Co-reporter:Fen Jiang, An-ping Guo, Jia-cheng Xu, Hui-Jie Wang, Xiao-fei Mo, Qi-Dong You, Xiao-Li Xu
European Journal of Medicinal Chemistry 2017 Volume 141(Volume 141) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.ejmech.2017.07.080
•A ROCS model was constructed to identify novel Hsp90 C-terminal inhibitors.•13 was identified as the lead compound and structure modification led to 69.•69 exhibited potent cytotoxicity and suitable physicochemical properties.•69 exhibited tumor growth inhibition and anti-metastasis effect in 4T1 mice models.In order to discover novel Hsp90 inhibitors targeting the C-terminal ATP binding pocket, a novobiocin derivative based ROCS model was constructed for virtual screening. Compound 13 was identified as the lead compound and then systematical structure activity relationship (SAR) study was conducted. These efforts led to compound 69, which exhibited potent anti-proliferative activities against MCF7 and SKBr3 breast cancer cell lines. In 4T1 mice breast cancer models, 69 exhibited potent tumor growth inhibition and anti-metastasis effect. Compound 69 as a potent antitumor agent targeting the Hsp90 C-terminal is worthy of further pre-clinical study.Download high-res image (194KB)Download full-size image
Co-reporter:Dong-Dong Li, Wei-Lin Chen, Zhi-Hui Wang, Yi-Yue Xie, Xiao-Li Xu, Zheng-Yu Jiang, Xiao-Jin Zhang, Qi-Dong You, Xiao-Ke Guo
European Journal of Medicinal Chemistry 2016 Volume 124() pp:480-489
Publication Date(Web):29 November 2016
DOI:10.1016/j.ejmech.2016.08.036
•Biphenyl compounds were designed and synthetized to disturb MLL1-WDR5 interaction.•30 effectively inhibited MLL1-dependent H3K4 methylation in vitro and in MV4-11 cells.•30 deregulated expression level of Hoxa9 and Meis-1 genes and induced MV4-11 cells apoptosis concentration-dependently.•42 (IC50 = 7.6 nM) was the most potent small molecule inhibitor, and showed most potent inhibitory activity in HTM assay.MLL1-WDR5 protein-protein interaction is essential for MLL1 H3K4 methyltransferase activity. Targeting MLL1 enzymatic activity to regulate expression level of MLL-dependent genes represents a therapeutic strategy for acute leukemia harboring MLL fusion proteins. Herein we reported a series of biphenyl compounds disturbed MLL1-WDR5 interaction. These compounds effectively inhibited MLL1 histone methyltransferase (HMT) activity in vitro and in MV4-11 cell line. The representative compound 30 (DDO-2084) inhibited proliferation and induced apoptosis of MV4-11 cells through deregulating expression level of Hoxa9 and Meis-1 genes, which emphasized our compounds were on-target. Optimization of compound 30 led to high-affinity inhibitors. Especially, compound 42 (DDO-2117, IC50 = 7.6 nM) bearing an amino and a 4-aminobutanamido group was the most potent inhibitor reported to-date, and showed the most potent inhibitory activity (IC50 = 0.19 μM) in HMT assay.
Co-reporter:Xiaoke Guo, Qian Yang, Jing Xu, Li Zhang, Hongxi Chu, Peng Yu, Yingying Zhu, Jinglian Wei, Weilin Chen, Yaozhong Zhang, Xiaojin Zhang, Haopeng Sun, Yiqun Tang, Qidong You
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 21) pp:6466-6476
Publication Date(Web):1 November 2013
DOI:10.1016/j.bmc.2013.08.041
Atrial fibrillation (AF) is one of the common arrhythmias that threaten human health. Kv1.5 potassium channel is reported as an efficacious and safe target for the treatment of AF. In this paper, we designed and synthesized three series of compounds through modifying the lead compound RH01617 that was screened out by the pharmacophore model we reported earlier. All of the compounds were evaluated by the whole-patch lamp technology and most of them possessed potent inhibitory activities against Kv1.5. Compounds IIIi and IIIl were evaluated for the target selectivity as well as the pharmacodynamic effects in an isolated rat model. Due to the promising pharmacological behavior, compound IIIl deserves further pharmacodynamic and pharmacokinetic evaluations.
Co-reporter:Zhengui Huang, Kejiang Lin, Qidong You
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 14) pp:4166-4171
Publication Date(Web):15 July 2013
DOI:10.1016/j.bmcl.2013.05.033
As increasing drug-resistance poses an emerging threat to public health, the development of novel antibacterial agents is critical. We developed a workflow consisting of various methods for de novo design. In the workflow, 2D-QSAR model based on molecular fingerprints was constructed to extract the bioactive molecular fingerprints from a data set of DNA–gyrase inhibitors with new structure and mechanism. These fingerprints were converted into molecular fragments which were recombined to generate compound library. The new compound library was virtually screened by LigandFit and Gold docking, and the results were further investigated by pharmacophore validation and binding mode analysis. The workflow successfully achieved a potential DNA–gyrase inhibitor. It could be applied to design more novel potential DNA–gyrase inhibitors and provide theoretical basis for further optimization of the hit compounds.
Co-reporter:Bin Qian, Qi-Dong You
Tetrahedron Letters 2012 Volume 53(Issue 29) pp:3750-3753
Publication Date(Web):18 July 2012
DOI:10.1016/j.tetlet.2012.05.016
A convenient and efficient method for preparing 2-hydroxy glycals was developed from thioglycosides by using 1,4-dioxane–bromine complex/DMAP as an efficient promoter with good yield (61–85%). In this synthetic method, a wide range of sugar thioglycosides could be used as substrates.A convenient and efficient method for preparing 2-hydroxy glycals was developed from thioglycosides by using 1,4-dioxane–bromine complex/DMAP as an efficient promoter with good yield (61–85%). In this synthetic method, a wide range of sugar thioglycosides could be used as substrates.
Co-reporter:Rong-geng Fu, Qi-dong You, Lei Yang, Wu-tong Wu, Cheng Jiang, Xiao-li Xu
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 22) pp:8035-8043
Publication Date(Web):15 November 2010
DOI:10.1016/j.bmc.2010.09.020
Four series of dihydropyrazolo[3,4-b]pyridines and benzo[4,5]imidazo[1,2-a]pyrimidines were designed and synthesized as dual KSP and Aurora-A kinase inhibitors for anti-cancer agents by introducing some fragments of Aurora-A kinase inhibitors into our KSP inhibitor CPUYL064. A total of 19 target compounds were evaluated by two related enzyme inhibition assays and a cytotoxicity assay in vitro. The results showed that some target compounds could inhibit both enzymes, and several of them showed significant inhibition activity against HCT116 cell line. Despite showing moderate KSP and Aurora-A kinase inhibition, the lead compounds 6a and 6e displayed significant cytotoxic activity in the micromolar range, especially against the HCT116 cell line and HepG2 cell line. The results may be useful for developing a new class of inhibitors having a dual function, KSP inhibition and Aurora-A kinase inhibition, for the treatment of cancer.Four series of dihydropyrazolo[3,4-b]pyridines and benzo[4,5]imidazo[1,2-a]pyrimidines were designed and synthesized as dual KSP and Aurora-A kinase inhibitors for anti-cancer agents by introducing some fragments of Aurora-A kinase inhibitors into KSP inhibitor CPUYL064.
Co-reporter:Liqin YU;Fei LIU;Yadong CHEN ;Qidong YOU
Chinese Journal of Chemistry 2009 Volume 27( Issue 3) pp:557-564
Publication Date(Web):
DOI:10.1002/cjoc.200990091
Abstract
