Co-reporter:Masayuki Iwasaki, Yasuhiro Araki, and Yasushi Nishihara
The Journal of Organic Chemistry June 16, 2017 Volume 82(Issue 12) pp:6242-6242
Publication Date(Web):May 18, 2017
DOI:10.1021/acs.joc.7b00848
A palladium-catalyzed benzannulation with o-bromobenzyl alcohols enabled the facile construction of phenanthrene skeletons via the sequential multiple carbon–carbon bond formations. A variety of multisubstituted phenanthrenes were synthesized by the reaction of (Z)-β-halostyrenes with o-bromobenzyl alcohols as well as by the three-component coupling of alkynes, aryl bromides, and o-bromobenzyl alcohols. The electron-deficient phosphine ligand played an important role to control the sequential oxidative addition of two different organic halides employed, which realized the selective formation of the desired phenanthrenes in good yields. This synthetic protocol was also applicable to the synthesis of the highly fused polycyclic aromatic hydrocarbons such as tetraphenes.
Co-reporter:Keita Hyodo, Ryota Toyama, Hiroki Mori, and Yasushi Nishihara
ACS Omega January 2017? Volume 2(Issue 1) pp:308-308
Publication Date(Web):January 30, 2017
DOI:10.1021/acsomega.7b00015
Efficient synthesis and characterization of several piceno[4,3-b:9,10-b′]dithiophene (PiDT) derivatives by Negishi coupling, epoxidation, and Lewis acid-catalyzed cycloaromatization sequences and their potential utility in organic field-effect transistors (OFETs) have been reported. PiDT derivatives, with extended π-electron systems, showed high air stability due to their deep highest occupied molecular orbital energy levels (around −5.6 eV). OFET devices based on 2,11-dioctylated PiDT (C8-PiDT) showed excellent hole mobility, as high as 2.36 cm2 V–1 s–1. Their structure–property relationships were investigated by X-ray diffraction and atomic force microscopy.Topics: Crystal structure; Electric transport processes and properties; Electronic structure; Microscopy; Microstructure; Molecular structure; Molecular structure-property relationship; Physical and chemical processes; Redox reaction; Redox reaction; Self-assembled monolayers; Solid state electrochemistry; Thin films; Thin films;
Co-reporter:Masayuki Iwasaki, Natsumi Miki, Yuta Tsuchiya, Kiyohiko Nakajima, and Yasushi Nishihara
Organic Letters 2017 Volume 19(Issue 5) pp:
Publication Date(Web):February 17, 2017
DOI:10.1021/acs.orglett.7b00116
Nickel-catalyzed direct selenation of benzamides bearing an 8-quinolyl auxiliary with elemental selenium provides benzoisoselenazolones in good yield via carbon–selenium and nitrogen–selenium bond formation under aerobic conditions. In addition to aryl C–H bonds, the method can also be applied to alkenyl C–H bonds, constructing an isoselenazolone skeleton. Simple mechanistic analysis shows that the reaction proceeds through a rate-determining C–H bond cleavage. The obtained benzoisoselenazolones are transformed into various organoselenium compounds and utilized as the catalyst for bromolactonization of alkenoic acids.
Co-reporter:Masayuki Iwasaki, Nikola Topolovčan, Hao Hu, Yugo Nishimura, Glwadys Gagnot, Rungsaeng Na nakorn, Ramida Yuvacharaskul, Kiyohiko Nakajima, and Yasushi Nishihara
Organic Letters 2016 Volume 18(Issue 7) pp:1642-1645
Publication Date(Web):March 21, 2016
DOI:10.1021/acs.orglett.6b00503
Palladium-catalyzed carbothiolation of terminal alkynes with azolyl sulfides affords various 2-(azolyl)alkenyl sulfides with perfect regio- and stereoselectivities. The present addition reaction proceeded through a direct cleavage of carbon–sulfur bonds in azolyl sulfides. The resulting adducts that are useful intermediates in organic synthesis are further transformed to multisubstituted olefins containing azolyl moieties.
Co-reporter:Hiroki Mori, Hikaru Nonobe and Yasushi Nishihara
Polymer Chemistry 2016 vol. 7(Issue 8) pp:1549-1558
Publication Date(Web):04 Jan 2016
DOI:10.1039/C5PY01878A
New PDT-based polymers were combined with two types of benzothiadiazole (BT) derivatives to improve their crystallinity and solar cell performance. These polymers show several advantages, including strong intermolecular interactions, deep HOMO energy levels, and a dense packing structure with a short π–π stacking distance of 3.5–3.6 Å. Combinations of PDT and BT units in polymers formed highly crystalline thin films with a long-range order, even in films blended with a fullerene derivative. This suggests that the introduction of optimal acceptor units may increase the regularity of the polymers, leading to effective π–π overlaps between the polymer backbones. However, although the present polymers also formed an appropriate phase separation structure in blended films, in fabricated solar cell devices they yielded low power conversion efficiencies (PCEs) not exceeding 3.8%. GI-WAXS analysis revealed that both polymers were present in a predominantly edge-on orientation. This unsuitable orientation for PSCs prevented efficient carrier transport and reduced charge collection efficiency, resulting in low Jsc and thus low PCE. On the other hand, these polymers also formed highly oriented edge-on structures on n+-Si/SiO2 substrates, which are suitable for high-performance field-effect transistors (FETs), and a fabricated FET device showed a hole mobility as high as 0.18 cm2 V−1 s−1.
Co-reporter:Takuya Ishitsuka, Yasuhiro Okuda, Robert K. Szilagyi, Seiji Mori and Yasushi Nishihara
Dalton Transactions 2016 vol. 45(Issue 18) pp:7786-7793
Publication Date(Web):24 Mar 2016
DOI:10.1039/C6DT00341A
Potential energy surface mapping was completed for the entire catalytic cycle of palladium-catalysed cyanoesterification onto norbornene (NBE) using density functional theory calculations. We found that after the oxidative addition step of the reagent methyl cyanoformate, the reaction proceeds through an insertion of olefin into a PdII–COOMe bond first. Subsequently, reductive elimination occurs by transferring the cyanide group from the Pd center to NBE. This rearrangement is triggered by the rotation of the ester group into a π-interaction with the PdII centre. The regioselectivity of olefin insertion is controlled by ionic and covalent interactions in the precursor π-complex formation step. Importantly, all of the intermediates and transition states along the exo pathway were found to be more stable than the corresponding structures of the endo pathway without any sign of crossing over between the two surfaces via isomerization. The rate-determining step is the reductive elimination despite the fact that the corresponding activation barrier is reduced by conformational changes via the rotation of a MeOOC–C(NBE) bond.
