Co-reporter:Katherine E. Peterson ; Maris A. Cinelli ; Andrew E. Morrell ; Akhil Mehta ; Thomas S. Dexheimer ; Keli Agama ; Smitha Antony ; Yves Pommier
Journal of Medicinal Chemistry 2011 Volume 54(Issue 14) pp:4937-4953
Publication Date(Web):June 28, 2011
DOI:10.1021/jm101338z
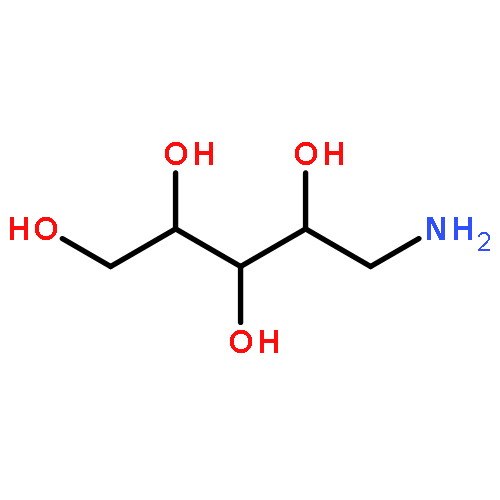
The DNA-relaxing enzyme topoisomerase I (Top1) can be inhibited by heterocyclic compounds such as indolocarbazoles and indenoisoquinolines. Carbohydrate and hydroxyl-containing side chains are essential for the biological activity of indolocarbazoles. The current study investigated how similar functionalities could be “translated” to the indenoisoquinoline system and how stereochemistry and hydrogen bonding affect biological activity. Herein is described the preparation and assay of indenoisoquinolines substituted with short-chain alcohols, diols, and carbohydrates. Several compounds (including those derived from sugars) display potent Top1 poisoning and antiproliferative activities. The Top1 poisoning activity of diol-substituted indenoisoquinolines is dependent upon stereochemistry. Although the effect is striking, molecular modeling and docking studies do not indicate any reason for the difference in activity due to similar calculated interactions between the ligand and Top1–DNA complex and ambiguity about the binding mode. A stereochemical dependence was also observed for carbohydrate-derived indenoisoquinolines. Although similar trends were observed in other classes of Top1 inhibitors, the exact nature of this effect has yet to be elucidated.
Co-reporter:Juma Hoshino ; Eun-Jung Park ; Tamara P. Kondratyuk ; Laura Marler ; John M. Pezzuto ; Richard B. van Breemen ; Shunyan Mo ; Yongchao Li
Journal of Medicinal Chemistry 2010 Volume 53(Issue 13) pp:5033-5043
Publication Date(Web):June 8, 2010
DOI:10.1021/jm100274c
Five resveratrol sulfate metabolites were synthesized and assessed for activities known to be mediated by resveratrol: inhibition of tumor necrosis factor (TNF) α induced NFκB activity, cylcooxygenases (COX-1 and COX-2), aromatase, nitric oxide production in endotoxin-stimulated macrophages, proliferation of KB or MCF7 cells, induction of quinone reductase 1 (QR1), accumulation in the sub-G1 phase of the cell cycle, and quenching of 2,2-diphenyl-1-picrylhydrazyl (DPPH) free radical. Two metabolites showed activity in these assays; the 3-sulfate exhibited QR1 induction, DPPH free radical scavenging, and COX-1 and COX-2 inhibitory activities and the 4′-sulfate inhibited NFκB induction, as well as COX-1 and COX-2 activities. Resveratrol and its 3′-sulfate and 4-sulfate inhibit NO production by NO scavenging and down-regulation of iNOS expression in RAW 264.7 cells. Resveratrol sulfates displayed low antiproliferative activity and negligible uptake in MCF7 cells.
Co-reporter:Arindam Talukdar, Ekaterina Morgunova, Jianxin Duan, Winfried Meining, Nicolas Foloppe, Lennart Nilsson, Adelbert Bacher, Boris Illarionov, Markus Fischer, Rudolf Ladenstein, Mark Cushman
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 10) pp:3518-3534
Publication Date(Web):15 May 2010
DOI:10.1016/j.bmc.2010.03.072
Virtual screening of a library of commercially available compounds versus the structure of Mycobacterium tuberculosis lumazine synthase identified 2-(2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamido)acetic acid (9) as a possible lead compound. Compound 9 proved to be an effective inhibitor of M. tuberculosis lumazine synthase with a Ki of 70 μM. Lead optimization through replacement of the carboxymethylsulfonamide sidechain with sulfonamides substituted with alkyl phosphates led to a four-carbon phosphate 38 that displayed a moderate increase in enzyme inhibitory activity (Ki 38 μM). Molecular modeling based on known lumazine synthase/inhibitor crystal structures suggests that the main forces stabilizing the present benzindolone/enzyme complexes involve π–π stacking interactions with Trp27 and hydrogen bonding of the phosphates with Arg128, the backbone nitrogens of Gly85 and Gln86, and the side chain hydroxyl of Thr87.Virtual screening of compounds versus Mycobacterium tuberculosis lumazine synthase put forth a possible lead compound. Lead optimization through replacement of carboxymethylsulfonamide side chain led to compound that display modest enzyme inhibitory activity.
Co-reporter:Maris A. Cinelli, Andrew E. Morrell, Thomas S. Dexheimer, Keli Agama, Surbhi Agrawal, Yves Pommier, Mark Cushman
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 15) pp:5535-5552
Publication Date(Web):1 August 2010
DOI:10.1016/j.bmc.2010.06.040
Aromathecins are inhibitors of human topoisomerase I (Top1). These compounds are composites of several heteroaromatic systems, namely the camptothecins and indenoisoquinolines, and they possess notable Top1 inhibition and cytotoxicity when substituted at position 14. The SAR of these compounds overlaps with indenoisoquinolines, suggesting that they may intercalate into the Top1-DNA complex similarly. Nonetheless, the proposed binding mode for aromathecins is purely hypothetical, as an X-ray structure is unavailable. In the present communication, we have synthesized eight novel series of A-ring-substituted (positions 1–3) aromathecins, through a simple, modular route, as part of a comprehensive SAR study. Certain groups (such as 2,3-ethylenedioxy) moderately improve Top1 inhibition, and, often, antiproliferative activity, whereas other groups (2,3-dimethoxy and 3-substituents) attenuate bioactivity. Strikingly, these trends are very similar to those previously observed for the A-ring of camptothecins, and this considerable SAR overlap lends further support (in the absence of crystallographic data) to the hypothesis that aromathecins bind in the Top1 cleavage complex as interfacial inhibitors in a ‘camptothecin-like’ pose.Novel aromathecins, substituted at four different positions, were prepared and evaluated against Top1 and human tumor cells. SAR trends mimic those of camptothecin and support our proposed camptothecin-like binding mode.
