63-2_b.png)
![4-(2-CHLORO-11H-DIBENZO[B,E][1,4]OXATHIEPIN-11-YL)-1-METHYLPIPERI<WBR />DINE (2E)-2-BUTENEDIOATE (1:1)](http://img.cochemist.com/ccimg/72800/72782-05-9.png)
![4-(2-CHLORO-11H-DIBENZO[B,E][1,4]OXATHIEPIN-11-YL)-1-METHYLPIPERI<WBR />DINE (2E)-2-BUTENEDIOATE (1:1)](http://img.cochemist.com/ccimg/72800/72782-05-9_b.png)




































![9,10-Anthracenedione, 1-[(3-methylphenyl)amino]-](http://img.cochemist.com/ccimg/33200/33175-69-8.png)
![9,10-Anthracenedione, 1-[(3-methylphenyl)amino]-](http://img.cochemist.com/ccimg/33200/33175-69-8_b.png)

























![5-Methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-amine](http://img.cochemist.com/ccimg/17900/17899-48-8.png)
![5-Methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-amine](http://img.cochemist.com/ccimg/17900/17899-48-8_b.png)
































![9,10-Anthracenedione, 1-[(3-chlorophenyl)amino]-](http://img.cochemist.com/ccimg/3000/2944-21-0.png)
![9,10-Anthracenedione, 1-[(3-chlorophenyl)amino]-](http://img.cochemist.com/ccimg/3000/2944-21-0_b.png)




![Propanamide,2,2-dimethyl-N-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/2000/1939-19-1.png)
![Propanamide,2,2-dimethyl-N-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/2000/1939-19-1_b.png)









![N-[2-(CYCLOPENTYLAMINO)-2-OXOETHYL]-N-(4-FLUOROBENZYL)-1,2,3-THIA<WBR />DIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/800/739-88-8.png)
![N-[2-(CYCLOPENTYLAMINO)-2-OXOETHYL]-N-(4-FLUOROBENZYL)-1,2,3-THIA<WBR />DIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/800/739-88-8_b.png)


![8-Oxa-3-azabicyclo[3.2.1]octane](http://img.cochemist.com/ccimg/300/280-13-7.png)
![8-Oxa-3-azabicyclo[3.2.1]octane](http://img.cochemist.com/ccimg/300/280-13-7_b.png)
























![4-tert-butyl-1-[(2,2-dichlorocyclopropyl)methyl]-2-nitrobenzene](http://img.cochemist.com/ccimg/5300/5216-30-8.png)
![4-tert-butyl-1-[(2,2-dichlorocyclopropyl)methyl]-2-nitrobenzene](http://img.cochemist.com/ccimg/5300/5216-30-8_b.png)






![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)