A three-dimensional pharmacophore model was established based on 24 hydroxamate histone deacetylase (HDAC) inhibitors by HypoGen algorithm embedded in Catalyst software. The best pharmacophore hypothesis (Hypo1), consisting of four chemical features (one hydrogen-bond acceptor, one aromatic ring and two hydrophobic groups), has a correlation coefficient of 0.946. The Hypol was also validated by a test set consisting of 20 other compounds. Compared with the prior studies towards HDAC inhibitors the detailed chemical features of the "CAP" region in the reported HDAC inhibitors were for the first time depicted, which would be helpful in the further designing of novel HDAC inhibitors.
Co-reporter:Lupei Du, Minyong Li, Qian Yang, Yiqun Tang, Qidong You, Lin Xia
Bioorganic & Medicinal Chemistry Letters 2009 19(5) pp: 1477-1480
Publication Date(Web):
DOI:10.1016/j.bmcl.2009.01.022
Co-reporter:Qian Yang, Lupei Du, Xiaojian Wang, Minyong Li, Qidong You
Journal of Molecular Graphics and Modelling 2008 Volume 27(Issue 2) pp:178-187
Publication Date(Web):September 2008
DOI:10.1016/j.jmgm.2008.04.002
The ultra-rapid delayed rectifier potassium current (IKur), encoded by Kv1.5 gene, is the critical determinant of Phase I repolarization of action potential duration (APD). The evidences that Kv1.5 gene expresses more extensively in human atrial myocytes than in ventricle and the IKur currents has not been recorded in the human ventricle, suggest Kv1.5 potassium channel as a selective target for the treatment of atrial fibrillation (AF). Recent mutagenesis studies have provided us some evidences that are useful in designing Kv1.5 blockers. In order to further evaluate these molecular biological information, the homology model of Kv1.5 potassium channel was established based on the Kv1.2 crystal structure (PDB entry: 2A79) using MODELLER 9v2 program. After the molecular dynamics refinement, the optimized homology model was assessed as a reliable structure by PROCHECK, ERRAT, WHAT-IF, PROSA2003 and DOPE graph. The results of molecular docking studies on different Kv1.5 inhibitors are in agreement with the published mutagenesis data. Based on the docking conformations, a pharmacophore model was developed by HipHop algorithm in order to probe the common features of blockers. By analyzing the results, active site architecture, certain key residues and pharmacophore common-features that are responsible for substrate specificity were identified on the Kv1.5 potassium channel, which would be very helpful in understanding the blockade mechanism of Kv1.5 potassium channel and providing insights into rational design of novel Kv1.5 blockers.
Co-reporter:Lü-Pei Du, Keng-Chang Tsai, Min-Yong Li, Qi-Dong You, Lin Xia
Bioorganic & Medicinal Chemistry Letters 2004 Volume 14(Issue 18) pp:4771-4777
Publication Date(Web):20 September 2004
DOI:10.1016/j.bmcl.2004.06.070
Predictive pharmacophore models were developed for a large series of IKr potassium channel blockers as class III antiarrhythmic agents using HypoGen in Catalyst software. The pharmacophore hypotheses were generated using a training set consisting of 34 compounds carefully selected from documents. Their biological data, expressed as IC50, spanned from 1.5 nM to 2.8 mM with 7 orders difference. The most predictive hypothesis (Hypo1), consisting of four features (one positive ionizable feature, two aromatic rings and one hydrophobic group), had a best correlation coefficient of 0.825, a lowest rms deviation of 1.612, and a highest cost difference (null cost−total cost) of 77.552, which represents a true correlation and a good predictivity. The hypothesis Hypo1 was then validated by a test set consisting of 21 compounds and by a cross-validation of 95% confidence level with randomizing the data using CatScramble program. Accordingly, our model has strong predictivity to identify structural diverse IKr potassium channel blockers with desired biological activity by virtual screeningThis paper reports a pharmacophore hypothesis for IKr potassium channel blockers using HypoGen module in Catalyst software. The pharmacophore can be used to in predicting biological activity of compounds by virtual screening.
Co-reporter:Li Zhao, Zhen Chen, Qing Zhao, Daidi Wang, Rong Hu, Qidong You, Qinglong Guo
Regulatory Toxicology and Pharmacology (July 2011) Volume 60(Issue 2) pp:212-217
Publication Date(Web):1 July 2011
DOI:10.1016/j.yrtph.2011.03.008
We studied the developmental toxicities and genotoxic potency of a widely bioactive plant medicine-wogonin in vivo and in vitro. In the in vivo developmental experiments, high dose of wogonin (40 mg/kg, intravenous injection) significantly induced the maternal weight gains and affected fetus including bodyweight, resorptions, live birth index and fetal skeletal alterations. In Ames test, no concentration-dependently increased TA98, TA100, and TA102 revertants were detected in wogonin groups whether in presence of metabolic activating enzymes or not. In the chromosome aberration test, wogonin dose-dependently increased structural chromosomal aberrations in CHL cells both with and without S9, even the effect was all judged (−). In micronucleus assay, no significant changes of MNPCE/PCE and PCE/NCE were found on mouse bone marrow micronucleus in wogonin groups. We concluded that wogonin induced developmental toxicities on pregnant mice and fetus, and the genotoxicities were positive. However no significant malformation was observed and only in vitro potency of chromosome aberration was weak, which suggested us wogonin could be a relatively safe drug in clinic.
Co-reporter:Qinglong Guo, Li Zhao, Qidong You, Yong Yang, Hongyan Gu, Guoliang Song, Na Lu, Jian Xin
Antiviral Research (April 2007) Volume 74(Issue 1) pp:16-24
Publication Date(Web):April 2007
DOI:10.1016/j.antiviral.2007.01.002
Co-reporter:Qing Zhao, Jia Wang, Mei-Juan Zou, Rong Hu, Li Zhao, Lei Qiang, Jing-Jing Rong, Qi-Dong You, Qing-Long Guo
Toxicology Letters (1 September 2010) Volume 197(Issue 3) pp:201-210
Publication Date(Web):1 September 2010
DOI:10.1016/j.toxlet.2010.05.019
Traditional Chinese medicines have been recognized as a new source of anticancer drugs or chemotherapy adjuvant to enhance the efficacy of chemotherapy and to ameliorate the side effects. Wogonin (WOG) has a potential for therapeutic use in the treatment of antitumor and chemoprophylaxis. 5-Fluorouracil (5-FU) is a key systemic chemotherapy drug and widely use in the treatment of solid tumors. In this study, we found that combination of WOG and 5-FU inhibited the viability of MGC-803 cells in a concentration-dependent manner and exhibited a synergistic anticancer effect (CI < 1) when 5-FU was used at relatively low concentrations. The pro-apoptotic activity of two-drug combination was much stronger than single. Furthermore, WOG could decrease the mRNA levels of dihydropyrimidine dehydrogenase (DPD), the metabolic enzymes of 5-FU. WOG could inhibit the NF-κB nuclear translocation and I-κB phosphorylation. Moreover, combined treatment caused significantly growth inhibition of human tumor xenografts. In addition, WOG markedly enhanced the antitumor activity of low dose 5-FU (i.p. 10 mg/kg/day), however there is no toxicity and influence on diet consumption in experimental animals. Taken together, our data's showed that WOG increased 5-FU retention for a prolonged catabolism by modulating 5-FU metabolic enzymes and sensitized the MGC-803 cells to 5-FU induced apoptosis by inhibiting the NF-κB nuclear translocation. The anti-gastric cancer effect of two-drug combination was much stronger than that of WOG or 5-FU alone. These results may be relevant to design new clinical therapeutic strategies against gastric cancer in future.