Co-reporter:Masayuki Iwasaki
The Chemical Record 2016 Volume 16( Issue 4) pp:2031-2045
Publication Date(Web):
DOI:10.1002/tcr.201600017
Co-reporter:Shuhei Nishinaga, Hiroki Mori, and Yasushi Nishihara
Macromolecules 2015 Volume 48(Issue 9) pp:2875-2885
Publication Date(Web):April 29, 2015
DOI:10.1021/acs.macromol.5b00622
The synthesis, characterization, and solar cell application of newly developed semiconducting polymers containing phenanthro[1,2-b:8,7-b′]dithiophene (PDT) combined with a bis(thienyl)isoindigo (IID) unit are described. The polymers with longer alkyl chains are sufficiently soluble to be compatible with the processes required to manufacture solar cells. In conventional solar cell devices, polymers with all branched alkyl chains tend to form a higher proportion of a well-ordered face-on crystallite in the π-stack direction than those with both linear and branched alkyl chains, which significantly improves the fill factor (FF), resulting in higher power conversion efficiency (PCE). In terms of optimizing the alkyl chain lengths, the installation of longer alkyl side chains on the polymer backbone leads to low molecular weight polymer, which may promote a large phase separation. As a result, the polymers 12OD and BOBO, bearing shorter alkyl groups, performed better, and a BOBO polymer-based solar cell (PSC) showed the best PCE value up to 3.83%. In the inverted PSCs, the polymers with all branched alkyl chains have a higher face-on ratio than those with both linear and branched alkyl chains. Because of their improved Jsc, inverted PSCs with BOBO/PC71BM gave the best performance, with a PCE up to 5.28%. Although an obvious dependence of photovoltaic properties on molecular order was observed in conventional solar cell devices, no trend was observed in inverted cells, possibly attributable to their amorphous nature, which arises from the axisymmetrical structure of PDT, leading to less effective π–π overlap and low crystallinity.
Co-reporter:Hiroki Mori;Masato Suetsugu;Shuhei Nishinaga;Ning-hui Chang;Hikaru Nonobe;Yasuhiro Okuda
Journal of Polymer Science Part A: Polymer Chemistry 2015 Volume 53( Issue 5) pp:709-718
Publication Date(Web):
DOI:10.1002/pola.27500
ABSTRACT
Synthesis, characterization, and polymer solar cell and transistor application of a series of phenanthro[1,2-b:8,7-b′]dithiophene-based donor–acceptor (D–A)-type semiconducting polymers combined with a diketopyrrolopyrrole unit are reported. The present polymers showed some unique features such as strong aggregation behavior, high thermal stability, and short π–π stacking distance (3.5–3.6 Å), which are suitable for high performance organic materials. In addition, they have a significantly extended absorption up to 1000 nm with a band gap of ca. 1.2 eV. However, such strong intermolecular interaction reduced their solubility and molecular weights, which resulted in low crystalline nature and moderate field-effect mobility of 0.01 cm2 V−1 s−1. Furthermore, such strong aggregation behavior led to the large-scale phase separation in the blend films, which may prevent the effective photocurrent generation, limiting Jsc and power conversion efficiency of 2.0%. © 2015 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2015, 53, 709–718
Co-reporter:Masayuki Iwasaki, Yasuhiro Araki, Shohei Iino, and Yasushi Nishihara
The Journal of Organic Chemistry 2015 Volume 80(Issue 18) pp:9247-9263
Publication Date(Web):August 31, 2015
DOI:10.1021/acs.joc.5b01693
o-Bromobenzyl alcohol has been developed as a novel annulating reagent, bearing both nucleophilic and electrophilic substituents, for the facile synthesis of polycyclic aromatic hydrocarbons. A palladium/electron-deficient phosphine catalyst efficiently coupled o-iodobiphenyls or (Z)-β-halostyrenes with o-bromobenzyl alcohols to afford triphenylenes and phenanthrenes, respectively. The present cascade reaction proceeded through deacetonative cross-coupling and sequential intramolecular cyclization. An array of experimental data suggest that the reaction mechanism involves the equilibrium of 1,4-palladium migration.
Co-reporter:Keita Hyodo, Masato Suetsugu, and Yasushi Nishihara
Organic Letters 2014 Volume 16(Issue 2) pp:440-443
Publication Date(Web):December 23, 2013
DOI:10.1021/ol403326z
Platinum-catalyzed diborylation of phenylethynyl MIDA boronate with Bpin–Bpin proceeds to yield 1,1,2-triboryl-2-phenylethene with two different classes of the boron functionalities. Sequentially, the obtained 1,1,2-triboryl-2-phenylethene are subjected to Suzuki–Miyaura coupling to introduce a series of aryl groups chemoselectively to afford 1,1-boryl-2,2-diarylethenes.
Co-reporter:Masayuki Iwasaki, Yuta Tsuchiya, Kiyohiko Nakajima, and Yasushi Nishihara
Organic Letters 2014 Volume 16(Issue 18) pp:4920-4923
Publication Date(Web):September 10, 2014
DOI:10.1021/ol502439m
A direct selenation of inert C–H bonds of benzamide derivatives and their related compounds with diselenides has been achieved with the palladium catalyst. The reaction was compatible with a variety of functional groups, including a bromo group. Primitive mechanistic insights revealed that the reaction proceeded through a C–H bond cleavage and the sequential oxidative addition of diselenides. The present synthetic method can be applied to the facile synthesis of selenoxanthone which can be regarded as promising heterocyclic materials.
Co-reporter:Jing Li, Yasuhiro Okuda, Jiaji Zhao, Seiji Mori, and Yasushi Nishihara
Organic Letters 2014 Volume 16(Issue 19) pp:5220-5223
Publication Date(Web):September 24, 2014
DOI:10.1021/ol5026519
An efficient method for the direct conversion of cyano-substituted iminoisobenzofurans into their corresponding alkyl 2-cyanobenzoates has been developed. This transformation proceeds via cleavage of C–C, C–O, and C–N bonds in starting iminoisobenzofurans. DFT study revealed that intermediate α-iminonitriles are produced in situ via C–C bond formation between 2-iminium benzoates and a cyanide ion. Generation of isocyanide as the byproduct in a more thermodynamic manner in DFT calculations also supports the experimental results.
Co-reporter:Yasuhiro Okuda, Robert K. Szilagyi, Seiji Mori and Yasushi Nishihara
Dalton Transactions 2014 vol. 43(Issue 25) pp:9537-9548
Publication Date(Web):11 Apr 2014
DOI:10.1039/C4DT00839A