Co-reporter:Matthew D. Cullen ; William C. Ho ; Joseph D. Bauman ; Kalyan Das ; Eddy Arnold ; Tracy L. Hartman ; Karen M. Watson ; Robert W. Buckheit ; Jr.; Christophe Pannecouque ; Erik De Clercq
Journal of Medicinal Chemistry 2009 Volume 52(Issue 20) pp:6467-6473
Publication Date(Web):September 23, 2009
DOI:10.1021/jm901167t
Two crystal structures have been solved for separate complexes of alkenyldiarylmethane (ADAM) nonnucleoside reverse transcriptase inhibitors (NNRTI) 3 and 4 with HIV-1 reverse transcriptase (RT). The structures reveal inhibitor binding is exclusively hydrophobic in nature and the shape of the inhibitor-bound NNRTI binding pocket is unique among other reported inhibitor−RT crystal structures. Primarily, ADAMs 3 and 4 protrude from a large gap in the back side of the binding pocket, placing portions of the inhibitors unusually close to the polymerase active site and allowing 3 to form a weak hydrogen bond with Lys223. The lack of additional stabilizing interactions, beyond the observed hydrophobic surface contacts, between 4 and RT is quite perplexing given the extreme potency of the compound (IC50 ≤ 1 nM). ADAM 4 was designed to be hydrolytically stable in blood plasma, and an investigation of its hydrolysis in rat plasma demonstrated it has a significantly prolonged half-life in comparison to ADAM lead compounds 1 and 2.
Co-reporter:Arup Maiti, P. V. Narasimha Reddy, Megan Sturdy, Laura Marler, Scott D. Pegan, Andrew D. Mesecar, John M. Pezzuto and Mark Cushman
Journal of Medicinal Chemistry 2009 Volume 52(Issue 7) pp:1873-1884
Publication Date(Web):March 6, 2009
DOI:10.1021/jm801335z
An efficient method has been developed to synthesize casimiroin (1), a component of the edible fruit of Casimiroa edulis, on a multigram scale in good overall yield. The route was versatile enough to provide an array of compound 1 analogues that were evaluated as QR2 and aromatase inhibitors. In addition, X-ray crystallography studies of QR2 in complex with compound 1 and one of its more potent analogues has provided insight into the mechanism of action of this new series of QR2 inhibitors. The initial biological investigations suggest that compound 1 and its analogues merit further investigation as potential chemopreventive or chemotherapeutic agents.
Co-reporter:Bo-Liang Deng, Yujie Zhao, Tracy L. Hartman, Karen Watson, Robert W. Buckheit Jr., Christophe Pannecouque, Erik De Clercq, Mark Cushman
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 3) pp:1210-1214
Publication Date(Web):March 2009
DOI:10.1016/j.ejmech.2008.09.013
As a continuation of efforts to replace the metabolically labile methyl esters of lead alkenyldiarylmethanes (ADAMs) with stable bioisosteres, compounds bearing benzo[d]isoxazole and oxazolidine-2-one rings were designed and evaluated as a new series of potent HIV-1 non-nucleoside reverse transcriptase inhibitors with anti-HIV activity. All of the resulting ADAMs were found to inhibit HIV-1 RT with poly(rC)·oligo(dG) as the template primer. The most promising compound in this series was ADAM 3, with EC50 values of 40 nM (vs HIV-1RF) and 20 nM (vs HIV-1IIIB). Compound 3 also inhibited HIV-1 reverse transcriptase with an IC50 of 0.91 μM. ADAM 4 has an antiviral EC50 of 0.6 μM in CEM-SS cells and a plasma half-life of 51.4 min.ADAMs incorporating a benzo[d]isoxazole ring were synthesized and evaluated as NNRTIs. Compound 3 [IC50 0.91 μM; EC50 40 nM (HIV-1RF) and 20 nM (HIV-1IIIB)] displayed the most potent activity.
Co-reporter:Arindam Talukdar, Meghan Breen, Adelbert Bacher, Boris Illarionov, Markus Fischer, Gunda Georg, Qi-Zhuang Ye and Mark Cushman
The Journal of Organic Chemistry 2009 Volume 74(Issue 15) pp:5123-5134
Publication Date(Web):June 24, 2009
DOI:10.1021/jo900238q
(E)-5-Nitro-6-(2-hydroxystyryl)pyrimidine-2,4(1H,3H)-dione (9) was identified as a novel inhibitor of Schizosaccharomyces pombe lumazine synthase by high-throughput screening of a 100000 compound library. The Ki of 9 vs Mycobacterium tuberculosis lumazine synthase was 95 μM. Compound 9 is a structural analogue of the lumazine synthase substrate 5-amino-6-(d-ribitylamino)-2,4-(1H,3H)pyrimidinedione (1). This indicates that the ribitylamino side chain of the substrate is not essential for binding to the enzyme. Optimization of the enzyme inhibitory activity through systematic structure modification of the lead compound 9 led to (E)-5-nitro-6-(4-nitrostyryl)pyrimidine-2,4(1H,3H)-dione (26), which has a Ki of 3.7 μM vs M. tuberculosis lumazine synthase.
Co-reporter:Maris A. Cinelli, Brenda Cordero, Thomas S. Dexheimer, Yves Pommier, Mark Cushman
Bioorganic & Medicinal Chemistry 2009 17(20) pp: 7145-7155
Publication Date(Web):
DOI:10.1016/j.bmc.2009.08.066
Co-reporter:Ze Li ; Mansoora Khaliq ; Zhigang Zhou ; Carol Beth Post ; Richard J. Kuhn
Journal of Medicinal Chemistry 2008 Volume 51(Issue 15) pp:4660-4671
Publication Date(Web):July 9, 2008
DOI:10.1021/jm800412d
Flavivirus envelope proteins (E proteins) have been shown to play a pivotal role in virus assembly, morphogenesis, and infection of host cells. Inhibition of flavivirus infection of a host cell by means of a small molecule envelope protein antagonist is an attractive strategy for the development of antiviral agents. Virtual screening of the NCI chemical database using the dengue virus envelope protein structure revealed several hypothetical hit compounds. Bioassay results identified a class of thiazole compounds with antiviral potency in cell-based assays. Modification of these lead compounds led to a series of analogues with improved antiviral activity and decreased cytotoxicity. The most active compounds 11 and 36 were effective in the low micromolar concentration range in a cellular assay system.
