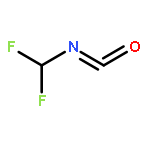
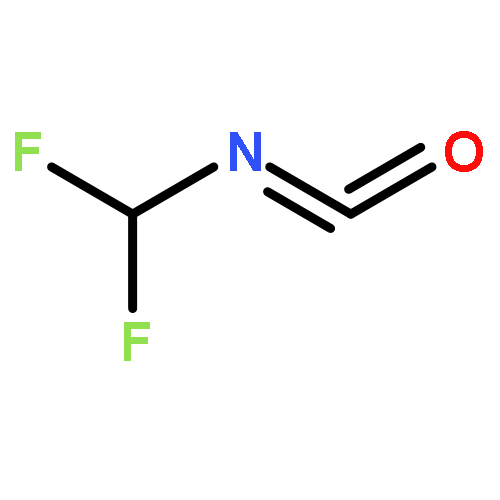
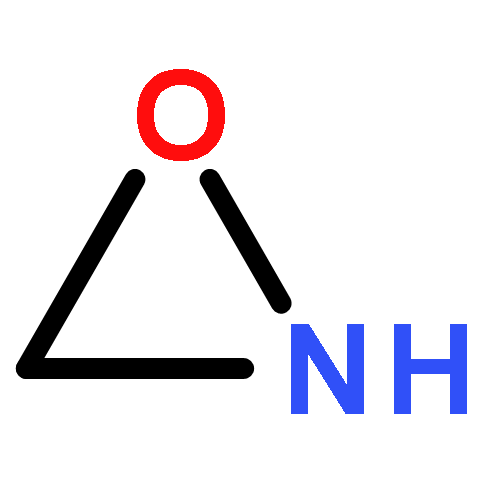
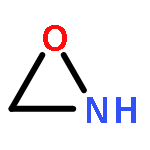
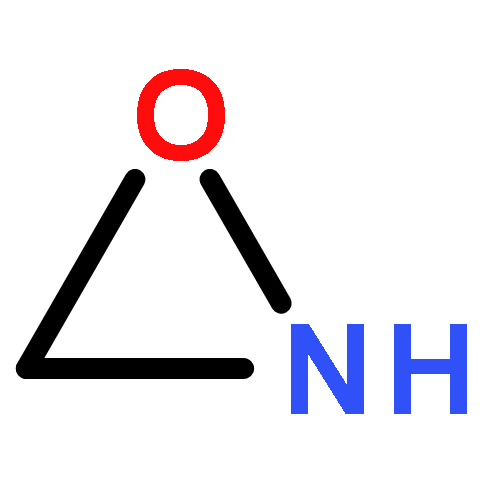
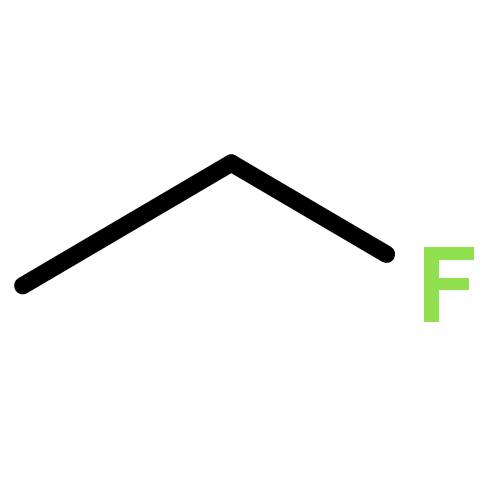
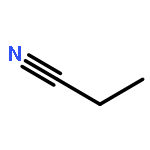
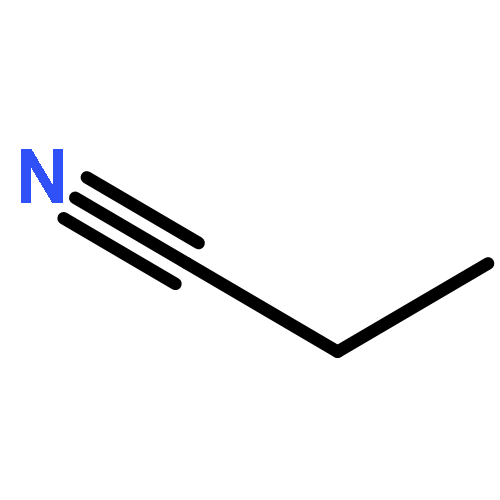
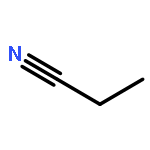
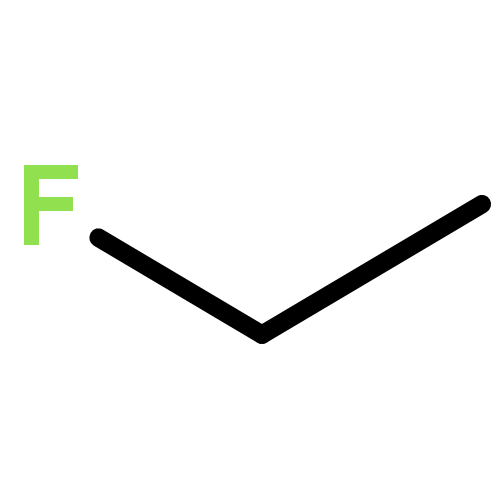
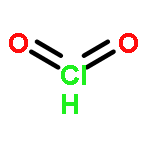
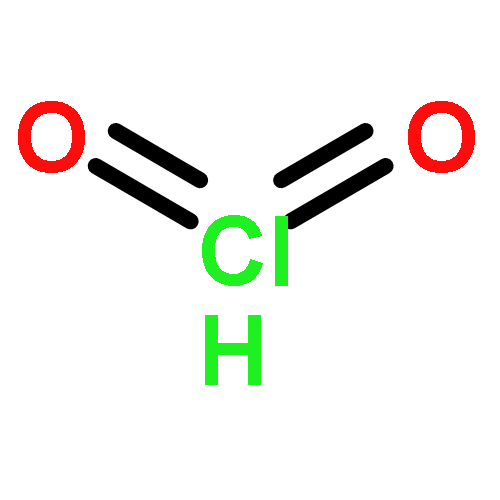
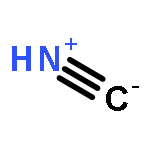
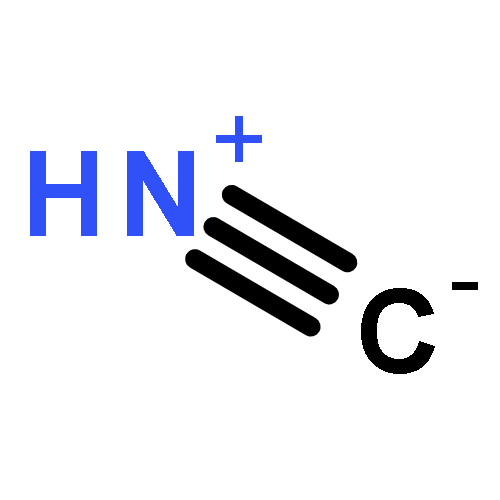
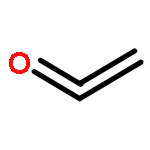
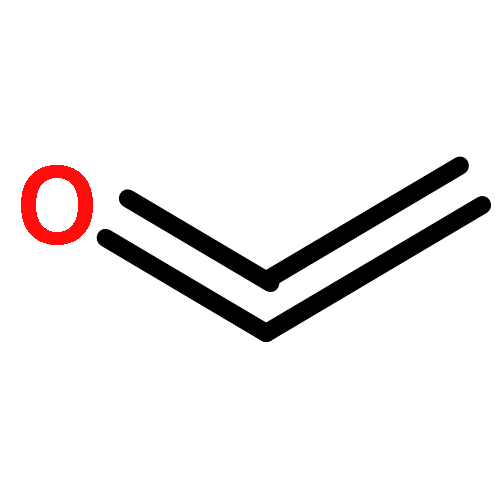
Co-reporter:Xiaoying Hou, Jing Liang, Tong Zhang, Yuhan Li, Shuwei Tang, Hao Sun, Jingping Zhang, and Haiming Xie
The Journal of Physical Chemistry C October 19, 2017 Volume 121(Issue 41) pp:22656-22656
Publication Date(Web):September 26, 2017
DOI:10.1021/acs.jpcc.7b06814
With the aid of first principle calculations, structural characteristics, mechanical stability, and electronic and electrochemical properties of two polymorphs of manganese-based pyrophosphate α- and β-phases of Li2MnP2O7 and their relevant delithiated structures are explored for comparison. Our results indicate that, although these two polymorphs of Li2MnP2O7 belong to the monoclinic space group, considerable differences are discovered in Mn local environment of crystal structures. The cell voltage vs Li/Li+ are 4.68 and 4.16 V for α- and β-phases of the Li2MnP2O7/LiMnP2O7 platform, respectively, comparable to the experimental values (4.45 and 4.00 V) for first voltage plateaus. All of the lithium atoms are practically fully ionized in the α- and β-Li2MnP2O7 and their relative half delithiated states, charge transfer mainly concentrated upon Mn and O, which leads to the oxidization state of Mn from Mn2+ to Mn3+ and then from Mn3+ to Mn4+. The band gaps of delithiated configurations decrease gradually with removing lithium ions, and the conductivity changed from insulator nature to conductor characteristic. By the elastic properties calculations, the Pugh ratios (B/G) are 3.28 and 2.86 for the α- and β-Li2MnP2O7, respectively, indicating their high mechanical stability. However, small B/G values are observed for the relevant delithiated phases. In addition, Young’s modulus (E) and Poisson’s ratio (ν) for α- and β-phases of Li2MnP2O7 and their delithiated configurations are also presented to explore the hardness and bond characteristics.
Co-reporter:Jing Liang, Yuhan Li, Xiaoying Hou, Fengdi Wang, Wanqiao Zhang, Jingping Zhang, Shuwei Tang, Hao Sun
Electrochimica Acta 2017 Volume 251(Volume 251) pp:
Publication Date(Web):10 October 2017
DOI:10.1016/j.electacta.2017.08.123
Li- and Na-transition metal fluorosulfate materials (AFeSO4F, A = Li, Na) are proposed as highly promising novel cathode materials for Li- and Na-ion batteries. In this study, the electrochemical performance and elastic properties of AFe1-xMxSO4F (A = Li, Na; M = Co, Ni, Mg; x = 0, 0.5, 1) are investigated from the view of first-principles calculations. The computational results reveal that the substitutions of Fe by Ni, Co and Mg enhance the intercalation voltage of the fluorosulphate materials. The density of states analysis shows that transition metal-doping AFeSO4F, especially for Ni doping case, could achieve better electronic conductivity in comparison with the pure phase and Mg-doped AFeSO4F. Further bader charges calculations give a confirmation that the Fe and Co in AFe0.5Co0.5SO4F play significant role in the charge transfer during delithiated or desodiated progresses, but the relatively inert character of Ni and Mg is discovered in AFe0.5Ni0.5SO4F and AFe0.5Mg0.5SO4F. LiFeSO4F and NaFeSO4F are found to be ductile from the exploration of elastic constants, whereas their delithiated and desodiated configurations have brittle character. In addition, Young’s Modulus (E), and Poisson's Ratio (ν) for LiFeSO4F, NaFeSO4F and 50% metal-doped AFe1-xMxSO4F (A = Li, Na; M = Co, Ni, Mg) are also presented to explore the hardness, bond characteristic and stability against shear.