A computational investigation has been carried out to elucidate the origin of the exclusive exo-selectivity in the Pd-catalyzed cyanoesterification of strained cyclic olefins, norbornene and norbornadiene. A hybrid density functional was selected for the level of theory with a triple-ζ quality basis set, which was proposed in an earlier study to provide an experimentally sound ground state electronic structure description for palladium(II) and palladium(IV) complexes from multi-edge X-ray absorption spectroscopic measurements. Given that the product of oxidative addition can be isolated, we focused on the olefin coordination as the earliest possible origin of exo-selectivity. The calculated geometric structure for the trans-Pd(CN)(COOR)(PPh3)2 complex at the BHandHLYP/def2TZVP/PCM(toluene) level is in an excellent agreement with its experimental structure from crystallographic measurements. Upon dissociation of one of its phosphane ligands, the coordinatively unsaturated trans-isomer is only 17 kJ mol−1 away from the isomerization transition state, leading to the 14-electron cis-isomers that are 17 to 37 kJ mol−1 lower in energy than the trans-isomers. Regardless of the initial complex for olefin coordination, the exo-isomer for the norbornene complex is at least 8 kJ mol−1 lower than the corresponding endo-isomer. The origin of this considerable difference in Gibbs free energy can be attributed to the remarkably different steric and agostic hydrogen interactions between the methylene and the ethylene bridges of the norbornene and the adjacent cis-ligands at the PdII center.
Co-reporter:Keita Hyodo, Hikaru Nonobe, Shuhei Nishinaga, Yasushi Nishihara
Tetrahedron Letters 2014 Volume 55(Issue 29) pp:4002-4005
Publication Date(Web):16 July 2014
DOI:10.1016/j.tetlet.2014.05.035
Phenanthro[1,2-b:8,7-b′]dithiophene (PDT) was prepared via Suzuki–Miyaura or Negishi cross-coupling of a 2-thienylboron or -zinc compound with 1,4-dibromobenzene, followed by Lewis acid-catalyzed regioselective cycloaromatization of the epoxide. A series of 2,9-dialkylated phenanthro[1,2-b:8,7-b′]dithiophene (PDT) derivatives could also be synthesized in good yields by Suzuki–Miyaura coupling of the brominated PDT with alkylboranes by introducing linear alkyl substituents.
Co-reporter:Masayuki Iwasaki, Wataru Kaneshika, Yuta Tsuchiya, Kiyohiko Nakajima, and Yasushi Nishihara
The Journal of Organic Chemistry 2014 Volume 79(Issue 23) pp:11330-11338
Publication Date(Web):November 15, 2014
DOI:10.1021/jo502274t
A palladium-catalyzed and picolinamide-directed C–H thiolation of naphthylamine derivatives with diaryl disulfides has been developed to provide a convenient route to 8-sulfenyl-1-naphthylamines. The reaction proceeds via a 5-membered palladacycle intermediates to afford the peri-thiolated products exclusively, in contrast to the conventional ortho-functionalization. Moreover, the related direct selenation was also achieved with diaryl diselenides, giving the corresponding selenated products with perfect site-selectivity.
Co-reporter:Jiao Jiao, Keita Hyodo, Hao Hu, Kiyohiko Nakajima, and Yasushi Nishihara
The Journal of Organic Chemistry 2014 Volume 79(Issue 1) pp:285-295
Publication Date(Web):December 9, 2013
DOI:10.1021/jo4024057
The synthesis of a series of gem- and vic-diborylated vinylsilanes was accomplished via highly selective transition-metal-catalyzed syn-dimetalation to the alkynylmetal species. This protocol served as a general synthetic method toward regio- and stereodefined multisubstituted olefins. The key steps are the diastereoselective Suzuki–Miyaura cross-coupling reactions of gem- and vic-diborylated vinylsilanes, in which the two boron groups showed discrete reactivities to afford diverse precursors of multisubstituted olefins.
Co-reporter:Dr. Masayuki Iwasaki;Miki Iyanaga;Yuta Tsuchiya;Yugo Nishimura;Wenjuan Li;Dr. Zhiping Li;Dr. Yasushi Nishihara
Chemistry - A European Journal 2014 Volume 20( Issue 9) pp:2459-2462
Publication Date(Web):
DOI:10.1002/chem.201304717
Abstract
A catalytic variant of the direct thiolation of arenes, bearing directing groups, with disulfides or thiols has been developed under palladium and copper co-catalysis. Both sulfenyl moieties of the disulfide could be incorporated into the thiolated products, therefore, the reactions reached completion with only half an equivalent of disulfide, with respect to the starting arene. Experimental evidence suggested that the reaction proceeds through a PdII/PdIV mechanism.
Co-reporter:Yasuhiro Okuda, Yuya Ishiguro, Seiji Mori, Kiyohiko Nakajima, and Yasushi Nishihara
Organometallics 2014 Volume 33(Issue 7) pp:1878-1889
Publication Date(Web):March 27, 2014
DOI:10.1021/om5002588
A series of cis-alkynyl(silyl)platinum(II) complexes was prepared via the chemoselective C(sp)–Si bond cleavage of alkynylsilanes by a platinum(0) complex ligated with the P–N hemilabile bidentate ligand. The coordination of the triple bond to the platinum center triggers selective C(sp)–Si bond cleavage. Hammett plots of the 31P{1H} NMR spectroscopic properties (δ and J values) reflect an electronic effect on platinum(II) complexes; trans substituents of arylethynyl groups are influenced, but cis-positioned silyl groups are not affected, as evidenced by 29Si{1H} NMR. In comparison, Hammett plots show that C(sp)–Si bond cleavage rates are accelerated by electron-rich alkynylsilanes, which is opposite to the ordinary oxidative addition of aryl halides to transition metals often observed in catalytic cross-coupling reactions. A DFT calculation reveals that intermediates and transition states are stabilized by electron-rich alkynylsilanes and that the five-membered hemilabile P–N ligand is essential, in which a reactive electron-deficient 14-electron platinum(0) species is produced via the dissociation of nitrogen, giving rise to a monodentate phosphine coordination. Electron-rich alkynylsilanes allow decreased π back-donation from the platinum center to the ligand, accelerating the dissociation of the more labile nitrogen. Steric congestion between diisopropylphosphino and silyl groups thermodynamically disfavors C(sp)–Si bond cleavage.
Co-reporter:Jing Li, Shintaro Noyori, Kiyohiko Nakajima, and Yasushi Nishihara
Organometallics 2014 Volume 33(Issue 13) pp:3500-3507
Publication Date(Web):June 23, 2014
DOI:10.1021/om500408h