Co-reporter:Maris A. Cinelli ; Andrew Morrell ; Thomas S. Dexheimer ; Evan S. Scher ; Yves Pommier
Journal of Medicinal Chemistry 2008 Volume 51(Issue 15) pp:4609-4619
Publication Date(Web):July 17, 2008
DOI:10.1021/jm800259e
The aromathecin or “rosettacin” class of topoisomerase I (top1) inhibitors is effectively a “composite” of the natural products camptothecin and luotonin A and the synthetic indenoisoquinolines. The aromathecins have aroused considerable interest following the isolation and total synthesis of 22-hydroxyacuminatine, a rare cytotoxic natural product containing the 12H-5,11a-diazadibenzo[b,h]fluoren-11-one system. We have developed two novel syntheses of this system and prepared a series of 14-substituted aromathecins as novel antiproliferative topoisomerase I poisons. These inhibitors are proposed to act via an intercalation and “poisoning” mechanism identical to camptothecin and the indenoisoquinolines. Many of these compounds possess greater antiproliferative activity and anti-top1 activity than the parent unsubstituted compound (rosettacin) and previously synthesized aromathecins, as well as greater top1 inhibitory activity than 22-hydroxyacuminatine. In addition to potentially aiding solubility and localization to the DNA−enzyme complex, nitrogenous substituents located at the 14-position of the aromathecin system have been proposed to project into the major groove of the top1−DNA complex and hydrogen-bond to major-groove amino acids, thereby stabilizing the ternary complex.
Co-reporter:Tsyr-Yan Yu, Robert D. O’Connor, Astrid C. Sivertsen, Colby Chiauzzi, Barbara Poliks, Markus Fischer, Adelbert Bacher, Ilka Haase, Mark Cushman and Jacob Schaefer
Biochemistry 2008 Volume 47(Issue 52) pp:13942-13951
Publication Date(Web):December 3, 2008
DOI:10.1021/bi8015789
Lumazine synthase catalyzes the reaction of 5-amino-6-d-ribitylamino-2,4(1H,3H)-pyrimidinedione (1) with (S)-3,4-dihydroxybutanone 4-phosphate (2) to afford 6,7-dimethyl-8-d-ribityllumazine (3), the immediate biosynthetic precursor of riboflavin. The overall reaction implies a series of intermediates that are incompletely understood. The 15N{31P} REDOR NMR spectra of three metabolically stable phosphonate reaction intermediate analogues complexed to Saccharomyces cerevisiae lumazine synthase have been obtained at 7 and 12 T. Distances from the phosphorus atoms of the ligands to the side chain nitrogens of Lys92, His97, Arg136, and His148 have been determined. These distances were used in combination with the X-ray crystal coordinates of one of the intermediate analogues complexed with the enzyme in a series of distance-restrained molecular dynamics simulations. The resulting models indicate mobility of the Lys92 side chain, which could facilitate the exchange of inorganic phosphate eliminated from the substrate in one reaction, with the organic phosphate-containing substrate necessary for the next reaction.
Co-reporter:Matthew D. Cullen, Taradas Sarkar, Ernest Hamel, Tracy L. Hartman, Karen M. Watson, Robert W. Buckheit Jr., Christophe Pannecouque, Erik De Clercq, Mark Cushman
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 2) pp:469-473
Publication Date(Web):15 January 2008
DOI:10.1016/j.bmcl.2007.11.114
During studies on the alkenyldiarylmethane (ADAM) class of non-nucleoside reverse transcriptase inhibitors (NNRTIs), analogues were discovered that exhibit low micromolar and submicromolar cytotoxicities. Since the ADAMs are structurally related to the tubulin polymerization inhibitor CC-5079, a set of 14 ADAMs were tested for inhibition of tubulin polymerization in an attempt to identify the biological target responsible for their cytotoxicity. The results indicate that, overall, the ADAMs are poor inhibitors of tubulin polymerization. However, the two most cytotoxic compounds, 15 and 16, are in fact active as inhibitors of tubulin assembly with IC50 values of 3.7 ± 0.3 and 2.8 ± 0.2 μM, respectively, and they both inhibit the binding of colchicine to tubulin. Both compounds were investigated for anticancer activity in the National Cancer Institute’s panel of 60 human cancer cell lines, and both compounds consistently displayed submicromolar cytotoxicities with mean-graph midpoint (MGM) values of 0.31 ± 0.08 and 0.47 ± 0.09 μM, respectively.
Co-reporter:Matthew D. Cullen, York-Fong Cheung, Miles D. Houslay, Tracy L. Hartman, Karen M. Watson, Robert W. Buckheit Jr., Christophe Pannecouque, Erik De Clercq, Mark Cushman
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 4) pp:1530-1533
Publication Date(Web):15 February 2008
DOI:10.1016/j.bmcl.2007.12.015
The alkenyldiarylmethanes (ADAMs) are currently being investigated as non-nucleoside HIV-1 reverse transcriptase inhibitors (NNRTIs) of potential value in the treatment of HIV infection and AIDS. During the course of these studies, a number of ADAM analogues have been identified that protect HIV-infected cells from the cytopathic effects of the virus by an unknown, HIV-1 RT-independent mechanism. Since the phosphodiesterase 4 family is required for HIV infection, the effect of various ADAMs on the activity of PDE4B2 was investigated in an effort to determine if the ADAMs could possibly be targeting phosphodiesterases. Six compounds representative of the ADAM class were tested for inhibition of cAMP hydrolysis by PDE4B2 enzymatic activity. Four ADAMs were found to be weak inhibitors of PDE4B2 and two of them were inactive. The experimental results are consistent with an antiviral mechanism that does not include inhibition of PDE4 isoforms.
