Co-reporter:Hao Su, Xiang Sheng, Wenyou Zhu, Guangcai Ma, and Yongjun Liu
ACS Catalysis August 4, 2017 Volume 7(Issue 8) pp:5534-5534
Publication Date(Web):July 14, 2017
DOI:10.1021/acscatal.7b01606
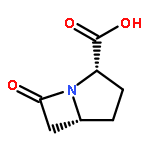
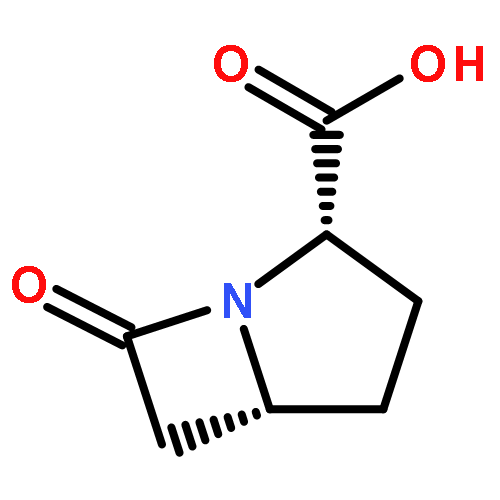
AsqJ from Aspergillus nidulans is a nonheme FeII/α-ketoglutarate-dependent dioxygenase that catalyzes the conversion of benzodiazepinedione into 4′-methoxyviridicatin, which is a key step in the biosynthesis of quinolone alkaloids. A series of recent experiments have demonstrated that AsqJ is able to perform the decoupled desaturation and epoxidation reactions. Herein, on the basis of the published crystal structures, combined quantum mechanics and molecular mechanics (QM/MM) calculations have been performed to explore both the desaturation and epoxidation processes. Our calculations reveal that the quintet state of the FeIV–O complex is the ground state, and the catalytic reaction occurs on the quintet-state surface. The FeIV–oxo species should first undergo an isomerization to initiate the reactions. In the desaturation process, the abstraction of the first hydrogen atom is suggested to follow the σ-channel mechanism. This step is calculated to be rate-limiting with an energy barrier of 19.3 kcal/mol. The abstraction of the second hydrogen atom is found to be quite easy. After the desaturation process, the regenerated FeIV–oxo species first attacks the C═C bond of the desaturated intermediate to form a carbon-based radical intermediate, corresponding to an energy barrier of 18.1 kcal/mol, then the radical intermediate completes the ring closure with a barrier of 3.9 kcal/mol. Besides, the calculations using the substrate analogous that lacks the N4-methyl reveal that the H atom abstraction by FeIV–oxo is still accessible, which suggests that the absence of N4-methyl does not affect the desaturation process itself but may influence the other processes that occur prior to the desaturation.Keywords: AsqJ; desaturation; epoxidation; nonheme dioxygenase; QM/MM;
Co-reporter:Ge Tian
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 11) pp:7733-7742
Publication Date(Web):2017/03/15
DOI:10.1039/C6CP08811B
Ubiquinone plays a pivotal role in the aerobic cellular respiratory electron transport chain, whereas ferulic acid decarboxylase (FDC) is involved in the biosynthesis of ubiquinone precursor. Recently, the complete crystal structure of FDC (based on the co-expression of the A. niger fdc1 gene in E. coli with the associated ubix gene from E. coli) at high resolution was reported. Herein, the detailed catalytic non-oxidative decarboxylation mechanism of FDC has been investigated by a combined quantum mechanics/molecular mechanics (QM/MM) approach. Calculation results indicate that, after the 1,3-dipolar cycloaddition of the substrate and cofactor, the carboxylic group can readily split off from the adduct, and the overall energy barrier of the whole catalytic reaction is 23.5 kcal mol−1. According to the energy barrier analysis, the protonation step is rate-limiting. The conserved protonated Glu282 is suggested to be the proton donor through a “water bridge”. Besides, two cases, that is, the generated CO2 escapes from the active site or remains in the active site, were considered. It was found that the prolonged leaving of CO2 can facilitate the protonation of the intermediate. In particular, our calculations shed light on the detailed function of both cofactors prFMNiminium and prFMNketamine in the decarboxylation step. The cofactor prFMNiminium is the catalytically relevant species compared with prFMNketamine.
Co-reporter:Xiya Wang;Hao Su
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 11) pp:7668-7677
Publication Date(Web):2017/03/15
DOI:10.1039/C7CP00313G
Fumitremorgin B endoperoxidase (FtmOx1) from Aspergillus fumigatus is the first reported α-ketoglutarate-dependent mononuclear non-haem iron enzyme that catalyzes the endoperoxide formation reaction, converting Fumitremorgin B to verruculogen. Experiments reveal that the molecular oxygen (O2) is incorporated into verruculogen without O–O bond scission, which differs from the currently known non-haem iron enzymes, but the mechanistic details are still unclear. In this paper, on the basis of the crystal structures of FtmOx1 in complex with either the co-substrate (α-ketoglutarate) or the substrate (fumitremorgin B), a ternary complex model of the enzyme-α-ketoglutarate-substrate has been constructed, and combined quantum mechanics and molecular mechanics (QM/MM) calculations have been performed to unravel the novel mechanism of FtmOx1. Our calculations indicate the quintet of the FeIVO complex as the ground state. The FeIVO complex firstly abstracts a hydrogen from the hydroxyl of Tyr228 to initiate the reaction, which corresponds to a lower energy barrier (9.1 kcal mol−1). If the FeIVO complex directly abstracts a hydrogen from C21 of the substrate, the energy barrier would increase to 33.9 kcal mol−1. When Tyr228 was mutated to Ala228, this energy barrier decreases to 24.0 kcal mol−1. In the subsequent reaction, the generated Tyr228 radical abstracts the hydrogen (H2) from C21 of the substrate with an energy barrier of 23.8 kcal mol−1. The second molecular oxygen binds to the C21 radical of the substrate in the active pocket and further completes the epoxidation with an energy barrier of 4.8 kcal mol−1. These results may provide useful information for understanding the reaction mechanism of FtmOx1 and provide guidance for further experimental investigations.
Co-reporter:Shujun Zhang;Xiya Wang
Catalysis Science & Technology (2011-Present) 2017 vol. 7(Issue 4) pp:911-922
Publication Date(Web):2017/02/20
DOI:10.1039/C6CY02553F
2,4′-Dihydroxyacetophenone dioxygenase (DAD) is a bacterial non-heme enzyme responsible for the oxygenative cleavage of the aliphatic C–C bond, which catalyzes the conversion of 2,4′-dihydroxyacetophenone to 4-hydroxybenzoic acid and formic acid. On the basis of the crystal structure and studies on two synthesized biomimetic model compounds, two possible reaction pathways that involve a dioxacyclic or alkylperoxo intermediate have been previously suggested. However, little is currently known about the mechanistic detail and the proposed intermediates have not been experimentally confirmed yet. To elucidate the reaction mechanism at the atomistic level, on the basis of the recently obtained crystal structure, the reactant enzyme–substrate complex has been constructed, and the reaction details have been studied using a quantum mechanics/molecular mechanics (QM/MM) approach. Our calculations reveal the triplet of the iron(III)-superoxide radical complex as the ground state, but the quintet state which is higher than the triplet by 11.2 kcal mol−1 corresponds to a lower energy barrier in the first step. Thus, the reactant complex may firstly undergo a triplet–quintet crossing to initiate the reaction and then the subsequent chemistry mainly occurs on the quintet state surface. The previously proposed key dioxacyclic or alkylperoxo intermediate was calculated to be energetically unreachable, and the corresponding mechanism has been revised, which contains eight elementary steps, and the key C–C bond cleavage is accompanied by an insertion reaction of the adjacent oxygen radical. Two elementary steps are calculated to be possible rate-determining steps. Our results may provide useful information for further understanding the cleavage mechanism of the aliphatic C–C bond catalyzed by DAD and other dioxygenase enzymes.
Co-reporter:Xiya Wang;Wenyou Zhu
Catalysis Science & Technology (2011-Present) 2017 vol. 7(Issue 13) pp:2846-2856
Publication Date(Web):2017/07/03
DOI:10.1039/C7CY00573C
L-Tryptophan lyase (NosL), a member of the radical S-adenosyl-L-methionine (SAM)-dependent superfamily, catalyzes the conversion of L-tryptophan to 3-methylindolic acid (MIA). In this article, on the basis of the recently obtained crystal structure of NosL (PDB code 4R34) in 2014, a combined quantum mechanical/molecular mechanical (QM/MM) approach has been employed to elucidate the reaction details, involving substrate amine dehydrogenation, C–C bond cleavage and carboxyl fragment migration. Our results show that the hydrogen in the amino group of L-tryptophan is suitable for abstraction by the Ado radical, and this step corresponds to an energy barrier of 12.4 kcal mol−1. Two possible modes of C–C cleavage (path1 and path2) have been considered. The cleavage of the Cα–Cβ bond is thermodynamically more favorable than the cleavage of the Cα–C bond with their energy barriers being 7.2 and 15.0 kcal mol−1, respectively. And the easy breaking of Cα–Cβ may be attributed to the electron hole delocalization in the amine radical of the substrate. The intermediate derived from the cleavage of the Cα–C bond is calculated to be a stable species, and the cleavage of the Cα–C bond is accompanied by the migration of the ˙COO− fragment. These conclusions are basically in accordance with EPR-trapped analysis and can account for the absence of the ˙COO− fragment. The shunt product (3-methylindole) is obtained from the cleavage of the Cα–Cβ bond with an energy barrier of 19.5 kcal mol−1. However, the rate limiting step is the formation of 3-methylindolic acid (MIA), which corresponds to an energy barrier of 26.9 kcal mol−1. Our investigations thus give a better comprehension of the NosL reaction mechanism and may contribute to the understanding of the SAM superfamily.
Co-reporter:Jing Zhang
Theoretical Chemistry Accounts 2017 Volume 136( Issue 4) pp:
Publication Date(Web):2017 April
DOI:10.1007/s00214-017-2079-x
Glyoxylate carboligase (GCL) catalyzes the ligation of two molecules of glyoxylate to form tartronate semialdehyde (TSA) and carbon dioxide. GCL is unique among ThDP-dependent enzymes because it lacks the canonical glutamate in the active site which is thought to be highly necessary for other ThDP enzymes. In this paper, the catalytic reactions of GCL and its mutant V51E have been explored using a combined QM/MM approach. On the basis of our calculations, the following important points have been obtained: (1) although the glutamate residue is absent in the active site of GCL, the activation of cofactor ThDP is still quite easy with a very low energy barrier of 5.0 kcal/mol; (2) the catalytic cycle can still proceed in the case where no other potential acid–base side chain exists in the active site; (3) for the wild-type GCL, the energy barrier of the most energy-requiring step is 17.9 kcal/mol, which agrees well with the experimental observations; (4) for V51E mutant, the concerted formation of the product TSA and regeneration of the ThDP ylide is calculated to be the rate-limiting step with an energy barrier of 19.5 kcal/mol. It is slightly higher than that of the wild-type GCL (19.5 vs. 17.9 kcal/mol), which can well explain the relatively lower activity of the V51E mutant than the wild-type enzyme. Our results may provide help for further understanding the catalytic mechanism of GCL.
Co-reporter:Jing Zhang
Theoretical Chemistry Accounts 2017 Volume 136( Issue 4) pp:
Publication Date(Web):2017 April
DOI:10.1007/s00214-017-2079-x
Glyoxylate carboligase (GCL) catalyzes the ligation of two molecules of glyoxylate to form tartronate semialdehyde (TSA) and carbon dioxide. GCL is unique among ThDP-dependent enzymes because it lacks the canonical glutamate in the active site which is thought to be highly necessary for other ThDP enzymes. In this paper, the catalytic reactions of GCL and its mutant V51E have been explored using a combined QM/MM approach. On the basis of our calculations, the following important points have been obtained: (1) although the glutamate residue is absent in the active site of GCL, the activation of cofactor ThDP is still quite easy with a very low energy barrier of 5.0 kcal/mol; (2) the catalytic cycle can still proceed in the case where no other potential acid–base side chain exists in the active site; (3) for the wild-type GCL, the energy barrier of the most energy-requiring step is 17.9 kcal/mol, which agrees well with the experimental observations; (4) for V51E mutant, the concerted formation of the product TSA and regeneration of the ThDP ylide is calculated to be the rate-limiting step with an energy barrier of 19.5 kcal/mol. It is slightly higher than that of the wild-type GCL (19.5 vs. 17.9 kcal/mol), which can well explain the relatively lower activity of the V51E mutant than the wild-type enzyme. Our results may provide help for further understanding the catalytic mechanism of GCL.
Co-reporter:Na Cheng;Changqiao Zhang
Journal of Molecular Modeling 2017 Volume 23( Issue 8) pp:225
Publication Date(Web):14 July 2017
DOI:10.1007/s00894-017-3388-7
Donor–acceptor conjugated polymers have been successfully applied in bulk heterojunction solar cell devices. Tuning their donor and acceptor units allows the design of new polymers with desired electronic and optical properties. Here, to screen new candidate polymers based on a newly synthesized donor unit, dithieo[2,3-d:2′,3′-d′]naphtho[1,2-b:3,4-b′]dithiophene (NDT), a series of model polymers with different acceptor units were designed and denoted NDT-A0 to NDT-A12, and the structures and optical properties of those polymers were investigated using DFT and TDDFT calculations. The results of the calculations revealed that the electronic and optical properties of these polymers depend on the acceptor unit present; specifically, their HOMO energies ranged from −4.89 to −5.38 eV, their HOMO–LUMO gaps ranged from 1.30 to 2.80 eV, and their wavelengths of maximum absorption ranged from 538 to 1212 nm. The absorption spectra of NDT–A1 to NDT–A6, NDT–A8, NDT–A9, and NDT–A12 occur within the visible region (<900 nm), indicating that these polymers are potential candidates for use in solar cells. On the other hand, the absorption spectra of NDT–A7, NDT–A10, and NDT–A11 extend much further into the near-infrared region, implying that they absorb near-infrared light. These polymers could meet the requirements of donor units for use in tandem and ternary solar cells.