![2-[3-(trifluoromethyl)phenyl]-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione](http://img.cochemist.com/ccimg/6100/6052-93-3.png)
![2-[3-(trifluoromethyl)phenyl]-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione](http://img.cochemist.com/ccimg/6100/6052-93-3_b.png)










![10H-Phenothiazine,10-[2-(1-methyl-2-piperidinyl)ethyl]-2-(methylsulfinyl)-](http://img.cochemist.com/ccimg/5600/5588-33-0.png)
![10H-Phenothiazine,10-[2-(1-methyl-2-piperidinyl)ethyl]-2-(methylsulfinyl)-](http://img.cochemist.com/ccimg/5600/5588-33-0_b.png)


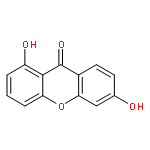
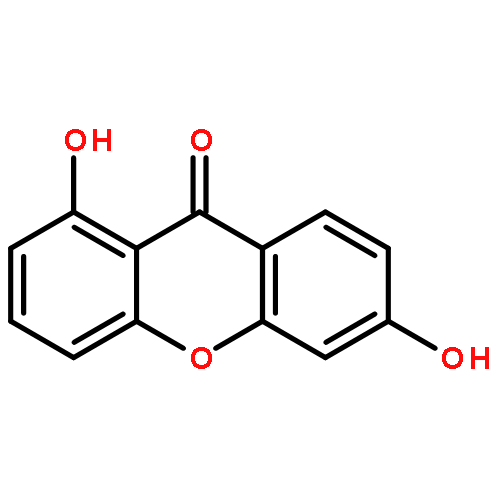
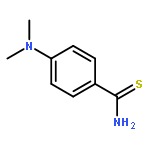
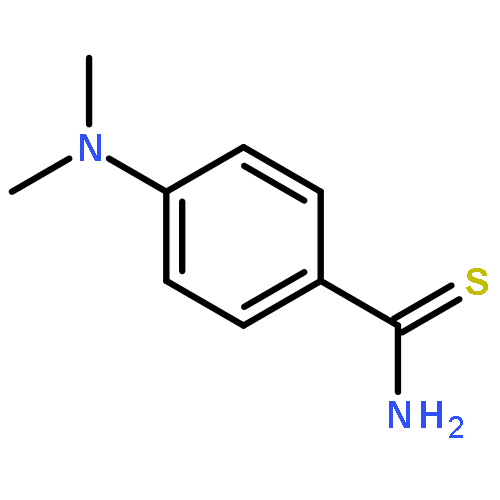
![2,2-dimethyl-3,4-dihydro-2H-benzo[g]chromene-5,10-dione](http://img.cochemist.com/ccimg/4800/4707-33-9.png)
![2,2-dimethyl-3,4-dihydro-2H-benzo[g]chromene-5,10-dione](http://img.cochemist.com/ccimg/4800/4707-33-9_b.png)






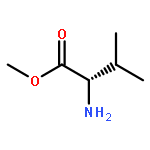
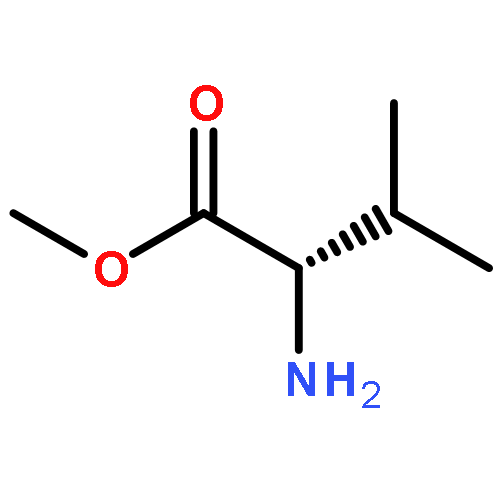
![4-hydroxy-3-[(1E)-3-methylbut-1-en-1-yl]naphthalene-1,2-dione](http://img.cochemist.com/ccimg/4100/4042-39-1.png)
![4-hydroxy-3-[(1E)-3-methylbut-1-en-1-yl]naphthalene-1,2-dione](http://img.cochemist.com/ccimg/4100/4042-39-1_b.png)


![N-[(2,6-DICHLOROPHENYL)SULFONYL]ALANINE](http://img.cochemist.com/ccimg/3900/3898-66-6.png)
![N-[(2,6-DICHLOROPHENYL)SULFONYL]ALANINE](http://img.cochemist.com/ccimg/3900/3898-66-6_b.png)
![10H-Phenothiazine-2-carbonitrile,10-[3-(dimethylamino)-2-methylpropyl]-](http://img.cochemist.com/ccimg/3600/3546-03-0.png)
![10H-Phenothiazine-2-carbonitrile,10-[3-(dimethylamino)-2-methylpropyl]-](http://img.cochemist.com/ccimg/3600/3546-03-0_b.png)