Co-reporter:Yunlong Song and Mark Cushman
The Journal of Physical Chemistry B 2008 Volume 112(Issue 31) pp:9484-9489
Publication Date(Web):July 18, 2008
DOI:10.1021/jp8005603
High level ab initio quantum chemical studies have shown that the binding orientations of topoisomerase I (top1) inhibitors such as camptothecins and indenoisoquinolines are primarily governed by π−π stacking. However, a recently discovered norindenoisoquinoline antitumor compound was observed by X-ray crystallography to adopt a “flipped” orientation (relative to indenoisoquinolines), which facilitates the formation of a characteristic hydrogen bond with the Arg364 of top1 in its binding with the top1−DNA complex. This observation raises the possibility that hydrogen bonding between the norindenoisoquinoline nitrogen and the Arg364 side chain of top1 might be responsible for the “flip”. It also brings into question whether π−π stacking, as opposed to hydrogen bonding, is primarily responsible for the binding orientations of indenoisoquinolines and norindenoisoquinolines. In this study, the forces responsible for the binding orientation of a norindenoisoquinoline in the DNA cleavage site were systematically investigated using MP2 methods. The theoretical calculation of the preferred binding orientation based solely on π−π stacking was completely consistent with the actual orientation observed by X-ray crystallography, indicating that the binding of the norindenoisoquinoline in the top1−DNA complex is mainly governed by π−π stacking forces and that the “flip” can occur independently from hydrogen bonding.
Co-reporter:Allison B. Edsall ; Gregory E. Agoston ; Anthony M. Treston ; Stacy M. Plum ; Robert H. McClanahan ; Tian-Sheng Lu ; Wei Song
Journal of Medicinal Chemistry 2007 Volume 50(Issue 26) pp:6700-6705
Publication Date(Web):December 6, 2007
DOI:10.1021/jm070639e
A prodrug strategy was investigated to address the problem of limited aqueous solubility and the resulting limited bioavailability of the antitumor agent 2-methoxyestradiol. The 3-phosphate, 17-phosphate, and 3,17-diphosphate of 2-methoxyestradiol were synthesized. 2-Methoxyestradiol 3-phosphate was metabolized more efficiently to the parent compound in vivo than 2-methoxyestradiol 17-phosphate, and it was also more cytotoxic in cancer cell cultures than either the 17-phosphate or the 3,17-diphosphate. These results agree with the in vivo anticancer activity of 2-methoxyestradiol 3-phosphate in a mouse Lewis lung carcinoma experimental metastasis model as opposed to the 17-phosphate and 3,17-diphosphate, both of which were inactive. The in vivo antitumor activity of 2-methoxyestradiol 3-phosphate at a dose of 200 mg/kg per day was comparable to that of a maximally tolerated dose of cyclophosphamide.
Co-reporter:Ha Young Kim, Richard J. Kuhn, Chinmay Patkar, Ranjit Warrier, Mark Cushman
Bioorganic & Medicinal Chemistry 2007 Volume 15(Issue 7) pp:2667-2679
Publication Date(Web):1 April 2007
DOI:10.1016/j.bmc.2007.01.040
The crystal structure of the Sindbis virus capsid protein contains one or two solvent-derived dioxane molecules in the hydrophobic binding pocket. A bis-dioxane antiviral agent was designed by linking the two dioxane molecules with a three-carbon chain having R,R connecting stereochemistry, and a stereospecific synthesis was performed. This resulted in an effective antiviral agent that inhibited Sindbis virus replication with an EC50 of 14 μM. The synthesis proceeded through an intermediate (R)-2-hydroxymethyl-[1,4]dioxane, which unexpectedly proved to be a more effecting antiviral agent than the target compound, as evidenced by its EC50 of 3.4 μM as an inhibitor of Sindbis virus replication. Both compounds were not cytotoxic in uninfected BHK cells at concentrations of 1 mM.
Co-reporter:Bo-Liang Deng, Matthew D. Cullen, Zhigang Zhou, Tracy L. Hartman, Robert W. Buckheit Jr., Christophe Pannecouque, Erik De Clercq, Phillip E. Fanwick, Mark Cushman
Bioorganic & Medicinal Chemistry 2006 Volume 14(Issue 7) pp:2366-2374
Publication Date(Web):1 April 2006
DOI:10.1016/j.bmc.2005.11.014
The HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) constitute a large and structurally diverse set of compounds, several of which are currently used in the treatment of AIDS. A series of novel alkenyldiarylmethanes (ADAMs) were designed and synthesized as part of an ongoing investigation to replace the metabolically labile methyl ester moieties found in the ADAM pharmacophore with stable modifications that retain the potent anti-HIV activity of the parent compounds. Unsurprisingly, the rat plasma half-lives of the new ADAMs were not improved when compared to the parent compounds, but all of the synthesized ADAMs inhibited the cytopathic effect of HIV-1 in cell culture. The most potent compound identified was (E)-5-[1-(3,7-dimethyl-2-oxo-2,3-dihydro-benzoxazol-5-yl)-5-methoxycarbonyl-pent-1-enyl]-2-methoxy-3-methylbenzoic acid methyl ester (7), which inhibited the cytopathic effects of both HIV-1RF and HIV-1IIIB strains in cell cultures with EC50 values of 30 and 90 nM, respectively, and inhibited HIV-1 reverse transcriptase with an IC50 of 20 nM.The syntheses and antiviral activities for a new set of alkenyldiarylmethane (ADAM) HIV-1 reverse transcriptase inhibitors bearing benzoxazolone and benzisoxazole rings are described.
Co-reporter:Andrew Morrell, Smitha Antony, Glenda Kohlhagen, Yves Pommier, Mark Cushman
Bioorganic & Medicinal Chemistry Letters 2006 Volume 16(Issue 7) pp:1846-1849
Publication Date(Web):1 April 2006
DOI:10.1016/j.bmcl.2006.01.008
A method has been developed that relies on a two-step, one-pot condensation between phthalide and 2-carboxybenzaldehydes to provide benz[d]indeno[1,2-b]pyran-5,11-diones in a multi-gram fashion. Treatment of these compounds with a primary amine allows rapid access to various N-substituted indenoisoquinolines, whose in vitro anticancer activity and topoisomerase I inhibition have been evaluated.A one-pot, two-step synthesis of indenopyrans and their conversion to indenoisoquinoline topoisomerase I inhibitors are reported.