Co-reporter:Tong Zhang, Hao Sun, Fengdi Wang, Wanqiao Zhang, Shuwei Tang, Junmei Ma, Hongwei Gong, Jingping Zhang
Applied Surface Science 2017 Volume 425(Volume 425) pp:
Publication Date(Web):15 December 2017
DOI:10.1016/j.apsusc.2017.06.229
•Doping transition metal atoms (Zr, Mo, Ti, Mn, Fe, Co) can change the electronic properties of graphene sheet system.•The Mn-doped graphene sheet exhibits the strongest adsorption effect for the phosgene molecule.•The electronic and optical properties of TM-doped graphene are significantly changed upon phosgene adsorption.•Mn-doped graphene is theoretically predicted to be a potential sensor and scavenger of phosgene.The adsorption of the phosgene molecule on pristine graphene and transition metal doped (TM = Zr, Mo, Ti, Mn, Fe, Co) graphene is investigated using the first principles method based on density functional theory. The nature of interaction between the phosgene molecule and pristine graphene or transition metal doped graphene (TM-doped graphene) is discovered by geometries, adsorption energies, Mulliken charge distribution, density of states analysis and UV spectrum. Computational results show that the interaction between the phosgene molecule and pristine graphene is a weak physisorption. But doping with transition metals results in stronger chemical adsorption. This is due to the formation of a chemical bond between the metal atom and oxygen atom of phosgene, which makes TM-doped graphene really promising material for phosgene removal. TM-doped graphene also exhibits different electronic properties after adsorbing phosgene, compared with pristine graphene. Furthermore, the calculations reveal that UV spectrum of the TM-doped graphene is modified by the phosgene adsorption. Thus, the significant variations in electronic and optical properties of the TM-doped graphene sheet as interacting with the phosgene can be utilized to detect the phosgene.Download high-res image (208KB)Download full-size image
Co-reporter:Tong Zhang;Fengdi Wang;Wanqiao Zhang
Theoretical Chemistry Accounts 2017 Volume 136( Issue 12) pp:134
Publication Date(Web):07 November 2017
DOI:10.1007/s00214-017-2166-z
Adsorption of formaldehyde molecule on the pristine or transition metal doped graphene is theoretically investigated using density functional theory method. The most stable adsorption structures, adsorption energy, Mulliken charge, and the electronic property are analyzed in details. The results show that the interaction between the formaldehyde and pristine graphene is weak physisorption, but the introduction of metal atom in graphene strengthens the adsorption of formaldehyde molecule on the material. As we know, overmuch adsorption makes the adsorbent hard to recover and recycle. It is found in the present work that the recovery of graphene substrate can be achieved by controlling the direction of the external electric field. In addition, electronic property of the substrate has a significant change after formaldehyde molecule adsorption, which makes transition metal doped graphene material a potential sensor for formaldehyde. The effect of humid environment on the interaction between the formaldehyde molecule and Mn-doped graphene sheet is also explored. The calculated results reveal that the adsorption strength of the formaldehyde molecule is weakened when the water molecules exist in the environment. However, this negative effect can be ameliorated by controlling the electric field of the system. These conclusions would provide some beneficial guidance to the related experiments and application in future.
