Co-reporter:Lina Ye, Yuanyuan Fang, Zhongping Ou, Songlin Xue, and Karl M. Kadish
Inorganic Chemistry November 6, 2017 Volume 56(Issue 21) pp:13613-13613
Publication Date(Web):October 24, 2017
DOI:10.1021/acs.inorgchem.7b02405
Three series of cobalt tetraarylporphyrins were synthesized and characterized by electrochemistry and spectroelectrochemistry. The investigated compounds have the general formula (TpYPP)Co, butano(TpYPP)CoII, and benzo(TpYPP)CoII, where TpYPP represents the dianion of the meso-substituted porphyrin, Y is a CH3, H, or Cl substituent on the para position of the four phenyl rings, and butano and benzo are respectively the β- and β′-substituted groups on the four pyrrole rings of the compound. Each porphyrin undergoes one or two reductions depending upon the meso substituent and solvent utilized. Two irreversible reductions are observed for (TpYPP)CoII and butano(TpYPP)CoII in CH2Cl2 containing 0.1 M tetra-n-butylammonium perchlorate; the first leads to the formation of a highly reactive cobalt(I) porphyrin, which can then rapidly react with a solvent to give a CoIIICH2Cl as the product. Only one reversible reduction is seen for benzo(TpYPP)CoII under the same solution conditions, and the one-electron-reduction product is assigned as a cobalt(II) porphyrin π-anion radical. Three oxidations can be observed for each examined compound in CH2Cl2. The first oxidation is metal-centered for the (TpYPP)Co and benzo(TpYPP)CoII derivatives, leading to generation of a cobalt(III) porphyrin with an intact π-ring system, but this redox process is ring-centered in the case of butano(TpYPP)CoII and gives a CoII π-cation radical product. Each porphyrin was also examined as a catalyst for oxygen reduction reactions (ORRs) when adsorbed on a graphite electrode in 1.0 M HClO4. The number of electrons transferred (n) during ORRs is 2.0 for the butano(TpYPP)CoII derivatives, consistent with only H2O2 being produced as a product for the reaction with O2. However, the reduction of O2 using the cobalt benzoporphyrins as catalysts gave n values between 2.6 and 3.1 under the same solution conditions, thus producing a mixture of H2O and H2O2 as the reduction product. This result indicates that the β and β′ substituents have a significant effect on the catalytic properties of the cobalt porphyrins for ORRs in acid media.
Co-reporter:Hong-Guang Jin, Xiaoqin Jiang, Irina A. Kühne, Sylvain Clair, Valérie Monnier, Christophe Chendo, Ghenadie Novitchi, Annie K. Powell, Karl M. Kadish, and Teodor Silviu Balaban
Inorganic Chemistry May 1, 2017 Volume 56(Issue 9) pp:4864-4864
Publication Date(Web):April 13, 2017
DOI:10.1021/acs.inorgchem.6b03056
Five heteroleptic lanthanide porphyrin–bis-phthalocyanine triple-decker complexes with bulky peripheral groups were prepared via microwave-assisted synthesis and characterized in terms of their spectroscopic, electrochemical, and magnetic properties. These compounds, which were easily obtained under our preparative conditions, would normally not be accessible in large quantities using conventional synthetic methods, as a result of the low yield resulting from steric congestion of bulky groups on the periphery of the phthalocyanine and porphyrin ligands. The electrochemically investigated triple-decker derivatives undergo four reversible one-electron oxidations and three reversible one-electron reductions. The sites of oxidation and reduction were assigned on the basis of redox potentials and UV–vis spectral changes during electron-transfer processes monitored by thin-layer spectroelectrochemistry, in conjunction with assignments of electronic absorption bands of the neutral compounds. Magnetic susceptibility measurements on two derivatives containing TbIII and DyIII metal ions reveal the presence of ferromagnetic interactions, probably resulting from magnetic dipolar interactions. The TbIII derivative shows SMM behavior under an applied field of 0.1 T, where the direct and Orbach process can be determined, resulting in an energy barrier of Ueff = 132.0 K. However, Cole–Cole plots reveal the presence of two relaxation processes, the second of which takes place at higher frequencies, with the data conforming to a 1/t ∝ T7 relation, thus suggesting that it can be assigned to a Raman process. Attempts were made to form two-dimensional (2D) self-assembled networks on a highly oriented pyrolytic graphite (HOPG) surface but were unsuccessful due to bulky peripheral groups on the two Pc macrocycles.
Co-reporter:Xiangyi Ke, Pinky Yadav, Lei Cong, Ravi Kumar, Muniappan Sankar, and Karl M. Kadish
Inorganic Chemistry July 17, 2017 Volume 56(Issue 14) pp:8527-8527
Publication Date(Web):July 5, 2017
DOI:10.1021/acs.inorgchem.7b01262
The first examples for the facile, reversible, and stepwise electrogeneration of triply ring-reduced porphyrin macrocycles are presented. The investigated compounds are represented as MTPP(NO2)(PE)6, MTTP(PE)8, NiTPP(NO2)(Ph)4, and MTPP(CN)4, where TTP and TPP are the dianions of tetratolylporphyrin and tetraphenylporphyrin, respectively, NO2, phenylethynyl (PE), and CN are substituents at the β-pyrrole positions of the macrocycle, and M = CuII, NiII, ZnII, CoII, or 2H. Each porphyrin undergoes three or four reductions within the negative potential limit of the electrochemical solvent. The UV–visible spectra of the first three reduction products were characterized by means of thin-layer UV–vis spectroelectrochemistry, and the generation of multianionic porphyrins is interpreted in terms of extensive stabilization of the LUMOs due to the electron-withdrawing and/or extended π-conjugation of the β-substituents.
Co-reporter:Jijun Tang, Zhongping Ou, Rui Guo, Yuanyuan Fang, Dong Huang, Jing Zhang, Jiaoxia Zhang, Song Guo, Frederick M. McFarland, and Karl M. Kadish
Inorganic Chemistry August 7, 2017 Volume 56(Issue 15) pp:8954-8954
Publication Date(Web):July 25, 2017
DOI:10.1021/acs.inorgchem.7b00936
A cobalt triphenylcorrole (CorCo) was covalently bonded to graphene oxide (GO), and the resulting product, represented as GO-CorCo, was characterized by UV–vis, FT-IR, and micro-Raman spectroscopy as well as by HRTEM, TGA, XRD, XPS, and AFM. The electrocatalytic activity of GO-CorCo toward the oxygen reduction reaction (ORR) was then examined in air-saturated 0.1 M KOH and 0.5 M H2SO4 solutions by cyclic voltammetry and linear sweep voltammetry using a rotating disk electrode and/or a rotating ring-disk electrode. An overall 4-electron reduction of O2 is obtained in alkaline media while under acidic conditions a 2-electron process is seen. The ORR results thus indicate that covalently bonded GO-CoCor can be used as a selective catalyst for either the 2- or 4-electron reduction of oxygen, the prevailing reaction depending upon the acidity of the solution.
Co-reporter:Pinky Yadav;Muniappan Sankar;Xiangyi Ke;Lei Cong
Dalton Transactions 2017 vol. 46(Issue 30) pp:10014-10022
Publication Date(Web):2017/08/01
DOI:10.1039/C7DT01814B
Di- and octa-phenylethynyl (PE) substituted π-extended copper corroles were synthesized and characterized as to their structural, electrochemical and spectroscopic properties. The addition of two or eight PE groups to the β-pyrrole positions of the corrole results in dramatic red shifts in the electronic absorption spectra and new reductions which are not seen for the parent compound lacking PE substituents. CuCor(PE)8 is reduced in four reversible one-electron transfer steps to give derivatives of [CuCor(PE)8]n− where n = 1, 2, 3 or 4. Variable temperature 1H NMR and EPR measurements were carried out and suggest that the octa- and di-PE substituted Cu-corroles can both be described as an antiferromagnetically coupled CuII corrole cation radical which is in equilibrium with a triplet state, possibly due to a lower singlet–triplet energy gap as compared to 1 and 2 at room temperature. The EPR spectra of one-electron oxidized and one electron reduced species exhibited the characteristics of Cu(II) corroles. The products generated in the first two reductions of each π-extended corrole were characterized by thin-layer spectroelectrochemistry, thus providing new insights into how UV-vis spectra of highly reduced corroles vary as a function of the number of PE groups and overall charge on the molecule. The singly reduced and singly oxidized copper corroles were also chemically generated in CH3CN and shown to have UV-visible spectra almost identical to the spectra obtained by electroreduction or electrooxidation in PhCN or THF containing 0.1 M tetrabutylammonium perchlorate.
Co-reporter:Yuanyuan Fang, Zhongping Ou, and Karl M. Kadish
Chemical Reviews 2017 Volume 117(Issue 4) pp:
Publication Date(Web):December 23, 2016
DOI:10.1021/acs.chemrev.6b00546
This review describes the known electrochemistry of corroles in nonaqueous media from 1980 until the present. The outline of the review is grouped according to the periodic table, proceeding from left to right, describing first monomeric free-base derivatives and then transition-metal compounds, followed by main-group corroles, before ending with a brief description of lanthanide and actinide corroles. Many similarities exist between the redox properties of metallocorroles and metalloporphyrins, but there are also many differences due, in part, to the different charges of the two conjugated macrocycles and the noninnocence of the corrole ligand in a variety of compounds. One part of this review will focus on describing redox behavior as a function of metal ion and axial ligands, while another will focus on how changes in structure of the macrocycle are associated with changes in redox behavior. It is hoped that this review will answer the majority of the readers’ questions as to what has been electrochemically observed for corroles in the past while at the same time enabling the reader to utilize data in the literature to predict and “tune” what might be observed in future electrochemical studies of corroles that have yet to be synthesized and characterized.
Co-reporter:Katarzyna Rybicka-Jasińska, Wenqian Shan, Katarzyna Zawada, Karl M. Kadish, and Dorota Gryko
Journal of the American Chemical Society 2016 Volume 138(Issue 47) pp:15451-15458
Publication Date(Web):November 2, 2016
DOI:10.1021/jacs.6b09036
Metalloporphyrins not only are vital in biological systems but also are valuable catalysts in organic synthesis. On the other hand, catalytic properties of free base porphyrins have been less explored. They are mostly known as efficient photosensitizers for the generation of singlet oxygen via photoinduced energy transfer processes, but under light irradiation, they can also participate in electron transfer processes. Indeed, we have found that free base tetraphenylporphyrin (H2TPP) is an efficient photoredox catalyst for the reaction of aldehydes with diazo compounds leading to α-alkylated derivatives. The performance of a porphyrin catalyst can be optimized by tailoring various substituents at the periphery of the macrocycle at both the β and meso positions. This allows for the fine tuning of their optical and electrochemical properties and hence their catalytic activity.