Co-reporter:Andrew Morrell, Muthusamy Jayaraman, Muthukaman Nagarajan, Brian M. Fox, Marintha Rae Meckley, Alexandra Ioanoviciu, Yves Pommier, Smitha Antony, Melinda Hollingshead, Mark Cushman
Bioorganic & Medicinal Chemistry Letters 2006 Volume 16(Issue 16) pp:4395-4399
Publication Date(Web):15 August 2006
DOI:10.1016/j.bmcl.2006.05.048
The indenoisoquinolines are a novel class of non-camptothecin topoisomerase I (Top1) inhibitors whose mechanism of action involves trapping the covalent complex formed between DNA and Top1 during cellular processes. As an ongoing evaluation of the indenoisoquinolines for Top1 inhibition and anticancer activity, indenoisoquinoline analogs have been screened in the National Cancer Institute’s hollow fiber assay (HFA). Some of the derivatives demonstrated significant activity at intraperitoneal and subcutaneous fiber placement sites, along with net cancer cell kill in one or more cell lines.
Co-reporter:Xiangshu Xiao, Ze-Hong Miao, Smitha Antony, Yves Pommier, Mark Cushman
Bioorganic & Medicinal Chemistry Letters 2005 Volume 15(Issue 11) pp:2795-2798
Publication Date(Web):2 June 2005
DOI:10.1016/j.bmcl.2005.03.101
Dihydroindenoisoquinolines are analogs of cytotoxic indenoisoquinoline topoisomerase I (Top1) inhibitors, exhibiting potent cytotoxicity but weak inhibitory activity toward Top1. Through COMPARE analysis, cytotoxicity studies in Top1-deficient cells, chemical synthesis and biological evaluation of methylated dihydroindenoisoquinoline 5, we demonstrated that dihydroindenoisoquinolines function as prodrugs of indenoisoquinolines in cancer cells.
Co-reporter:Ha Young Kim, Chinmay Patkar, Ranjit Warrier, Richard Kuhn, Mark Cushman
Bioorganic & Medicinal Chemistry Letters 2005 Volume 15(Issue 13) pp:3207-3211
Publication Date(Web):1 July 2005
DOI:10.1016/j.bmcl.2005.05.013
Dioxane-based antiviral agents targeted to the hydrophobic binding pocket of Sindbis virus capsid protein were designed by computer graphics molecular modeling and synthesized. Virus production using SIN-IRES-Luc and capsid assembly were monitored to evaluate antiviral activity. A compound with a three-carbon linker chain connecting two dioxane moieties inhibited virus production by 50% at a concentration of 40 μM, while (R)-hydroxymethyldioxane inhibited virus production by 50% at a concentration of 1 μM. Both compounds were not cytotoxic in uninfected BHK cells at concentrations of 1 mM.Dioxane-based antiviral agents targeted to the hydrophobic binding pocket of Sindbis virus capsid protein were designed by computer graphics molecular modeling and synthesized.
Co-reporter:Jinhua Chen, Boris Illarionov, Adelbert Bacher, Markus Fischer, Ilka Haase, Gunda Georg, Qi-zhuang Ye, Zeqiang Ma, Mark Cushman
Analytical Biochemistry 2005 Volume 338(Issue 1) pp:124-130
Publication Date(Web):1 March 2005
DOI:10.1016/j.ab.2004.11.033
A high-throughput screening method based on the competitive binding of a lumazine synthase inhibitor and riboflavin to the active site of Schizosaccharomyces pombe lumazine synthase was developed. This assay is sensitive, simple, and robust. During assay development, all of the known active inhibitors tested were positively identified. Preliminary high-throughput screening in 384-well format resulted in a Z factor of 0.7. The approach utilizes a thermodynamic assay to bypass the problems associated with the instabilities of both lumazine synthase substrates that complicate the use of a kinetic assay in a high-throughput format, and it removes the time element from the assay, thus simplifying the procedure.
Co-reporter:Xiangshu Xiao, Smitha Antony, Glenda Kohlhagen, Yves Pommier, Mark Cushman
Bioorganic & Medicinal Chemistry 2004 Volume 12(Issue 19) pp:5147-5160
Publication Date(Web):1 October 2004
DOI:10.1016/j.bmc.2004.07.027
The cytotoxic indenoisoquinolines are a novel class of noncamptothecin topoisomerase I inhibitors having certain features that compare favorably with the camptothecins. A new strategy was adopted to attach aminoalkenyl substituents at C-11 of the indenoisoquinoline ring system, which, according to molecular modeling, would orient the side chains toward the DNA minor groove. All of the newly synthesized compounds were more cytotoxic than the parent indenoisoquinoline NSC 314622. Despite an imperfect correlation between cytotoxicities and topoisomerase I inhibition results, the hypothetical structural model of the cleavage complex presented here provides a conceptual framework to explain the structure–activity relationships.
Co-reporter:Andrew Morrell, Smitha Antony, Glenda Kohlhagen, Yves Pommier, Mark Cushman
Bioorganic & Medicinal Chemistry Letters 2004 Volume 14(Issue 14) pp:3659-3663
Publication Date(Web):16 July 2004
DOI:10.1016/j.bmcl.2004.05.022
Indenoisoquinolines and dihydroindenoisoquinolines have been synthesized possessing a nitro-substituted isoquinoline ring in an effort to explore the effects of electron-withdrawing substituents on biological activity. The in vitro anticancer activities of these molecules have been tested in the National Cancer Institute's screen of 55 cell lines. The compounds have also been tested for topoisomerase I (top1) inhibition. The results indicate that these substances are a potent class of top1 inhibitors with sub-micromolar cytotoxicity mean graph midpoints (MGM) and top1 inhibition equal to camptothecin.Substitution of the indenoisoquinoline system with an electron-withdrawing nitro group has been investigated as a strategy for increasing cytotoxicity and topoisomerase I inhibitory activity.