Co-reporter:Lihua Dong, Shujun Zhang, Yongjun Liu
Journal of Molecular Graphics and Modelling 2017 Volume 76(Volume 76) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.jmgm.2017.07.016
•The catalytic mechanism of BphD has been elucidated by using QM/MM calculations.•The hydrolysis of CC bond contains two half-reactions: acylation and deacylation.•An active site water molecule is suggested to play important roles for the deprotonation of Ser112.•The mechanism of BphD may highlight the versatility of Ser-His-Asp triad.As members of the α/β-hydrolase superfamily, Meta-cleavage product (MCP) hydrolases generally utilize a Ser-His-Asp catalytic triad to hydrolyze the cleavage of CC bond during the aerobic catabolism of aromatic compounds by bacteria. BphD is one kind of MCP hydrolase that catalyzes the hydrolysis of 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoic acid (HOPDA) to 2-hydroxypenta-2,4-dienoic acid (HPD) and benzoate. In this article, a combined quantum mechanics and molecule mechanics (QM/MM) approach has been employed to explore the reaction mechanism of BphD from Burkholderia xenovorans LB400. On the basis of the recently resolved crystal structures, three computational models have been constructed. Our calculation results reveal that BphD utilizes a water-assisted nucleophilic mechanism, which contains acylation and deacylation stages. In acylation reaction, an active site water molecule assists the proton transfer from Ser112 to the carbanion intermediate (substrate) by forming hydrogen bonds with Ser112 and His265, and this proton transfer is in concert with the nucleophilic attack of deprotonated Ser112 on the C6-carbonyl of substrate to form the acylated intermediate. In deacylation, the Asp237-His265 dyad acts as a general base to activate the hydrolytic water, whose nucleophilic attack leads to the collapses of acyl-enzyme intermediate. The acylation and deacylation process correspond to the highest energy barriers of 21.0 and 23.9 kcal/mol, respectively. During the catalytic reaction, the active site water and Asp237-His265 dyad play an important role for each elementary steps.Download high-res image (103KB)Download full-size image
Co-reporter:Beibei Lin, Guangcai Ma, and Yongjun Liu
ACS Catalysis 2016 Volume 6(Issue 10) pp:7010
Publication Date(Web):September 7, 2016
DOI:10.1021/acscatal.6b01417
Ethylmalonic encephalopathy protein 1 (ETHE1) is a β-lactamase fold-containing protein, which is related to the increased cellular levels of hydrogen sulfide. ETHE1 is essential for the survival of a range of organisms and catalyzes the oxidation of glutathione persulfide (GSSH). Currently, the catalytic mechanism of ETHE1 still remains unclear, despite a catalytic cycle that has been suggested from the crystal structure and a proposal for the mechanistically related cysteine dioxygenase (CDO). In this Article, we performed a series of quantum mechanical/molecular mechanical (QM/MM) calculations on the substrate GSSH oxidation by human ETHE1. Our calculation results reveal that the ground state of the iron(II)-superoxo reactant is quintet, which can be described as GSS+•–Fe(II)–O2•, and the most feasible reaction channel was found to start from the cleavage of dioxygen and a concerted attack of distal oxygen on the sulfur atom of the substrate, forming the metal-bound activated oxygen and a sulfite intermediate. Moreover, the reaction starts from a quintet ground-state reactant, undergoes a triplet intermediate, and finally generates the septet product rather than the reaction of CDO, which starts from a singlet–quintet crossing.Keywords: ethylmalonic encephalopathy protein 1(ETHE1); glutathione persulfide; GSSH; oxidation; QM/MM; reaction mechanism
Co-reporter:Hao Su, Xiang Sheng and Yongjun Liu
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 13) pp:3432-3442
Publication Date(Web):04 Mar 2016
DOI:10.1039/C6OB00320F
The peptidoglycan (PG) metabolic process is essential for bacterial growth. β-N-Acetylglucosaminidases (NagZ enzymes) are involved in the PG process and they catalyze the removal of terminal N-acetylglucosamine in PG fragments. According to the amino acid sequence and secondary structures, NagZ enzymes should belong to the glycoside hydrolase family GH3. However, a recent experimental study revealed that NagZ enzymes are glycoside phosphorylases rather than glycoside hydrolases. To further understand the catalytic process of NagZs at the atomistic level, the reaction mechanism of NagZ from Bacillus subtilis (BsNagZ) has been studied by using a QM/MM approach. Our calculation results show that the glycosylation of the substrate is the rate limiting step of the whole catalytic cycle with an energy barrier of 19.3 kcal mol−1, which is close to the free energy barrier (16.4 kcal mol−1) estimated from the experimental rate constant. For deglycosylation, both the hydrolysis and phosphorylation of the glycosyl-enzyme intermediate were explored. The phosphorylation corresponds to the lower energy barrier than hydrolysis (1.8 vs. 17.7 kcal mol−1), giving theoretical support to the previously suggested phosphorylase activity of NagZ enzymes. In both the glycosylation and deglycosylation steps, the oxocarbenium-ion-like transition states are always involved, and the substrate distortion in the active site can significantly facilitate the reaction, in which residue D123 plays a key role in this distortion. This is the first computational report for understanding the phosphorylase activity of NagZ enzymes.
Co-reporter:Beibei Lin, Hao Su, Guangcai Ma, Yongjun Liu and Qianqian Hou
RSC Advances 2016 vol. 6(Issue 65) pp:60376-60384
Publication Date(Web):17 Jun 2016
DOI:10.1039/C6RA09735A
Serum paraoxonase 1 (PON1) is a calcium-dependent enzyme that can catalyze the hydrolysis of multiple substrates, including lactones, thiolactones, carbonates, esters and phosphotriesters, as well as the formation of a variety of lactones. To better understand the lactonase mechanism of PON1, the hydrolysis of dihydrocoumarin, which is considered as a native substrate of PON1, has been investigated by using a combined quantum mechanics and molecular mechanics (QM/MM) approach. Two possible reaction pathways with either Glu53 or His115 acts as the general base have been considered. On the basis of our calculations, these two pathways correspond to the overall energy barriers of 12.5 and 9.0 kcal mol−1, respectively. During the catalytic reaction, if one of the two residues (Glu53 and His115) acts as the catalytic base, the other one forms strong hydrogen bonding interaction with the attacking hydroxide to facilitate the hydrolysis. However, mutation studies reveal that Glu53 is necessary for hydrolysis, whereas His115 is not essential but can promote the activity of PON1. Natural population analysis indicates that the catalytic Ca2+ does not act as Lewis acid but plays structural role in fixing the orientations of the substrate and related residues. In addition, Asp269 is found to coordinate with Ca2+ cation and facilitate the protonation of the alkoxide leaving group by forming hydrogen bond with lactone. These results can explain the fact that mutation of Glu53 results in the loss of activity of PON1, and the hydrolysis of dihydrocoumarin is unaffected by mutation of H115.
Co-reporter:Shujun Zhang, Hao Su, Guangcai Ma and Yongjun Liu
RSC Advances 2016 vol. 6(Issue 28) pp:23396-23402
Publication Date(Web):25 Feb 2016
DOI:10.1039/C6RA00328A
N-Acyl-homoserine lactonase from Ochrobactrum sp. strain (AidH) is a novel AHL (N-acyl-homoserine lactone)-lactonase that hydrolyzes the ester bond of the homoserine lactone ring of AHLs. In this article, on the basis of the high-resolution crystal structure of the mutated AidH in complex with substrate, a combined quantum mechanics/molecular mechanics (QM/MM) approach has been employed to study the detailed catalytic mechanism of AidH using C6-HSL (N-hexanoyl homoserine lactone) as the substrate. The calculation results reveal that the catalytic reaction starts from the abstraction of the hydroxyl hydrogen of Ser102 by His248. Both the formation and cleavage of the covalent bond between Ser102 and the substrate are possible rate-limiting steps, corresponding to energy barriers of 19.2 and 21.7 kcal mol−1, respectively. The ring-opening of the covalent intermediate is calculated to be quite easy. During the catalysis, His248 acts as a dual Lewis acid/base, whereas Glu219 is not directly involved in the chemical process and Tyr160 only plays a role in stabilizing the transition state and orienting the position of the hydrolytic water molecule. In addition, the distant surrounding residues were found to have different influences on the reaction by their electrostatic interaction with the substrate. These results may provide useful information for the novel treatment of plant and animal infections that rely on AHL signaling.
Co-reporter:Na Cheng, Yuchen Ma, Yongjun Liu, Changqiao Zhang, Chengbu Liu
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2016 Volume 159() pp:262-268
Publication Date(Web):15 April 2016
DOI:10.1016/j.saa.2016.02.003
•The optical properties of BTT-based donor–acceptor copolymers were studied.•The positions of sulfur atoms in BTT units have great influence on the properties.•The maximum shift of main absorption peaks of these polymers can reach ~ 150 nm.In this paper, we have investigated the structures, electronic and optical properties of five conjugated copolymers (BTT1–BTz, BTT2–BTz, BTT3–BTz, BTT4–BTz and BTT5–BTz) featuring benzotrithiophene (BTT) isomers as donor units and benzothiadiazole (BTz) as acceptor units, linked through thiophene spacers, employing many-body perturbation theory (MBPT). We have explored the isomer effects by configuration of the sulfur atoms in BTT units, aimed to get insight into how the structural modifications to the conjugated backbone can influence the molecular structures and electronic properties of conjugated polymers. Using the trimer as the computational model, the calculated low and high energy absorption bands (660 and 413 nm) for BTT1–BTz agree well with the experimental ones (645 and 430 nm) with a small offset of ~ 15 nm. On the basis of our calculations, it is found that the backbones of these polymers display different coplanarities, with the dihedral angles between the two neighboring rings varying from 12.3° to 79.0°. Importantly, both BTT1–BTz and BTT2–BTz exhibit intense adsorption around 660 and 623 nm, indicating their promising application in solar cells, whereas BTT3–BTz and BTT4–BTz display the intense adsorption at 569 and 551 nm, which are also usable in the tandem solar cells. BTT5–BTz has narrow and weak adsorption in the visible and infrared region, implying it is not conducive to the sunlight absorption. The blue shift of about 150 nm from BTT1–BTz to BTT5–BTz is suggested to be originated from the shorter effective conjugation lengths.
Co-reporter:Wenyou Zhu and Yongjun Liu
ACS Catalysis 2015 Volume 5(Issue 7) pp:3953
Publication Date(Web):May 21, 2015
DOI:10.1021/acscatal.5b00156
7-Carboxy-7-deazaguanine synthase (QueE) is a radical S-adenosylmethionine (SAM) enzyme that catalyzes the conversion of 6-carboxy-5,6,7,8-tetrahydropterin (CPH4) to 7-carboxy-7-deazaguanine (CDG). QueE also shows a clear dependence on Mg2+ ion and is considered a new feature for a radical SAM enzyme. The catalytic mechanism of QueE from B. multivorans has been studied using a combined quantum mechanics and molecular mechanics (QM/MM) method. The results of our calculations reveal that the key ring-contraction step involves a bridged intermediate rather than a ring-opening one. For the QueE–Mg2+ system, the elimination of ammonia is calculated to be rate limiting with a free energy barrier of 18.8 kcal/mol, which is basically in accordance with the estimated value (20.9 kcal/mol) from the experiment. For QueE–Na+ complex, the rate-limiting step switches to the formation of the bridged intermediate with an energy barrier of 29.3 kcal/mol. Natural population analysis indicates that the metal ions do not act as Lewis acids; therefore, they mainly play a role in fixing the substrate in its reactive conformation. The different coordination of Mg2+ and Na+ with the substrate is suggested to be the main reason for leading to the different activities of QueE–Mg2+ and QueE–Na+ complexes.Keywords: 6-carboxy-5,6,7,8-tetrahydropterin (CPH4); 7-carboxy-7-deazaguanine (CDG); 7-carboxy-7-deazaguanine synthase (QueE); QM/MM; reaction mechanism; SAM radical enzyme
Co-reporter:Guangcai Ma, Wenyou Zhu, Hao Su, Na Cheng, and Yongjun Liu
ACS Catalysis 2015 Volume 5(Issue 9) pp:5556
Publication Date(Web):August 18, 2015
DOI:10.1021/acscatal.5b01275
Author: Carbapenem antibiotics possess a broad spectrum of antibacterial activity and high resistance to hydrolytic inactivation by β-lactamases. Carbapenem synthase (CarC), an iron(II) and 2-(oxo)glutarate-dependent oxygenase, catalyzes the epimerization and desaturation of (3S,5S)-carbapenam to produce (5R)-carbapenem in the last step of the simple carbapenem biosynthesis. Recently, the complete crystal structure of CarC was reported, allowing us to perform accurate quantum mechanics/molecular mechanics calculations to explore the detailed reaction mechanism. We first analyzed the dioxygen binding site on metal and identified that the FeIV–oxo species has two potential orientations with the oxo group trans to either His101 or His251. The former is energetically unstable, which can rapidly isomerize into the latter by rotation of the oxo group. Arg279 plays important roles in regulating the dioxygen binding and assisting the isomerization of FeIV–oxo species. The calculation results clearly support the stepwise C5-epimerization and C2/3-desaturation processes, involving two complete oxidative cycles. The epimerization process converts (3S,5S)-carbapenam to the initial product (3S,5R)-carbapenam, undergoing H5 atom abstraction by FeIV═O species, inversion of the C5-radical, and reconstitution of the inverted C5–H bond by Tyr165. In the desaturation process, (3S,5R)-carbapenam rebinds the CarC active site with a new orientation different from what (3S,5S)-carbapenam does in the epimerization. In addition, the desaturation across C2–C3 occurs without involving any active site residue other than the FeIV═O center. Whereas Tyr165 is not involved in the desaturation reaction, it plays a key role in binding (3S,5R)-carbapenam. (3S,5R)-Carbapenam is a substrate superior to its epimer (3S,5S)-carbapenam for CarC to produce (5R)-carbapenem by efficient desaturation. In addition, the substrate hydroxylations compete with the target epimerization and desaturation reaction.Keywords: carbapenem synthase; desaturation; dioxygen binding; epimerization; QM/MM
Co-reporter:Jing Zhang and Yongjun Liu
RSC Advances 2015 vol. 5(Issue 123) pp:101672-101682
Publication Date(Web):20 Nov 2015
DOI:10.1039/C5RA21535H
Succinic semialdehyde dehydrogenase (SSADH) belongs to the aldehyde dehydrogenase (ALDH) superfamily, which oxidizes succinic semialdehyde (SSA) to succinate (SA) in the final step of the degradation of the inhibitory neurotransmitter γ-aminobutyric acid (GABA). In this article, the catalytic mechanism of SSADH has been studied using a combined quantum mechanics and molecular mechanics (QM/MM) approach on the basis of the crystal structures of SSADH from Synechococcus sp. PCC 7002 (SySSADH) and Salmonella typhimurium (StSSADH). Our calculations reveal that, for SySSADH, the acylation process of substrate SSA is relatively difficult owing to the fact that the catalytic cysteine residue has already formed an adduct with the cofactor (NADP+), which corresponds to an overall energy barrier of 18.2 kcal mol−1. However for StSSADH, the cysteine residue exists as the thiolate ion and the acylation process is easily occurs, corresponding to an overall energy barrier of 9.6 kcal mol−1. In the subsequent deacylation process, using SySSADH to construct the computational model, the activation of the hydrolytic water molecule is concerted with the formation of a thioester intermediate, which is the rate-limiting step for the deacylation process, corresponding to an energy barrier of 18.2 kcal mol−1. Thus, for SySSADH, both the acylation and deacylation are possible rate-limiting steps. The pocket residues such as S261, C262 and S419/S425 play an important role in stabilizing the substrate and involved intermediates. Our calculation results may provide useful information for further understanding the catalytic mechanism of SSADH.