![1-Piperazineacetamide,4-[4,4-bis(4-fluorophenyl)butyl]-N-(2,6-dimethylphenyl)-](http://img.cochemist.com/ccimg/3500/3416-26-0.png)
![1-Piperazineacetamide,4-[4,4-bis(4-fluorophenyl)butyl]-N-(2,6-dimethylphenyl)-](http://img.cochemist.com/ccimg/3500/3416-26-0_b.png)



![1-[2-HYDROXY-3,4-BIS(PHENYLMETHOXY)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/2700/2652-27-9.png)
![1-[2-HYDROXY-3,4-BIS(PHENYLMETHOXY)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/2700/2652-27-9_b.png)













![3-(Dibenzo[b,e]oxepin-11(6H)-ylidene)-N,N-dimethylpropan-1-amine](http://img.cochemist.com/ccimg/1700/1668-19-5.png)
![3-(Dibenzo[b,e]oxepin-11(6H)-ylidene)-N,N-dimethylpropan-1-amine](http://img.cochemist.com/ccimg/1700/1668-19-5_b.png)


![Benzeneethanol, b-[(2S)-2-(dimethylamino)propyl]-a-ethyl-b-phenyl-, 1-acetate, (aS)-](http://img.cochemist.com/ccimg/1500/1477-40-3.png)
![Benzeneethanol, b-[(2S)-2-(dimethylamino)propyl]-a-ethyl-b-phenyl-, 1-acetate, (aS)-](http://img.cochemist.com/ccimg/1500/1477-40-3_b.png)
![[1,1'-Biphenyl]-2,2'-dicarboxaldehyde](http://img.cochemist.com/ccimg/1300/1210-05-5.png)
![[1,1'-Biphenyl]-2,2'-dicarboxaldehyde](http://img.cochemist.com/ccimg/1300/1210-05-5_b.png)
![2-Butenal,2-methyl-4-[(1R,3aS,5S,14aS)-3a,4,5,7-tetrahydro-8-hydroxy-3,3,11,11-tetramethyl-13-(3-methyl-2-buten-1-yl)-7,15-dioxo-1,5-methano-1H,3H,11H-furo[3,4-g]pyrano[3,2-b]xanthen-1-yl]-,(2Z)-](http://img.cochemist.com/ccimg/1200/1183-12-6.png)
![2-Butenal,2-methyl-4-[(1R,3aS,5S,14aS)-3a,4,5,7-tetrahydro-8-hydroxy-3,3,11,11-tetramethyl-13-(3-methyl-2-buten-1-yl)-7,15-dioxo-1,5-methano-1H,3H,11H-furo[3,4-g]pyrano[3,2-b]xanthen-1-yl]-,(2Z)-](http://img.cochemist.com/ccimg/1200/1183-12-6_b.png)

METHANONE](http://img.cochemist.com/ccimg/1100/1013-77-0.png)
METHANONE](http://img.cochemist.com/ccimg/1100/1013-77-0_b.png)





![7-AMINO-2-CHLORODIBENZO[B,F][1,4]OXAZEPIN-11(10H)-ONE](http://img.cochemist.com/ccimg/800/775-33-7.png)
![7-AMINO-2-CHLORODIBENZO[B,F][1,4]OXAZEPIN-11(10H)-ONE](http://img.cochemist.com/ccimg/800/775-33-7_b.png)


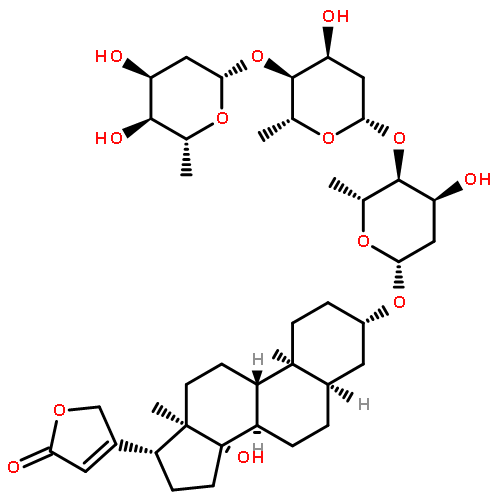
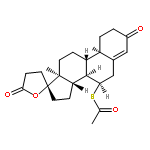
![Card-20(22)-enolide,3-[(6-deoxy-a-L-mannopyranosyl)oxy]-1,5,11,14,19-pentahydroxy-,(1b,3b,5b,11a)-](http://img.cochemist.com/ccimg/700/630-60-4.png)
![Card-20(22)-enolide,3-[(6-deoxy-a-L-mannopyranosyl)oxy]-1,5,11,14,19-pentahydroxy-,(1b,3b,5b,11a)-](http://img.cochemist.com/ccimg/700/630-60-4_b.png)
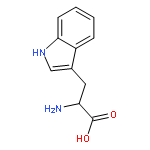
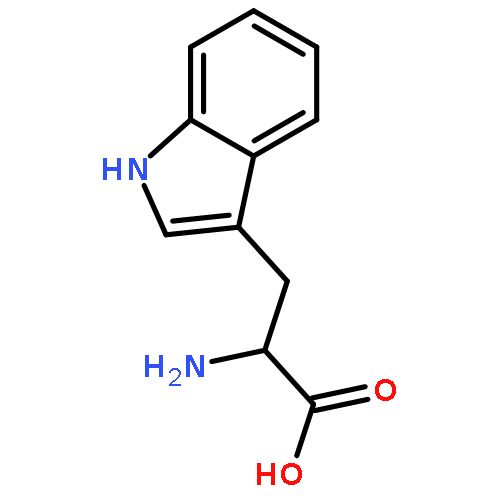
![L-Homoserine,O-[(aminoiminomethyl)amino]-](http://img.cochemist.com/ccimg/600/543-38-4.png)
![L-Homoserine,O-[(aminoiminomethyl)amino]-](http://img.cochemist.com/ccimg/600/543-38-4_b.png)


![8-Azabicyclo[3.2.1]octane-2-carboxylicacid, 3-(benzoyloxy)-8-methyl-, ethyl ester, (1R,2R,3S,5S)-](http://img.cochemist.com/ccimg/600/529-38-4.png)
![8-Azabicyclo[3.2.1]octane-2-carboxylicacid, 3-(benzoyloxy)-8-methyl-, ethyl ester, (1R,2R,3S,5S)-](http://img.cochemist.com/ccimg/600/529-38-4_b.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6_b.png)