Co-reporter:Zhaoli Xue, Yemei Wang, John Mack, Scebi Mkhize, Tebello Nyokong, Yuanyuan Fang, Zhongping Ou and Karl M. Kadish
RSC Advances 2016 vol. 6(Issue 48) pp:41919-41926
Publication Date(Web):22 Apr 2016
DOI:10.1039/C6RA03028A
A thermal reaction using a series of [14]tribenzotriphyrins(2.1.1) (TriPs, 1a–d) with Rh2(C8H12)Cl2 provides RhIII–TriP complexes (2a–d) in 40−52% yields. The complexes were characterized by mass spectrometry, UV-visible absorption and 1H NMR spectroscopy. Single crystal X-ray analysis reveals that 2b adopts a dome-shaped conformation. The rhodium(III) ion is coordinated by the three pyrrole nitrogen atoms, two chloride ions and the nitrogen atom of an acetonitrile (CH3CN) solvent molecule. The optical spectra can be assigned using Michl's perimeter model. The L and B bands of the 2a–d complexes lie at ca. 600 and 500 nm, respectively, and are markedly red shifted relative to those of 1a–d. A reversible one-electron oxidation and two reversible one-electron reductions are observed in the cyclic voltammograms of 2a–d in CH2Cl2. The redox potentials are consistent with the optical data and the relatively narrow HOMO–LUMO gaps that are predicted in DFT calculations. TD-DFT calculations have been used to assign a third intense spectral band at 375 nm to a higher energy π → π* transition of the [14]tribenzotriphyrin(2.1.1) π-system.
Co-reporter:Guifen Lu, Sen Yan, Mengying Shi, Wenhan Yu, Jing Li, Weihua Zhu, Zhongping Ou and Karl M. Kadish
Chemical Communications 2015 vol. 51(Issue 12) pp:2411-2413
Publication Date(Web):22 Dec 2014
DOI:10.1039/C4CC09755F
The first europium triple-decker tetrapyrrole with mixed corrole and phthalocyanine macrocycles was synthesized and characterized by spectroscopic and electrochemical methods. The molecular structure was characterized by single-crystal X-ray diffraction and showed the corrole to be in the middle of the sandwich with phthalocyanine macrocycles at each extreme.
Co-reporter:Christina M. Davis, Kei Ohkubo, I-Ting Ho, Zhan Zhang, Masatoshi Ishida, Yuanyuan Fang, Vincent M. Lynch, Karl M. Kadish, Jonathan L. Sessler and Shunichi Fukuzumi
Chemical Communications 2015 vol. 51(Issue 31) pp:6757-6760
Publication Date(Web):12 Mar 2015
DOI:10.1039/C5CC00903K
Photoexcitation of dichloromethane solutions of an uranyl macrocyclic complex with cyclo[1]furan[1]pyridine[4]-pyrrole (1) at the near-infrared (NIR) band (1177 nm) in the presence of electron donors and acceptors resulted in NIR-induced electron transfer without producing singlet oxygen via energy transfer.
Co-reporter:Guifen Lu; Jing Li; Xiaoqin Jiang; Zhongping Ou
Inorganic Chemistry 2015 Volume 54(Issue 18) pp:9211-9222
Publication Date(Web):September 11, 2015
DOI:10.1021/acs.inorgchem.5b01713
A series of europium triple-decker complexes containing phthalocyanine and nitrophenyl–corrole macrocycles were synthesized and characterized by spectroscopic and electrochemical methods in nonaqueous media. The examined compounds are represented as Eu2[Pc(OC4H9)8]2[Cor(Ph)n(NO2Ph)3–n], where n varies from 0 to 3, Pc(OC4H9)8 represents the phthalocyanine macrocycle, and Cor indicates the corrole macrocycle having phenyl (Ph) or nitrophenyl (NO2Ph) meso substituents. Three different methods were used for syntheses of the target complexes, two of which are reported here for the first time. Each examined compound undergoes five reversible one-electron oxidations and 3–5 one-electron reductions depending upon the number of NO2Ph substituents. The nitrophenyl groups on the meso positions of the corrole are highly electron-withdrawing, and this leads to a substantial positive shift in potential for the five oxidations and first reduction in CH2Cl2, PhCN, or pyridine as compared to the parent triple-decker compound with a triphenylcorrole macrocycle. The measured E1/2 values are linearly related to the number of NO2Ph groups on the corrole, and the relative magnitude of the shift in potential for each redox reaction was used in conjunction with the results from thin-layer spectro-electrochemistry to assign the initial site of oxidation or reduction on the molecule. The nitrophenyl substituents are also redox-active, and each is reduced to [C6H4NO2]− in a separate one-electron transfer step at potentials between −1.12 and −1.42 V versus saturated calomel electrode.
Co-reporter:Yuanyuan Fang; Yulia G. Gorbunova; Ping Chen; Xiaoqin Jiang; Machima Manowong; Anna A. Sinelshchikova; Yulia Yu. Enakieva; Alexander G. Martynov; Aslan Yu. Tsivadze; Alla Bessmertnykh-Lemeune; Christine Stern; Roger Guilard
Inorganic Chemistry 2015 Volume 54(Issue 7) pp:3501-3512
Publication Date(Web):March 19, 2015
DOI:10.1021/acs.inorgchem.5b00067
Two series of diphosphoryl-substituted porphyrins were synthesized and characterized by electrochemistry and spectroelectrochemistry in nonaqueous media containing 0.1 M tetra-n-butylammonium perchlorate (TBAP). The investigated compounds are 5,15-bis(diethoxyphosphoryl)-10,20-diphenylporphyrins (Ph)2(P(O)(OEt)2)2PorM and 5,15-bis(diethoxyphosphoryl)-10,20-di(para-carbomethoxyphenyl)porphyrins (PhCOOMe)2(P(O)(OEt)2)2PorM where M = 2H, Co(II), Ni(II), Cu(II), Zn(II), Cd(II), or Pd(II). The free-base and five metalated porphyrins with nonredox active centers undergo two ring-centered oxidations and two ring-centered reductions, the latter of which is followed by a chemical reaction of the porphyrin dianion to give an anionic phlorin product. The phlorin anion is electroactive and can be reoxidized by two electrons to give back the starting porphyrin, or it can be reversibly reduced by one electron at more negative potentials to give a phlorin dianion. The chemical conversion of the porphyrin dianion to a phlorin anion proceeds at a rate that varies with the nature of the central metal ion and the solvent. This rate is slowest in the basic solvent pyridine as compared to CH2Cl2 and PhCN, giving further evidence for the involvement of protons in the chemical reaction leading to phlorin formation. Calculations of the electronic structure were performed on the Ni(II) porphyrin dianion, and the most favorable atoms for electrophilic attack were determined to be the two phosphorylated carbon atoms. Phlorin formation was not observed after the two-electron reduction of the cobalt porphyrins due to the different oxidation state assignment of the doubly reduced species, a Co(I) π anion radical in one case and an M(II) dianion for all of the other derivatives. Each redox reaction was monitored by thin-layer UV–visible spectroelectrochemistry, and an overall mechanism for each electron transfer is proposed on the basis of these data.
Co-reporter:Guifen Lu, Jing Li, Sen Yan, Weihua Zhu, Zhongping Ou, and Karl M. Kadish
Inorganic Chemistry 2015 Volume 54(Issue 12) pp:5795-5805
Publication Date(Web):May 28, 2015
DOI:10.1021/acs.inorgchem.5b00477
We recently reported the first example of a europium triple-decker tetrapyrrole with mixed corrole and phthalocyanine macrocycles and have now extended the synthetic method to prepare a series of rare earth corrole–phthalocyanine heteroleptic triple-decker complexes, which are characterized by spectroscopic and electrochemical methods. The examined complexes are represented as M2[Pc(OC4H9)8]2[Cor(ClPh)3], where Pc = phthalocyanine, Cor = corrole, and M is Pr(III), Nd(III), Sm(III), Eu(III), Gd(III), or Tb(III). The Y(III) derivative with OC4H9 Pc substituents was obtained in too low a yield to characterize, but for the purpose of comparison, Y2[Pc(OC5H11)8]2[Cor(ClPh)3] was synthesized and characterized in a similar manner. The molecular structure of Eu2[Pc(OC4H9)8]2[Cor(ClPh)3] was determined by single-crystal X-ray diffraction and showed the corrole to be the central macrocycle of the triple-decker unit with a phthalocyanine on each end. Each triple-decker complex undergoes up to eight reversible or quasireversible one-electron oxidations and reductions with E1/2 values being linearly related to the ionic radius of the central ions. The energy (E) of the main Q-band is also linearly related to the radius of the metal. Comparisons are made between the physicochemical properties of the newly synthesized mixed corrole–phthalocyanine complexes and previously characterized double- and triple-decker derivatives with phthalocyanine and/or porphyrin macrocycles.
Co-reporter:Giuseppe Pomarico; Pierluca Galloni; Federica Mandoj; Sara Nardis; Manuela Stefanelli; Andrea Vecchi; Sara Lentini; Daniel O. Cicero; Yan Cui; Lihan Zeng; Karl M. Kadish;Roberto Paolesse
Inorganic Chemistry 2015 Volume 54(Issue 21) pp:10256-10268
Publication Date(Web):October 13, 2015
DOI:10.1021/acs.inorgchem.5b01575
Complexes of 5,10,15-triferrocenylcorrole were synthesized from the crude free-base corrole product obtained by the reaction of ferrocenyl aldehyde and pyrrole. Direct formation of the complex in this manner leads to an increase of the reaction yield by protecting the corrole ring toward oxidative decomposition. The procedure was successful and gave the expected product in the case of the copper and triphenylphosphinecobalt complexes, but an unexpected result was obtained in the case of the nickel derivative, where metal insertion led to a ring opening of the macrocycle at the 5 position, giving as a final product a linear tetrapyrrole nickel complex bearing two ferrocenyl groups. The purified 5,10,15-triferrocenylcorrole complexes have been fully characterized by a combination of spectroscopic methods, electrochemistry, spectroelectrochemistry, and density functional theory calculations. Copper derivatives of 10-monoferrocenyl- and 5,15-diferrocenylcorrole were prepared to investigate how the number and position of the ferrocenyl groups influenced the spectroscopic and electrochemical properties of the resulting complexes. A complete assignment of resonances in the 1H and 13C NMR spectra was performed for the cobalt and nickel complexes, and detailed electrochemical characterization was carried out to provide additional insight into the degree of communication between the meso-ferrocenyl groups on the conjugated macrocycle and the central metal ion of the ferrocenylcorrole derivatives.