A variety of α-iminonitriles were formed as the minor products of three-component coupling reactions of arynes, isocyanides, and cyanoformates in the presence of the cationic palladium complex [Pd(NCPh)2(dppf)](BF4)2 as the catalyst, along with cyano-substituted iminoisobenzofurans as the major products. α-Iminonitriles obtained from this process are hardly accessible by conventional methods. In addition, when the isolated iminoisobenzofurans were treated with diisobutylaluminum hydride (DIBAL-H) or AlMe3, the transformation of cyano-substituted iminoisobenzofurans into α-iminonitriles was observed.
Co-reporter:Dr. Masayuki Iwasaki;Tomoya Fujii;Arisa Yamamoto;Dr. Kiyohiko Nakajima;Dr. Yasushi Nishihara
Chemistry – An Asian Journal 2014 Volume 9( Issue 1) pp:58-62
Publication Date(Web):
DOI:10.1002/asia.201301295
Abstract
Chlorothiolation of terminal alkynes with sulfenyl chlorides yields anti-adducts without transition-metal catalysts. In sharp contrast, transition-metal-catalyzed chlorothiolation has not been developed to date, possibly because organosulfur compounds can poison catalyst. Herein, the regio- and stereoselective palladium-catalyzed chlorothiolation of terminal alkynes with sulfenyl chlorides is described. syn-Chlorothiolation offers a complementary synthetic route to chloroalkenyl sulfides. 2-Chloroalkenyl sulfides can easily be transformed into various sulfur-containing products, most of which are often found in natural products and pharmaceuticals.
Co-reporter:Dr. Masayuki Iwasaki;Tomoya Fujii;Arisa Yamamoto;Dr. Kiyohiko Nakajima;Dr. Yasushi Nishihara
Chemistry – An Asian Journal 2014 Volume 9( Issue 1) pp:
Publication Date(Web):
DOI:10.1002/asia.201390048
Co-reporter:Dr. Masayuki Iwasaki;Tomoya Fujii;Dr. Kiyohiko Nakajima;Dr. Yasushi Nishihara
Angewandte Chemie International Edition 2014 Volume 53( Issue 50) pp:13880-13884
Publication Date(Web):
DOI:10.1002/anie.201408121
Abstract
The radical addition of the ClS σ-bond in sulfenyl chlorides to various CC triple bonds has been achieved with excellent regio- and stereoselectivity in the presence of a catalytic amount of a common iron salt. The reaction is compatible with a variety of functional groups and can be scaled up to the gram-scale with no loss in yield. As well as terminal alkynes, internal alkynes underwent stereodefined chlorothiolation to provide tetrasubstituted alkynes. Preliminary mechanistic investigations revealed a plausible radical process involving a sulfur-centered radical intermediate via iron-mediated homolysis of the ClS bond. The resulting chlorothiolation adducts can be readily transformed to the structurally complex alkenyl sulfides by cross-coupling reactions. The present reaction can also be applied to the complementary synthesis of the potentially useful bis-sulfoxide ligands for transition-metal-catalyzed reactions.
Co-reporter:Dr. Masayuki Iwasaki;Tomoya Fujii;Dr. Kiyohiko Nakajima;Dr. Yasushi Nishihara
Angewandte Chemie 2014 Volume 126( Issue 50) pp:14100-14104
Publication Date(Web):
DOI:10.1002/ange.201408121
Abstract
The radical addition of the ClS σ-bond in sulfenyl chlorides to various CC triple bonds has been achieved with excellent regio- and stereoselectivity in the presence of a catalytic amount of a common iron salt. The reaction is compatible with a variety of functional groups and can be scaled up to the gram-scale with no loss in yield. As well as terminal alkynes, internal alkynes underwent stereodefined chlorothiolation to provide tetrasubstituted alkynes. Preliminary mechanistic investigations revealed a plausible radical process involving a sulfur-centered radical intermediate via iron-mediated homolysis of the ClS bond. The resulting chlorothiolation adducts can be readily transformed to the structurally complex alkenyl sulfides by cross-coupling reactions. The present reaction can also be applied to the complementary synthesis of the potentially useful bis-sulfoxide ligands for transition-metal-catalyzed reactions.
Co-reporter:Hiroki Mori, Xi-chao Chen, Ning-hui Chang, Shino Hamao, Yoshihiro Kubozono, Kiyohiko Nakajima, and Yasushi Nishihara
The Journal of Organic Chemistry 2014 Volume 79(Issue 11) pp:4973-4983
Publication Date(Web):May 2, 2014
DOI:10.1021/jo500543h
A series of picenes having methoxy groups was synthesized through Pd-catalyzed Suzuki–Miyaura couplings or Wittig reaction/intramolecular cyclization sequences, and their physicochemical properties and single-crystal structures were evaluated. The substitution position effects between the outer 1,12-, 2,11-, and 4,9-position and the inner 3,10-position are quite different; the former showed the same electronic structure as that of picene, but the latter results in a HOMO geometry different from those of picene and other methoxy picenes. In addition, crystal structures of four types of methoxy-substituted picenes 4a–c,e strongly depend on their substitution position and number of methoxy groups, which dramatically changes the structures from the fully anisotropic 1D π-stacked structure to a unique 3D herringbone structure due to steric hindrance of methoxy groups. The calculations of transfer integrals based on their single-crystal structures reveal that the methoxy picenes have intermolecular overlaps less effective than that of the parent nonsubstituted picene. These results are attributed not only to the packing structure but also to electronic structures such as the HOMO distribution. The preliminary OFET of the representative 4c,e showed hole mobilities significantly lower than that of picene due to their less effective intermolecular overlaps, as predicted by the calculated transfer integrals.
Co-reporter:Jiao Jiao, Kiyohiko Nakajima, and Yasushi Nishihara
Organic Letters 2013 Volume 15(Issue 13) pp:3294-3297
Publication Date(Web):June 18, 2013
DOI:10.1021/ol401333j
A highly regio- and stereoselective silylborylation of an alkynylboronate is disclosed. PhMe2Si–Bpin undergoes a Pd(OAc)2/tOctNC-catalyzed syn-addition to the alkynylboronate to yield 1-phenyl-1-silyl-2,2-diborylethene with high regioselectivity. The product 1-phenyl-1-silyl-2,2-diborylethene is then chemoselectively arylated by Suzuki–Miyaura coupling to afford (Z)-1-silyl-2-borylstilbene derivatives. This approach is extended to the synthesis of a tetraarylated olefin with four different substituents.
Co-reporter:Yasushi Nishihara, Masato Suetsugu, Daisuke Saito, Megumi Kinoshita, and Masayuki Iwasaki
Organic Letters 2013 Volume 15(Issue 10) pp:2418-2421
Publication Date(Web):April 30, 2013
DOI:10.1021/ol400896u