Co-reporter:Guozhang Xu, Tracy L. Hartman, Heather Wargo, Jim A. Turpin, Robert W. Buckheit Jr., Mark Cushman
Bioorganic & Medicinal Chemistry 2002 Volume 10(Issue 2) pp:283-290
Publication Date(Web):February 2002
DOI:10.1016/S0968-0896(01)00282-6
The existing methods for the synthesis of alkenyldiarylmethane (ADAM) non-nucleoside reverse transcriptase inhibitors proceed from symmetrical benzophenones and therefore result in products with identical aromatic rings. New methods have therefore been devised for the preparation of stereochemically defined ADAMs with non-identical aromatic rings. The new routes rely on palladium-catalyzed reactions, including Sonogashira, Suzuki, Stille, and hydroarylation methodology. Several of the new ADAMs inhibited the cytopathic effect of HIV-1 in cell culture and HIV-1 reverse transcriptase at submicromolar concentrations.Graphic
Co-reporter:Agustin Casimiro-Garcia, Erik De Clercq, Christophe Pannecouque, Myriam Witvrouw, Tracy L Loftus, Jim A Turpin, Robert W Buckheit Jr., Phillip E Fanwick, Mark Cushman
Bioorganic & Medicinal Chemistry 2001 Volume 9(Issue 11) pp:2827-2841
Publication Date(Web):November 2001
DOI:10.1016/S0968-0896(01)00152-3
A new series of cosalane analogues incorporating two fragments of the dichlorodisalicylmethane pharmacophore has been synthesized. In order to identify the position for the attachment of the pharmacophore fragments to the steroid ring that results in the most potent analogues, two types of compounds were designed. In the first type, the two pharmacophore fragments were attached at C-3 and C-17 of the steroid ring by using appropriate linker units. In the second type, both pharmacophore groups were connected to C-3 of the steroid through an alkenyl chain containing an amide moiety. All of the new compounds displayed antiviral activity versus HIV-1RF, HIV-1IIIB, and HIV-2ROD in cell culture. The relative potencies of the compounds resulting from the two attachment strategies were found to depend on the viral strain as well as the cell type. Overall, the attachment of the second pharmacophore did not result in either a large gain or a large loss in anti-HIV activity, and the results are therefore consistent with the hypothesis that the two pharmacophores act independently, and one at a time, with positively charged amino acid side chains present on the surface of gp120 and CD4.Graphic
Co-reporter:O.M.Zack Howard, Hui Fang Dong, Joost J. Oppenheim, Shabana Insaf, Kalpathy C. Santhosh, Gitendra Paul, Mark Cushman
Bioorganic & Medicinal Chemistry Letters 2001 Volume 11(Issue 1) pp:59-62
Publication Date(Web):8 January 2001
DOI:10.1016/S0960-894X(00)00601-6
The anti-HIV agent cosalane and several of its analogues inhibited RANTES-induced migration of human monocytes, but they did not inhibit migration induced by MIP1α or MIP1β. RANTES-induced migration of single receptor CCR1-HEK transfectants was also inhibited by the cosalanes. Acetylation of the reactive amino groups of RANTES abrogated the inhibitory activity of cosalane. The data suggest that cosalane and its structural analogues may interfere with the RANTES/CCR1 interaction by binding to RANTES.
Co-reporter:Agustin Casimiro-Garcia, Erik De Clercq, Christophe Pannecouque, Myriam Witvrouw, Tracy L. Stup, Jim A. Turpin, Robert W. Buckheit Jr., Mark Cushman
Bioorganic & Medicinal Chemistry 2000 Volume 8(Issue 1) pp:191-200
Publication Date(Web):1 January 2000
DOI:10.1016/S0968-0896(99)00269-2
Introduction of an amido group or an amino moiety into the alkenyl linker chain of cosalane (1) provided a new series of analogues 3–8. The new compounds were evaluated as inhibitors of the cytopathic effect of HIV-1 and HIV-2 in cell culture. The replacement of the 1′ and 2′ carbons in the linker chain of 1 by an amido group was generally tolerated. The length of the linker chain and the stereochemistry of the substituent at C-3 of the steroidal ring had significant effects on the antiviral activity and potency. Incorporation of an amino moiety into the linker completely abolished the anti-HIV activity. There are several steps in the HIV replication cycle that have been proposed as targets for the development of therapeutic agents (De Clercq, E. J. Med. Chem.1995, 38, 2491; De Clercq, E. Pure Appl. Chem.1998, 70, 567). However, currently approved anti-HIV drugs are only directed against the viral enzymes reverse transcriptase or protease (Carpenter, C. C. J.; Fischl, M. A.; Hammer, S. M.; Hirsch, M. S.; Jacobsen, D. M.; Katzenstein, D. A.; Montaner, J. S. G.; Richman, D. D.; Saag, M. S.; Schooley, R. T.; Thompson, M. A.; Vella, S.; Yeni, P. G.; Volberding, P. A. JAMA1998, 280, 78). Drugs capable of interfering with other steps of the virus life cycle will be highly valuable in the antiretroviral therapy of AIDS, as they will have different patterns of resistance mutations than the drugs currently used clinically. In addition, their utilization in combination with other therapeutic agents could provide more potent drug ‘cocktails’ capable of completely suppressing virus replication. Consequently, there is an urgent need for the discovery of clinically useful anti-HIV agents possessing novel mechanisms of action.
Co-reporter:Gitendra C Paul, Erik De Clercq, Christophe Pannecouque, Myriam Witvrouw, Tracy L Loftus, Jim A Turpin, Robert W Buckheit Jr., Mark Cushman
Bioorganic & Medicinal Chemistry Letters 2000 Volume 10(Issue 18) pp:2149-2152
Publication Date(Web):September 2000
DOI:10.1016/S0960-894X(00)00417-0
The binding of the anti-HIV agent cosalane to CD4 is thought to involve ionic interactions of negatively charged carboxylates of the ligand with positively charged residues on the surface of the protein. An investigation of the optimal anion distances for anti-HIV activity in a series of cosalane tetracarboxylate analogues has been completed, and maximal activity results when the two proximal and the two distal carboxylates are separated by eight atoms.
Co-reporter:Kalpathy C Santhosh, Erik De Clercq, Christophe Pannecouque, Myriam Witvrouw, Tracy L Loftus, Jim A Turpin, Robert W Buckheit Jr., Mark Cushman
Bioorganic & Medicinal Chemistry Letters 2000 Volume 10(Issue 22) pp:2505-2508
Publication Date(Web):20 November 2000
DOI:10.1016/S0960-894X(00)00515-1
The binding of the anti-HIV agent cosalane to CD4 is thought to involve ionic interactions of negatively charged carboxylates of the ligand with positively charged residues on the surface of the protein. The purpose of the present study was to examine the hypothesis that the two carboxyl groups of cosalane could be sacrificed through conjugation to amino acids, and the anti-HIV activity still be retained, provided that at least two new carboxyl groups are contributed by the amino acid residues.