Co-reporter:Zhaoli Xue; Yemei Wang; John Mack; Yuanyuan Fang; Zhongping Ou; Weihua Zhu
Inorganic Chemistry 2015 Volume 54(Issue 24) pp:11852-11858
Publication Date(Web):November 25, 2015
DOI:10.1021/acs.inorgchem.5b02093
Metalation of 6,13,20,21-tetrakis-aryl-22H-[14]tribenzotriphyrin(2.1.1) (TriPs) with PdCl2 provides PdII–TriP complexes in 45–56% yields. The complexes were characterized by mass spectrometry, and UV–visible absorption, magnetic circular dichroism, and 1H NMR spectroscopy. A single crystal X-ray analysis reveals that the PdII–TriPs adopts a deeply saddled conformation. The palladium(II) ion is coordinated by two pyrrole nitrogen atoms and two chloride ions to form the square-planar coordination environment. The redox properties of the PdII–TriPs were studied by cyclic voltammetry. Each compound undergoes one irreversible and two reversible one-electron reductions. There is a marked red-shift of the main spectral bands, relative to those of the free-base TriP ligand, due to a marked relative stabilization of the LUMO upon coordination by PdCl2.
Co-reporter:Guifen Lu, Jing Li, Sen Yan, Cheng He, Mengying Shi, Weihua Zhu, Zhongping Ou, Karl M. Kadish
Dyes and Pigments 2015 Volume 121() pp:38-45
Publication Date(Web):October 2015
DOI:10.1016/j.dyepig.2015.05.008
•Nanostructures of two mixed sandwich complexes were first prepared.•One is belt-like and the other is sheet-like.•Both complexes exhibit good third-order NLO properties.Two heteroleptic corrole–phthalocyanine europium triple-decker complexes were fabricated into organic nanostructures by the standard phase-transfer method. The investigated compounds are represented by Eu2(Pc)2[Cor(ClPh)3] (1) and Eu2[Pc(OC8H17)8]2[Cor(ClPh)3] (2), where Pc represents the dianion of phthalocyanine and Cor(ClPh)3 the trianion of 5,10,15-tri(4-chlorophenyl)corrole. Both compounds 1 and 2 show typical electronic absorption spectra for a non-aggregated molecular structure in chloroform or toluene. However, in methanol compound 1 self-assembled into belt-like nanostructures while compound 2, which possesses long OC8H17 alkoxy side chains, self-assembled into large scale sheet-like nanostructures. The third-order nonlinear optical properties of both triple-decker compounds were also investigated in toluene by the Z-scan technique and characterized as showing nonlinear reverse saturation absorption and self-defocusing behavior. A third-order nonlinear optical susceptibility, χ(3), of 3.18 × 10−10 and 3.35 × 10−10 esu was obtained for 1 and 2, respectively, thus indicating the potential application of these compounds in the field of optical devices.Nanostructures of two heteroleptic corrole–phthalocyanine europium triple-decker complexes with different phthalocyanine substituents were prepared and characterized. The examined compounds self-assembled into two different nanostructures; one is belt-like and the other is sheet-like. The third-order nonlinear optical properties of both triple-decker compounds were investigated in toluene by the Z-scan technique and characterized as showing nonlinear reverse saturation absorption and self-defocusing behavior. The third-order nonlinear optical susceptibility values indicate that the examined compounds have potential applications in the fields of organic semiconductors and optical limiting devices.
Co-reporter:Ru Feng, Zhongping Ou, Zhaoli Xue, Yuanyuan Fang, Yang Song and Karl M. Kadish
RSC Advances 2015 vol. 5(Issue 117) pp:96769-96776
Publication Date(Web):04 Nov 2015
DOI:10.1039/C5RA19603E
Three meso-substituted pyrrole-terminated tripyrrins were isolated for the first time as side products in the synthesis of triarylcorroles and characterized by spectroscopic and electrochemical techniques. The examined compounds are represented as (Ar)2TriPyH, where the TriPy is the conjugated tripyrrin monoanion and Ar a 2,6-diFPh, 2,6-diClPh or 2,4-diClPh substituent. A single crystal X-ray structure of (2,6-diFPh)2TriPyH is also presented. This is the first X-ray structure of a meso-aryl substituted tripyrrin. Each tripyrrin undergoes two reductions and three oxidations in CH2Cl2. The first one-electron addition and first one-electron abstraction lead to formation of π–anion and π–cation radicals with a potential separation between the two processes of 1.71 to 1.76 V. However, both electrogenerated products are unstable and undergo a rapid chemical reaction to give new electroactive species which are identified as the deprotonated and protonated compounds, respectively. The reaction products were characterized by spectroelectrochemistry and comparisons are made with spectroscopic data obtained during base and acid titrations in CH2Cl2.
Co-reporter:Lina Ye, Zhongping Ou, Yuanyuan Fang, Songlin Xue, Yang Song, Liping Wang, Mengli Wang and Karl M. Kadish
RSC Advances 2015 vol. 5(Issue 94) pp:77088-77096
Publication Date(Web):04 Sep 2015
DOI:10.1039/C5RA15593B
Two series of copper tetraarylporphyrins containing β,β′-fused tetrabutano or tetrabenzo groups were synthesized and characterized as to their electrochemistry and spectroelectrochemistry in nonaqueous media. The examined compounds are represented as butano-(TpYPP)CuII and benzo-(TpYPP)CuII, where TpYPP is the porphyrin dianion and Y is a CH3, H or Cl substituent on the para-position of the four meso-phenyl rings of the compound. Each neutral porphyrin in the two series is ESR active and shows a typical d9 Cu(II) signal in frozen CH2Cl2 solution. Each Cu(II) porphyrin also undergoes two reversible one-electron reductions and two reversible one-electron oxidations in DMF or CH2Cl2 containing 0.1 M tetra-n-butylammonium perchlorate to give a π-anion radical and dianion upon reduction and a π-cation radical and dication upon oxidation. A third one-electron oxidation is also observed for butano-(TpYPP)Cu (Y = CH3 and H) and benzo-(TPP)Cu in PhCN and this process is assigned to the CuII/CuIII transition. The reversible half-wave potential for the first oxidation of each compound in both series is shifted negatively by about 500 mV as compared to E1/2 values for oxidation of the related copper tetraarylporphyrin without the four fused benzo or butano rings while smaller positive shifts of 60 and 300 mV are seen for reduction of the tetrabenzotetraarylporphyrins and tetrabutaotetraarylporphyrins, respectively, as compared to the same redox reactions of the related tetraarylporphyrins. The electrochemically measured HOMO–LUMO gap averages 1.76 ± 0.05 V for benzo-(TpYPP)CuII, 2.04 ± 0.06 V for butano-(TpYPP)CuII and 2.33 ± 0.03 for (TpYPP)Cu in CH2Cl2.
Co-reporter:R. G. Waruna Jinadasa, Yuanyuan Fang, Siddhartha Kumar, Allen J. Osinski, Xiaoqin Jiang, Christopher J. Ziegler, Karl M. Kadish, and Hong Wang
The Journal of Organic Chemistry 2015 Volume 80(Issue 24) pp:12076-12087
Publication Date(Web):November 18, 2015
DOI:10.1021/acs.joc.5b01906
The synthesis of a series of β-functionalized push–pull dibenzoporphyrins was realized. These porphyrins display subtle push–pull effects, demonstrating the exceptional tunability of their electronic and electrochemical properties. The UV–vis spectra of these porphyrins show unique absorption patterns with shouldered Soret bands and extra absorptions in the Q-band region. Stronger electron-withdrawing groups display more significant bathochromic shifts of the Soret bands. The fluorescence spectra of these porphyrins show strong near-IR emission bands (600–850 nm). In particular, fluorescence quenching effect was observed for pyridyl carrying push–pull porphyrin 4c in the presence of an acid. TFA titration study of 4c using UV–vis and fluorescence spectroscopy reveals that the fluorescence quenching can be mainly attributed to the protonation of the pyridyl groups of 4c. The versatile synthetic methods developed in this work may open a door to access a large number of functionalized organic materials that are currently unavailable. The structure–property studies provided in this work may provide useful guidelines for the design of new generations of materials in dye-sensitized solar cells, in nonlinear optical applications, as fluorescence probes, as well as sensitizers for photodynamic therapy.
Co-reporter:Songlin Xue; Zhongping Ou;Lina Ye;Guifen Lu;Yuanyuan Fang;Xiaoqin Jiang; Karl M. Kadish
Chemistry - A European Journal 2015 Volume 21( Issue 6) pp:2651-2661
Publication Date(Web):
DOI:10.1002/chem.201405570
Abstract
A series of N-confused free-base meso-substituted tetraarylporphyrins was investigated by electrochemistry and spectroelectrochemistry in nonaqueous media containing 0.1 M tetra-n-butylammonium perchlorate (TBAP) and added acid or base. The investigated compounds are represented as (XPh)4NcpH2, in which “Ncp” is the N-confused porphyrin macrocycle and X is a OCH3, CH3, H, or Cl substituent on the para position of each meso-phenyl ring of the macrocycle. Two distinct types of UV/Vis spectra are initially observed depending upon solvent, one corresponding to an inner-2H form and the other to an inner-3H form of the porphyrin. Both forms have an inverted pyrrole with a carbon inside the cavity and a nitrogen on the periphery of the π-system. Each porphyrin undergoes multiple irreversible reductions and oxidations. The first one-electron addition and first one-electron abstraction are located on the porphyrin π-ring system to give π-anion and π-cation radicals with a potential separation of 1.52 to 1.65 V between the two processes, but both electrogenerated products are unstable and undergo a rapid chemical reaction to give new electroactive species, which were characterized in the present study. The effect of the solvent and protonation/deprotonation reactions on the UV/Vis spectra, redox potentials and reduction/oxidation mechanisms is discussed with comparisons made to data and mechanisms for the structurally related free-base corroles and porphyrins.
Co-reporter:Dr. Kolle E. Thomas;Dr. Hugo Vazquez-Lima;Dr. Yuanyuan Fang;Yang Song;Dr. Kevin J. Gagnon;Dr. Christine M. Beavers;Dr. Karl M. Kadish;Dr. Abhik Ghosh
Chemistry - A European Journal 2015 Volume 21( Issue 47) pp:16839-16847
Publication Date(Web):
DOI:10.1002/chem.201502150
Abstract
A silver β-octabromo-meso-triarylcorrole has been found to exhibit a strongly saddled geometry, providing the first instance of a strongly saddled corrole complex involving a metal other than copper. The Soret maxima of the Ag octabromocorroles also redshift markedly in response to increasingly electron-donating para substituents on the meso-aryl groups. In both these respects, the Ag octabromocorroles differ from simple Ag triarylcorrole derivatives, which exhibit only mild saddling and substituent-insensitive Soret maxima. These results have been rationalized in terms of an innocent MIII-corrole3− description for the simple Ag corroles and a noninnocent MII-corrole.2− description for the Ag octabromocorroles. In contrast, all copper corroles are thought to be noninnocent, while all gold corroles are innocent. Uniquely among metallocorroles, silver corroles thus seem poised on a knife-edge, so to speak, between innocent and noninnocent electronic structures and may tip either way, depending on the exact nature of the corrole ligand.