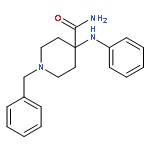
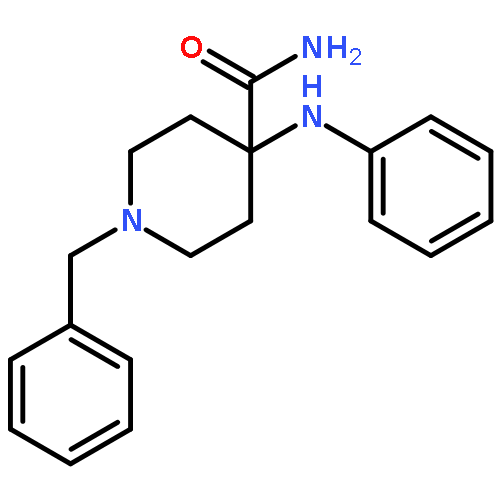
![N-phenyl-n-[1-(2-phenylethyl)piperidin-4-yl]propanamide](http://img.cochemist.com/ccimg/500/437-38-7.png)
![N-phenyl-n-[1-(2-phenylethyl)piperidin-4-yl]propanamide](http://img.cochemist.com/ccimg/500/437-38-7_b.png)
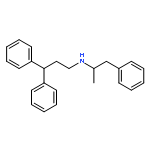
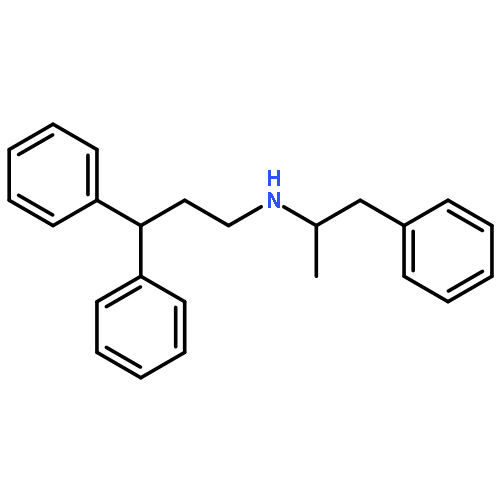


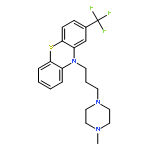
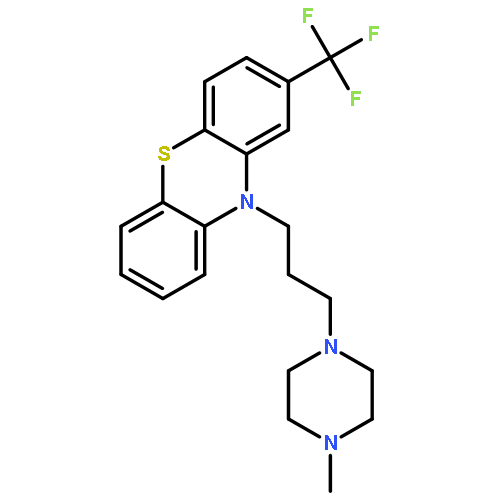















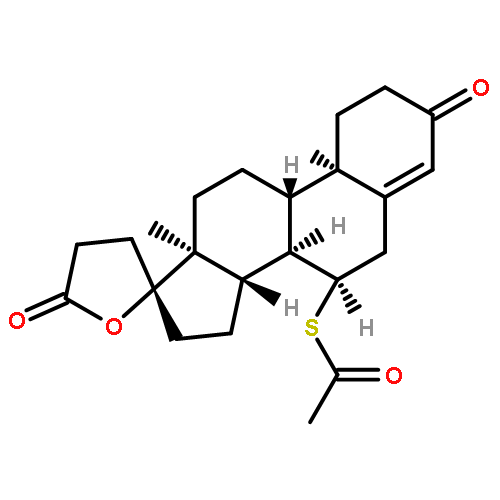




![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2_b.png)


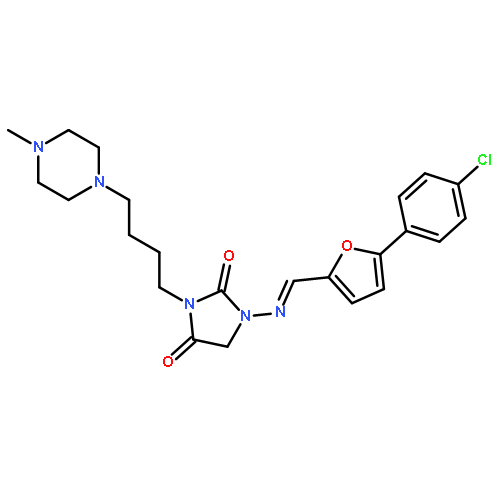
![2,4-Imidazolidinedione, 1-amino-3-[3-(4-methyl-1-piperazinyl)propyl]-](http://img.cochemist.com/ccimg/915500/915404-28-3.png)
![2,4-Imidazolidinedione, 1-amino-3-[3-(4-methyl-1-piperazinyl)propyl]-](http://img.cochemist.com/ccimg/915500/915404-28-3_b.png)
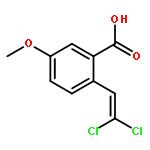
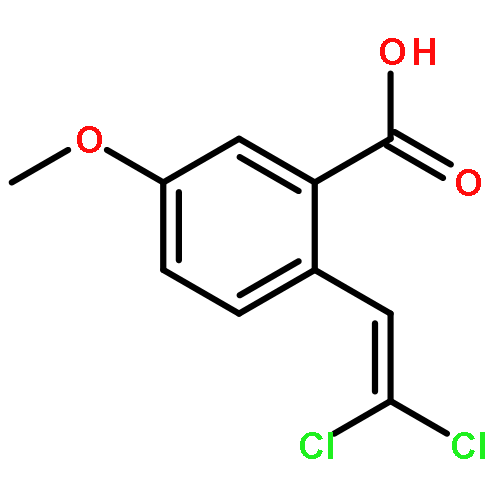




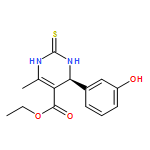






![Naphtho[1,2-b]furan-4,5-dione,2,3-dihydro-2-methyl-](http://img.cochemist.com/ccimg/195200/195156-60-6.png)
![Naphtho[1,2-b]furan-4,5-dione,2,3-dihydro-2-methyl-](http://img.cochemist.com/ccimg/195200/195156-60-6_b.png)














![Benzenesulfonamide,N-[2-[[[3-(4-chlorophenyl)-2-propen-1-yl]methylamino]methyl]phenyl]-N-(2-hydroxyethyl)-4-methoxy-](http://img.cochemist.com/ccimg/139300/139298-40-1.png)
![Benzenesulfonamide,N-[2-[[[3-(4-chlorophenyl)-2-propen-1-yl]methylamino]methyl]phenyl]-N-(2-hydroxyethyl)-4-methoxy-](http://img.cochemist.com/ccimg/139300/139298-40-1_b.png)