Co-reporter:Wenyou Zhu, Yongjun Liu, and Baoping Ling
Biochemistry 2015 Volume 54(Issue 33) pp:
Publication Date(Web):August 10, 2015
DOI:10.1021/acs.biochem.5b00527
Deubiquitinating enzymes (DUBs) catalyze the cleavage of the isopeptide bond in polyubiquitin chains to control and regulate the deubiquitination process in all known eukaryotic cells. The human AMSH-LP DUB domain specifically cleaves the isopeptide bonds in the Lys63-linked polyubiquitin chains. In this article, the catalytic mechanism of AMSH-LP has been studied using a combined quantum mechanics and molecular mechanics method. Two possible hydrolysis processes (Path 1 and Path 2) have been considered. Our calculation results reveal that the activation of Zn2+-coordinated water molecule is the essential step for the hydrolysis of isopeptide bond. In Path 1, the generated hydroxyl first attacks the carbonyl group of Gly76, and then the amino group of Lys63 is protonated, which is calculated to be the rate limiting step with an energy barrier of 13.1 kcal/mol. The energy barrier of the rate limiting step and the structures of intermediate and product are in agreement with the experimental results. In Path 2, the protonation of amino group of Lys63 is prior to the nucleophilic attack of activated hydroxyl. The two proton transfer processes in Path 2 correspond to comparable overall barriers (33.4 and 36.1 kcal/mol), which are very high for an enzymatic reaction. Thus, Path 2 can be ruled out. During the reaction, Glu292 acts as a proton transfer mediator, and Ser357 mainly plays a role in stabilizing the negative charge of Gly76. Besides acting as a Lewis acid, Zn2+ also influences the reaction by coordinating to the reaction substrates (W1 and Gly76).
Co-reporter:Hao Su, Lihua Dong and Yongjun Liu
RSC Advances 2015 vol. 5(Issue 11) pp:8507-8508
Publication Date(Web):06 Jan 2015
DOI:10.1039/C4RA90062F
Correction for ‘A QM/MM study of the catalytic mechanism of α-1,4-glucan lyase from the red seaweed Gracilariopsis lemaneiformis’ by Hao Su et al., RSC Adv., 2014, 4, 54398–54408.
Co-reporter:Guangcai Ma, Na Cheng, Hao Su and Yongjun Liu
RSC Advances 2015 vol. 5(Issue 10) pp:7781-7788
Publication Date(Web):22 Dec 2014
DOI:10.1039/C4RA13278E
WlbB, one of the enzymes required for the biosynthesis of UDP-2,3-diacetamido-2,3-dideoxy-D-mannuronic acid (UDP-ManNAc3NAcA), is an N-acetyltransferase that catalyzes the N-acetylation of UDP-2-acetamido-3-amino-2,3-dideoxy-D-glucuronic acid (UDP-GlcNAc3NA) to form UDP-2,3-diacetamido-2,3-dideoxy-D-glucuronic acid (UDP-GlcNAc3NAcA). In this paper, based on the crystal structure, the detailed reaction mechanism of WlbB has been studied by using a combined QM/MM method. In particular, six snapshots taken from MD trajectories were used as the computational models to investigate how the starting geometries influence the calculation results. Our calculations suggest that the WlbB-catalyzed process involves two sequential steps. The nucleophilic attack of the C3-amino group of the substrate on the carbonyl carbon of acetyl-CoA occurs in concert with the departure of CoA from acetyl-CoA, generating a negatively charged CoA and a positively charged intermediate, which is inconsistent with the previous proposals that the catalytic reaction undergoes an oxyanion tetrahedral intermediate. Subsequently, the sulfur anion of CoA accepts the proton of the positively charged intermediate to yield the final product. Although Asn84 is not essential, it is important for promoting the catalysis by forming a hydrogen bond with the C3-amino group to position the lone pair of the electrons of the C3-amino group in an ideal orientation for nucleophilic attack and stabilize the transition states and intermediate. The cautious selection of initial geometries was found to be important for exploring the enzymatic mechanism and getting reliable energy barriers of the reaction pathways.
Co-reporter:Shujun Zhang, Guangcai Ma, Yongjun Liu, Baoping Ling
Journal of Molecular Graphics and Modelling 2015 Volume 61() pp:21-29
Publication Date(Web):September 2015
DOI:10.1016/j.jmgm.2015.06.011
•The catalytic mechanism of LigI has been studied by using QM/MM method.•Asp248 acts as the general base to activate the hydrolytic water molecule.•The intermediate has two intramolecular proton transfer pathways.•His31, His33, His180 play assistant roles by forming electrostatic interactions with substrate.2-Pyrone-4,6-dicarboxylate lactonase (LigI) is the first identified enzyme from amidohydrolase superfamily that does not require a divalent metal ion for catalytic activity. It catalyzes the reversible hydrolysis of 2-pyrone-4,6-dicarboxylate (PDC) to 4-oxalomesaconate (OMA) and 4-carboxy-2-hydroxymuconate (CHM) in the degradation of lignin. In this paper, a combined quantum mechanics and molecule mechanics (QM/MM) approach was employed to study the reaction mechanism of LigI from Sphingomonas paucimobilis. According to the results of our calculations, the whole catalytic reaction contains three elementary steps, including the nucleophilic attack, the cleavage of CO of lactone (substrate) and the intramolecular proton transfer. The intermediate has two intramolecular proton transfer pathways, due to which, two final hydrolysis products can be obtained. The energy profile indicates that 4-carboxy-2-hydroxymuconate (CHM) is the main hydrolysis product, therefore, the isomerization between 4-carboxy-2-hydroxymuconate (CHM) and 4-oxalomesaconate (OMA) is suggested to occur in solvent. During the catalytic reaction, residue Asp248 acts as a general base to activate the hydrolytic water molecule. Although His31, His33 and His180 do not directly participate in the chemical process, they play assistant roles by forming electrostatic interactions with the substrate and its involved species in activating the carbonyl group of the substrate and stabilizing the intermediates and transition states.
Co-reporter:Xueli Cheng;Yanyun Zhao;Feng Li
Journal of Molecular Modeling 2015 Volume 21( Issue 9) pp:
Publication Date(Web):2015 September
DOI:10.1007/s00894-015-2780-4
The oxidation of CO catalyzed by clusters of Au11, Au10Pt and Au9Pt2 was investigated using the M06 functional suite of the density functional theory. Au and Pt atoms were described with the double-ζ valence basis set Los Alamos National Laboratory 2-double-z (LanL2DZ), whereas the standard 6-311++G(d,p) basis set was employed for the C and O atoms. Our theoretical model showed that (1) after coordination to Au and Au-Pt cluster, O2 and CO are apparently activated, and Mulliken charges show that the gold atoms in the active sites of Au11 are negatively charged; (2) Au-Pt clusters with 11 atoms can effectively catalyze the oxidation of CO by O2; (3) Au11 exhibits good catalytic performance for the oxidation of CO; (4) oxidation of CO occurs preferably on the Au–Pt active sites in Pt-doped clusters, and the single-center mechanisms are more favorable energetically than the two-center mechanisms; (5) after adsorption, an O2 molecule oxidates two CO molecules via stepwise mechanisms; and (6) the catalytic processes are highly exothermic.
Co-reporter:Xiaohua Chen ; Guangcai Ma ; Weichao Sun ; Hongjing Dai ; Dong Xiao ; Yanfang Zhang ; Xin Qin ; Yongjun Liu ;Yuxiang Bu
Journal of the American Chemical Society 2014 Volume 136(Issue 12) pp:4515-4524
Publication Date(Web):March 6, 2014
DOI:10.1021/ja406340z
The proton/electron transfer reactions between cysteine residue (Cys) and tyrosinyl radical (Tyr•) are an important step for many enzyme-catalyzed processes. On the basis of the statistical analysis of protein data bank, we designed three representative models to explore the possible proton/electron transfer mechanisms from Cys to Tyr• in proteins. Our ab initio calculations on simplified models and quantum mechanical/molecular mechanical (QM/MM) calculations on real protein environment reveal that the direct electron transfer between Cys and Tyr• is difficult to occur, but an inserted water molecule can greatly promote the proton/electron transfer reactions by a double-proton-coupled electron transfer (dPCET) mechanism. The inserted H2O plays two assistant roles in these reactions. The first one is to bridge the side chains of Tyr• and Cys via two hydrogen bonds, which act as the proton pathway, and the other one is to enhance the electron overlap between the lone-pair orbital of sulfur atom and the π-orbital of phenol moiety and to function as electron transfer pathway. This water-mediated dPCET mechanism may offer great help to understand the detailed electron transfer processes between Tyr and Cys residues in proteins, such as the electron transfer from Cys439 to Tyr730• in the class I ribonucleotide reductase.
Co-reporter:Qianqian Hou, Xiang Sheng and Yongjun Liu
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 23) pp:11366-11373
Publication Date(Web):17 Mar 2014
DOI:10.1039/C3CP55263B
Archaeal fructose-1,6-bisphosphate aldolase/phosphatase (FBPA/P) is a newly identified unusual bifunctional enzyme (Nature, 2010, 464, 1077), which contains one single catalytic domain but catalyzes two chemically distinct reactions of gluconeogenesis. It is different from the ordinary enzymes whose active sites are responsible for a specific reaction. To explore the catalytic characteristic of FBPA/P, the aldol condensation mechanism of bifunctional FBPA/P has been investigated using quantum mechanics/molecular mechanics (QM/MM) method. The whole reaction process can be divided into two half-reactions involving seven elementary steps. A Schiff base intermediate is theoretically confirmed, agreeing well with the recently resolved crystal structures (Nature, 2011, 478, 538). The free energy barrier of the rate-limiting step is calculated to be 22.2 kcal mol−1, which is a concerted process of a nucleophilic attack by the enolic carbon to the ketonic carbon and a proton transfer from Tyr229 to the ketonic oxygen. Lys232 plays an important role in forming a Schiff base intermediate with the substrate (DHAP). Tyr229 functions as a proton shuttle during the catalysis. This is the first theoretical study on the aldol condensation mechanism of FBPA/P, which may provide useful information for understanding bifunctional enzymes.
Co-reporter:Xiang Sheng and Yongjun Liu
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 8) pp:1265-1277
Publication Date(Web):13 Jan 2014
DOI:10.1039/C3OB42182A
Nicotinamidase (Pnc1) is a member of Zn-dependent amidohydrolases that hydrolyzes nicotinamide (NAM) to nicotinic acid (NA), which is a key step in the salvage pathway of NAD+ biosynthesis. In this paper, the catalytic mechanism of Pnc1 has been investigated by using a combined quantum-mechanical/molecular-mechanical (QM/MM) approach based on the recently obtained crystal structure of Pnc1. The reaction pathway, the detail of each elementary step, the energetics of the whole catalytic cycle, and the roles of key residues and Zn-binding site are illuminated. Our calculation results indicate that the catalytic water molecule comes from the bulk solvent, which is then deprotonated by residue D8. D8 functions as a proton transfer station between C167 and NAM, while the activated C167 serves as the nucleophile. The residue K122 only plays a role in stabilizing intermediates and transition states. The oxyanion hole formed by the amide backbone nitrogen atoms of A163 and C167 has the function to stabilize the hydroxyl anion of nicotinamide. The Zn-binding site rather than a single Zn2+ ion acts as a Lewis acid to influence the reaction. Two elementary steps, the activation of C167 in the deamination process and the decomposition of catalytic water in the hydrolysis process, correspond to the large energy barriers of 25.7 and 28.1 kcal mol−1, respectively, meaning that both of them contribute a lot to the overall reaction barrier. Our results may provide useful information for the design of novel and efficient Pnc1 inhibitors and related biocatalytic applications.