![[1-(4-nitrophenyl)ethylidene]hydrazine](http://img.cochemist.com/ccimg/28200/28153-22-2.png)
![[1-(4-nitrophenyl)ethylidene]hydrazine](http://img.cochemist.com/ccimg/28200/28153-22-2_b.png)


![1H-BENZOTRIAZOLE, 1-[[[2-(2-PYRIDINYLDITHIO)ETHOXY]CARBONYL]OXY]-](http://img.cochemist.com/ccimg/913200/913168-10-2.png)
![1H-BENZOTRIAZOLE, 1-[[[2-(2-PYRIDINYLDITHIO)ETHOXY]CARBONYL]OXY]-](http://img.cochemist.com/ccimg/913200/913168-10-2_b.png)
![BENZ[D]INDENO[1,2-B]PYRAN-5,11-DIONE, 3-NITRO-](http://img.cochemist.com/ccimg/885000/884902-28-7.png)
![BENZ[D]INDENO[1,2-B]PYRAN-5,11-DIONE, 3-NITRO-](http://img.cochemist.com/ccimg/885000/884902-28-7_b.png)
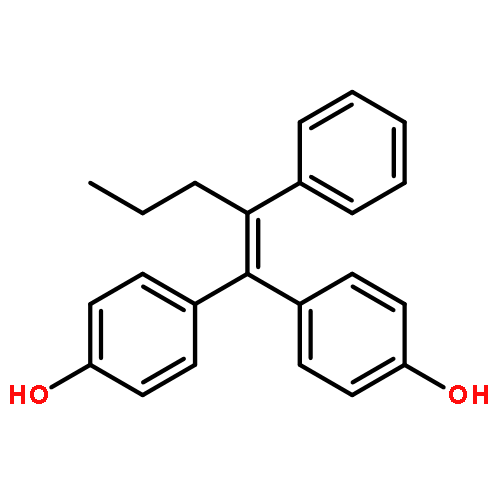
![PHENOL, 4-[2-(4-HYDROXYPHENYL)-3-METHYL-1-PHENYL-1-BUTENYL]-](http://img.cochemist.com/ccimg/851400/851342-39-7.png)
![PHENOL, 4-[2-(4-HYDROXYPHENYL)-3-METHYL-1-PHENYL-1-BUTENYL]-](http://img.cochemist.com/ccimg/851400/851342-39-7_b.png)











![Furo[3,4-b]pyridin-5(7H)-one,7-hydroxy-](http://img.cochemist.com/ccimg/252300/252289-75-1.png)
![Furo[3,4-b]pyridin-5(7H)-one,7-hydroxy-](http://img.cochemist.com/ccimg/252300/252289-75-1_b.png)





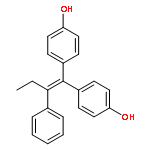
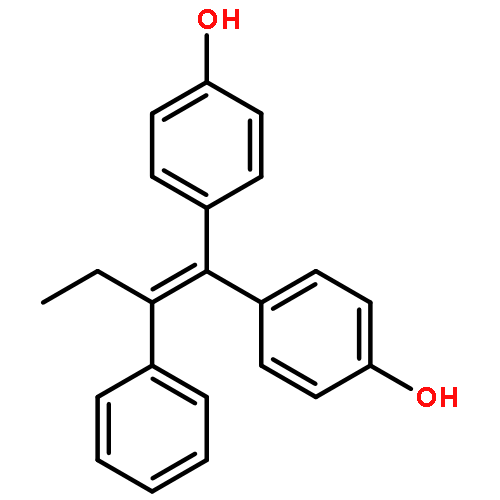

![2-[4-[(Z)-1,2-diphenylbut-1-enyl]phenoxy]ethanamine](http://img.cochemist.com/ccimg/80300/80234-20-4.png)
![2-[4-[(Z)-1,2-diphenylbut-1-enyl]phenoxy]ethanamine](http://img.cochemist.com/ccimg/80300/80234-20-4_b.png)
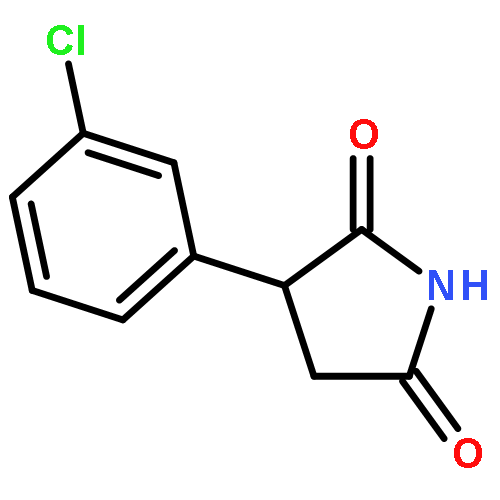
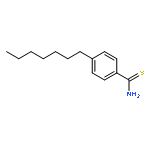
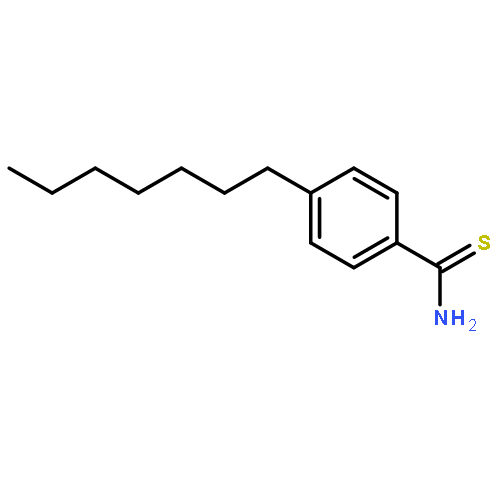


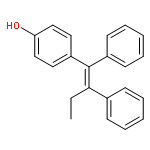


![Propanedioic acid, [[3-(phenylmethoxy)phenyl]methylene]-, diethyl ester](http://img.cochemist.com/ccimg/63000/62904-57-8.png)
![Propanedioic acid, [[3-(phenylmethoxy)phenyl]methylene]-, diethyl ester](http://img.cochemist.com/ccimg/63000/62904-57-8_b.png)
![2,5-Pyrrolidinedione, 3-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/60100/60050-38-6.png)
![2,5-Pyrrolidinedione, 3-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/60100/60050-38-6_b.png)
![1-[5-(3-NITROPHENYL)FURAN-2-YL]ETHANONE](http://img.cochemist.com/ccimg/56700/56656-35-0.png)
![1-[5-(3-NITROPHENYL)FURAN-2-YL]ETHANONE](http://img.cochemist.com/ccimg/56700/56656-35-0_b.png)











![3-[3-(Trifluoromethyl)phenyl]pyrrolidine HCl](http://img.cochemist.com/ccimg/21800/21767-35-1.png)
![3-[3-(Trifluoromethyl)phenyl]pyrrolidine HCl](http://img.cochemist.com/ccimg/21800/21767-35-1_b.png)




![[1,1'-Biphenyl]-4-carbothioamide](http://img.cochemist.com/ccimg/13400/13363-50-3.png)
![[1,1'-Biphenyl]-4-carbothioamide](http://img.cochemist.com/ccimg/13400/13363-50-3_b.png)
![Propanedioic acid, [(3-chlorophenyl)methylene]-, diethyl ester](http://img.cochemist.com/ccimg/6800/6768-21-4.png)