Synthesis of novel cyclic 1-alkenylboronates is accomplished through the zirconium-mediated regio- and stereoselective double functionalization of 1-alkynylboronates and the subsequent ruthenium-catalyzed ring-closing metathesis (RCM). The obtained substituted cyclic 1-alkenylboronates are transformed into o-terphenyl and triphenylene derivatives.
Co-reporter:Ning-hui Chang, Xi-chao Chen, Hikaru Nonobe, Yasuhiro Okuda, Hiroki Mori, Kiyohiko Nakajima, and Yasushi Nishihara
Organic Letters 2013 Volume 15(Issue 14) pp:3558-3561
Publication Date(Web):July 1, 2013
DOI:10.1021/ol401375n
A novel and versatile synthetic method for picene derivatives is developed using the Pd-catalyzed intramolecular double cyclization of the corresponding 2,3-bis[(1Z)-2-phenylethenyl]-1,4-dichlorobenzenes, which are readily prepared by Suzuki–Miyaura cross-coupling reactions of polyhalobenzenes with (Z)-arylethenylboronates. The physical properties of the obtained picenes can be modified via introducing a variety of functional groups to the picene framework. All compounds are investigated by UV–vis and fluorescence spectroscopic measurements, CV, and DFT calculations as well as X-ray diffraction analysis.
Co-reporter:Masayuki Iwasaki, Shohei Iino, and Yasushi Nishihara
Organic Letters 2013 Volume 15(Issue 20) pp:5326-5329
Publication Date(Web):October 3, 2013
DOI:10.1021/ol4025869
Treatment of o-iodobiphenyls with o-bromobenzyl alcohols in the presence of cesium carbonate under palladium catalysis affords a series of highly substituted triphenylenes. The reaction involves two C–C bond formations and C–C and C–H bond cleavages. A combination of palladium and an electron-deficient phosphine ligand proves to be effective for both decarbonylative cross-coupling and intramolecular cyclization.
Co-reporter:Daisuke Ogawa, Jing Li, Masato Suetsugu, Jiao Jiao, Masayuki Iwasaki, Yasushi Nishihara
Tetrahedron Letters 2013 Volume 54(Issue 6) pp:518-521
Publication Date(Web):6 February 2013
DOI:10.1016/j.tetlet.2012.11.069
A series of unsymmetrical diarylethynes have been synthesized by the copper-catalyzed cross-coupling reaction of alkynylboronates with aryl iodides in high to excellent yields under palladium-free conditions. A wide range of substrates bearing an electron-donating or an electron-withdrawing substituent on the aromatic ring are compatible with this reaction.
Co-reporter:Daisuke Ogawa, Keita Hyodo, Masato Suetsugu, Jing Li, Yoshiaki Inoue, Mamoru Fujisawa, Masayuki Iwasaki, Kentaro Takagi, Yasushi Nishihara
Tetrahedron 2013 69(12) pp: 2565-2571
Publication Date(Web):
DOI:10.1016/j.tet.2013.01.058
Co-reporter:Yasushi Nishihara, Eiji Inoue, Shintaro Noyori, Daisuke Ogawa, Yoshiaki Okada, Masayuki Iwasaki, Kentaro Takagi
Tetrahedron 2012 68(24) pp: 4869-4881
Publication Date(Web):
DOI:10.1016/j.tet.2012.03.093
Co-reporter:Jiao Jiao, Yasushi Nishihara
Journal of Organometallic Chemistry 2012 s 721–722() pp: 3-16
Publication Date(Web):
DOI:10.1016/j.jorganchem.2012.05.027
Co-reporter:Yasuhiro Okuda, Robert K. Szilagyi, Seiji Mori and Yasushi Nishihara
Dalton Transactions 2014 - vol. 43(Issue 25) pp:NaN9548-9548
Publication Date(Web):2014/04/11
DOI:10.1039/C4DT00839A
A computational investigation has been carried out to elucidate the origin of the exclusive exo-selectivity in the Pd-catalyzed cyanoesterification of strained cyclic olefins, norbornene and norbornadiene. A hybrid density functional was selected for the level of theory with a triple-ζ quality basis set, which was proposed in an earlier study to provide an experimentally sound ground state electronic structure description for palladium(II) and palladium(IV) complexes from multi-edge X-ray absorption spectroscopic measurements. Given that the product of oxidative addition can be isolated, we focused on the olefin coordination as the earliest possible origin of exo-selectivity. The calculated geometric structure for the trans-Pd(CN)(COOR)(PPh3)2 complex at the BHandHLYP/def2TZVP/PCM(toluene) level is in an excellent agreement with its experimental structure from crystallographic measurements. Upon dissociation of one of its phosphane ligands, the coordinatively unsaturated trans-isomer is only 17 kJ mol−1 away from the isomerization transition state, leading to the 14-electron cis-isomers that are 17 to 37 kJ mol−1 lower in energy than the trans-isomers. Regardless of the initial complex for olefin coordination, the exo-isomer for the norbornene complex is at least 8 kJ mol−1 lower than the corresponding endo-isomer. The origin of this considerable difference in Gibbs free energy can be attributed to the remarkably different steric and agostic hydrogen interactions between the methylene and the ethylene bridges of the norbornene and the adjacent cis-ligands at the PdII center.
Co-reporter:Takuya Ishitsuka, Yasuhiro Okuda, Robert K. Szilagyi, Seiji Mori and Yasushi Nishihara
Dalton Transactions 2016 - vol. 45(Issue 18) pp:NaN7793-7793
Publication Date(Web):2016/03/24
DOI:10.1039/C6DT00341A
Potential energy surface mapping was completed for the entire catalytic cycle of palladium-catalysed cyanoesterification onto norbornene (NBE) using density functional theory calculations. We found that after the oxidative addition step of the reagent methyl cyanoformate, the reaction proceeds through an insertion of olefin into a PdII–COOMe bond first. Subsequently, reductive elimination occurs by transferring the cyanide group from the Pd center to NBE. This rearrangement is triggered by the rotation of the ester group into a π-interaction with the PdII centre. The regioselectivity of olefin insertion is controlled by ionic and covalent interactions in the precursor π-complex formation step. Importantly, all of the intermediates and transition states along the exo pathway were found to be more stable than the corresponding structures of the endo pathway without any sign of crossing over between the two surfaces via isomerization. The rate-determining step is the reductive elimination despite the fact that the corresponding activation barrier is reduced by conformational changes via the rotation of a MeOOC–C(NBE) bond.
Co-reporter:Xi-Chao Chen, Shuhei Nishinaga, Yasuhiro Okuda, Jia-Ji Zhao, Jie Xu, Hiroki Mori and Yasushi Nishihara
Inorganic Chemistry Frontiers 2015 - vol. 2(Issue 5) pp:NaN541-541
Publication Date(Web):2015/03/16
DOI:10.1039/C5QO00039D
A series of 3,10-dialkylated picenes has been synthesized from 3,10-dimethoxypicene through a divergent approach involving Ni-catalyzed alkynylation of a C–O bond, alkylation, and hydrogenation.