Co-reporter:Fengdi Wang, Tong Zhang, Xiaoying Hou, Wanqiao Zhang, ... Jingping Zhang
International Journal of Hydrogen Energy 2017 Volume 42, Issue 15(Volume 42, Issue 15) pp:
Publication Date(Web):13 April 2017
DOI:10.1016/j.ijhydene.2017.01.121
•H2 adsorption on Li-decorated porous graphene (Li-PG) is investigated.•Li atoms are dispersed uniformly on PG film without clustering.•The H1-H'1 system stores 10.89 wt% H2 at T = 300 K (no external pressure).•Under the same conditions, the H2-H'2 system stores 10.79 wt% H2.Development of novel carbon-based nanoporous materials with high reversible capacity and excellent cycling stability is a hot topic in the field of hydrogen delivery and storage. In this work, first-principles calculations are carried out to discuss the hydrogen storage properties of Li-decorated porous graphene (Li-PG). The binding energies, electronic structures, storage capacities of hydrogen on different sites are investigated in details. The computational results show that with the increase of lithium doping concentration, the electron concentration of donor atoms exceeds the Nc value, and as a consequence, the PG changes from the p-type semiconductor to the n-type degenerate semiconductor. The maximum hydrogen adsorption configurations of H1a-H'1b and H2a-H'2b systems are obtained, and the average binding energy of per H2 molecule is 0.245 eV and 0.263 eV, respectively. Furthermore, ab inito MD simulation results show that the H1-H'1 and H2-H'2 systems can hold up to sixteen and fifteen H2 molecules, which corresponds to a hydrogen storage capacity of 10.89 wt% and 10.79 wt% at T = 300 K (no external pressure), respectively.Download high-res image (324KB)Download full-size image
Co-reporter:Haijie Shi, Fengdi Wang, Wei Chen, Shuwei Tang, Wanqiao Zhang, Wenliang Li, Hao Sun, Jingping Zhang, Rongshun Wang
Journal of Molecular Graphics and Modelling 2015 Volume 59() pp:31-39
Publication Date(Web):June 2015
DOI:10.1016/j.jmgm.2015.03.004
•Homodimer of DeAPb has more stability than other dimers in various conditions.•Electron donating substituents can provide favorable free energies of dimerization.•The dimerization can be favored in weakly polar solvents.•Electron transfers from acceptor to donor owing to hyperconjugative interactions.•UV absorption spectra show evident difference of λmax between monomers and dimers.The heterocyclic urea of deazapterin (DeAPa) and its protomeric conformers (b, c) with different substituents are selected as the building block for a series of dimers in different configurations. The stabilities of all dimers in various conditions have been investigated by density functional theory. Homodimer of b has more stability than other dimers. Topological analyses certify the coexistence of intermolecular with intramolecular H-bonds. Investigations into frequency demonstrate that all H-bonds show an evident red shift in their stretching vibrational frequencies. Electron donating substituents can provide favorable free energies of the dimer. Solvent effect computations suggest that the dimerization can be favored in weakly polar solvents, such as toluene and chloroform. UV–visible spectra exhibit obvious difference of maximum absorption wavelengths between monomers and dimers, thus may have