Co-reporter:Hao Su, Lihua Dong and Yongjun Liu
RSC Advances 2014 vol. 4(Issue 97) pp:54398-54408
Publication Date(Web):17 Oct 2014
DOI:10.1039/C4RA09758K
α-1,4-Glucan lyase (GLase, EC 4.2.2.13), a unique glycoside hydrolase family member, specifically cleaves the α-1,4-glucosidic linkages in glycogen, starch and malto-oligosaccharides to produce 1,5-anhydro-D-fructose from the non-reducing end. Previous studies have proved that GLase belongs to the retaining glycoside lyase, and the catalytic reaction contains both the glycosylation and deglycosylation/elimination steps, in which a covalent glycosyl–enzyme intermediate is involved. On the basis of the newly reported crystal structure of GLase (2X2I) and the speculated mechanism, the whole catalytic cycle of GLase has been studied by using a QM/MM method. Calculation results indicate that the whole catalytic cycle contains five elementary steps. Firstly, the aspartic acid residue D665 acts as an acid to protonate the glycoside oxygen, which is concerted with the cleavage of the glycoside bond. Then, the residue D553 functions as the nucleophile to attack the anomeric carbon to form the glycosyl–enzyme intermediate. Different from the retaining α-glucosidases whose glycosylation is a typical concerted process, the glycosylation process of glycosidic lyase follows a stepwise mechanism. For the deglycosylation/elimination step, two cases with or without the maltotriose group in the active site were considered. The departure of the maltotriose can facilitate the proceeding of this process. The deprotonated aspartic acid residue D553 further acts as a catalytic base to abstract the C2-proton of the glucosyl residue. The proton abstraction in the deglycosylation/elimination step is calculated to be the rate-limiting step of the whole catalytic reaction, which corresponds to the energy barriers of 20.69 and 18.53 kcal mol−1 for both of the two cases.
Co-reporter:Xiang Sheng, Yongjun Liu and Rui Zhang
RSC Advances 2014 vol. 4(Issue 67) pp:35777-35788
Publication Date(Web):05 Aug 2014
DOI:10.1039/C4RA03611E
Oxalate is harmful to many organisms by forming insoluble precipitates with some metal cations. In humans, calcium oxalate is a major constituent of kidney stones leading to urolithiasis. Oxalobacter formigenes is a bacterium in most vertebrates and can regulate the homeostasis of oxalate. Replacement therapies of O. formigenes or related-enzymes are new strategies for treating oxalate-related diseases. Oxalyl-CoA decarboxylase (OXC) is an enzyme involved in the oxalate degradation in O. formigenes. In this paper, the catalytic mechanism of OXC was investigated by using the density functional theory (DFT) method. The most likely reaction pathway, detail of elementary steps, and roles of key residues were determined. Our calculation results indicate that the decarboxylation process can proceed rapidly, which agrees well with the experimental observation. In the protonation of the HDC-ThDP intermediate, the 4-NH2 of ThDP is suggested to be the proton donor, which abstracts a proton from the nearby residue E121. The rate-limiting step is calculated to be the proton transfer from 4′-NH2 to the HDC-ThDP intermediate with an energy barrier of 21.8 kcal mol−1. However, if this pathway is blocked by mutating residue E121, the reaction may follow another mechanism, in which Y483 acts as the proton donor and uses a water molecule as a mediator. These findings can explain the experimental observation that replacement of residues Y483 or E121 significantly reduces but does not completely abolish the activity of the enzyme. Our results may provide useful information for exploring the enzymatic mechanism and developing biocatalytic applications for treating the oxalate-related diseases.
Co-reporter:Guangcai Ma, Lihua Dong and Yongjun Liu
RSC Advances 2014 vol. 4(Issue 67) pp:35449-35458
Publication Date(Web):30 Jul 2014
DOI:10.1039/C4RA04406A
dTDP-glucose 4,6-dehydratase catalyzes the biotransformation of dTDP-glucose into dTDP-4-keto-6-deoxy-glucose. We have utilized the quantum mechanical/molecular mechanical (QM/MM) approach to investigate the detailed mechanism of dTDP-glucose 4,6-dehydratase from Streptomyces venezuelae. On the basis of our calculation results, the previously proposed mechanism has been revised. The overall catalytic cycle can be divided into three sequential chemical steps: oxidation, dehydration and reduction, containing four enzymatic elementary reactions and one non-enzymatic enol–keto tautomerization reaction. The oxidation step proceeds through a concerted asynchronous mechanism with a calculated free energy barrier of 21.1 kcal mol−1, in which the hydride transfer lags behind the proton transfer. The dehydration step prefers a stepwise mechanism rather than a concerted mechanism, and involves an enolate intermediate. Two highly conserved residues Glu129 and Asp128 are involved in this step. In the reduction step, NADH returns the hydride back to glycosyl C6 and the phenolic hydroxyl of Tyr151 spontaneously donates its proton to the C4-keto group, forming an enol sugar as the enzymatic product. After dissociation from the dehydratase active site and diffusion into the solution, this enol sugar will facilely rearrange to give the more favorable dTDP-4-keto-6-dexoyglucose product. Although Thr127 is not directly involved in the whole enzymatic reaction, it is responsible for promoting the catalysis by forming hydrogen-bonding interactions with glycosyl. These calculation results may provide new insight and inspiration for the catalytic mechanism of dTDP-glucose 4,6-dehydratase, even though it is not fully consistent with the previous experimental proposals.
Co-reporter:Xiang Sheng and Yongjun Liu
Biochemistry 2014 Volume 53(Issue 27) pp:4455-4466
Publication Date(Web):June 25, 2014
DOI:10.1021/bi500020r
Pyruvate carboxylase (PC) catalyzes the carboxylation of pyruvate to produce oxaloacetate. Its activity is directly related to insulin release and thus PC has recently attracted great interest as a potential target for diabetes treatment. In this article, the catalytic mechanism of the carboxyl transferase domain of PC from Staphylococcus aureus was investigated by using a combined quantum-mechanical/molecular-mechanical approach. Our calculation results indicate that the catalytic reaction starts from the decarboxylation of carboxybiotin to generate an enol-BTI intermediate, followed by two consecutive proton-transfer processes (from T908 to enol-BTI and from PYR to T908). During the catalytic reaction, the main-chain amide of T908 plays a key role in catching CO2 and preventing its diffusion from the active center. A triad of residues, R571, Q575, and K741, contributes both to substrate binding and enol-pyruvate stabilization. The oxyanion hole, consisting of the side-chain hydroxyl of S911 and the side-chain amino of Q870, plays an important role in stabilizing the hydroxyl anion of BTI. The coordination of the metal cation by pyruvate is a second sphere, rather than an inner sphere, interaction, and the metal cation stabilizes the species through the medium of residue K741. The decarboxylation of carboxybiotin corresponds to the highest free energy barrier of 21.7 kcal/mol. Our results may provide useful information for both the regulation of enzyme activity and the development of related biocatalytic applications.
Co-reporter:Na Cheng, Fuzhen Bi, Yongjun Liu, Changqiao Zhang and Chengbu Liu
New Journal of Chemistry 2014 vol. 38(Issue 3) pp:1256-1263
Publication Date(Web):27 Jan 2014
DOI:10.1039/C3NJ01015E
To explore the nature of unconventional halogen bonds, the halogen bonds between a series of halides FXOn (X = Cl, Br; n = 0–3) and CH3CN have been studied at M05-2X/6-311++G(d,p), MP2/aug-cc-pVTZ and CCSD(T)/aug-cc-pVTZ levels. Our calculations reveal that the electrostatic potentials of these hypervalent halogen atoms are greatly different to those of monovalent halogen atoms and, accordingly, the strength and interaction modes of these halogen bonds show different characteristics. For complexes of monovalent and heptavalent halides, only one interaction mode was found, but for those of tervalent and pentavalent halides, three kinds of stable configurations with different interaction modes were recognized. Configuration I is a linear structure formed by a single halogen bond, while configurations II and III are cyclic structures formed by the cooperation of a halogen bond and a hydrogen bond. From an energy point of view, monovalent halides form the strongest halogen bonds, and heptavalent halides feature the weakest halogen bonds. For tervalent and pentavalent halides, the interaction energies of the three configurations are similar. In the two cyclic configurations of complexes formed by tervalent and pentavalent halides, due to the competition of the halogen bond with the hydrogen bond, the halogen bonds are weaker than that of configuration I. NBO analysis indicates that the three configurations of complexes display different donor–acceptor orbital interactions. AIM and LMO-EDA analysis reveal that the electrostatic interaction is the dominant driving force for the formation of the complexes, and all the halogen and hydrogen bonds in these complexes are closed-shell interactions.
Co-reporter:Yi Zhao, Yuxia Liu, Siwei Bi, Yongjun Liu
Journal of Organometallic Chemistry 2014 Volume 758() pp:45-54
Publication Date(Web):15 May 2014
DOI:10.1016/j.jorganchem.2014.02.008
•Reaction mechanisms were studied with DFT calculations.•Regioselectivity of the reaction has been elucidated.•Explained the inaccessible path involving oxidative coupling of the two CC bonds.Reaction mechanisms of the Ni(COD)2-catalyzed [2 + 2 + 2] cycloaddition of unsymmetric diynes and CO2 have been theoretically studied by using the density functional theory calculations. Three major steps are included, oxidative coupling of CO2 with Et-substituted CC bond, the second CC bond insertion and reductive elimination of the product from the Ni center, in which the CC bond insertion was found to be rate-determinant. The steric arrangement of the N,P-bidentate ligand was demonstrated to be influential on reaction barriers. Based on the mechanistic study, the regioselectivity of the catalytic reaction was elucidated. In addition, we also explained why the mechanisms involving oxidative coupling of both the CC bonds are unavailable.The reaction mechanisms and the regioselectivity of the Ni(COD)2-catalyzed [2 + 2 + 2] cycloaddition of unsymmetric diyne and CO2 were studied using density functional theory calculations.
Co-reporter:Jing Zhang, Yongjun Liu
Journal of Molecular Graphics and Modelling 2014 Volume 51() pp:113-119
Publication Date(Web):June 2014
DOI:10.1016/j.jmgm.2014.05.003
•The catalytic mechanism of aspartate ammonia lyase has been studied by using QM/MM method.•Ser318 functions as the catalytic base to abstract the Cβ proton of substrate l-aspartate.•The abstraction of Cβ proton of l-aspartate by Ser318 is the rate-limiting step.•His188 is a dispensable residue, but its protonation state can influence the active site structure.Aspartate ammonia lyase (Asp) is one of three types of ammonia lyases specific for aspartate or its derivatives as substrates, which catalyzes the reversible reaction of l-aspartate to yield fumarate and ammonia. In this paper, the catalytic mechanism of Asp has been studied by using combined quantum-mechanical/molecular-mechanical (QM/MM) approach. The calculation results indicate that the overall reaction only contains two elementary steps. The first step is the abstraction of Cβ proton of l-aspartate by Ser318, which is calculated to be rate limiting. The second step is the cleavage of CαN bond of l-aspartate to form fumarate and ammonia. Ser318 functions as the catalytic base, whereas His188 is a dispensable residue, but its protonation state can influence the active site structure and the existing form of leaving amino group, thereby influences the activity of the enzyme, which can well explain the pH dependence of enzymatic activity. Mutation of His188 to Ala only changes the active site structure and slightly elongates the distance of Cβ proton of substrate with Ser318, causing the enzyme to remain significant but reduced activity.
Co-reporter:Xiang Sheng and Yongjun Liu
Biochemistry 2013 Volume 52(Issue 45) pp:
Publication Date(Web):October 30, 2013
DOI:10.1021/bi400577f
Pyruvate dehydrogenase multienzyme complex (PDHc) is a member of a family of 2-oxo acid dehydrogenase (OADH) multienzyme complexes involved in several central points of oxidative metabolism, and the E1 subunit is the most important component in the entire PDHc catalytic system, which catalyzes the reversible transfer of an acetyl group from a pyruvate to the lipoyl group of E2 subunit lipoly domain. In this article, the catalytic mechanism of the E1 subunit has been systematically studied using density functional theory (DFT). Four possible pathways with different general acid/base catalysts in decarboxylation and reductive acylation processes were explored. Our calculation results indicate that the 4′-amino pyrimidine of ThDP and residue His128 are the most likely proton donors in the decarboxylation and reductive acylation processes, respectively. During the reaction, each C–C and C–S bond formation or cleavage process, except for the liberation of CO2, is always accompanied by a proton transfer between the substrates and proton donors. The liberation of CO2 is calculated to be the rate-limiting step for the overall reaction, with an energy barrier of 13.57 kcal/mol. The decarboxylation process is endothermic by 5.32 kcal/mol, whereas the reductive acylation process is exothermic with a value of 5.74 kcal/mol. The assignment of protonation states of the surrounding residues can greatly influence the reaction. Residues His128 and His271 play roles in positioning the first substrate pyruvate and second substrate lipoyl group, respectively.
Co-reporter:Xueli Cheng, Yanyun Zhao, Yongjun Liu and Feng Li
New Journal of Chemistry 2013 vol. 37(Issue 5) pp:1371-1377
Publication Date(Web):07 Feb 2013
DOI:10.1039/C3NJ41140K
The hydrolysis and condensation mechanisms of tetramethoxysilane aided by F− were investigated with Gaussian03 program package. The coordination of F− to tetramethoxysilane and the first-order hydrolysis of the fluorotetramethoxysilane anion were studied extensively with both CPCM full optimization and CPCM single-point energy (SPE) calculations, and the single-point energies agree well with those obtained from the full optimization with CPCM solvation model. The coordination of F− decreases the Mulliken charge on the Si atom and the energy gap between the HOMO and LUMO, and alters the shape of the frontier orbitals. Our calculations show that entropic effects elevate potential energy surface (PES) profiles distinctly, but have a minor influence on the free energy barriers. With the aid of F−, the hydrolysis barriers of fluorotetramethoxysilane anion and the dimerization free energy barriers before entropic corrections decrease from 104, 106, 109, 109 and 136, 104, 99, 94 to kJ mol−1 to 84.2, 88.5, 77.2, 81.9 and 82.2, 80.0, 87.7, 88.7 kJ mol−1, respectively, compared with the neutral hydrolysis and condensation barriers. These barriers are much lower than the corresponding neutral SN2 ones, but only slightly higher than the SN1 ones. The role of nucleophile F− is similar to HO−.