![Benzothiazole, 2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/59100/59066-61-4.png)
![Benzothiazole, 2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/59100/59066-61-4_b.png)
![Methanone, phenyl[4-(phenylmethyl)phenyl]-](http://img.cochemist.com/ccimg/58300/58280-04-9.png)
![Methanone, phenyl[4-(phenylmethyl)phenyl]-](http://img.cochemist.com/ccimg/58300/58280-04-9_b.png)








![Pyridine, 2-[2-(phenylthio)phenyl]-](http://img.cochemist.com/ccimg/897500/897440-13-0.png)
![Pyridine, 2-[2-(phenylthio)phenyl]-](http://img.cochemist.com/ccimg/897500/897440-13-0_b.png)
![6H-Dibenzo[b,d]pyran, 6,6-dimethyl-](http://img.cochemist.com/ccimg/29600/29574-51-4.png)
![6H-Dibenzo[b,d]pyran, 6,6-dimethyl-](http://img.cochemist.com/ccimg/29600/29574-51-4_b.png)
![Diselenide, bis[2-(trifluoromethyl)phenyl]](http://img.cochemist.com/ccimg/131800/131700-81-7.png)
![Diselenide, bis[2-(trifluoromethyl)phenyl]](http://img.cochemist.com/ccimg/131800/131700-81-7_b.png)