potential applications for identifying intermolecular H-bonds and calculating association constant of DeAP equilibrium systems in experiments.Intermolecular and intramolecular H-bonds coexist in dimers based on DeAP. Electron donating substituents and weakly polar solvents can promote the dimerization. Electron transfers from lone pairs of electron of O/N atoms in acceptor to NH bonds in donor as a result of hyperconjugative interactions. UV–visible spectra exhibit obvious difference of maximum absorption wavelengths between monomers and dimers.
Co-reporter:F.D. Wang, F. Wang, N.N. Zhang, Y.H. Li, S.W. Tang, H. Sun, Y.F. Chang, R.S. Wang
Chemical Physics Letters 2013 Volume 555() pp:212-216
Publication Date(Web):3 January 2013
DOI:10.1016/j.cplett.2012.11.015
Abstract
Based on density-functional theory, we found that a boron-substituted graphene uniformly decorated with Na atoms on double sides can serve as a high-capacity hydrogen storage medium. Otherwise, the semimetallic boron-substituted graphene change into conductor after the adsorption of Na atoms. While the adsorbed Na atoms forming a (2 × 2) pattern can capture nine H2 molecules corresponding to a storage capacity of 11.7 wt.% at T = 300 K and without external pressure.
Co-reporter:Yunju Zhang, Jingyu Sun, Kai Chao, Fang Wang, ShuWei Tang, Xiumei Pan, Jingping Zhang, Hao Sun, Rongshun Wang
Computational and Theoretical Chemistry 2012 Volume 981() pp:7-13
Publication Date(Web):1 February 2012
DOI:10.1016/j.comptc.2011.11.010
The complex potential energy surfaces for the reaction of atomic radical F with CH2CHCH2Cl (3-chloropropene) are explored at the CCSD(T)/cc-pVTZ//MP2(full)/6-311++G(d,p) level. There are various possible reaction pathways including the addition–elimination and H-abstraction reaction. Among them, the most feasible pathway should be to produce P1 (CH2CHCH2F + Cl), which is in good agreement with the experiment. Among the H-abstraction reactions, the most competitive pathway is the atomic radical F abstracting hydrogen atom from allylic group. Because all of the transition states and intermediates involved in the title reaction lie below the reactants, the F + CH2CHCH2Cl reaction is expected to be rapid. The present results could lead us to deeply understand the mechanism of the title reaction and may provide some useful information for future experimental investigation of the title reaction.Graphical abstractHighlights► The mechanism of the title reaction has been carried out at MP2 and CCSD(T) levels. ► Two kinds of hydrogen abstraction and addition/elimination channels were considered. ► The results are in good agreement with available experimental value.
Co-reporter:Fang Wang, Hao Sun, Jingyu Sun, Xiujuan Jia, Yunju Zhang, Yizhen Tang, Xiumei Pan, Zhongmin Su, Lizhu Hao and Rongshun Wang
The Journal of Physical Chemistry A 2010 Volume 114(Issue 10) pp:3516-3522
Publication Date(Web):February 23, 2010
DOI:10.1021/jp910754b
Both singlet and triplet potential energy surfaces for the reaction of ground-state formaldehyde (CH2O) and ozone (O3) are theoretically investigated at the BMC-CCSD//BHandHLYP/6-311+G(d,p) level. Various possible isomerization and dissociation pathways are probed. Hydrogen abstraction, oxygen abstraction, and C-addition/elimination are found on both the singlet and the triplet surfaces. The major products for the total reaction are HCO and HOOO, which are generated via hydrogen abstraction. The transition state theory (TST) and multichannel RRKM calculations have been carried out for the total and individual rate constants for determinant channels over a wide range of temperatures and pressures.