Co-reporter:Jing Zhang, Yongjun Liu
Computational and Theoretical Chemistry 2013 Volume 1025() pp:1-7
Publication Date(Web):1 December 2013
DOI:10.1016/j.comptc.2013.09.026
•The catalytic mechanism of phosphoketolase (PK) has been studied by DFT method.•The dehydration process is distinct from that of other THDP-dependent enzymes.•His553 plays intermediary role in the Keto–Enol tautomerism.•His97 is the probable B2 catalyst in the dehydration process.Phosphoketolase (PK) is a thiamine diphosphate (THDP) dependent enzyme which plays key roles in the metabolism of heterofermentative bacteria. By using density functional theory (DFT) method, the catalytic mechanism of PK has been studied on simplified models. The calculation results indicate that the formation of 2-α,β-dihydroxyethylidene-THDP (DHETHDP) and erythrose-4-phosphate (E4P) involves one C–C bond formation and one C–C bond cleavage process. Each C–C bond formation or cleavage is always accompanied by a proton transfer in a concerted but asynchronous way. The dehydration process in the reaction of PK is distinct from that of other THDP-dependent enzymes. The Keto–Enol tautomerism process is assisted with a mediator His553. His64, His553 and His97 are found to have the function to stabilize the transition states and intermediates. His64 is a better candidate of B1 catalyst. His553 acts as a proton donor to protonate the carbonyl oxygen, and plays intermediary role in the Keto–Enol tautomerism process. His97 is the probable B2 catalyst in the dehydration process.Graphical abstractBy using density functional theory (DFT) method, the main formation mechanism of acetyl phosphate (AcP) and erythrose-4-phosphate (E4P) from fructose-6-phosphate (F6P) and Pi catalyzed by phosphoketolase (PK) has been studied on simplified models. The calculation results indicate that His64 is a better candidate of B1 catalyst, and His97 is the probable B2 catalyst in the dehydration process. His553 acts as a proton donor to protonate the carbonyl oxygen, and plays intermediary role in the Keto–Enol tautomerism process.
Co-reporter:Xueli Cheng, Guofang He, Liqing Li, Yongjun Liu
Computational and Theoretical Chemistry 2013 Volume 1023() pp:19-23
Publication Date(Web):1 November 2013
DOI:10.1016/j.comptc.2013.09.004
•The methanolysis mechanisms of Si(OCH3)4 and Al(OCH3)3 were investigated systematically with the DFT method.•In the methanolysis of Si(OCH3)4, the silicone channels of the methanolysis products is energetically more favorable.•The methyl transfer channels are energetically more favorable than the hydrogen transfer ones in the Al(OCH3)3 methanolysis.•Aluminum alkoxides can be methanolyzed due to the relatively lower barriers.In the present work, the methanolysis mechanisms of Si(OCH3)4 and Al(OCH3)3 were investigated systematically with the density functional theory (DFT) method. Although the methanolysis barriers of Si(OCH3)4 are high in pure methanol, the silicone channels of the methanolysis products is favorable. In contrast with the methanolysis of Si(OCH3)4 where the hydrogen transfer channels bear relatively lower barriers, the methyl transfer channels are energetically more favorable than the hydrogen transfer ones in the Al(OCH3)3 methanolysis, and have the lowest free energy barriers.Graphical abstract
Co-reporter:Jinhu Wang, Salila Pengthaisong, James R. Ketudat Cairns, Yongjun Liu
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2013 Volume 1834(Issue 2) pp:536-545
Publication Date(Web):February 2013
DOI:10.1016/j.bbapap.2012.11.003
Nucleophile mutants of retaining β-glycosidase can act as glycosynthases to efficiently catalyze the synthesis of oligosaccharides. Previous studies proved that rice BGlu1 mutants E386G, E386S and E386A catalyze the oligosaccharide synthesis with different rates. The E386G mutant gave the fastest transglucosylation rate, which was approximately 3- and 19-fold faster than those of E386S and E386A. To account for the differences of their activities, in this paper, the X-ray crystal structures of BGlu1 mutants E386S and E386A were solved and compared with that of E386G mutant. However, they show quite similar active sites, which implies that their activities cannot be elucidated from the crystal structures alone. Therefore, a combined quantum mechanical/molecular mechanical (QM/MM) calculations were further performed. Our calculations reveal that the catalytic reaction follows a single-step mechanism, i.e., the extraction of proton by the acid/base, E176, and the formation of glycosidic bond are concerted. The energy barriers are calculated to be 19.9, 21.5 and 21.9 kcal/mol for the mutants of E386G, E386S and E386A, respectively, which is consistent with the order of their experimental relative activities. But based on the calculated activation energies, 1.1 kcal/mol energy difference may translate to nearly 100 fold rate difference. Although the rate limiting step in these mutants has not been established, considering the size of the product and the nature of the active site, it is likely that the product release, rather than chemistry, is rate limiting in these oligosaccharides synthesis catalyzed by BGlu1 mutants.Highlights► The mechanism of glycosynthase was explored by X-ray crystallographic and QM/MM methods. ► The catalytic reaction proceeds via a single-step mechanism. ► The energy barriers are sensitive to hindered interactions of the mutated residues. ► The incoming crystal water Wat1 was important to the reaction.
Co-reporter:Yi Zhao, Yuxia Liu, Siwei Bi, Yongjun Liu
Journal of Organometallic Chemistry 2013 s 745–746() pp: 166-172
Publication Date(Web):
DOI:10.1016/j.jorganchem.2013.07.065
Co-reporter:Wenyou Zhu;Rui Zhang
Theoretical Chemistry Accounts 2013 Volume 132( Issue 9) pp:
Publication Date(Web):2013 September
DOI:10.1007/s00214-013-1385-1
Methylornithine synthase (PylB) belongs to the family of radical SAM enzymes which converts (2S)-lysine to (2R,3R)-3-methylornithine in a radical mechanism. In this paper, the mechanism of lysine mutase reaction catalyzed by PylB has been studied by using quantum mechanics/molecular mechanics approach. The calculations reveal that the PylB-catalyzed reaction follows a fragmentation–recombination mechanism involving seven elementary reaction steps. Both the hemolytic cleavage of Cα–Cβ bond of lysine and the ligation of glycyl radical with aminobutene are possible rate limiting, corresponding to the calculated energy barriers of 23.0 and 24.1 kcal/mol, respectively. The intramolecular rotation of a fragment (aminobutene) can well explain the stereochemistry of the final product. Asp 279 functions as a general acid/base, and the other pocket residues such as Asp112, Arg235, and Ser277 form a network of hydrogen bonds responsible for orientation of the substrate.
Co-reporter:Xiang Sheng, Yongjun Liu, Chengbu Liu
Journal of Molecular Graphics and Modelling 2013 Volume 39() pp:23-28
Publication Date(Web):February 2013
DOI:10.1016/j.jmgm.2012.11.001
Transketolase is a convenient model system to study enzymatic thiamin catalysis. By using density functional theory (DFT) method, the transfer mechanism of a 2-carbon fragment between a donor ketose X5P and an acceptor aldose R5P catalyzed by transketolase has been studied on simplified models. The calculation results indicate that the whole reaction cycle contains several proton transfer processes as well as CC bond formation and cleavage steps. Each CC bond formation or cleavage step is always accompanied by a proton transfer process, which follows a concerted but asynchronous mechanism. The CC bond formation is always prior to the proton transfer, and the CC bond cleavage is always later than proton transfer, suggesting that the CC bond ligation facilitates the proton transfer, and proton transfer promotes the CC bond cleavage. In the first half- and second half-reactions, the energy barriers of CC bond formations are always higher than those of CC bond cleavages. The 4-amino group of cofactor ThDP and histidine residue can act as the proton donor/acceptor during the catalytic reaction.Graphical abstractBy using density functional theory (DFT) method, the transfer mechanism of a 2-carbon fragment between a donor ketose X5P and an acceptor aldose R5P catalyzed by transketolase has been studied on simplified models. The calculation results indicate that each CC bond formation or cleavage process is always accompanied by a proton transfer, and they follow a concerted but asynchronous mechanism.Highlights► We present the transfer mechanism of a 2-carbon fragment catalyzed by transketolase. ► Each CC bond formation or cleavage process is accompanied by a proton transfer. ► CC bond formation/cleavage process and proton transfer follow a concerted mechanism.
Co-reporter:Xiang Sheng, Jun Gao, Yongjun Liu, Chengbu Liu
Journal of Molecular Graphics and Modelling 2013 Volume 44() pp:17-25
Publication Date(Web):July 2013
DOI:10.1016/j.jmgm.2013.04.009
•The proton shuttle mechanism of saccharopine dehydrogenase was studied by DFT method.•The residues Lys77 and His96 act as the acid–base catalysts in catalytic reaction.•The hydride transfer step is rate limiting and the reaction is endothermic.•The reverse reaction is more favored from energy point of view.Saccharopine dehydrogenase (SDH) is the last enzyme in the AAA pathway of l-lysine biosynthesis. On the basis of crystal structures of SDH, the whole catalytic cycle of SDH has been studied by using density functional theory (DFT) method. Calculation results indicate that hydride transfer is the rate-limiting step with an energy barrier of 25.02 kcal/mol, and the overall catalytic reaction is calculated to be endothermic by 9.63 kcal/mol. Residue Lys77 is proved to be functional only in the process of saccharopine deprotonation until the formation of product l-lysine, and residue His96 is confirmed to take part in multiple proton transfer processes and can be described as a proton transfer station. From the point of view of energy, the SDH catalytic reaction for the synthesis of l-lysine is unfavorable compared with its reverse reaction for the synthesis of saccharopine. These results are essentially consistent with the experimental observations from pH dependence of kinetic parameters and isotope effects.
Co-reporter:Kang Wang, Qianqian Hou, Yongjun Liu
Journal of Molecular Graphics and Modelling 2013 Volume 46() pp:65-73
Publication Date(Web):November 2013
DOI:10.1016/j.jmgm.2013.09.010
•The mechanism of Taxus canadensis phenylalanine aminomutase was studied by DFT method.•The reaction prefers the amino-MIO adduct mechanism.•The isomeriazation of (S)-α-phenylalanine to (R)-β-isomer contains six elementary steps.•A C1Cα bond rotation of 180° of the cinnamate skeleton is proved in the active site.The Taxus canadensis phenylalanine aminomutase (TcPAM) catalyze the isomerization of (S)-α-phenylalanine to the (R)-β-isomer. The active site of TcPAM contains the signature 5-methylene-3,5-dihydroimidazol-4-one (MIO) prosthesis, observed in the ammonia lyase class of enzymes. Up to now, there are two plausible mechanisms for these MIO-dependent enzymes, i.e., the amino-MIO adduct mechanism and the Friedel–Crafts-type reaction mechanism. In response to this mechanistic uncertainty, the phenylalanine aminomutase mechanism was investigated by using density functional methods. The calculation results indicate that: (1) the reaction prefers the amino-MIO adduct mechanism where the 2,3-amine shift process contains six elementary steps; (2) the ammonia elimination step proceeds through an E2 mechanism; (3) a single C1Cα bond rotation of 180° in the cinnamate skeleton occurs in the active site prior to the rebinding of NH2 group to the cinnamate. This can be used to explain the stereochemistry of the TcPAM reaction product which is contrary to those of the PaPAM and SgTAM enzymes. Based on these calculations, the roles of important residues in the active site were also elucidated.
Co-reporter:Xueli Cheng;Wenchao Ding;Dairong Chen
Journal of Molecular Modeling 2013 Volume 19( Issue 4) pp:1565-1572
Publication Date(Web):2013 April
DOI:10.1007/s00894-012-1718-3
Aluminum aerogels have extremely low thermal conductivities, and are ideal candidates for use in thermal superinsulators, adsorbents, sensors, catalyst carriers, and inorganic fillers. In the present work, the oligomerization mechanisms of Al(OH)3 were investigated systematically with the Gaussian 03 package at the B3LYP/6-311++G(d,p) level in combination with CPCM single-point energy calculations. The results of our theoretical model showed that: (1) the Al atoms are tetracoordinate and pentacoordinate; (2) in alkaline solution, Al(OH)3 tends to condense into more soluble polyhydroxy compounds; (3) the neutral dimerization of Al(OH)3 and the transfer of the hydrogen on the bridging hydroxyl are energetically favorable, but the most stable geometry is a four-membered Al–O ring structure linked by two bridging hydroxyls; (4) Al(OH)3 is inclined to form tetracoordinate oligomers, which develop into three-dimensional structures connected by four-membered Al–O rings.
Co-reporter:Qianqian Hou, Xin Hu, Xiang Sheng, Yongjun Liu, Chengbu Liu
Journal of Molecular Graphics and Modelling 2013 Volume 42() pp:26-31
Publication Date(Web):May 2013
DOI:10.1016/j.jmgm.2013.02.010
Poly(ADP-ribose) glycohydrolase (PARG) is the only enzyme responsible for the degradation of ADP-ribose polymers. Very recently, the first crystal structure of PARG was reported (Dea Slade, et al., Nature 477 (2011) 616), and a possible SN1-type-like mechanism was proposed. In this work, we present a computational study on the hydrolysis of glycosidic ribose–ribose bond catalyzed by PARG using hybrid density functional theory (DFT) methods. Based on the crystal structure of PARG, three models of the active site were constructed. The calculation results suggest that the degradation of poly(ADP-ribose) follows an SN2 mechanism, and the oxocarbenium expected by Dea Slade is a possible transition state but not an intermediate. The calculated reaction pathway agrees with the proposed mechanism. According to the computational models with different sizes, the roles of key residues are elucidated. Our results may provide useful information for the subsequent experimental and theoretical studies on the structure and functional relationships of PARG.Graphical abstractHighlights► We present a computational study on the hydrolysis of glycosidic ribose–ribose bond catalyzed by PARG. ►The degradation of poly(ADP-ribose) follows a SN2 mechanism. ►The roles of key residues are also be elucidated.