Co-reporter:Dr. Kolle E. Thomas;Dr. Hugo Vazquez-Lima;Dr. Yuanyuan Fang;Yang Song;Dr. Kevin J. Gagnon;Dr. Christine M. Beavers;Dr. Karl M. Kadish;Dr. Abhik Ghosh
Chemistry - A European Journal 2015 Volume 21( Issue 47) pp:
Publication Date(Web):
DOI:10.1002/chem.201503939
Abstract
Invited for the cover of this issue are Abhik Ghosh and his collaborators at UiT (The Arctic University of Norway, Tromsø) and in the United States. The image depicts a silver corrole complex poised on the summit ridge of Mount Sir Alexander, a peak of the Canadian Rockies (image courtesy of Chris Goulet), symbolizing the balance between innocent and noninnocent ligands. Read the full text of the article at 10.1002/chem.201502150.
Co-reporter:Dr. Kolle E. Thomas;Dr. Hugo Vazquez-Lima;Dr. Yuanyuan Fang;Yang Song;Dr. Kevin J. Gagnon;Dr. Christine M. Beavers;Dr. Karl M. Kadish;Dr. Abhik Ghosh
Chemistry - A European Journal 2015 Volume 21( Issue 47) pp:
Publication Date(Web):
DOI:10.1002/chem.201584701
Co-reporter:Yuanyuan Fang, Dominik Koszelewski, Karl M. Kadish and Daniel T. Gryko
Chemical Communications 2014 vol. 50(Issue 64) pp:8864-8867
Publication Date(Web):16 Jun 2014
DOI:10.1039/C4CC02759K
The facile electrosynthesis of π-extended porphyrins is demonstrated for a series of Zn(II), In(III), Ir(III) and free-base meso-substituted derivatives containing the 4,7-dimethoxynaphthalen-1-yl substituent. Electrochemical data suggest that the overall process initially involves two stepwise one-electron oxidations, followed by an intramolecular oxidative aromatic coupling to give the electrooxidized π-extended porphyrin.
Co-reporter:Bin Sun, Zhongping Ou, Deying Meng, Yuanyuan Fang, Yang Song, Weihua Zhu, Pavlo V. Solntsev, Victor N. Nemykin, and Karl M. Kadish
Inorganic Chemistry 2014 Volume 53(Issue 16) pp:8600-8609
Publication Date(Web):July 28, 2014
DOI:10.1021/ic501210t
Cobalt porphyrins having 0–4 meso-substituted ferrocenyl groups were synthesized and examined as to their electrochemical properties in N,N′-dimethylformamide (DMF) containing 0.1 M tetra-n-butylammonium perchlorate as a supporting electrolyte. The examined compounds are represented as (Fc)n(CH3Ph)4–nPorCo, where Por is a dianion of the substituted porphyrin, Fc and CH3Ph represent ferrocenyl and/or p-CH3C6H4 groups linked at the four meso-positions of the macrocycle, and n varies from 0 to 4. Each porphyrin undergoes two reversible one-electron reductions and two to six one-electron oxidations in DMF, with the exact number depending upon the number of Fc groups on the compound. The first electron addition is metal-centered to generate a Co(I) porphyrin. The second is porphyrin ring-centered and leads to formation of a Co(I) π-anion radical. The first oxidation of each Co(II) porphyrin is metal-centered to generate a Co(III) derivative under the given solution conditions. Each ferrocenyl substituent can also be oxidized by one electron, and this occurs at more positive potentials. Each compound was investigated as a catalyst for the electoreduction of dioxygen when adsorbed on a graphite electrode in 1.0 M HClO4. The number of electrons transferred (n) during the catalytic reduction was 2.0 for the three ferrocenyl substituted compounds, consistent with only H2O2 being produced as a product of the reaction. Most monomeric cobalt porphyrins exhibit n values between 2.6 and 3.1 under the same solution conditions, giving a mixture of H2O and H2O2 as a reduction product, although some monomeric porphyrins can give an n value of 4.0. Our results in the current study indicate that appending ferrocene groups directly to the meso positions of a porphyrin macrocycle will increase the selectivity of the oxygen reduction, resulting in formation of only H2O2 as a reaction product. This selectivity of the electrocatalytic oxygen reduction reaction is explained on the basis of steric hindrance by the ferrocene substituents which prevent dimerization.
Co-reporter:Yuanyuan Fang, Mathias O. Senge, Eric Van Caemelbecke, Kevin M. Smith, Craig J. Medforth, Min Zhang, and Karl M. Kadish
Inorganic Chemistry 2014 Volume 53(Issue 19) pp:10772-10778
Publication Date(Web):September 25, 2014
DOI:10.1021/ic502162p
Electrochemical studies of the oxidation of dodecasubstituted and highly nonplanar nickel porphyrins in a noncoordinating solvent have previously revealed the first nickel(III) porphyrin dication. Herein, we investigate if these nonplanar porphyrins can also be used to detect the so far unobserved copper(III) porphyrin dication. Electrochemical studies of the oxidation of (DPP)Cu and (OETPP)Cu show three processes, the first two of which are macrocycle-centered to give the porphyrin dication followed by a CuII/CuIII process at more positive potential. Support for the assignment of the CuII/CuIII process comes from the linear relationships observed between E1/2 and the third ionization potential of the central metal ions for iron, cobalt, nickel, and copper complexes of (DPP)M and (OETPP)M. In addition, the oxidation behavior of additional nonplanar nickel porphyrins is investigated in a noncoordinating solvent, with nickel meso-tetraalkylporphyrins also being found to form nickel(III) porphyrin dications. Finally, examination of the nickel meso-tetraalkylporphyrins in a coordinating solvent (pyridine) reveals that the first oxidation becomes metal-centered under these conditions, as was previously noted for a range of nominally planar porphyrins.
Co-reporter:Machima Manowong, Baocheng Han, Thomas R. McAloon, Jianguo Shao, Ilia A. Guzei, Siyabonga Ngubane, Eric Van Caemelbecke, John L. Bear, and Karl M. Kadish
Inorganic Chemistry 2014 Volume 53(Issue 14) pp:7416-7428
Publication Date(Web):July 8, 2014
DOI:10.1021/ic5007605
Three related diruthenium complexes containing four symmetrical anionic bridging ligands were synthesized and characterized as to their electrochemical and spectroscopic properties. The examined compounds are represented as Ru2(dpb)4Cl, Ru2(dpb)4(CO), and Ru2(dpb)4(NO) in the solid state, where dpb = diphenylbenzamidinate anion. Different forms of Ru2(dpb)4Cl are observed in solution depending on the utilized solvent and the counteranion added to solution. Each Ru25+ form of the compound undergoes multiple redox processes involving the dimetal unit. The reversibility as well as potentials of these diruthenium-centered electrode reactions depends upon the solvent and the bound axial ligand. The Ru25+/4+ and Ru25+/6+ processes of Ru2(dpb)4Cl were monitored by UV–vis spectroscopy in both CH2Cl2 and PhCN. A conversion of Ru2(dpb)4Cl to [Ru2(dpb)4(CO)]+ was also carried out by simply bubbling CO gas through a CH2Cl2 solution of Ru2(dpb)4Cl at room temperature. The chemically generated [Ru2(dpb)4(CO)]+ complex undergoes several electron transfer processes in CH2Cl2 containing 0.1 M TBAClO4 under a CO atmosphere, and the same reactions were seen for a chemically synthesized sample of Ru2(dpf)4(CO) in CH2Cl2, 0.1 M TBAClO4 under a N2 atmosphere, where dpf = N,N′-diphenylformamidinate anion. Ru2(dpb)4(NO) undergoes two successive one-electron reductions and a single one-electron oxidation, all of which involve the diruthenium unit. The CO and NO adducts of Ru2(dpb)4 were further characterized by FTIR spectroelectrochemistry, and the IR spectral data of these compounds are discussed in light of results for previously characterized Ru2(dpf)4(CO) and Ru2(dpf)4(NO) derivatives under similar solution conditions.
Co-reporter:Yuanyuan Fang, Federica Mandoj, Sara Nardis, Giuseppe Pomarico, Manuela Stefanelli, Daniel O. Cicero, Sara Lentini, Andrea Vecchi, Yan Cui, Lihan Zeng, Karl M. Kadish, and Roberto Paolesse
Inorganic Chemistry 2014 Volume 53(Issue 14) pp:7404-7415
Publication Date(Web):June 30, 2014
DOI:10.1021/ic500757a
The reaction of 5,10,15-tris(4-tert-butylphenyl)corrole with 2,3-bis(bromomethyl)-5,6-dicyanopyrazine provides a new example of corrole ring expansion to a hemiporphycene derivative. The ring expansion is regioselective, with insertion of the pyrazine derivative at the 5-position of the corrole ring, affording the corresponding 5-hemiporphycene. Different macrocyclic products accompany formation of the 5-hemiporphycene, depending on the reaction experimental conditions. Br-substitued 5-hemiporphycenes and the 2-Br substituted corrole were obtained in 1,2,4-trichlorobenzene, while in refluxing toluene traces of an inner core substituted corrole were observed together with a significant amount of the unreacted corrole. These results provide an important indication of the reaction pathway. The coordination behavior of the 5-hemiporphycene, together with detailed electrochemical characterization of the free-base and some metal complexes, provides evidence for the reactivity of the peripheral pyrazino group.
Co-reporter:Bin Sun, Zhongping Ou, Shuibo Yang, Deying Meng, Guifen Lu, Yuanyuan Fang and Karl M. Kadish
Dalton Transactions 2014 vol. 43(Issue 28) pp:10809-10815
Publication Date(Web):02 May 2014
DOI:10.1039/C4DT01072H
Four cobalt(II) porphyrins, two of which contain a β-pyrrole nitro substituent, were synthesized and characterized by electrochemistry and spectroelectrochemistry. The investigated compounds are represented as (TRPP)Co and (NO2TRPP)Co, where TRPP is the dianion of a substituted tetraphenylporphyrin and R is a CH3 or OCH3 substituent on the four phenyl rings of the macrocycle. Two reductions and three oxidations are observed for each compound in CH2Cl2 containing 0.10 M tetra-n-butylammonium perchlorate. The first reduction of the compounds without a nitro substituent is metal-centered and leads to formation of a Co(I) porphyrin which then reacts with the CH2Cl2 solvent to generate a carbon σ-bonded CoIII–R complex. A further reduction then occurs at more negative potentials to generate an unstable Co(II) σ-bonded compound. In contrast to these reactions, the first reduction of the nitro-substituted porphyrins is macrocycle-centered under the same solution conditions and gives a Co(II) porphyrin π-anion radical product. This reversible electron transfer is then followed at more negative potentials by a second reversible one-electron addition to give a Co(II) dianion. Three reversible one-electron oxidations are also seen for each compound. The first is metal-centered and the next two involve the conjugated π-system of the macrocycle. Each neutral Co(II) porphyrin was also examined as to its catalytic activity for electroreduction of molecular oxygen when coated on an edge-plane pyrolytic graphite electrode in 1.0 M HClO4. The β-pyrrole nitro-substituted derivatives were shown to be better catalysts than the non-nitro substituted compounds under the utilized experimental conditions.