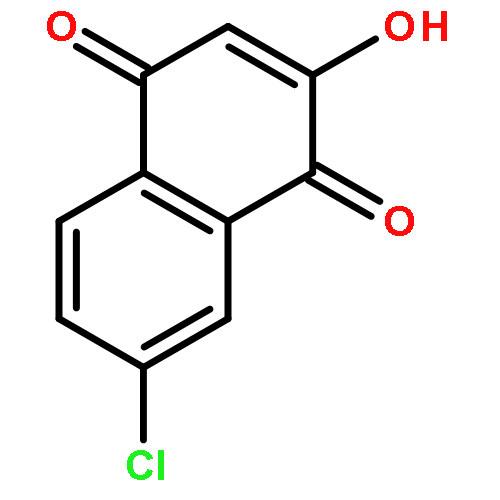
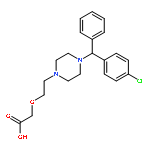
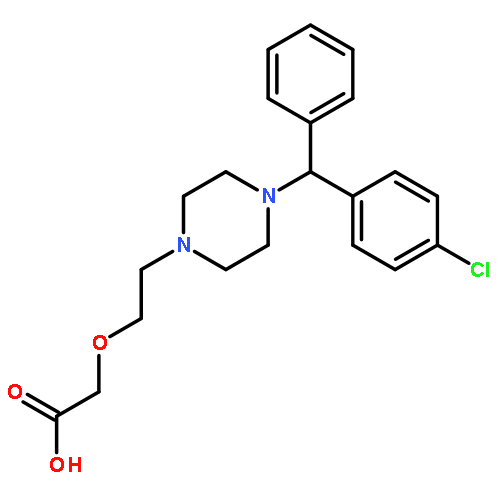


![(2R)-5-hydroxy-2,4-dimethyl-2-[(1E)-2-methylbuta-1,3-dienyl]thiophen-3-one](http://img.cochemist.com/ccimg/82100/82079-32-1.png)
![(2R)-5-hydroxy-2,4-dimethyl-2-[(1E)-2-methylbuta-1,3-dienyl]thiophen-3-one](http://img.cochemist.com/ccimg/82100/82079-32-1_b.png)

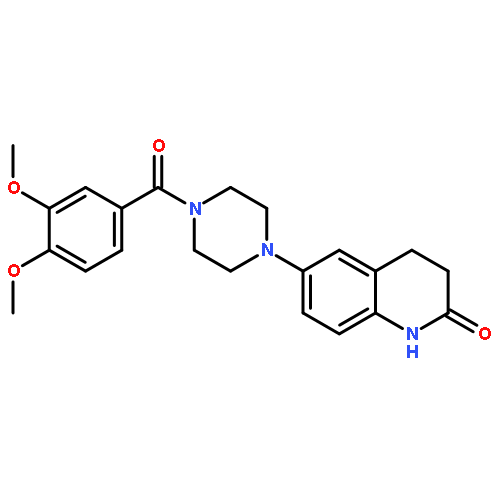

![1-(4-Fluorobenzyl)-N-(1-(4-methoxyphenethyl)piperidin-4-yl)-1H-benzo[d]imidazol-2-amine](http://img.cochemist.com/ccimg/68900/68844-77-9.png)
![1-(4-Fluorobenzyl)-N-(1-(4-methoxyphenethyl)piperidin-4-yl)-1H-benzo[d]imidazol-2-amine](http://img.cochemist.com/ccimg/68900/68844-77-9_b.png)
![N-[4-(4-chlorophenyl)butyl]-N,N-diethylheptan-1-aminium](http://img.cochemist.com/ccimg/68400/68379-02-2.png)
![N-[4-(4-chlorophenyl)butyl]-N,N-diethylheptan-1-aminium](http://img.cochemist.com/ccimg/68400/68379-02-2_b.png)
















![Benzamide,4-(acetylamino)-N-[2-(diethylamino)ethyl]-](http://img.cochemist.com/ccimg/32800/32795-44-1.png)
![Benzamide,4-(acetylamino)-N-[2-(diethylamino)ethyl]-](http://img.cochemist.com/ccimg/32800/32795-44-1_b.png)



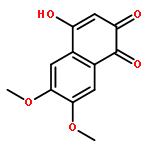
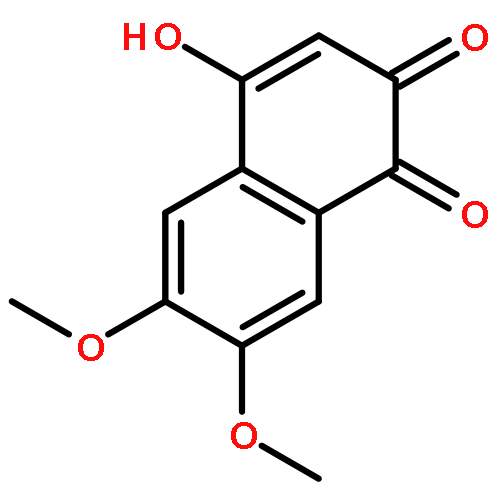


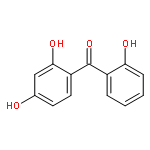
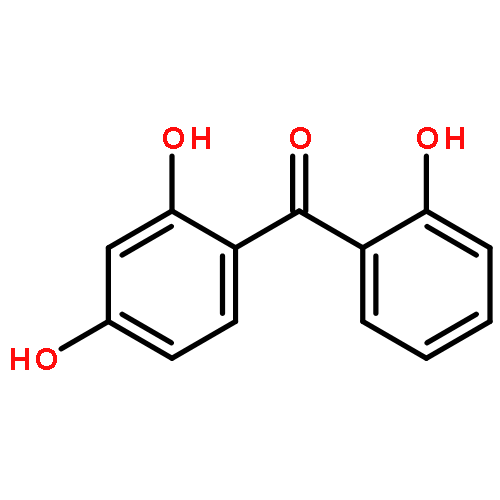

























![9H-Xanthen-9-one, 3,5,6-tris[(1,1-dimethyl-2-propynyl)oxy]-1-hydroxy-](http://img.cochemist.com/ccimg/668000/667914-53-6.png)
![9H-Xanthen-9-one, 3,5,6-tris[(1,1-dimethyl-2-propynyl)oxy]-1-hydroxy-](http://img.cochemist.com/ccimg/668000/667914-53-6_b.png)











![Naphtho[1,2-b]furan-4,5-dione, 2,3-dihydro-2,2-dimethyl-](http://img.cochemist.com/ccimg/52500/52436-88-1.png)
![Naphtho[1,2-b]furan-4,5-dione, 2,3-dihydro-2,2-dimethyl-](http://img.cochemist.com/ccimg/52500/52436-88-1_b.png)