Co-reporter:Jinhu Wang, Xiang Sheng, Yi Zhao, Yongjun Liu, Chengbu Liu
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2012 Volume 1824(Issue 5) pp:750-758
Publication Date(Web):May 2012
DOI:10.1016/j.bbapap.2012.03.005
Bacteroides thetaiotaomicron α-glucosidase BtGH97a is an inverting enzyme. In this paper, the hydrolysis mechanism of p-nitro-phenyl α-d-glucopyranoside (pNP-Glc) catalyzed by BtGH97a was firstly studied by using quantum mechanical/molecular mechanical (QM/MM) approach. Two possible reaction pathways were considered. In the first pathway, a water molecule deprotonated by a nucleophilic base (here E439 or E508) attacks firstly on the anomeric carbon of pNP-Glc, then a proton from an acid residue (E532) attacks on the glycosidic oxygen to finish the hydrolysis reaction (named as nucleophilic attack-first pathway). In the second pathway, the proton from E532 attacks firstly on the glycosidic oxygen, then the water deprotonated by the nucleophilic base attacks on the anomeric carbon of pNP-Glc (named as proton attack-first pathway). Our calculation results indicate that the nucleophilic attack-first pathway is favorable in energy, in which the nucleophilic attack process is the rate-determining step with an energy barrier of 15.4 kcal/mol in the case of residue E508 as nucleophilic base. In this rate-determining step, the deprotonation of water and the attack on the anomeric carbon are concerted. In the proton attack-first pathway, the proton attack on the glycosidic oxygen is the rate-determining step, and the energy barrier is 24.1 kcal/mol. We conclude that the hydrolysis mechanism would follow nucleophilic attack-first pathway.Highlights► We carried out QM/MM calculations to elucidate the catalytic mechanism of BtGH97a. ► Two possible reaction pathways were considered. ► The nucleophilic attack-first pathway is favorable in energy. ► E508 is the possible nucleophilic base.
Co-reporter:Q.Q. Hou, J.H. Wang, J. Gao, Y.J. Liu, C.B. Liu
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2012 Volume 1824(Issue 4) pp:533-541
Publication Date(Web):April 2012
DOI:10.1016/j.bbapap.2012.01.017
Epinephrine is a naturally occurring adrenomedullary hormone that transduces environmental stressors into cardiovascular actions. As the only route in the catecholamine biosynthetic pathway, Phenylethanolamine N-methyltransferase (PNMT) catalyzes the synthesis of epinephrine. To elucidate the detailed mechanism of enzymatic catalysis of PNMT, combined quantum-mechanical/molecular-mechanical (QM/MM) calculations were performed. The calculation results reveal that this catalysis contains three elementary steps: the deprotonation of protonated norepinphrine, the methyl transferring step and deprotonation of the methylated norepinphrine. The methyl transferring step was proved to be the rate-determining step undergoing a SN2 mechanism with an energy barrier of 16.4 kcal/mol. During the whole catalysis, two glutamic acids Glu185 and Glu219 were proved to be loaded with different effects according to the calculations results of the mutants. These calculation results can be used to explain the experimental observations and make a good complementarity for the previous QM study.Highlights► We carried out QM/MM calculations to elucidate the reaction mechanism of PNMT. ► The catalysis contains three elementary steps. ► Glu185, Glu219, and a water molecule play an important role in the catalysis.
Co-reporter:Baoping Ling, Min Sun, Siwei Bi, Zhihong Jing, Yongjun Liu
Journal of Molecular Graphics and Modelling 2012 Volume 35() pp:1-10
Publication Date(Web):May 2012
DOI:10.1016/j.jmgm.2012.01.005
Mycobacterium tuberculosisl-alanine dehydrogenase (l-MtAlaDH) catalyzes the NADH-dependent reversible oxidative deamination of l-alanine to pyruvate and ammonia. l-MtAlaDH has been proposed to be a potential target in the treatment of tuberculosis. Based on the crystal structures of this enzyme, molecular dynamics simulations were performed to investigate the conformational changes of l-MtAlaDH induced by coenzyme NADH. The results show that the presence of NADH in the binding domain restricts the motions and conformational distributions of l-MtAlaDH. There are two loops (residues 94–99 and 238–251) playing important roles for the binding of NADH, while another loop (residues 267–293) is responsible for the binding of substrate. The opening/closing and twisting motions of two domains are closely related to the conformational changes of l-MtAlaDH induced by NADH.Graphical abstractBased on the crystal structures, molecular dynamics simulations were performed to investigate the conformational changes of l-MtAlaDH induced by coenzyme NADH. The results show that the presence of coenzyme NADH in NAD-binding domain restricts the motions and conformational distributions of l-MtAlaDH. Two loops (residues 94–99 and 238–251) play important roles in the binding of NADH, while the other loop (residues 267–293) is responsible for the binding of the substrate; the opening/closing and twisting motions are directly related to conformational changes of l-MtAlaDH induced by NADH.Highlights► The conformational changes of l-MtAlaDH induced by coenzyme NADH were studied. ► The presence of NDAH restricts the motions and conformational changes of protein. ► The loops and α-helixes participate in the binding/releasing of the substrate/product. ► The opening/closing and twisting motions of interdomains of protein were directly related to the conformational changes of l-MtAlaDH.
Co-reporter:Xueli Cheng; Dr. Dairong Chen; Dr. Yongjun Liu
ChemPhysChem 2012 Volume 13( Issue 9) pp:2392-2404
Publication Date(Web):
DOI:10.1002/cphc.201200115
Abstract
Silica aerogels possess a variety of unique and remarkable properties, but the mechanisms of silicon alkoxide, Si(OR)4, hydrolyses and oligomerization in the initial stage of sol–gel processes are still not well understood. On the basis of density functional theory calculations at the B3LYP/6-31G(d,p)//B3LYP/6-311++G(d,p) basis set level, the hydrolysis and oligomerization reactions of Si(OR)4 in neutral, acidic, and alkaline solutions were systematically investigated and we found that in acidic solutions the precursor Si(OCH3)4 was inclined to hydrolyze rather than to condense and the hydrolysis processes were energetically more favorable than the neutral ones. In alkaline solutions, the hydrolysis products oligomerize through an SN1 dimerization mechanism and the condensation rates are fast to form denser colloidal aerogels. Our calculations also testify that the subsequent cyclization reactions are energetically unfavorable.
Co-reporter:Q.Q. Hou, X. Sheng, J.H. Wang, Y.J. Liu, C.B. Liu
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2012 Volume 1824(Issue 2) pp:263-268
Publication Date(Web):February 2012
DOI:10.1016/j.bbapap.2011.08.014
Limonene 1,2-epoxide hydrolase (LEH) is completely different from those of classic epoxide hydrolases (EHs) which catalyze the hydrolysis of epoxides to vicinal diols. A novel concerted general acid catalysis step involving the Asp101–Arg99–Asp132 triad is proposed to play an important role in the mechanism. Combined quantum-mechanical/molecular-mechanical (QM/MM) calculations gave activation barriers of 16.9 and 25.1 kcal/mol at the B3LYP/6-31G(d,p)//CHARMM level for nucleophilic attack on the more and less substituted epoxide carbons, respectively. Furthermore, the important roles of residues Arg99, Tyr53 and Asn55 on mutated LEH were evaluated by QM/MM-scanned energy mapping. These results may provide an explanation for site-directed mutagenesis.The final solvated model of LEH after QM/MM optimization is shown in the left panel, and the optimized structure of the reactive pocket is shown in the right panel. As can be seen from the right panel, there is a large hydrogen bonding network in the active pocket, and the substrate presents favorable direction for nucleophilic attack by the catalytic water molecule.Research Highlights►We carried out QM/MM calculations to elucidate the reaction mechanism of LEH. ►The nucleophilic attack on the more substituted carbon is favorable. ►Arg99, Tyr53 and Asn55 play an important role in the catalytic reaction.
Co-reporter:Junyou Shi;Guoliang Li;Honglun Wang;Jie Zheng;Yourui Suo;Jinmao You
Phytochemical Analysis 2011 Volume 22( Issue 5) pp:450-454
Publication Date(Web):
DOI:10.1002/pca.1301
ABSTRACT
Introduction
Owing to them having the same traditional name, the leaves of Apoacynum venetum and Poacynum hendersonii are used indiscriminately in some areas of China. Although a series of studies have been conducted on Apoacynum venetum, there are only a few studies on Poacynum hendersonii.
Objective
To develop an efficient method for the preparative isolation and purification of flavonoids from the leaves of Poacynum hendersonii by high-speed counter-current chromatography (HSCCC).
Methodology
Powdered Poacynum hendersonii lead was extracted three times with 75% ethanol at 60 °C for 3 h. The distribution constant (KD) was measured to select an optimal two-phase solvent system for HSCCC separation. The purities of the target compounds were tested using HPLC and their structures were identified by 1H-NMR and 13C-NMR.
Results
Using a two-phase solvent system composed of n-butanol–petroleum ether–0.5% acetic acid (5:3:5, v/v), three main flavonoids, i.e. isoquercitrin, quercetin-3-O-sophoroside and quercetin-3-O-(6′′-O-malonyl)-β-d-glucoside, were separated from 240 mg crude sample in a one-step separation by using HSCCC method. After further purification with a Sepdex-LH20 column, 5.7 mg isoquercitrin (LC purity 98.72%), 4.9 mg quercetin-3-O-sophoroside (LC purity 99.06%) and 7.4 mg quercetin-3-O-(6′′-O-malonyl)-β-d-glucoside (LC purity 99.31%) were obtained, respectively.
Conclusion
The optimised high-speed counter-current chromatography method is fast, simple and efficient for the preparative separation of flavonoids from the leave of Poacynum hendersonii. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:Jinhu Wang, Qianqian Hou, Lihua Dong, Yongjun Liu, Chengbu Liu
Journal of Molecular Graphics and Modelling 2011 30() pp: 148-152
Publication Date(Web):1 September 2011
DOI:10.1016/j.jmgm.2011.06.012
The quantum-mechanical/molecular-mechanical (QM/MM) method was used to study the glycosylation mechanism of rice BGlu1 β-glucosidase in complex with laminaribiose. The calculation results reveal that the glycosylation step experiences a concerted process from the reactant to the glycosyl-enzyme complex with an activation barrier of 15.7 kcal/mol, in which an oxocarbenium cation-like transition state (TS) is formed. At the TS, the terminal saccharide residue planarizes toward the half-chair conformation, and the glycosidic bond cleavage is promoted by the attacks of proton donor (E176) on glycosidic oxygen and nucleophilic residue (E386) on the anomeric carbon of laminaribiose. Both the nucleophilic glutamate (E386) and acid/base catalyst (E176) establish shorter hydrogen bridges with the C2-hydroxyl groups of sugar ring, which play an important role in the catalytic reaction of rice BGlu1 β-glucosidase.Graphical abstractThe quantum mechanical/molecular mechanical (QM/MM) method was used to study the general glycosylation mechanism of rice BGlu1 β-glucosidase. The reaction experiences an oxocarbenium cation-like transition state with a barrier of 15.7 kcal/mol. Both the nucleophilic glutamate (E386) and acid/base catalyst (E176) have hydrogen bonding interactions with the substrate, which is useful in stabilizing the transition state and lowering the barrier.Download high-res image (21KB)Download full-size imageHighlights► The glycosylation mechanism of rice β-glucosidases was studied by using QM/MM method. ► The catalytic reaction experiences an oxocarbenium cation-like transition state. ► The attacks of E176 on glycosidic oxygen and E386 on anomeric carbon are concerted. ► The energy barrier derived from QM/MM method is much lower than those from simplified models.
Co-reporter:Ke Tang, Jinhu Wang, Qianqian Hou, Xueli Cheng, Yongjun Liu
Tetrahedron: Asymmetry 2011 Volume 22(Issue 9) pp:942-947
Publication Date(Web):15 May 2011
DOI:10.1016/j.tetasy.2011.05.019
Recent theoretical studies on N-heterocyclic carbenes catalyzed Staudinger reactions revealed that the stereoselectivities are determined by an elementary reaction step, that is, the reaction of an imine with N-heterocyclic carbene-ketene zwitterionic intermediate (INTN-k). To better understand the stereoselectivities of some of the experimentally reported reaction systems, the corresponding transition states were studied further by using an ONIOM approach. A combination of B3LYP/6-31G(d) and PM3 levels of theory was used for transition state optimization, and M06-2X/6-31+G(d,p) level for single point calculations. The calculations were found to provide predictions that were in qualitative agreement with the sense and magnitude of experimental stereoselectivities. By analyzing the electronic structures of the transition states, the origin of divergent stereoselectivities in different N-heterocyclic carbenes catalyzed system can be attributed to the cooperation of the electrostatic and steric effects involved in the reaction of imines with N-heterocyclic carbene-ketene intermediates. These results may provide useful information for selecting the N-heterocyclic carbene catalysts in the synthesis of β-lactams.
Co-reporter:Xueli Cheng, Yongjun Liu, and Dairong Chen
The Journal of Physical Chemistry A 2011 Volume 115(Issue 18) pp:4719-4728
Publication Date(Web):April 18, 2011
DOI:10.1021/jp110848e
As one of the representative superinsulating materials, the aluminum trioxypropyl Al(OC3H7)3 aerogel may be applied in launch vehicles and manned spacecrafts. In this study, the structures and hydrolysis mechanisms of the monomer, dimers, and trimers of Al(OC3H7)3 in neutral and alkaline environments were studied at the B3LYP/6-31G(d,p) level by using the CPCM solvation model to understand the fundamental chemistry of Al(OC3H7)3 hydrolysis and oligomerization. Our calculation shows that the first-order hydrolyses of the monomer and oligomers are energetically favorable in both alkaline and neutral solutions. In alkaline solutions, they are more apt to oligomerize than to hydrolyze due to high energy barriers and large binding energies in the formation of anionic species. For the oligomers under neutral condition (1) Al(OC3H7)3 is linked by four-membered Al–O rings with pentacoordinated bridging and tetracoordinated Al atoms, (2) the hydrolyzed propoxy groups will be expelled by solvent molecules, and (3) partly hydrolyzed species can condense to oligomers with bridging OH groups or O atoms.