Co-reporter:Maria Pia Donzello;Giorgia De Mori;Elisa Viola;David Futur;Zhen Fu;Corrado Rizzoli;Luisa Mannina;Enrico Bodo;Maria Luisa Astolfi;Claudio Ercolani
European Journal of Inorganic Chemistry 2014 Volume 2014( Issue 22) pp:3572-3581
Publication Date(Web):
DOI:10.1002/ejic.201402282
Abstract
Electrochemical and UV/Vis spectral studies on the quinoxaline compound 2,3-di(2-pyridyl)-6,7-dicyano-1,4-quinoxaline, [(CN)2Py2Qx], its metallated derivatives [(CN)2Py2QxMCl2] (M = PdII, PtII) and their analogues sharing the dicyanopyrazine fragment are presented and discussed. X-ray work on the new quinoxaline complex [(CN)2Py2QxPtCl2] establishes that PtII is coordinated to the pyridine N atoms (“py–py” coordination). The spectra of the 1– charged quinoxaline and pyrazine compounds show new intense absorptions in the region of 500–900 nm, and redshifted bands in the 250–400 nm region. By using DFT/time-dependent DFT calculations, the spin density and spectral features of both couples of precursors and PtII derivatives were investigated in terms of single-electron excitations between the Kohn–Sham orbitals of the optimized structures in the gas and condensed phases. A detailed comparison is allowed of the experimental and calculated spectral features of the neutral and 1– charged species.
Co-reporter:Alexer Mitrofanov;Machima Manowong;Yoann Rousselin;Stéphane Brès;Roger Guilard;Alla Bessmertnykh-Lemeune;Ping Chen;Nataliya Goulioukina;Irina Beletskaya
European Journal of Inorganic Chemistry 2014 Volume 2014( Issue 21) pp:3370-3386
Publication Date(Web):
DOI:10.1002/ejic.201402161
Abstract
Two series of copper(I) complexes with diethoxyphosphoryl-substituted 1,10-phenanthroline ligands were synthesized and characterized in the solid state and in solution. The first comprised mixed-ligand CuI complexes with phenanthroline and triphenylphosphine. The second series includes bis-chelates with two phenanthroline ligands. According to the X-ray data for the six complexes, the ditopic phenanthroline ligands exhibit bidentate coordination to the copper(I) atom through two nitrogen atoms in both series. Solution equilibria involving different phenanthroline copper(I) species were studied by 1H and 31P NMR spectroscopy, electrochemistry, and spectroelectrochemistry. The solution speciation of these labile complexes is different for these two series and depends on the nature of solvent and the location of the phosphorus substituent on the phenanthroline backbone. Coordinating solvents can replace a bromide, triphenylphosphine, and even a phenanthroline ligand in the inner coordination sphere of the metal center. Copper(I) complexes with α-substituted phenanthrolines easily dissociate even in noncoordinating solvents such as CH2Cl2 and CHCl3. Ligand-exchange reactions leading to less sterically hindered species were observed under the utilized solution conditions. The coordination mode of the phenanthroline chelators does not change under any of the utilized solution conditions, and binding of the phosphoryl group to the metal center was never observed by spectroscopic or spectroelectrochemical methods.
Co-reporter:Bihong Li, Zhongping Ou, Deying Meng, Jijun Tang, Yuanyuan Fang, Rui Liu, Karl M. Kadish
Journal of Inorganic Biochemistry 2014 Volume 136() pp:130-139
Publication Date(Web):July 2014
DOI:10.1016/j.jinorgbio.2013.12.014
•Synthesis of new nitro corroles with different numbers of substituted nitro groups.•Effect of substituted nitro groups on redox potentials of cobalt corroles.•Effect of substituted nitro groups on UV-visible spectra of cobalt corroles.•The use of nitro-substituted cobalt corroles as catalyst for the reduction of oxygen.Cobalt(III) triarylcorroles containing 0–3 nitro groups on the para-position of the three meso-phenyl rings of the macrocycle were synthesized and characterized by electrochemistry, mass spectrometry, (UV–vis) and 1H NMR spectroscopy. The examined compounds are represented as (NO2Ph)nPh3 − nCorCo(PPh3), where n varies from 0 to 3 and Cor represents the core of the corrole. Each compound can undergo two metal-centered one-electron reductions leading to formation of Co(II) and Co(I) derivatives in CH2Cl2 or pyridine containing 0.1 M tetra-n-butylammonium perchlorate (TBAP). A stepwise two electron reduction of each NO2Ph group of the compound is also observed. The first is reversible and occurs in a single overlapping step at the same potential which involves an overall one-, two- or three-electron transfer process for compounds 2–4, respectively. This indicates the lack of an interaction between these redox active sites on the corroles. The second reduction of the NO2Ph groups is irreversible and located at a potential which overlaps the Co(II)/Co(I) process of the compounds. Thin-layer UV–visible spectroelectrochemical measurements in CH2Cl2, 0.1 M TBAP demonstrate the occurrence of an equilibrium between a Co(III) π-anion radical and a Co(II) derivative with an uncharged macrocycle after the first controlled potential reduction of the nitro-substituted corroles. All four cobalt corroles were also examined as catalysts for the electroreduction of O2 when coated on an edge-plane pyrrolytic graphite electrode in 1.0 M HClO4. This study indicates that the larger the number of nitro-substituents on the cobalt corrole, the better the compound acts as a catalyst.Cobalt(III) triarylcorroles containing one, two or three NO2 groups on the para-position of the three meso-phenyl rings of the macrocycle were synthesized and characterized by electrochemistry and spectroscopic techniques. Each corrole was also examined as a catalyst for the electroreduction of O2 in 1.0 M HClO4 when coated on the surface of an edge-plane pyrrolytic graphite electrode.
Co-reporter:Yuanyuan Fang; P. Bhyrappa; Zhongping Ou; Karl M. Kadish
Chemistry - A European Journal 2014 Volume 20( Issue 2) pp:524-532
Publication Date(Web):
DOI:10.1002/chem.201303141
Abstract
A series of planar and nonplanar free-base β-pyrrole substituted meso-tetraarylporphyrins were characterized by electrochemistry, spectroelectrochemistry, and protonation or deprotonation reactions in neutral, acidic, and basic solutions of CH2Cl2. The neutral compounds are represented as H2(P), in which P represents a porphyrin dianion with one of several different sets of electron-withdrawing or -donating substituents at the messo and/or β-pyrrole positions of the macrocycle. The conversion of H2(P) to [H4(P)]2+ in CH2Cl2 was accomplished by titration of the neutral porphyrin with trifluoroacetic acid (TFA) while the progress of the protonation was monitored by UV/Vis spectroscopy, which was also used to calculate logβ2 for proton addition to the core nitrogen atoms of the macrocycle. Cyclic voltammetry was performed after each addition of TFA or TBAOH to CH2Cl2 solutions of the porphyrin and half-wave potentials for reduction were evaluated as a function of the added acid or base concentration. Thin-layer spectroelectrochemistry was used to obtain UV/Vis spectra of the neutral and protonated or deprotonated porphyrins under the application of an applied reducing potential. The magnitude of the protonation constants, the positions of λmax in the UV/Vis spectra and the half-wave or peak potentials for reduction are then related to the electronic properties of the porphyrin and the data evaluated as a function of the planarity or nonplanarity of the porphyrin macrocycle. Surprisingly, the electroreduction of the diprotonated nonplanar porphyrins in acid media leads to H2(P), whereas the nonplanar H2(P) derivatives are reduced to [(P)]2− in CH2Cl2 containing 0.1 M tetra-n-butylammonium perchlorate (TBAP). Thus, in both cases an electrochemically initiated deprotonation is observed.
Co-reporter:Zhongping Ou, Tony Khoury, Yuanyuan Fang, Weihua Zhu, Paul J. Sintic, Maxwell J. Crossley, and Karl M. Kadish
Inorganic Chemistry 2013 Volume 52(Issue 5) pp:2474-2483
Publication Date(Web):February 20, 2013
DOI:10.1021/ic302380z
Gold(III) porphyrins containing two, three, or four β,β′-fused quinoxalines were synthesized and examined as to their electrochemical properties in tetrahydrofuran (THF), pyridine, CH2Cl2, and CH2Cl2 containing added acid in the form of trifluoroacetic acid (TFA). The investigated porphyrins are represented as Au(PQ2)PF6, Au(PQ3)PF6, and Au(PQ4)PF6, where P is the dianion of the 5,10,15,20-tetrakis(3,5-di-tert-butylphenyl)porphyrin and Q is a quinoxaline group fused to a β,β′-pyrrolic position of the porphyrin macrocycle. In the absence of added acid, all three gold(III) porphyrins undergo a reversible one-electron oxidation and several reductions. The first reduction is characterized as a AuIII/AuII process which is followed by additional porphyrin- and quinoxaline-centered redox reactions at more negative potentials. However, when 3–5 equivalents of acid are added to the CH2Cl2 solution, the initial AuIII/AuII process is followed by a series of internal electron transfers and protonations, leading ultimately to triply reduced and doubly protonated AuII(PQ2H2) in the case of AuIII(PQ2)+, quadruply reduced and triply protonated AuII(PQ3H3) in the case of AuIII(PQ3)+, and AuII(PQ4H4) after addition of five electrons and four protons in the case of AuIII(PQ4)+. Under these solution conditions, the initial Au(PQ2)PF6 compound is shown to undergo a total of three AuIII/AuII processes while Au(PQ3)PF6 and Au(PQ4)PF6 exhibit four and five metal-centered one-electron reductions, respectively, prior to the occurrence of additional reductions at the conjugated macrocycle and fused quinoxaline rings. Each redox reaction was monitored by cyclic voltammetry and thin-layer spectroelectrochemistry, and an overall mechanism for reduction in nonaqueous media with and without added acid is proposed. The effect of the number of Q groups on half-wave potentials for reduction and UV–visible spectra of the electroreduced species are analyzed using linear free energy relationships.