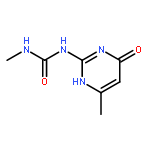
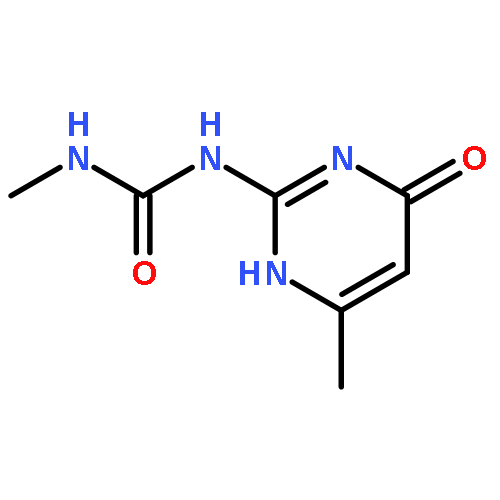
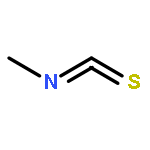
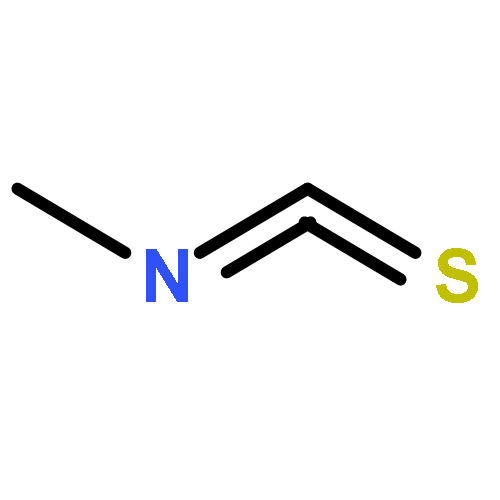
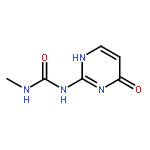
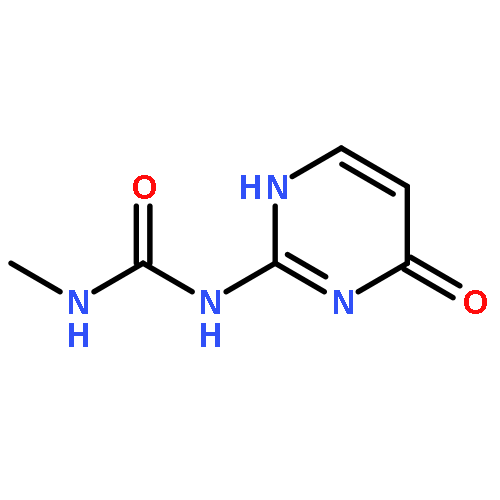