Co-reporter:Ke Tang;Jinhu Wang;Xueli Cheng;Qianqian Hou
European Journal of Organic Chemistry 2010 Volume 2010( Issue 32) pp:6249-6255
Publication Date(Web):
DOI:10.1002/ejoc.201000774
Abstract
N-Heterocyclic carbenes (NHCs) have experimentally proved to be powerful catalysts for the Staudinger reaction ([2+2] cycloaddition of a ketene with an imine) but without giving a clear catalytic mechanism. According to different experimental results, the “ketene-first” and the “imine-first” mechanisms, arguing which reactant should be initially activated by the NHC catalyst, have been proposed. Our theoretical investigation by employing density functional theory (DFT) reveals that the reaction mechanism of the NHC-catalyzed Staudinger reaction is exclusively the “ketene-first” mechanism, but the competitive reactions of NHC catalysts with ketenes or imines will lead to different experimental observations. On the basis of this conclusion, we found that the NHC-catalyzed Staudinger reaction would exhibit different stereoselectivities by appropriate choice of the nitrogen substituent of the imines. Furthermore, these results are supposed to be applicable for other nucleophile-catalyzed Staudinger reactions.
Co-reporter:Xueli Cheng, Jinhu Wang, Ke Tang, Yongjun Liu, Chengbu Liu
Chemical Physics Letters 2010 Volume 496(1–3) pp:36-41
Publication Date(Web):20 August 2010
DOI:10.1016/j.cplett.2010.07.052
Abstract
Decarboxylation is normally a dissociative process, commonly catalyzed by proton or enzymes. The decarboxylation mechanism of pyrrole-2-carboxylic acid involves the addition of water to the carboxyl group, and the C–C bond cleavage leading to the protonated carbonic acid. The direct decarboxylation and decarboxylation aided with water were also investigated to consider the functions of proton and water. Our calculations with Gaussian 03 package show that, with the assistance of H3O+, the potential energy of the C–C rupture decreases significantly to 9.77 kcal/mol, and the total energy barrier decreases to 33.99 kcal/mol.
Co-reporter:Baoping Ling, Lihua Dong, Rui Zhang, Zhiguo Wang, Yongjun Liu, Chengbu Liu
Journal of Molecular Graphics and Modelling 2010 Volume 29(Issue 3) pp:354-362
Publication Date(Web):November 2010
DOI:10.1016/j.jmgm.2010.09.011
X-linked IAP can bind caspase-9 and inhibit its activity. Mitochondrial protein Smac/DIABLO can also interact with XIAP and relieve the inhibition on caspase-9 to induce apoptosis. A series of artificial Smac mimetics have been used to mimic the Smac N-terminal tetrapeptide AVPI to bind to XIAP-BIR3, but these structural diverse mimetics exhibited distinct binding affinities. To get an insight into the binding nature and optimize the structures, molecular docking and dynamics simulations were used to study the binding of XIAP-BIR3 with three groups of Smac mimetics. The docking results reveal that these Smac mimetics anchored on the surface groove of XIAP-BIR3 and superimposed well with AVPI. The modifications on the seven-membered ring of bicyclic core segment do not strengthen the binding affinity, while a benzyl introduced to the five-membered ring is favorable to the binding. Molecular dynamics simulations on three typical systems show that these complexes are very stable. Hydrogen bonds between the bicyclic core segment and Thr308 play critical roles in maintaining the stability of complex. The binding free energies calculated by MM_PBSA method are consistent with the experimental results.Graphical abstract. The interactions of XIAP-BIR3 domain with bicyclic and tricyclic core monovalent Smac mimetics were studied by using molecular docking and dynamics simulations. The calculated binding free energies by MM_PBSA method are consistent with the experimental results. These results give useful information about the relations between binding affinities and structures of the Smac mimetics.Research highlights▶ Mitochondrial protein Smac/DIABLO can also interact with XIAP and relieve the inhibition on caspase-9 to induce apoptosis. A series of artificial Smac mimetics have been used to mimic the Smac N-terminal tetrapeptide AVPI to bind to XIAP-BIR3, but these structural diverse mimetics exhibited distinct binding affinities. In this paper, the interactions between XIAP-BIR3 domain and Smac mimetics were studied by using molecular docking and dynamics simulations. The calculation results can well illustrate the relationship between binding affinities and structures of mimetics.
Co-reporter:Xueli Cheng, Yanyun Zhao, Ke Tang, Jinhu Wang, Yongjun Liu
Journal of Molecular Structure: THEOCHEM 2010 Volume 945(1–3) pp:53-56
Publication Date(Web):15 April 2010
DOI:10.1016/j.theochem.2010.01.008
The catalysis of N2O + CO is an important reaction system for understanding the performance of catalysts, and can serve as a prototype to investigate a variety of reaction mechanisms of catalytic processes. The reaction of N2O + CO catalyzed by Ir+ and Co+ was studied by using density functional theory (B3LYP). A large basis set of 6-311++G∗∗ was used for H, C, N, O and Co atoms, and the effective core potentials (ECPs) of Hay and Wadt with a double-ζ basis set (LanL2DZ) was employed to describe Ir atom. On the basis of full optimization, the reaction mechanism was elucidated with vibrational analysis, and a new process of CO seizing the O atom directly, which has relatively low energy barrier, was reported; the reaction mechanism of N2O to N2 catalyzed by Co+ is very similar to that of Ir+, but in the quintet reaction pathways of CoO+ + CO to Co+ + CO2, there is only a CO-capture channel.
Co-reporter:Qianqian Hou, Likai Du, Jun Gao, Yongjun Liu, and Chengbu Liu
The Journal of Physical Chemistry B 2010 Volume 114(Issue 46) pp:15296-15300
Publication Date(Web):November 1, 2010
DOI:10.1021/jp106714m
Combined quantum-mechanical/molecular-mechanical (QM/MM) approaches have been applied to investigate the detailed reaction mechanism of human O6-alkylguanine−DNA alkyltransferase (AGT). AGT is a direct DNA repair protein that is capable of repairing alkylated DNA by transferring the methyl group to the thiol group of a cysteine residue (Cys145) in the active site in an irreversible and stoichiometric reaction. Our QM/MM calculations reveal that the methyl group transferring step is expected to occur through two steps, in which the methyl carbocation generating step is the rate-determining step with an energy barrier of 14.4 kcal/mol at the QM/MM B3LYP/6-31G(d,p)//CHARMM22 level of theory. It is different from the previous theoretical studies based on QM calculations by using a cluster model in which the methyl group transferring step is a one-step process with a higher energy barrier.
Co-reporter:Baoping Ling, Zhiguo Wang, Rui Zhang, Xianghua Meng, Yongjun Liu, Changqiao Zhang, Chengbu Liu
Journal of Molecular Graphics and Modelling 2009 Volume 28(Issue 1) pp:37-45
Publication Date(Web):August 2009
DOI:10.1016/j.jmgm.2009.03.005
Recent experimental study [S.D. Gilbert, S.J. Mediatore, R.T. Batey, Modified pyrimidine specifically bind the purine riboswitch, J. Am. Chem. Soc. 128 (2006) 14214–14215] demonstrated that the purine riboswitch could specifically bind some ligands other than purines such as amino-pyrimidines, and the authors proposed that the five-membered ring of purine was not required for recognition. To get insight into the interaction details, we used molecular docking method to investigate the interactions of a mutant form of guanine riboswitch with a series of amino-purines, amino-pyrimidines and imidazole derivatives, and employed molecular simulation method to study the dynamic behavior of the selected complexes. The calculation results reveal that (1) all the amino-purines and amino-pyrimidines bind in a same cavity composed of four nucleobases including U22, U47, U51 and U74, which is consistent with the experimental results, while the two imidazole derivatives adopt other binding modes; (2) the purines are engulfed within three-way junction motifs, but most pyrimidines only form two-way junctions with the riboswitch; (3) the number and position of amino substituents could seriously affect the binding of pyrimidines. As riboswitches are potentially excellent candidates for antibiotic therapeutics, these findings may be useful for understanding the range of compounds that riboswitch can specifically recognize.
Co-reporter:Zhiguo Wang, Baoping Ling, Rui Zhang, Yourui Suo, Yongjun Liu, Zhangyu Yu, Chengbu Liu
Journal of Molecular Graphics and Modelling 2009 Volume 28(Issue 2) pp:162-169
Publication Date(Web):September 2009
DOI:10.1016/j.jmgm.2009.06.003
Phenolic marine natural product is a kind of new potential aldose reductase inhibitors (ARIs). In order to investigate the binding mode and inhibition mechanism, molecular docking and dynamics studies were performed to explore the interactions of six phenolic inhibitors with human aldose reductase (hALR2). Considering physiological environment, all the neutral and other two ionized states of each phenolic inhibitor were adopted in the simulation. The calculations indicate that all the inhibitors are able to form stable hydrogen bonds with the hALR2 active pocket which is mainly constructed by residues TYR48, HIS110 and TRP111, and they impose the inhibition effect by occupying the active space. In all inhibitors, only La and its two ionized derivatives La_ion1 and La_ion2, in which neither of the ortho-hydrogens of 3-hydroxyl is substituted by Br, bind with hALR2 active residues using the terminal 3-hydroxyl. While, all the other inhibitors, at least one of whose ortho-sites of 3- and 6-hydroxyls are substituted by Br substituent which take much electron-withdrawing effect and steric hindrance, bind with hALR2 through the lactone group. This means that the Br substituent can effectively regulate the binding modes of phenolic inhibitors. Although the lactone bound inhibitors have relatively high RMSD values, our dynamics study shows that both binding modes are of high stability. For each inhibitor molecule, the ionization does not change its original binding mode, but it does gradually increase the binding free energy, which reveals that besides hydrogen bonds, the electrostatic effect is also important to the inhibitor–hALR2 interaction.
Co-reporter:Zhiguo Wang, Honglun Wang, Yongjun Liu, Yourui Suo
Journal of Molecular Structure: THEOCHEM 2008 Volume 850(1–3) pp:72-78
Publication Date(Web):15 February 2008
DOI:10.1016/j.theochem.2007.10.023
In this paper, the reactions of nitrone, N-methyl nitrone, N-phenyl nitrone and their hydroxylamine tautomers (vinyl-hydroxylamine, N-methyl-vinyl-hydroxylamine and N-phenyl-vinyl-hydroxylamine) on the reconstructed C(1 0 0)-2 × 1 surface have been investigated using hybrid density functional theory (B3LYP), Møller–Plesset second-order perturbation (MP2) and multi-configuration complete-active-space self-consistent-field (CASSCF) methods. The calculations showed that all the nitrones can react with the surface “dimer” via facile 1,3-dipolar cycloaddition with small activation barriers (less than 12.0 kJ/mol at B3LYP/6-31g(d) level). The [2+2] cycloaddition of hydroxylamine tautomers on the C(1 0 0) surface follows a diradical mechanism. Hydroxylamine tautomers first form diradical intermediates with the reconstructed C(1 0 0)-2 × 1 surface by overcoming a large activation barrier of 50–60 kJ/mol (B3LYP), then generate [2+2] cycloaddition products via diradical transition states with negligible activation barriers. The surface reactions result in hydroxyl or amino-terminated diamond surfaces, which offers new opportunity for further modifications.
Co-reporter:Ge Tian and Yongjun Liu
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 11) pp:NaN7742-7742
Publication Date(Web):2017/02/20
DOI:10.1039/C6CP08811B
Ubiquinone plays a pivotal role in the aerobic cellular respiratory electron transport chain, whereas ferulic acid decarboxylase (FDC) is involved in the biosynthesis of ubiquinone precursor. Recently, the complete crystal structure of FDC (based on the co-expression of the A. niger fdc1 gene in E. coli with the associated ubix gene from E. coli) at high resolution was reported. Herein, the detailed catalytic non-oxidative decarboxylation mechanism of FDC has been investigated by a combined quantum mechanics/molecular mechanics (QM/MM) approach. Calculation results indicate that, after the 1,3-dipolar cycloaddition of the substrate and cofactor, the carboxylic group can readily split off from the adduct, and the overall energy barrier of the whole catalytic reaction is 23.5 kcal mol−1. According to the energy barrier analysis, the protonation step is rate-limiting. The conserved protonated Glu282 is suggested to be the proton donor through a “water bridge”. Besides, two cases, that is, the generated CO2 escapes from the active site or remains in the active site, were considered. It was found that the prolonged leaving of CO2 can facilitate the protonation of the intermediate. In particular, our calculations shed light on the detailed function of both cofactors prFMNiminium and prFMNketamine in the decarboxylation step. The cofactor prFMNiminium is the catalytically relevant species compared with prFMNketamine.
Co-reporter:Xiya Wang, Hao Su and Yongjun Liu
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 11) pp:NaN7677-7677
Publication Date(Web):2017/02/17
DOI:10.1039/C7CP00313G
Fumitremorgin B endoperoxidase (FtmOx1) from Aspergillus fumigatus is the first reported α-ketoglutarate-dependent mononuclear non-haem iron enzyme that catalyzes the endoperoxide formation reaction, converting Fumitremorgin B to verruculogen. Experiments reveal that the molecular oxygen (O2) is incorporated into verruculogen without O–O bond scission, which differs from the currently known non-haem iron enzymes, but the mechanistic details are still unclear. In this paper, on the basis of the crystal structures of FtmOx1 in complex with either the co-substrate (α-ketoglutarate) or the substrate (fumitremorgin B), a ternary complex model of the enzyme-α-ketoglutarate-substrate has been constructed, and combined quantum mechanics and molecular mechanics (QM/MM) calculations have been performed to unravel the novel mechanism of FtmOx1. Our calculations indicate the quintet of the FeIVO complex as the ground state. The FeIVO complex firstly abstracts a hydrogen from the hydroxyl of Tyr228 to initiate the reaction, which corresponds to a lower energy barrier (9.1 kcal mol−1). If the FeIVO complex directly abstracts a hydrogen from C21 of the substrate, the energy barrier would increase to 33.9 kcal mol−1. When Tyr228 was mutated to Ala228, this energy barrier decreases to 24.0 kcal mol−1. In the subsequent reaction, the generated Tyr228 radical abstracts the hydrogen (H2) from C21 of the substrate with an energy barrier of 23.8 kcal mol−1. The second molecular oxygen binds to the C21 radical of the substrate in the active pocket and further completes the epoxidation with an energy barrier of 4.8 kcal mol−1. These results may provide useful information for understanding the reaction mechanism of FtmOx1 and provide guidance for further experimental investigations.