![Dibenzo[c,m]picene](http://img.cochemist.com/ccimg/13200/13109-47-2.png)
![Dibenzo[c,m]picene](http://img.cochemist.com/ccimg/13200/13109-47-2_b.png)













![Silane, trimethyl[3-[(trimethylsilyl)ethynyl]phenyl]-](http://img.cochemist.com/ccimg/16200/16116-86-2.png)
![Silane, trimethyl[3-[(trimethylsilyl)ethynyl]phenyl]-](http://img.cochemist.com/ccimg/16200/16116-86-2_b.png)






![Phenanthro[1,2-b:8,7-b']dithiophene](/data/chemimg/2271600/1491133-64-2.png)
![Phenanthro[1,2-b:8,7-b']dithiophene](/data/chemimg/2271600/1491133-64-2_b.png)











![Benzo[c]picene](http://img.cochemist.com/ccimg/300/217-37-8.png)
![Benzo[c]picene](http://img.cochemist.com/ccimg/300/217-37-8_b.png)












![Methanesulfonic acid, trifluoro-, [1,1'-biphenyl]-2-yl ester](http://img.cochemist.com/ccimg/17800/17763-65-4.png)
![Methanesulfonic acid, trifluoro-, [1,1'-biphenyl]-2-yl ester](http://img.cochemist.com/ccimg/17800/17763-65-4_b.png)
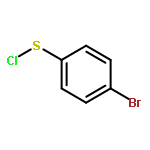
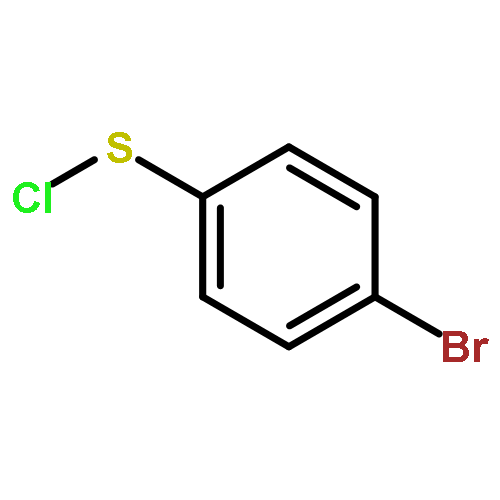








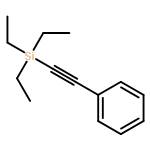


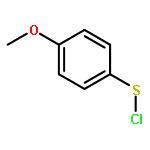
![Benzene, 1-methoxy-4-[(4-methylphenyl)ethynyl]-](http://img.cochemist.com/ccimg/24800/24785-38-4.png)
![Benzene, 1-methoxy-4-[(4-methylphenyl)ethynyl]-](http://img.cochemist.com/ccimg/24800/24785-38-4_b.png)





















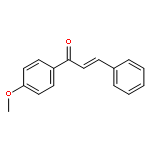
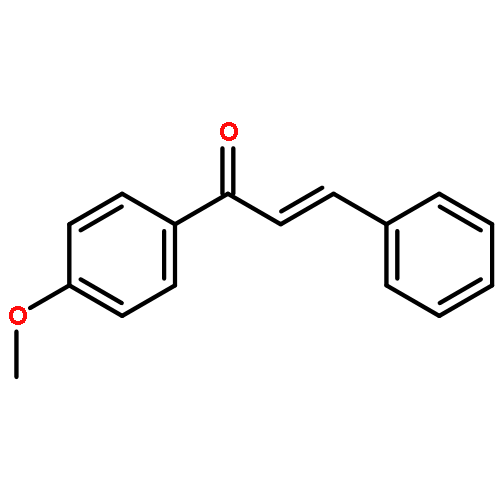
![Phosphonium, [(2-bromo-5-methoxyphenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/263900/263840-63-7.png)
![Phosphonium, [(2-bromo-5-methoxyphenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/263900/263840-63-7_b.png)
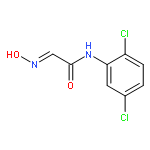