Co-reporter:Mingzhu Yuan, Zhongping Ou, Yuanyuan Fang, Shi Huang, Zhaoli Xue, Guifen Lu, and Karl M. Kadish
Inorganic Chemistry 2013 Volume 52(Issue 11) pp:6664-6673
Publication Date(Web):May 16, 2013
DOI:10.1021/ic4006955
Open-chain pentapyrroles were isolated as side-products from the synthesis of triaryl-corroles and then converted to the corresponding sapphyrins by catalytic oxidation in acidic media. The investigated compounds were characterized by UV–vis and 1H NMR spectroscopy, mass spectrometry, electrochemistry, and spectroelectrochemistry and are represented as (Ar)4PPyH3 and (Ar)4SH3, where Ar is a F– or Cl– substituted phenyl group, PPy is a trianion of the open-chain pentapyrrole, and S is a trianion of the sapphyrin. Cyclic voltammetry and thin-layer UV–vis spectroelectrochemistry measurements were carried out in PhCN and CH2Cl2 containing 0.1 M tetra-n-butylammonium perchlorate. The open-chain pentapyrroles undergo two reversible one-electron reductions and two reversible one-electron oxidations to generate [(Ar)PPyH3]−, [(Ar)PPyH3]2–, [(Ar)PPyH3]+, and [(Ar)PPyH3]2+ which were spectroscopically characterized. The corresponding sapphyrins exhibit two or three reversible one-electron oxidations in PhCN, but the reductions of these compounds are irreversible because of coupled chemical reactions following electron transfer. Comparisons are made between redox potentials and spectral properties of the open-chain pentapyrroles, sapphyrins, and structurally related corroles. Protonation of the open-chain pentapyrroles and sapphyrins was also carried out in CH2Cl2, and equilibrium constants were calculated by monitoring the spectral changes during titrations with trifluoroacetic acid. The pentapyrroles undergo a simultaneous two-proton addition to generate [(Ar)4PPyH5]2+ while the sapphyrins undergo two stepwise single proton additions to give [(Ar)4SH4]+ and [(Ar)4SH5]2+, respectively.
Co-reporter:Ping Chen, Yuanyuan Fang, Karl M. Kadish, Jan P. Lewtak, Dominik Koszelewski, Anita Janiga, and Daniel T. Gryko
Inorganic Chemistry 2013 Volume 52(Issue 16) pp:9532-9538
Publication Date(Web):July 29, 2013
DOI:10.1021/ic401214e
A Ni(II) complex of a π-extended porphyrin bearing three mesityl substituents and one electron-rich naphthalene moiety has been prepared via electrochemical oxidation. It was proven that the whole oxidative process starts from electrochemical generation of a radical-cation on the porphyrin core. Electrochemistry and spectroelectrochemistry of both a naphthalenyl-substituted porphyrin and a porphyrin with a fused naphthalenyl group on the π-ring system provide clear distinction between metal- and ring-centered processes. The redox reactivity of the naphthalenyl-substituted metalloporphyrin in nonaqueous media is presented while outlining the most important structural factors which influence the reversible half-wave potentials for oxidation and reduction of this complex and the following chemical reactions which lead to an extended π-system.
Co-reporter:Siyabonga Ngubane, Karl M. Kadish, John L. Bear, Eric Van Caemelbecke, Antoine Thuriere and Kevin P. Ramirez
Dalton Transactions 2013 vol. 42(Issue 10) pp:3571-3580
Publication Date(Web):17 Dec 2012
DOI:10.1039/C2DT32715E
A mixed-ligand metal–metal bonded diruthenium complex having the formula Ru2(2,4,6-(CH3)3ap)3(O2CCH3)Cl where ap is the anilinopyridinate anion was synthesized from the reaction of Ru2(O2CCH3)4Cl and H(2,4,6-(CH3)3ap), after which the isolated product was structurally, spectroscopically and electrochemically characterized. The crystal structure reveals an unusual arrangement of the bridging ligands around the dimetal unit where one ruthenium atom is coordinated to one anilino and two pyridyl nitrogen atoms while the other ruthenium atom is coordinated to one pyridyl and two anilino nitrogen atoms. To our knowledge, Ru2(2,4,6-(CH3)3ap)3(O2CCH3)Cl is the only example of a mixed-ligand diruthenium complex of the type [Ru2L3(O2CCH3)]+, where L is an unsymmetrical anionic bridging ligand that has been structurally characterized with a “(2,1)” geometric conformation of the bridging ligands, all others being “(3,0)”. The initial Ru25+ compound in CH2Cl2 or CH3CN containing 0.1 M tetra-n-butylammonium perchlorate (TBAP) undergoes up to four one-electron redox processes involving the dimetal unit. The Ru25+/4+ and Ru25+/6+ processes were characterized under N2 using thin-layer UV-visible spectroelectrochemistry and this data is compared to UV-visible spectral changes obtained during similar electrode reactions for related diruthenium compounds having the formula Ru2L4Cl or Ru2L3(O2CCH3)Cl where L is an anionic bridging ligand. Ru2(2,4,6-(CH3)3ap)3(O2CCH3)Cl was also examined by UV-visible and FTIR spectroelectrochemistry under a CO atmosphere and two singly reduced Ru24+ species, [Ru2(2,4,6-(CH3)3ap)3(O2CCH3)(CO)Cl]− and Ru2(2,4,6-(CH3)3ap)3(O2CCH3)(CO) were in situ generated for further characterization. The CO-bound complexes could be further reduced and exhibited additional reductions to their Ru23+ and Ru22+ oxidation states.
Co-reporter:Zhongping Ou, Deying Meng, Mingzhu Yuan, Wenhao Huang, Yuanyuan Fang, and Karl M. Kadish
The Journal of Physical Chemistry B 2013 Volume 117(Issue 43) pp:13646-13657
Publication Date(Web):September 26, 2013
DOI:10.1021/jp408905a
Electrochemical and spectroelectrochemical properties of three open-chain pentapyrroles and the corresponding sapphyrins were examined in pyridine containing 0.1 M tetra-n-butylammonium perchlorate and dichloromethane (CH2Cl2) or benzonitrile (PhCN) containing tetra-n-butylammonium hydroxide (TBAOH). The investigated compounds are represented as (Ar)4PPyH3 and (Ar)4SH3, where Ar is a F– or Cl– substituted phenyl group, PPy is a trianion of the open-chain pentapyrrole, and S is a trianion of the sapphyrin. The pentapyrroles, (Ar)4PPyH3, undergo two reversible one-electron reductions in pyridine, while the structurally related sapphyrins exhibit four reductions in this solvent, the first two of which are irreversible due to coupled chemical reactions following the electron transfers. Both series of neutral compounds could be deprotonated in CH2Cl2 or PhCN by addition of TBAOH to solution, and the progress of these reactions was monitored as a function of the base concentration by cyclic voltammetry and UV–vis spectroscopy. The neutral pentapyrroles were spectroscopically shown to undergo a loss of two protons in a single step to generate the [(Ar)4PPyH]2– dianion while the sapphyrins could only be monodeprotonated, leading to formation of the [(Ar)4SH2]− monoanion under the same solution conditions. The deprotonation constants were measured for each series of compounds in benzonitrile, and oxidation–reduction mechanisms are examined as a function of the solution ‘basicity’.
Co-reporter:Maria Pia Donzello, Elisa Viola, Claudio Ercolani, Zhen Fu, David Futur, and Karl M. Kadish
Inorganic Chemistry 2012 Volume 51(Issue 22) pp:12548-12559
Publication Date(Web):November 2, 2012
DOI:10.1021/ic301989a
Heteropentanuclear porphyrazines having the formula [(PtCl2)4LM] where L = tetrakis-2,3-[5,6-di(2-pyridyl)pyrazino]porphyrazinato dianion and M = ZnII, MgII(H2O), PdII, CuII or CoII were characterized by elemental analyses, IR–UV–visible spectroscopy and electrochemistry and the data compared to new and previously published results for the corresponding homopentanuclear compound [(PtCl2)4LPt]. This latter species has four external N2(py)PtCl2 coordination sites which closely resemble cis-platin, (NH3)2PtCl2, the potent chemotherapeutic anticancer drug, and is able to act as a photosensitizer for the generation of 1O2, the cytotoxic agent in photodynamic therapy (PDT). UV–visible spectra and half wave potentials for reduction of [(PtCl2)4LM], [(PtCl2)4LPt], the parallel series of mononuclear [LM] compounds and the pentanuclear [(PdCl2)4LM] compounds were examined in the nonaqueous solvents dimethyl sulfoxide, pyridine, and dimethylformamide. The complete set of available data indicate that external coordination of the PtCl2 and PdCl2 units significantly increases the level of the electron-deficiency of the entire molecular framework despite the fact that these groups are far away from the central porphyrazine π-ring system and have coordination sites nearly orthogonal to the plane of the macrocycle. The pentanuclear species [(M′Cl2)4LM] (M′ = PtII, PdII) undergo multiple one-electron transfers and exhibit an easier reducibility as compared to related electrode reactions of the parent compounds [LM] having the same central metal. Aggregation phenomena and reducibility of the porphyrazines to their monoanionic form (prevalently in DMF) are observed for some of the examined compounds and were analyzed and accurately taken into account. Quantum yields of 1O2 (ΦΔ), of interest in PDT, were measured for [(PtCl2)4LM] with M = ZnII, MgII(H2O), or PdII and the related macrocycles [(PdCl2)4LM] and [LM] in dimethylformamide (DMF) and/or DMF preacidified with HCl (DMF/HCl, [HCl]: 1–2 × 10–4 M). Excellent ΦΔ values (0.5–0.6) which qualify the compounds as potent photosensitizers in PDT were obtained for the pentanuclear species having ZnII or PdII as central metal ions. The [(PtCl2)4LZn] and [(PtCl2)4LPd] complexes are of special interest as potential bimodal anticancer agents because of the incorporated four cis-platin-like functionalities.
Co-reporter:Ping Chen, Olga S. Finikova, Zhongping Ou, Sergei A. Vinogradov, and Karl M. Kadish
Inorganic Chemistry 2012 Volume 51(Issue 11) pp:6200-6210
Publication Date(Web):May 24, 2012
DOI:10.1021/ic3003367
Fourteen platinum(II) porphyrins with different π-conjugated macrocycles and different electron-donating or electron-withdrawing substituents were investigated as to their electrochemical and spectroscopic properties in nonaqueous media. Eight compounds have the formula (Ar4P)PtII, where Ar4P = the dianion of a tetraarylporphyrin, while six have π-extented macrocycles with four β,β′-fused benzo or naphtho groups and are represented as (TBP)PtII and (TNP)PtII where TBP and TNP are the dianions of tetrabenzoporphyrin and tetranaphthoporphyrin, respectively. Each Pt(II) porphyrin undergoes two reversible one-electron reductions and one to three reversible one-electron oxidations in nonaqueous media. These reactions were characterized by cyclic voltammetry, UV–visible thin-layer spectroelectrochemistry and in some cases by ESR spectroscopy. The two reductions invariably occur at the conjugated π-ring system to yield relatively stable Pt(II) π-anion radicals and dianions. The first oxidation leads to a stable π-cation radical for each investigated porphyrin; but in the case of tetraarylporphyrins containing electron-withdrawing substituents, the product of the second oxidation may undergo an internal electron transfer to give a Pt(IV) porphyrin with an unoxidized macrocycle. The effects of macrocycle structure on UV–visible spectra, oxidation/reduction potentials, and site of electron transfer are discussed.