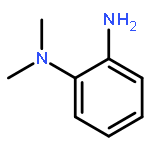
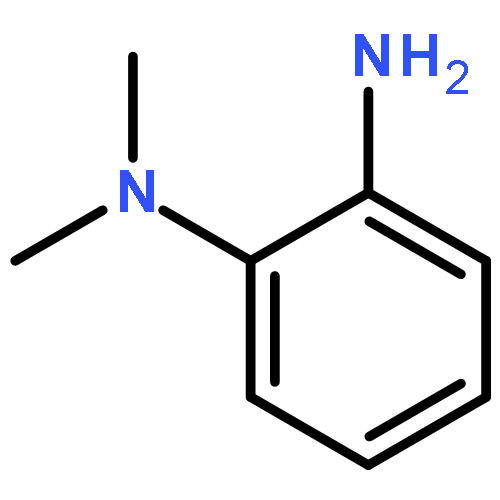
![1,2-Ethanediamine,N1-[(4-methoxyphenyl)methyl]-N2,N2-dimethyl-N1-2-pyridinyl-](http://img.cochemist.com/ccimg/100/91-84-9.png)
![1,2-Ethanediamine,N1-[(4-methoxyphenyl)methyl]-N2,N2-dimethyl-N1-2-pyridinyl-](http://img.cochemist.com/ccimg/100/91-84-9_b.png)


![9H-XANTHEN-9-ONE, 3,5,6-TRIS[(1,1-DIMETHYL-2-PROPENYL)OXY]-1-HYDROXY-](http://img.cochemist.com/ccimg/668000/667914-52-5.png)
![9H-XANTHEN-9-ONE, 3,5,6-TRIS[(1,1-DIMETHYL-2-PROPENYL)OXY]-1-HYDROXY-](http://img.cochemist.com/ccimg/668000/667914-52-5_b.png)




![2,2-Dimethyl-3,4-dihydro-2H-benzo[h]chromene-5,6-dione](/data/chemimg/41800/4707-32-8.png)
![2,2-Dimethyl-3,4-dihydro-2H-benzo[h]chromene-5,6-dione](/data/chemimg/41800/4707-32-8_b.png)

















![2-Methyl-5,6,7,8-tetrahydro-pyrido[4,3-d]pyrimidine](http://img.cochemist.com/ccimg/677000/676994-65-3.png)
![2-Methyl-5,6,7,8-tetrahydro-pyrido[4,3-d]pyrimidine](http://img.cochemist.com/ccimg/677000/676994-65-3_b.png)

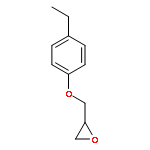
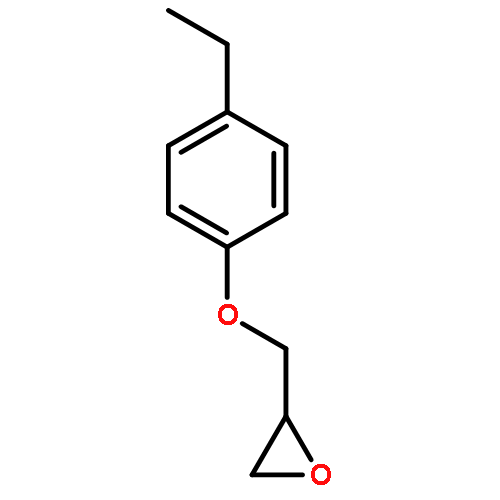
![2-{[2-(4-Ethylphenoxy)ethoxy]methyl}oxirane](http://img.cochemist.com/ccimg/540800/540760-63-2.png)
![2-{[2-(4-Ethylphenoxy)ethoxy]methyl}oxirane](http://img.cochemist.com/ccimg/540800/540760-63-2_b.png)
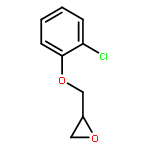
![2-{[2-(4-chlorophenoxy)ethoxy]methyl}oxirane](http://img.cochemist.com/ccimg/540800/540760-62-1.png)
![2-{[2-(4-chlorophenoxy)ethoxy]methyl}oxirane](http://img.cochemist.com/ccimg/540800/540760-62-1_b.png)


![1-[[[[2-[2-[2-[2-Methoxyethoxy]ethoxy]ethoxy]ethoxy]carbonyl]oxy]methyl]-4-[N'-cyano-N''-[6-[4-chlorophenoxy]hexyl]guanidino]pyridinium chloride](http://img.cochemist.com/ccimg/432100/432037-57-5.png)
![1-[[[[2-[2-[2-[2-Methoxyethoxy]ethoxy]ethoxy]ethoxy]carbonyl]oxy]methyl]-4-[N'-cyano-N''-[6-[4-chlorophenoxy]hexyl]guanidino]pyridinium chloride](http://img.cochemist.com/ccimg/432100/432037-57-5_b.png)










![tert-Butyl 2-methyl-7,8-dihydropyrido[4,3-d]pyrimidine-6(5H)-carboxylate](http://img.cochemist.com/ccimg/210600/210538-72-0.png)
![tert-Butyl 2-methyl-7,8-dihydropyrido[4,3-d]pyrimidine-6(5H)-carboxylate](http://img.cochemist.com/ccimg/210600/210538-72-0_b.png)
![2-[6-(4-chlorophenoxy)hexyl]-1-cyano-3-pyridin-4-ylguanidine](http://img.cochemist.com/ccimg/200500/200484-11-3.png)
![2-[6-(4-chlorophenoxy)hexyl]-1-cyano-3-pyridin-4-ylguanidine](http://img.cochemist.com/ccimg/200500/200484-11-3_b.png)










![Pyrido[4,3-d]pyrimidin-2-amine,5,6,7,8-tetrahydro-](http://img.cochemist.com/ccimg/124500/124458-31-7.png)
![Pyrido[4,3-d]pyrimidin-2-amine,5,6,7,8-tetrahydro-](http://img.cochemist.com/ccimg/124500/124458-31-7_b.png)


![Piperazine, 1-[2-(1-methylethyl)phenyl]-](http://img.cochemist.com/ccimg/119700/119695-82-8.png)
![Piperazine, 1-[2-(1-methylethyl)phenyl]-](http://img.cochemist.com/ccimg/119700/119695-82-8_b.png)