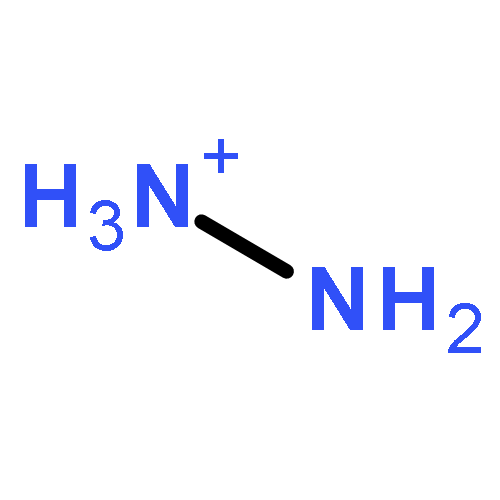


![Benzo[1,2-b:4,5-b']dipyrrole-4,8(1H,5H)-dione](http://img.cochemist.com/ccimg/176600/176507-55-4.png)
![Benzo[1,2-b:4,5-b']dipyrrole-4,8(1H,5H)-dione](http://img.cochemist.com/ccimg/176600/176507-55-4_b.png)
![1H-Pyrrolo[2,3-f]indolizine-4,9-dione](http://img.cochemist.com/ccimg/176600/176507-54-3.png)
![1H-Pyrrolo[2,3-f]indolizine-4,9-dione](http://img.cochemist.com/ccimg/176600/176507-54-3_b.png)




![2-Thiophenesulfonamide, 5-bromo-N-[(dimethylamino)methylene]-](/data/chemimg/132700/104438-09-7.png)
![2-Thiophenesulfonamide, 5-bromo-N-[(dimethylamino)methylene]-](/data/chemimg/132700/104438-09-7_b.png)


![5H,10H-Diimidazo[1,2-a:1',5'-d]pyrazine-5,10-dione](http://img.cochemist.com/ccimg/99600/99560-60-8.png)
![5H,10H-Diimidazo[1,2-a:1',5'-d]pyrazine-5,10-dione](http://img.cochemist.com/ccimg/99600/99560-60-8_b.png)






![DIIMIDAZO[1,3-B:1',3'-E]PYRAZINE-5,10-DIONE](http://img.cochemist.com/ccimg/79800/79711-73-2.png)
![DIIMIDAZO[1,3-B:1',3'-E]PYRAZINE-5,10-DIONE](http://img.cochemist.com/ccimg/79800/79711-73-2_b.png)
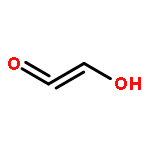




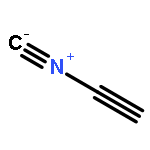
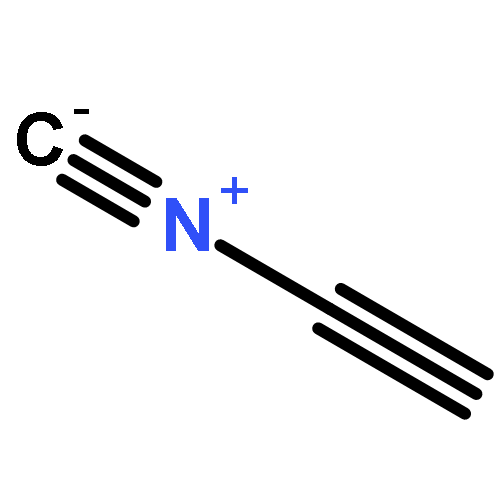

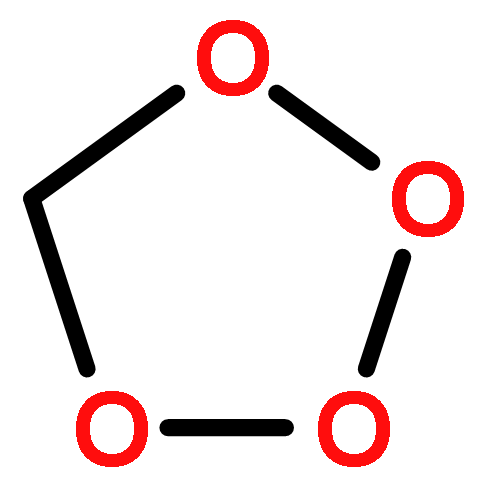

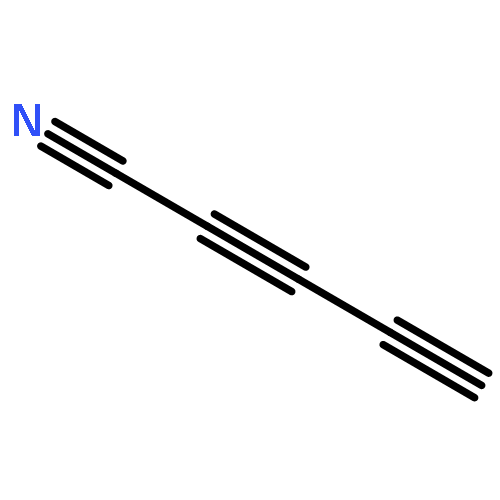



![Arsonous dichloride, [(1E)-2-chloroethenyl]-](http://img.cochemist.com/ccimg/50400/50361-05-2.png)
![Arsonous dichloride, [(1E)-2-chloroethenyl]-](http://img.cochemist.com/ccimg/50400/50361-05-2_b.png)