Co-reporter:Sara Nardis, Manuela Stefanelli, Pruthviraj Mohite, Giuseppe Pomarico, Luca Tortora, Machima Manowong, Ping Chen, Karl M. Kadish, Frank R. Fronczek, Gregory T. McCandless, Kevin M. Smith, and Roberto Paolesse
Inorganic Chemistry 2012 Volume 51(Issue 6) pp:3910-3920
Publication Date(Web):March 6, 2012
DOI:10.1021/ic3002459
Two different methods for the regioselective nitration of different meso-triarylcorroles leading to the corresponding β-substituted nitrocorrole iron complexes have been developed. A two-step procedure affords three Fe(III) nitrosyl products—the unsubstituted corrole, the 3-nitrocorrole, and the 3,17-dinitrocorrole. In contrast, a one-pot synthetic approach drives the reaction almost exclusively to formation of the iron nitrosyl 3,17-dinitrocorrole. Electron-releasing substituents on the meso-aryl groups of the triarylcorroles induce higher yields and longer reaction times than what is observed for the synthesis of similar triarylcorroles with electron-withdrawing functionalities, and these results can be confidently attributed to the facile formation and stabilization of an intermediate iron corrole π-cation radical. Electron-withdrawing substituents on the meso-aryl groups of triarylcorrole also seem to labilize the axial nitrosyl group which, in the case of the pentafluorophenylcorrole derivative, results in the direct formation of a disubstituted iron μ-oxo dimer complex. The influence of meso-aryl substituents on the progress and products of the nitration reaction was investigated. In addition, to elucidate the most important factors which influence the redox reactivity of these different iron nitrosyl complexes, selected compounds were examined by cyclic voltammetry and thin-layer UV–visible or FTIR spectroelectrochemistry in CH2Cl2.
Co-reporter:Manuela Stefanelli, Giuseppe Pomarico, Luca Tortora, Sara Nardis, Frank R. Fronczek, Gregory T. McCandless, Kevin M. Smith, Machima Manowong, Yuanyuan Fang, Ping Chen, Karl M. Kadish, Angela Rosa, Giampaolo Ricciardi, and Roberto Paolesse
Inorganic Chemistry 2012 Volume 51(Issue 12) pp:6928-6942
Publication Date(Web):June 5, 2012
DOI:10.1021/ic3007926
Functionalization of the β-pyrrolic positions of the corrole macrocycle with −NO2 groups is limited at present to metallocorrolates due to the instability exhibited by corrole free bases under oxidizing conditions. A careful choice of the oxidant can limit the transformation of corroles into decomposition products or isocorrole species, preserving the corrole aromaticity, and thus allowing the insertion of nitro groups onto the corrole framework. Here we report results obtained by reacting 5,10,15-tritolylcorrole (TTCorrH3) with the AgNO2/NaNO2 system, to give mono- and dinitrocorrole derivatives when stoichiometry is carefully controlled. Reactions were found to be regioselective, affording the 3-NO2TTCorrH3 and 3,17-(NO2)2TTCorrH3 isomers as the main products in the case of mono- and disubstitution, in 53 and 20% yields, respectively. In both cases, traces of other mono- and disubstituted isomers were detected, which were structurally characterized by X-ray crystallography. The influence of the β-nitro substituents on the corrole properties is studied in detail by UV–visible, electrochemical, and spectroelectrochemical characterization of these functionalized corroles. Density functional theory (DFT) and time-dependent DFT (TDDFT) calculations of the ground and excited state properties of these β-nitrocorrole derivatives also afforded significant information, closely matching the experimental observations. It is found that the β-NO2 substituents conjugate with the π-aromatic system of the macrocycle, which initiates significant changes in both the spectroscopic and redox properties of the so functionalized corroles. This effect is more pronounced when the nitro group is introduced at the 2-position, because in this case the conjugation is, for steric reasons, more efficient than in the 3-nitro isomer.
Co-reporter:Zhongping Ou, Aixiang Lü, Deying Meng, Shi Huang, Yuanyuan Fang, Guifen Lu, and Karl M. Kadish
Inorganic Chemistry 2012 Volume 51(Issue 16) pp:8890-8896
Publication Date(Web):August 3, 2012
DOI:10.1021/ic300886s
Five meso-substituted cobalt(III) corroles were examined as to their catalytic activity for the electoreduction of O2 when coated on an edge-plane pyrolytic graphite electrode in 1.0 M HClO4. The investigated compounds are represented as (TpRPCor)Co(PPh3), where TpRPCor is the trianion of a para-substituted triphenylcorrole and R = OMe, Me, H, F, or Cl. Three electrochemical techniques, cyclic voltammetry, linear sweep voltammetry with a rotating disk electrode (RDE), and voltammetry at a rotating ring disk electrode (RRDE), were utilized to evaluate the catalytic activity of the corroles in the reduction of O2. Cobalt corroles containing electron-withdrawing substituents were shown to be better catalysts than those having electron-donating groups on the three meso-phenyl rings of the triarylcorroles.
Co-reporter:Andrei Kozyrev, Manivannan Ethirajan, Ping Chen, Kei Ohkubo, Byron C. Robinson, Kathleen M. Barkigia, Shunichi Fukuzumi, Karl M. Kadish, and Ravindra K. Pandey
The Journal of Organic Chemistry 2012 Volume 77(Issue 22) pp:10260-10271
Publication Date(Web):October 22, 2012
DOI:10.1021/jo301895p
A series of new bacteriochlorins was synthesized using 132-oxo-bacteriopyropheophorbide a (derived from bacteriochlorophyll a) as a starting material, which on reacting with o-phenylenediamine and 1,10-diaminonaphthalene afforded highly conjugated annulated bacteriochlorins with fused quinoxaline, benzimidazole, and perimidine rings, respectively. The absorption spectra of these novel bacteriochlorins demonstrated remarkably red-shifted intense Qy absorption bands observed in the range of 816–850 nm with high molar extinction coefficients (89,900–136,800). Treatment of 132-oxo-bacteriopyropheophorbide a methyl ester with diazomethane resulted in the formation of bacterioverdins containing a fused six-membered methoxy-substituted cyclohexenone (verdin) as an isomeric mixture. The pure isomers which exhibit long-wavelength absorptions in the near-IR region (865–890 nm) are highly stable at room temperature with high reactivity with O2 at the triplet photoexcited state and favorable redox potential and could be potential candidates for use as photosensitizers in photodynamic therapy (PDT).
Co-reporter:Zhen Fu, Min Zhang, Weihua Zhu, Elizabeth Karnas, Kentaro Mase, Kei Ohkubo, Jonathan L. Sessler, Shunichi Fukuzumi, and Karl M. Kadish
The Journal of Physical Chemistry A 2012 Volume 116(Issue 41) pp:10063-10073
Publication Date(Web):September 18, 2012
DOI:10.1021/jp3074706
The electroreduction and acid–base properties of dipyrrolylquinoxalines of the form H2DPQ, H2DPQ(NO2), and H2DPQ(NO2)2 were investigated in benzonitrile (PhCN) containing 0.1 M tetra-n-butylammonium perchlorate (TBAP). This study focuses on elucidating the complete electrochemistry, spectroelectrochemistry, and acid–base properties of H2DPQ(NO2)n (n = 0, 1, or 2) in PhCN before and after the addition of trifluoroacetic acid (TFA), tetra-n-butylammonium hydroxide (TBAOH), tetra-n-butylammonium fluoride (TBAF), or tetra-n-butylammonium acetate (TBAOAc) to solution. Electrochemical and spectroelectrochemical data provide support for the formation of a monodeprotonated anion after disproportionation of a dipyrrolylquinoxaline radical anion produced initially. The generated monoanion is then further reduced in two reversible one-electron-transfer steps at more negative potentials in the case of H2DPQ(NO2) and H2DPQ(NO2)2. Electrochemically monitored titrations of H2DPQ(NO2)n with OH–, F–, or OAc– (in the form of TBA+X– salts) give rise to the same monodeprotonated H2DPQ(NO2)n produced during electroreduction in PhCN. This latter anion can then be reduced in two additional one-electron-transfer steps in the case of H2DPQ(NO2) and H2DPQ(NO2)2. Spectroscopically monitored titrations of H2DPQ(NO2)n with X– show a 1:2 stoichiometry and provide evidence for the production of both [H2DPQ(NO2)n]− and XHX–. The spectroscopically measured equilibrium constants range from log β2 = 5.3 for the reaction of H2DPQ with TBAOAc to log β2 = 8.8 for the reaction of H2DPQ(NO2)2 with TBAOH. These results are consistent with a combined deprotonation and anion binding process. Equilibrium constants for the addition of one H+ to each quinoxaline nitrogen of H2DPQ, H2DPQ(NO2), and H2DPQ(NO2)2 in PhCN containing 0.1 M TBAP were also determined via electrochemical and spectroscopic means; this gave rise to log β2 values ranging from 0.7 to 4.6, depending upon the number of nitro substituents present on the H2DPQ core. The redox behavior of the H2DPQ(NO2)n compounds of the present study were further analyzed through comparisons with simple quinoxalines that lack the two linked pyrrole groups, i.e., Q(NO2)n where n = 0, 1, or 2. It is concluded that the pyrrolic substituents play a critical role in regulating the electrochemical and spectroscopic features of DPQs.
Co-reporter:Shun Sugawara;Yusuke Hirata;Dr. Satoshi Kojima; Yohsuke Yamamoto;Dr. Eigo Miyazaki; Kazuo Takimiya;Dr. Shiro Matsukawa;Dr. Daisuke Hashizume;Dr. John Mack; Nagao Kobayashi;Zhen Fu; Karl M. Kadish;Young Mo Sung;Dr. Kil Suk Kim; Dongho Kim
Chemistry - A European Journal 2012 Volume 18( Issue 12) pp:3566-3581
Publication Date(Web):
DOI:10.1002/chem.201101846
Abstract
A two-electron oxidation of the CuII (9) and ZnII (12) complexes of tetraphenyltetrabenzoporphyrin (TPTBP) results in the formation of stable antiaromatic [(TPTBP)CuII(H2O)]2+⋅2 [SbF6]− (10) and [(TPTBP)ZnII(H2O)2]2+⋅2 [SbF6]− (13) with 16π electrons on the inner ligand perimeter. X-ray structures of the parent TPTBP complexes, the dications, and singly oxidized species [(TPTBP)CuII]⋅+[SbF6]− (11) reveal that the use of TPTBP rather than a porphyrin ligand reduces the degree of nonplanarity in the 16π-electron species relative to the parent 18π complex. Significant high-field shifts of the 1H NMR signals of the outer ring protons and large positive values in calculations of nucleus-independent chemical shifts on the central cavity of the porphyrin ring provide unambiguous evidence for the antiaromatic character of the 16π ZnII species. A combination of magnetic circular dichroism spectroscopic studies and TD-DFT calculations on both the ZnII and CuII species demonstrates that the main electronic bands of the dicationic species can be readily assigned by using Michl’s 4N perimeter model. Femtosecond transient absorption studies clearly demonstrated that the number of π electrons on the inner ligand perimeter and the configuration of the central metal ion play a critical role in the excited-state relaxation dynamics. Redox potentials for conversion between the 16π, 17π, and 18π systems were measured by cyclic voltammetry in dichloromethane and benzonitrile, and UV/Vis spectra of each oxidation/reduction product were monitored by thin-layer spectroelectrochemistry.