Co-reporter:Qianqian Hou, Xiang Sheng and Yongjun Liu
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 23) pp:NaN11373-11373
Publication Date(Web):2014/03/17
DOI:10.1039/C3CP55263B
Archaeal fructose-1,6-bisphosphate aldolase/phosphatase (FBPA/P) is a newly identified unusual bifunctional enzyme (Nature, 2010, 464, 1077), which contains one single catalytic domain but catalyzes two chemically distinct reactions of gluconeogenesis. It is different from the ordinary enzymes whose active sites are responsible for a specific reaction. To explore the catalytic characteristic of FBPA/P, the aldol condensation mechanism of bifunctional FBPA/P has been investigated using quantum mechanics/molecular mechanics (QM/MM) method. The whole reaction process can be divided into two half-reactions involving seven elementary steps. A Schiff base intermediate is theoretically confirmed, agreeing well with the recently resolved crystal structures (Nature, 2011, 478, 538). The free energy barrier of the rate-limiting step is calculated to be 22.2 kcal mol−1, which is a concerted process of a nucleophilic attack by the enolic carbon to the ketonic carbon and a proton transfer from Tyr229 to the ketonic oxygen. Lys232 plays an important role in forming a Schiff base intermediate with the substrate (DHAP). Tyr229 functions as a proton shuttle during the catalysis. This is the first theoretical study on the aldol condensation mechanism of FBPA/P, which may provide useful information for understanding bifunctional enzymes.
Co-reporter:Xiang Sheng and Yongjun Liu
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 8) pp:NaN1277-1277
Publication Date(Web):2014/01/13
DOI:10.1039/C3OB42182A
Nicotinamidase (Pnc1) is a member of Zn-dependent amidohydrolases that hydrolyzes nicotinamide (NAM) to nicotinic acid (NA), which is a key step in the salvage pathway of NAD+ biosynthesis. In this paper, the catalytic mechanism of Pnc1 has been investigated by using a combined quantum-mechanical/molecular-mechanical (QM/MM) approach based on the recently obtained crystal structure of Pnc1. The reaction pathway, the detail of each elementary step, the energetics of the whole catalytic cycle, and the roles of key residues and Zn-binding site are illuminated. Our calculation results indicate that the catalytic water molecule comes from the bulk solvent, which is then deprotonated by residue D8. D8 functions as a proton transfer station between C167 and NAM, while the activated C167 serves as the nucleophile. The residue K122 only plays a role in stabilizing intermediates and transition states. The oxyanion hole formed by the amide backbone nitrogen atoms of A163 and C167 has the function to stabilize the hydroxyl anion of nicotinamide. The Zn-binding site rather than a single Zn2+ ion acts as a Lewis acid to influence the reaction. Two elementary steps, the activation of C167 in the deamination process and the decomposition of catalytic water in the hydrolysis process, correspond to the large energy barriers of 25.7 and 28.1 kcal mol−1, respectively, meaning that both of them contribute a lot to the overall reaction barrier. Our results may provide useful information for the design of novel and efficient Pnc1 inhibitors and related biocatalytic applications.
Co-reporter:Hao Su, Xiang Sheng and Yongjun Liu
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 13) pp:NaN3442-3442
Publication Date(Web):2016/03/04
DOI:10.1039/C6OB00320F
The peptidoglycan (PG) metabolic process is essential for bacterial growth. β-N-Acetylglucosaminidases (NagZ enzymes) are involved in the PG process and they catalyze the removal of terminal N-acetylglucosamine in PG fragments. According to the amino acid sequence and secondary structures, NagZ enzymes should belong to the glycoside hydrolase family GH3. However, a recent experimental study revealed that NagZ enzymes are glycoside phosphorylases rather than glycoside hydrolases. To further understand the catalytic process of NagZs at the atomistic level, the reaction mechanism of NagZ from Bacillus subtilis (BsNagZ) has been studied by using a QM/MM approach. Our calculation results show that the glycosylation of the substrate is the rate limiting step of the whole catalytic cycle with an energy barrier of 19.3 kcal mol−1, which is close to the free energy barrier (16.4 kcal mol−1) estimated from the experimental rate constant. For deglycosylation, both the hydrolysis and phosphorylation of the glycosyl-enzyme intermediate were explored. The phosphorylation corresponds to the lower energy barrier than hydrolysis (1.8 vs. 17.7 kcal mol−1), giving theoretical support to the previously suggested phosphorylase activity of NagZ enzymes. In both the glycosylation and deglycosylation steps, the oxocarbenium-ion-like transition states are always involved, and the substrate distortion in the active site can significantly facilitate the reaction, in which residue D123 plays a key role in this distortion. This is the first computational report for understanding the phosphorylase activity of NagZ enzymes.
Co-reporter:Xiya Wang, Wenyou Zhu and Yongjun Liu
Catalysis Science & Technology (2011-Present) 2017 - vol. 7(Issue 13) pp:NaN2856-2856
Publication Date(Web):2017/05/30
DOI:10.1039/C7CY00573C
L-Tryptophan lyase (NosL), a member of the radical S-adenosyl-L-methionine (SAM)-dependent superfamily, catalyzes the conversion of L-tryptophan to 3-methylindolic acid (MIA). In this article, on the basis of the recently obtained crystal structure of NosL (PDB code 4R34) in 2014, a combined quantum mechanical/molecular mechanical (QM/MM) approach has been employed to elucidate the reaction details, involving substrate amine dehydrogenation, C–C bond cleavage and carboxyl fragment migration. Our results show that the hydrogen in the amino group of L-tryptophan is suitable for abstraction by the Ado radical, and this step corresponds to an energy barrier of 12.4 kcal mol−1. Two possible modes of C–C cleavage (path1 and path2) have been considered. The cleavage of the Cα–Cβ bond is thermodynamically more favorable than the cleavage of the Cα–C bond with their energy barriers being 7.2 and 15.0 kcal mol−1, respectively. And the easy breaking of Cα–Cβ may be attributed to the electron hole delocalization in the amine radical of the substrate. The intermediate derived from the cleavage of the Cα–C bond is calculated to be a stable species, and the cleavage of the Cα–C bond is accompanied by the migration of the ˙COO− fragment. These conclusions are basically in accordance with EPR-trapped analysis and can account for the absence of the ˙COO− fragment. The shunt product (3-methylindole) is obtained from the cleavage of the Cα–Cβ bond with an energy barrier of 19.5 kcal mol−1. However, the rate limiting step is the formation of 3-methylindolic acid (MIA), which corresponds to an energy barrier of 26.9 kcal mol−1. Our investigations thus give a better comprehension of the NosL reaction mechanism and may contribute to the understanding of the SAM superfamily.
Co-reporter:Shujun Zhang, Xiya Wang and Yongjun Liu
Catalysis Science & Technology (2011-Present) 2017 - vol. 7(Issue 4) pp:NaN922-922
Publication Date(Web):2017/01/23
DOI:10.1039/C6CY02553F
2,4′-Dihydroxyacetophenone dioxygenase (DAD) is a bacterial non-heme enzyme responsible for the oxygenative cleavage of the aliphatic C–C bond, which catalyzes the conversion of 2,4′-dihydroxyacetophenone to 4-hydroxybenzoic acid and formic acid. On the basis of the crystal structure and studies on two synthesized biomimetic model compounds, two possible reaction pathways that involve a dioxacyclic or alkylperoxo intermediate have been previously suggested. However, little is currently known about the mechanistic detail and the proposed intermediates have not been experimentally confirmed yet. To elucidate the reaction mechanism at the atomistic level, on the basis of the recently obtained crystal structure, the reactant enzyme–substrate complex has been constructed, and the reaction details have been studied using a quantum mechanics/molecular mechanics (QM/MM) approach. Our calculations reveal the triplet of the iron(III)-superoxide radical complex as the ground state, but the quintet state which is higher than the triplet by 11.2 kcal mol−1 corresponds to a lower energy barrier in the first step. Thus, the reactant complex may firstly undergo a triplet–quintet crossing to initiate the reaction and then the subsequent chemistry mainly occurs on the quintet state surface. The previously proposed key dioxacyclic or alkylperoxo intermediate was calculated to be energetically unreachable, and the corresponding mechanism has been revised, which contains eight elementary steps, and the key C–C bond cleavage is accompanied by an insertion reaction of the adjacent oxygen radical. Two elementary steps are calculated to be possible rate-determining steps. Our results may provide useful information for further understanding the cleavage mechanism of the aliphatic C–C bond catalyzed by DAD and other dioxygenase enzymes.
Co-reporter:Hao Su, Xiang Sheng and Yongjun Liu
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 40) pp:NaN27938-27938
Publication Date(Web):2016/09/16
DOI:10.1039/C6CP04918D
Imidazolonepropionase (HutI, EC 3.5.2.7) catalyzes the hydrolytic cleavage of carbon–nitrogen bond in 4-imidazolone-5-propionic acid (IPA) to yield L-formiminoglutamic acid, which is the third step in the universal histidine degradation pathway. In this article, using a combined quantum mechanics and molecular mechanics (QM/MM) approach, the specificity and catalytic mechanism of HutI from Bacillus subtilis have been explored by considering the four isomers of (S)- and (R)-enantiomers of IPA (SIPA-1, SIPA-2, RIPA-1 and RIPA-2). Our calculations reveal that the activation of hydrolytic water (a zinc-bound water) is performed by residue E252 via a “bridging” water molecule, which occurs before binding of the substrate. After the substrate binding, this activation channel is blocked by the substrate, and the other two residues (D324 and H272) cannot act as the general base to activate the hydrolytic water. For the two (S)-enantiomers of IPA, HutI can specifically convert one isomer of (S)-enantiomer (SIPA-1) to L-formiminoglutamic acid with an energy barrier of 16.6 kcal mol−1. The conversion of another (S)-enantiomer (SIPA-2) corresponds to the energy barrier of 21.9 kcal mol−1. However, for the two isomers of (R)-enantiomer, RIPA-1 corresponds to a higher energy barrier (21.8 kcal mol−1), and RIPA-2 is associated with a weak binding in the active site compared to SIPA-2. Thus, on the basis of our calculations, SIPA-1 is suggested to be the most favorable substrate for HutI, whereas the hydrolytic cleavage of SIPA-2 may require a preliminary isomerization to SIPA-1.

![S-[2-[3-[[4-[[[(2R,3S,4R,5R)-5-(6-AMINOPURIN-9-YL)-4-HYDROXY-3-PHOSPHONOOXYOXOLAN-2-YL]METHOXY-HYDROXYPHOSPHORYL]OXY-HYDROXYPHOSPHORYL]OXY-2-HYDROXY-3,3-DIMETHYLBUTANOYL]AMINO]PROPANOYLAMINO]ETHYL] METHANETHIOATE](http://img.cochemist.com/ccimg/13200/13131-49-2.png)
![S-[2-[3-[[4-[[[(2R,3S,4R,5R)-5-(6-AMINOPURIN-9-YL)-4-HYDROXY-3-PHOSPHONOOXYOXOLAN-2-YL]METHOXY-HYDROXYPHOSPHORYL]OXY-HYDROXYPHOSPHORYL]OXY-2-HYDROXY-3,3-DIMETHYLBUTANOYL]AMINO]PROPANOYLAMINO]ETHYL] METHANETHIOATE](http://img.cochemist.com/ccimg/13200/13131-49-2_b.png)
![Poly[imino[(1S)-1-(1H-imidazol-4-ylmethyl)-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26900/26854-81-9.png)
![Poly[imino[(1S)-1-(1H-imidazol-4-ylmethyl)-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26900/26854-81-9_b.png)




















![Uridine 5'-(trihydrogendiphosphate), P'-[2-(acetylamino)-2-deoxy-a-D-glucopyranosyl] ester](http://img.cochemist.com/ccimg/600/528-04-1.png)
![Uridine 5'-(trihydrogendiphosphate), P'-[2-(acetylamino)-2-deoxy-a-D-glucopyranosyl] ester](http://img.cochemist.com/ccimg/600/528-04-1_b.png)



![2-amino-4,7-dihydro-4-oxo-3H-Pyrrolo[2,3-d]pyrimidine-5-carboxylic acid](http://img.cochemist.com/ccimg/92700/92636-62-9.png)
![2-amino-4,7-dihydro-4-oxo-3H-Pyrrolo[2,3-d]pyrimidine-5-carboxylic acid](http://img.cochemist.com/ccimg/92700/92636-62-9_b.png)
![1-Azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid, 7-oxo-, (5R)-](http://img.cochemist.com/ccimg/78900/78854-41-8.png)
![1-Azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid, 7-oxo-, (5R)-](http://img.cochemist.com/ccimg/78900/78854-41-8_b.png)









![1-Azabicyclo[3.2.0]heptane-2-carboxylicacid, 7-oxo-, (2S,5S)-](http://img.cochemist.com/ccimg/123500/123409-86-9.png)
![1-Azabicyclo[3.2.0]heptane-2-carboxylicacid, 7-oxo-, (2S,5S)-](http://img.cochemist.com/ccimg/123500/123409-86-9_b.png)