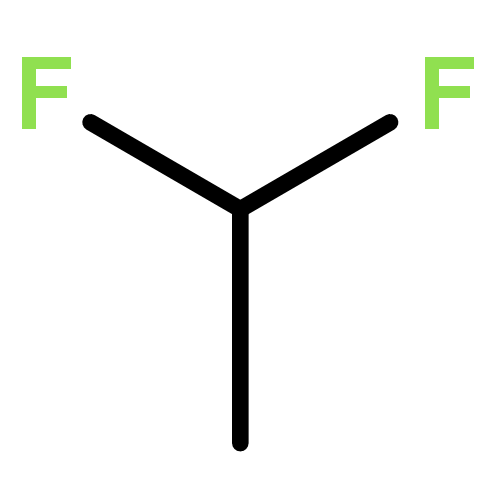
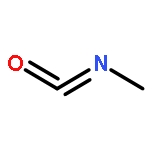

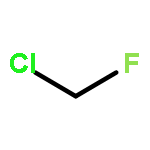
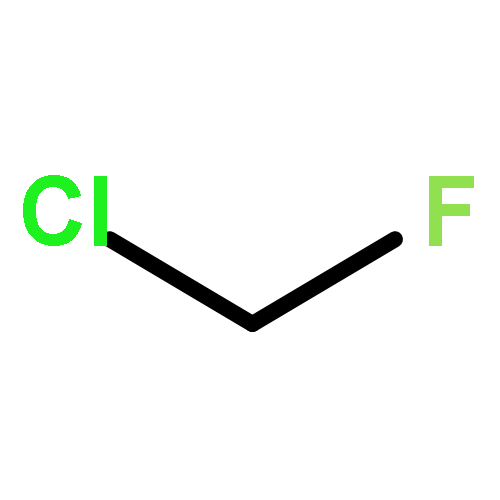
![Benzenesulfonamide, 4-chloro-N-[(dimethylamino)methylene]-](http://img.cochemist.com/ccimg/29700/29619-30-5.png)
![Benzenesulfonamide, 4-chloro-N-[(dimethylamino)methylene]-](http://img.cochemist.com/ccimg/29700/29619-30-5_b.png)
![Benzenesulfonamide, N-[(diethylamino)methylene]-4-methyl-](http://img.cochemist.com/ccimg/29400/29397-20-4.png)
![Benzenesulfonamide, N-[(diethylamino)methylene]-4-methyl-](http://img.cochemist.com/ccimg/29400/29397-20-4_b.png)
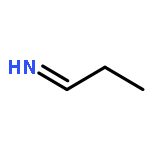
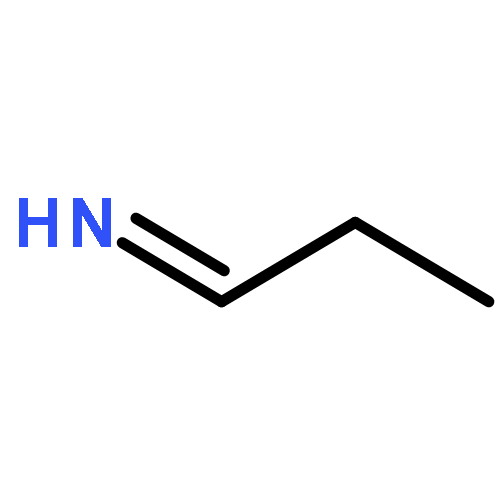


![N-[(e)-(dimethylamino)methylidene]-4-methylbenzenesulfonamide](http://img.cochemist.com/ccimg/25800/25770-53-0.png)
![N-[(e)-(dimethylamino)methylidene]-4-methylbenzenesulfonamide](http://img.cochemist.com/ccimg/25800/25770-53-0_b.png)