![Benzo[g]chrysene](http://img.cochemist.com/ccimg/200/196-78-1.png)
![Benzo[g]chrysene](http://img.cochemist.com/ccimg/200/196-78-1_b.png)







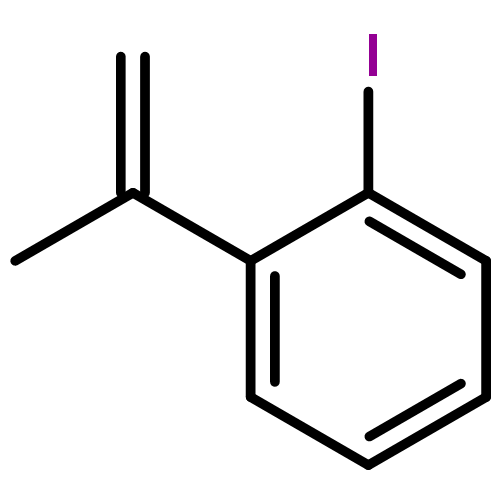
![2'-BROMO-[1,1',2',1']TERPHENYL](http://img.cochemist.com/ccimg/75300/75295-57-7.png)
![2'-BROMO-[1,1',2',1']TERPHENYL](http://img.cochemist.com/ccimg/75300/75295-57-7_b.png)










![Dichloro[bis(1,3-diphenylphosphino)propane]palladium(II)](http://img.cochemist.com/ccimg/59900/59831-02-6.png)
![Dichloro[bis(1,3-diphenylphosphino)propane]palladium(II)](http://img.cochemist.com/ccimg/59900/59831-02-6_b.png)


![Tris[3,5-bis(trifluoromethyl)phenyl]phosphine](http://img.cochemist.com/ccimg/175200/175136-62-6.png)
![Tris[3,5-bis(trifluoromethyl)phenyl]phosphine](http://img.cochemist.com/ccimg/175200/175136-62-6_b.png)







![[4-(TRIFLUOROMETHYL)PHENYL] THIOHYPOCHLORITE](http://img.cochemist.com/ccimg/72000/71947-62-1.png)
![[4-(TRIFLUOROMETHYL)PHENYL] THIOHYPOCHLORITE](http://img.cochemist.com/ccimg/72000/71947-62-1_b.png)




















![Carbamic acid,N-[(4-methylphenyl)sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/14500/14437-03-7.png)
![Carbamic acid,N-[(4-methylphenyl)sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/14500/14437-03-7_b.png)

























![Ethanone, 1-[4-[(4-methoxyphenyl)ethynyl]phenyl]-](http://img.cochemist.com/ccimg/123800/123770-67-2.png)
![Ethanone, 1-[4-[(4-methoxyphenyl)ethynyl]phenyl]-](http://img.cochemist.com/ccimg/123800/123770-67-2_b.png)












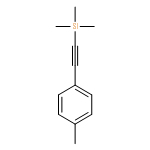

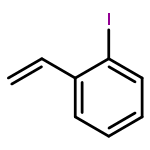
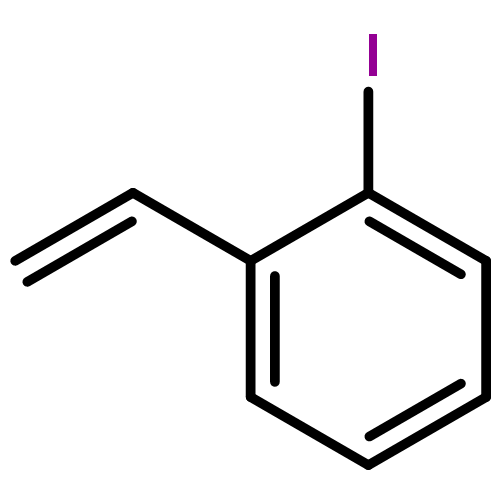


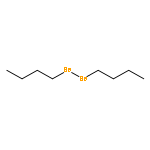



![Benzene,[[(1E)-2-chloro-2-phenylethenyl]sulfonyl]-](http://img.cochemist.com/ccimg/31600/31598-92-2.png)
![Benzene,[[(1E)-2-chloro-2-phenylethenyl]sulfonyl]-](http://img.cochemist.com/ccimg/31600/31598-92-2_b.png)




![3'H-Cyclopropa[1,2][5,6]fullerene-C60-Ih-3'-butanoic acid, 3'-phenyl-, methyl ester](/data/chemimg/102800/824404-45-7.png)
![3'H-Cyclopropa[1,2][5,6]fullerene-C60-Ih-3'-butanoic acid, 3'-phenyl-, methyl ester](/data/chemimg/102800/824404-45-7_b.png)






![Pyrrolo[3,4-c]pyrrole-1,4-dione, 2,5-dihydro-](/data/chemimg/100900/114482-12-1.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 2,5-dihydro-](/data/chemimg/100900/114482-12-1_b.png)











![DICHLORO-[1,3-BIS(DIISOPROPYLPHENYL)-2-IMIDAZOLIDINYLIDENE]-(3-CHLOROPYRIDYL)PALLADIUM(II)](http://img.cochemist.com/ccimg/927800/927706-57-8.png)
![DICHLORO-[1,3-BIS(DIISOPROPYLPHENYL)-2-IMIDAZOLIDINYLIDENE]-(3-CHLOROPYRIDYL)PALLADIUM(II)](http://img.cochemist.com/ccimg/927800/927706-57-8_b.png)
![2-Propen-1-one, 1-phenyl-3-[4-(trifluoromethyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/144100/144017-74-3.png)
![2-Propen-1-one, 1-phenyl-3-[4-(trifluoromethyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/144100/144017-74-3_b.png)