![Piperazine,1-[2-(phenylmethoxy)phenyl]-](http://img.cochemist.com/ccimg/96300/96221-84-0.png)
![Piperazine,1-[2-(phenylmethoxy)phenyl]-](http://img.cochemist.com/ccimg/96300/96221-84-0_b.png)
![N-[2-(1-piperazinyl)phenyl]-Acetamide](http://img.cochemist.com/ccimg/91700/91646-29-6.png)
![N-[2-(1-piperazinyl)phenyl]-Acetamide](http://img.cochemist.com/ccimg/91700/91646-29-6_b.png)
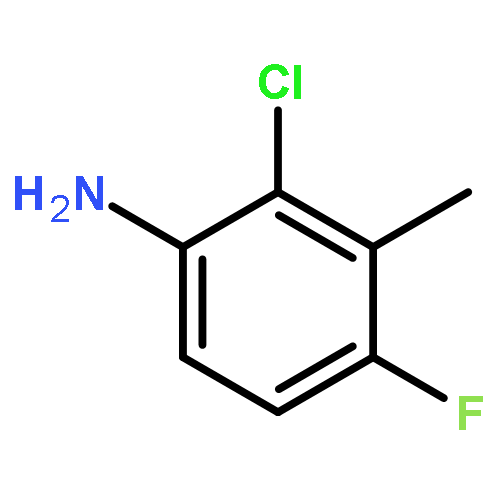
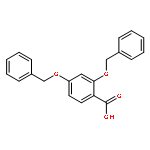
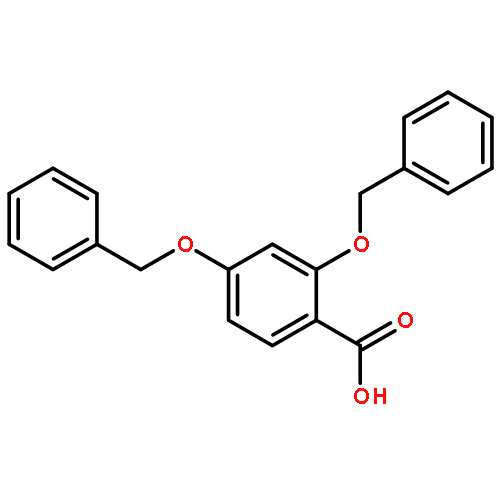
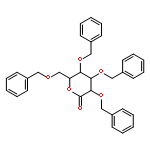





![4-methyl-N-[4-(4-methylbenzenesulfonamido)phenyl]benzene-1-sulfonamide](http://img.cochemist.com/ccimg/41600/41595-29-3.png)
![4-methyl-N-[4-(4-methylbenzenesulfonamido)phenyl]benzene-1-sulfonamide](http://img.cochemist.com/ccimg/41600/41595-29-3_b.png)
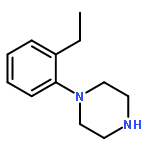
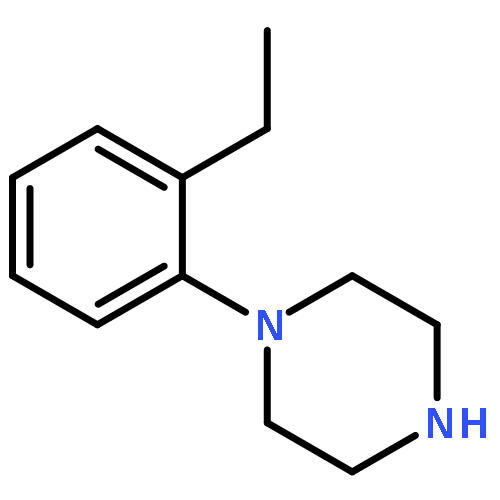
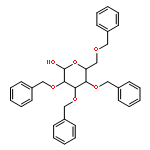
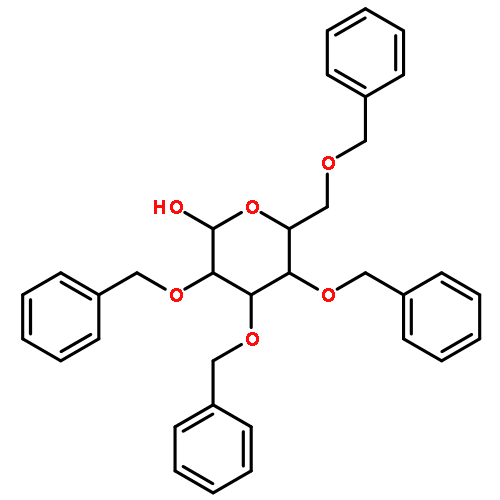




















![Oxirane,2-[(2-naphthalenyloxy)methyl]-](http://img.cochemist.com/ccimg/5300/5234-06-0.png)
![Oxirane,2-[(2-naphthalenyloxy)methyl]-](http://img.cochemist.com/ccimg/5300/5234-06-0_b.png)
![Oxirane,2-[([1,1'-biphenyl]-4-yloxy)methyl]-](http://img.cochemist.com/ccimg/4700/4698-96-8.png)
![Oxirane,2-[([1,1'-biphenyl]-4-yloxy)methyl]-](http://img.cochemist.com/ccimg/4700/4698-96-8_b.png)



![Oxirane,2-[(4-bromophenoxy)methyl]-](http://img.cochemist.com/ccimg/2300/2212-06-8.png)
![Oxirane,2-[(4-bromophenoxy)methyl]-](http://img.cochemist.com/ccimg/2300/2212-06-8_b.png)
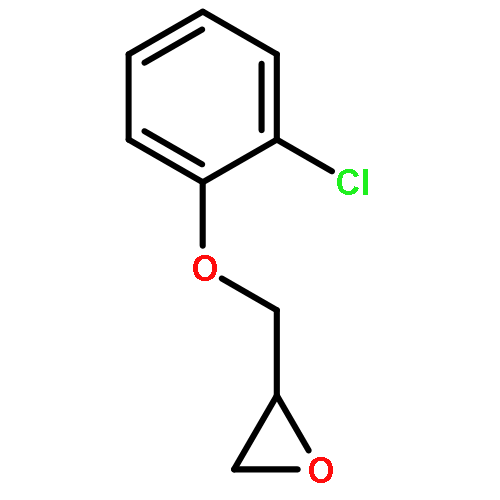
![Oxirane, [(3-methoxyphenoxy)methyl]-](http://img.cochemist.com/ccimg/2300/2210-75-5.png)
![Oxirane, [(3-methoxyphenoxy)methyl]-](http://img.cochemist.com/ccimg/2300/2210-75-5_b.png)
![Oxirane,2-[(3-methylphenoxy)methyl]-](http://img.cochemist.com/ccimg/2200/2186-25-6.png)
![Oxirane,2-[(3-methylphenoxy)methyl]-](http://img.cochemist.com/ccimg/2200/2186-25-6_b.png)
![Oxirane,2-[(4-methylphenoxy)methyl]-](http://img.cochemist.com/ccimg/2200/2186-24-5.png)
![Oxirane,2-[(4-methylphenoxy)methyl]-](http://img.cochemist.com/ccimg/2200/2186-24-5_b.png)