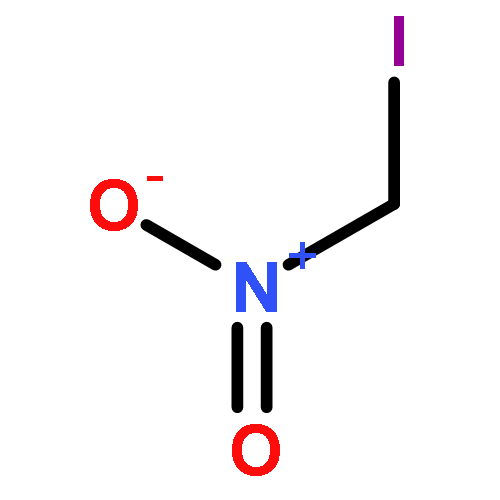












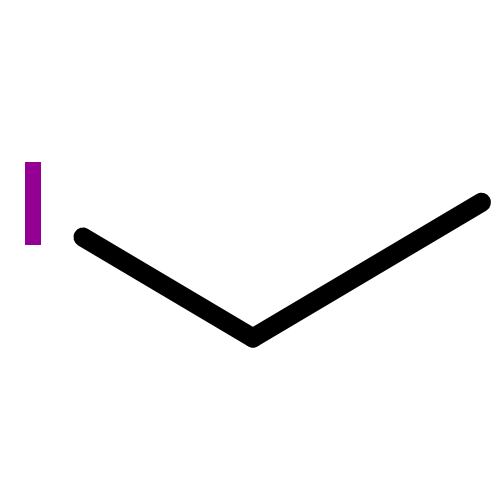











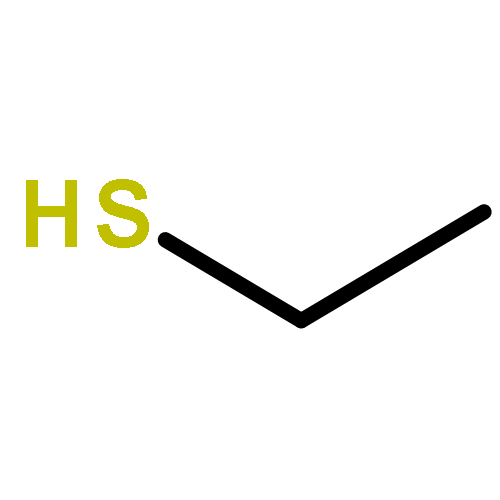
![Benzenesulfonamide, N-[(dimethylamino)methylene]-](http://img.cochemist.com/ccimg/13800/13707-43-2.png)
![Benzenesulfonamide, N-[(dimethylamino)methylene]-](http://img.cochemist.com/ccimg/13800/13707-43-2_b.png)






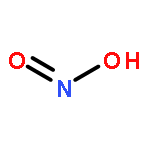
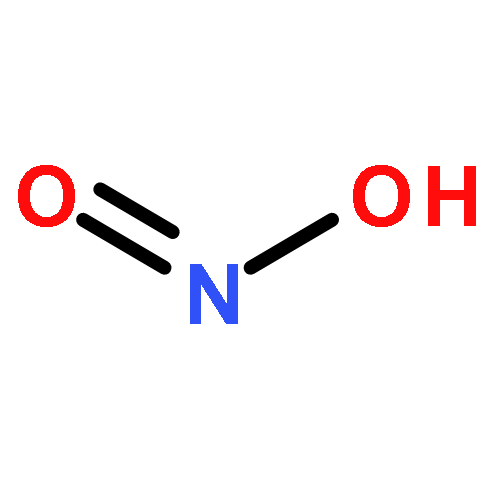














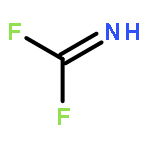
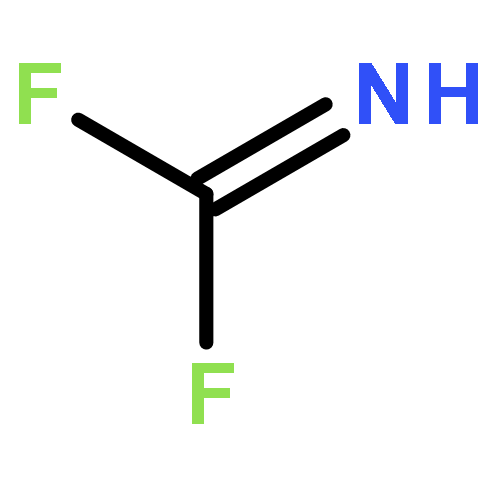

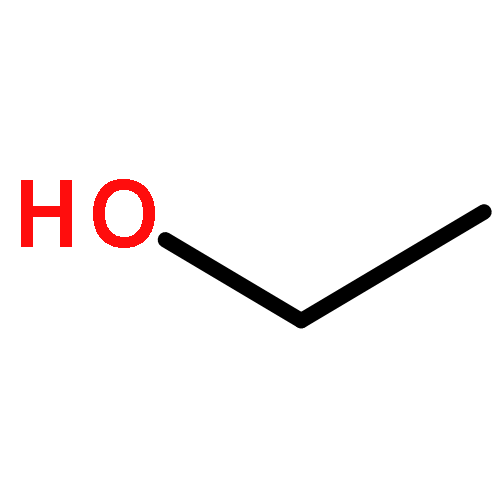



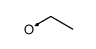


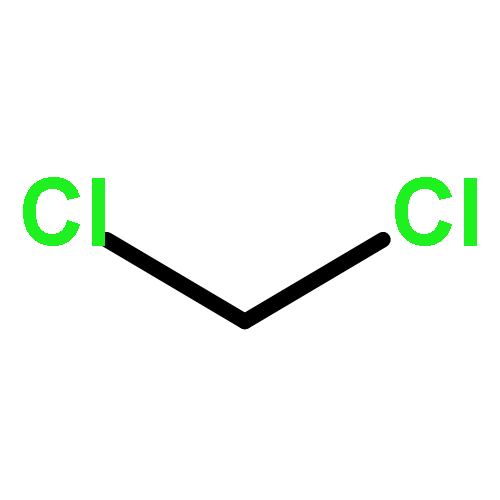




![5H,10H-Dipyrrolo[1,2-a:1',2'-d]pyrazine-5,10-dione](http://img.cochemist.com/ccimg/500/484-73-1.png)
![5H,10H-Dipyrrolo[1,2-a:1',2'-d]pyrazine-5,10-dione](http://img.cochemist.com/ccimg/500/484-73-1_b.png)