![Propanedioic acid, [(3-chlorophenyl)methylene]-, diethyl ester](http://img.cochemist.com/ccimg/6800/6768-21-4_b.png)
![1,3-dimethyl-5-{[2-methyl-1-(prop-2-en-1-yl)-1H-indol-3-yl]methylidene}-2-thioxodihydropyrimidine-4,6(1H,5H)-dione](http://img.cochemist.com/ccimg/6400/6331-45-9.png)
![1,3-dimethyl-5-{[2-methyl-1-(prop-2-en-1-yl)-1H-indol-3-yl]methylidene}-2-thioxodihydropyrimidine-4,6(1H,5H)-dione](http://img.cochemist.com/ccimg/6400/6331-45-9_b.png)



![6-methyl-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione](http://img.cochemist.com/ccimg/5300/5291-18-9.png)
![6-methyl-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione](http://img.cochemist.com/ccimg/5300/5291-18-9_b.png)
![Ethanone, 1-(3'-nitro[1,1'-biphenyl]-4-yl)-](http://img.cochemist.com/ccimg/5100/5002-11-9.png)
![Ethanone, 1-(3'-nitro[1,1'-biphenyl]-4-yl)-](http://img.cochemist.com/ccimg/5100/5002-11-9_b.png)
![Furo[3,4-c]pyridin-1(3H)-one](http://img.cochemist.com/ccimg/4800/4741-42-8.png)
![Furo[3,4-c]pyridin-1(3H)-one](http://img.cochemist.com/ccimg/4800/4741-42-8_b.png)
![furo[3,4-b]pyridin-5(7H)-one](http://img.cochemist.com/ccimg/4800/4733-69-1.png)
![furo[3,4-b]pyridin-5(7H)-one](http://img.cochemist.com/ccimg/4800/4733-69-1_b.png)
![N-[(2,6-DICHLOROPHENYL)SULFONYL]ALANINE](http://img.cochemist.com/ccimg/3900/3898-66-6.png)
![N-[(2,6-DICHLOROPHENYL)SULFONYL]ALANINE](http://img.cochemist.com/ccimg/3900/3898-66-6_b.png)
![3-AMINO-3-[5-(3-CHLORO-4-METHOXY-PHENYL)-2-FURYL]PROPANAMIDE](http://img.cochemist.com/ccimg/3500/3464-18-4.png)
![3-AMINO-3-[5-(3-CHLORO-4-METHOXY-PHENYL)-2-FURYL]PROPANAMIDE](http://img.cochemist.com/ccimg/3500/3464-18-4_b.png)










![Furo[3,4-c]pyridin-3(1H)-one](http://img.cochemist.com/ccimg/5700/5657-52-3.png)
![Furo[3,4-c]pyridin-3(1H)-one](http://img.cochemist.com/ccimg/5700/5657-52-3_b.png)
![Furo[3,4-f]-1,3-benzodioxol-5(7H)-one](http://img.cochemist.com/ccimg/4800/4792-36-3.png)
![Furo[3,4-f]-1,3-benzodioxol-5(7H)-one](http://img.cochemist.com/ccimg/4800/4792-36-3_b.png)
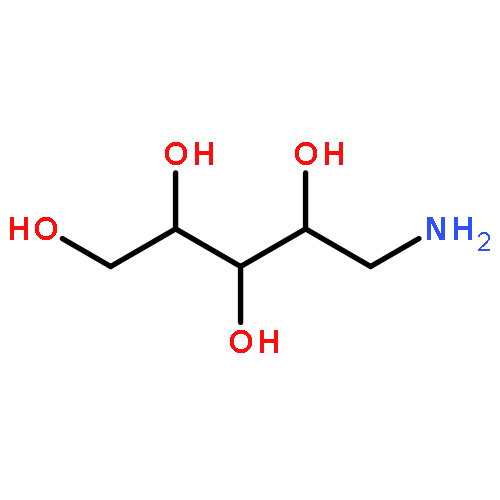


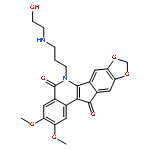
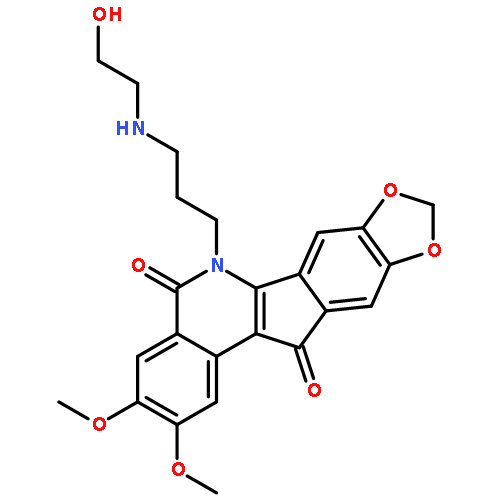
![4-(5,11-DIOXOINDENO[1,2-C]ISOQUINOLIN-6-YL)BUTANOIC ACID](http://img.cochemist.com/ccimg/220700/220627-87-2.png)
![4-(5,11-DIOXOINDENO[1,2-C]ISOQUINOLIN-6-YL)BUTANOIC ACID](http://img.cochemist.com/ccimg/220700/220627-87-2_b.png)


![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 6-[(2,6-dimethoxybenzoyl)amino]-3,3-dimethyl-7-oxo-, (2S,5R,6R)-](http://img.cochemist.com/ccimg/100/61-32-5.png)
![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 6-[(2,6-dimethoxybenzoyl)amino]-3,3-dimethyl-7-oxo-, (2S,5R,6R)-](http://img.cochemist.com/ccimg/100/61-32-5_b.png)