Co-reporter:Shunichi Fukuzumi ; Kentaro Mase ; Kei Ohkubo ; Zhen Fu ; Elizabeth Karnas ; Jonathan L. Sessler
Journal of the American Chemical Society 2011 Volume 133(Issue 19) pp:7284-7287
Publication Date(Web):April 21, 2011
DOI:10.1021/ja200925e
Disproportionation of dipyrrolylquinoxaline radical anions occurs via hydrogen atom transfer from the pyrrole moiety to the quinoxaline moiety to produce monodeprotonated dipyrrolylquinoxaline anions and monohydrodipyrrolylquinoxaline anions. In contrast, simple quinoxaline radical anions without pyrrole moieties are stable, and disproportionation occurs only in the presence of external protons.
Co-reporter:Ping Chen ; Maya El Ojaimi ; Claude P. Gros ; Philippe Richard ; Jean-Michel Barbe ; Roger Guilard ; Jing Shen
Inorganic Chemistry 2011 Volume 50(Issue 8) pp:3479-3489
Publication Date(Web):March 15, 2011
DOI:10.1021/ic102399g
A series of homobimetallic manganese cofacial porphyrin-corrole dyads were synthesized and investigated as to their electrochemistry, spectroelectrochemistry, and ligand binding properties in nonaqueous media. Four dyads were investigated, each of which contained a Mn(III) corrole linked in a face-to-face arrangement with a Mn(III) porphyrin. The main difference between compounds in the series is the type of spacer, 9,9-dimethylxanthene, anthracene, dibenzofuran, or diphenylether, which determines the distance and interaction between the metallomacrocycles. Each redox process of the porphyrin-corrole dyads was assigned on the basis of spectroscopic and electrochemical data and by comparison with reactions and properties of the monocorrole and the monoporphyrin which were examined under the same solution conditions. The Mn(III) porphyrin part of the dyad undergoes two major one-electron reductions in pyridine and benzonitrile, the first of which involves a Mn(III)/Mn(II) process and the second the addition of an electron to the conjugated π-ring system of the macrocycle. The Mn(III) corrole part of the dyads also exhibits two major redox processes, one involving Mn(III)/Mn(II) and the other Mn(III) to Mn(IV) under the same solution conditions. The potentials and reversibility of each electron transfer reaction were shown to depend upon the solvent, type of spacer separating the two macrocycles, and the presence or absence of axial ligation, the latter of which was investigated in detail for the case of acetate ion which was found to bind within the cavity of the dyad to both manganese centers, both before and after the stepwise electroreduction to the Mn(II) forms of the two macrocycles. An intramolecular chloride ion exchange between the porphyrin part of the dyads which contain MnIIICl and the singly oxidized corrole in the dyad is observed after the Mn(III)/Mn(IV) reaction of the corrole, suggesting that chloride is coordinated inside the cavity in the neutral compound.
Co-reporter:Manuela Stefanelli, Federica Mandoj, Marco Mastroianni, Sara Nardis, Pruthviray Mohite, Frank R. Fronczek, Kevin M. Smith, Karl M. Kadish, Xiao Xiao, Zhongping Ou, Ping Chen, and Roberto Paolesse
Inorganic Chemistry 2011 Volume 50(Issue 17) pp:8281-8292
Publication Date(Web):July 28, 2011
DOI:10.1021/ic2008073
Copper and germanium complexes of β-substituted nitrocorroles were reacted with 4-amino-4H-1,2,4-triazole to give the corresponding β-amino-β-nitro derivatives, in moderate to good yields. This is the first successful example of a vicarious nucleophilic substitution performed on corrole derivatives, because the same reaction carried out on silver complexes afforded the corresponding 6-azahemiporphycenes by way of corrole ring expansion. The first step of this work is related to the modification of a synthetic protocol for preparation of the β-substituted nitro corroles. The nitration reaction was carried out on a copper corrole using NaNO2 as the primary source of NO2– coupled with AgNO2 used as oxidant. By variation of the molar ratio of the reagents it was possible to direct the product distribution toward mono- and dinitro derivatives. The reaction between mono- and dinitro derivatives of (TtBuCorrCu) with 4-amino-4H-1,2,4-triazole gave good results, leading to the isolation of 2-(NH2)-3-(NO2)-TtBuCorrCu and 2,18-(NH2)2-3,17-(NO2)2-TtBuCorrCu in moderate yields. To elucidate factors that influence the reaction, and to highlight the different behavior observed for different metal complex substrates, the electrochemistry of three copper complexes, TtBuPCorrCu, (NO2)TtBuPCorrCu, and (NO2)2TtBuPCorrCu, was studied by cyclic voltammetry and thin-layer UV–visible spectroelectrochemistry. The nitro groups on (NO2)xTtBuPCorrCu are highly electron-withdrawing, which leads not only to a substantial positive shift of all redox potentials but also to a unique redox behavior and UV–vis spectrum of the singly reduced product as compared to the parent compound, TtBuPCorrCu. Finally, the amination reaction was carried out on a Ge(IV) nitrocorrolate, giving in good yield the 2-amino-3-nitroderivative, which was structurally characterized by single crystal X-ray crystallography.
Co-reporter:Giorgia De Mori, Zhen Fu, Elisa Viola, Xiaohui Cai, Claudio Ercolani, Maria Pia Donzello, and Karl M. Kadish
Inorganic Chemistry 2011 Volume 50(Issue 17) pp:8225-8237
Publication Date(Web):July 27, 2011
DOI:10.1021/ic2007556
A series of pyrazinoporphyrazine macrocycles carrying externally appended 2-thienyl rings, represented as [Th8TPyzPzM], where Th8TPyzPz = tetrakis-2,3-[5,6-di(2-thienyl)pyrazino]porphyrazinato anion and M = MgII(H2O), ZnII, CoII, CuII, or 2H1, were prepared and isolated as solid air-stable hydrated species. All of the compounds, completely insoluble in water, were characterized by their UV–visible spectra and electrochemical behavior in solutions of dimethylformamide (DMF), dimethyl sulfoxide, and pyridine. Molecular aggregation occurs at concentrations of ca. 10–4 M, but monomers are formed in more dilute solutions of 10–5 M or less. The examined octathienyl compounds [Th8TPyzPzM] behave as electron-deficient macrocycles, and UV–visible spectral measurements provide useful information about how the peripheral thienyl rings influence the electronic distribution over the entire macrocyclic framework. Cyclic voltammetric and spectroelectrochemical data confirm the easier reducibility of the compounds as compared to the related phthalocyanine analogues, and the overall redox behavior and thermodynamic potentials for the four stepwise one-electron reductions of the compounds are similar to those of the earlier examined octapyridinated analogues [Py8TPyzPzM]. Quantum yields (ΦΔ) for the generation of singlet oxygen, 1O2, the cytotoxic agent active in photodynamic therapy (PDT), and fluorescence quantum yields (ΦF) were measured for the ZnII and MgII complexes, [Th8TPyzPzZn] and [Th8TPyzPzMg(H2O)], and the data were compared to those of corresponding octapyridino macrocycles [Py8TPyzPzZn] and [Py8TPyzPzMg(H2O)] and their related octacations [(2-Mepy)8TPyzPzZn]8+ and [(2-Mepy)8TPyzPzMg(H2O)]8+. These measurements were carried out in DMF and in DMF preacidified with HCl (ca. 10–4 M). All of the examined ZnII compounds behave as excellent photosensitizers (ΦΔ = 0.4–0.6) both in DMF and DMF/HCl solutions, whereas noticeable fluorescence activity (ΦF = 0.36–0.43) in DMF/HCl solutions is shown by the MgII derivatives; these data might provide perspectives for applications in PDT (ZnII) and imaging response and diagnosis (MgII).
Co-reporter:Zhongping Ou, Weihua Zhu, Yuanyuan Fang, Paul J. Sintic, Tony Khoury, Maxwell J. Crossley, and Karl M. Kadish
Inorganic Chemistry 2011 Volume 50(Issue 24) pp:12802-12809
Publication Date(Web):November 9, 2011
DOI:10.1021/ic2019567
The electrochemistry of gold(III) mono- and bis-quinoxalinoporphyrins was examined in CH2Cl2 or PhCN containing 0.1 M tetra-n-butylammonium perchlorate (TBAP) before and after the addition of trifluoroacetic acid to solution. The investigated porphyrins are represented as Au(PQ)PF6 and Au(QPQ)PF6, where P is the dianion of the 5,10,15,20-tetrakis(3,5-di-tert-butylphenyl)porphyrin and Q is a quinoxaline group fused to a β,β′-pyrrolic position of the porphyrin macrocycle; in Au(QPQ)PF6 there is a linear arrangement where the quinoxalines are fused to pyrrolic positions that are opposite each other. The porphyrin without the fused quinoxaline groups, Au(P)PF6, was also investigated under the same solution conditions. In the absence of acid, all three gold(III) porphyrins undergo a single reversible AuIII/AuII process leading to the formation of a Au(II) porphyrin which can be further reduced at more negative potentials to give stepwise the Au(II) porphyrin π-anion radical and dianion, respectively. However, in the presence of acid, the initial AuIII/AuII processes of Au(PQ)PF6 and Au(QPQ)PF6 are followed by an internal electron transfer and protonation to regenerate new Au(III) porphyrins assigned as AuIII(PQH)+ and AuIII(QPQH)+. Both protonated gold(III) quinoxalinoporphyrins then undergo a second AuIII/AuII process at more negative potentials. The electrogenerated monoprotonated monoquinoxalinoporphyrin, AuII(PQH), is then further reduced to its π-anion radical and dianion forms, but this is not the case for the monoprotonated bis-quinoxalinoporphyrin, AuII(QPQH), which accepts a second proton and is rapidly converted to AuIII(HQPQH)+ before undergoing a third AuIII/AuII process to produce AuII(HQPQH) as a final product. Thus, Au(P)PF6 undergoes one metal-centered reduction while Au(PQ)PF6 and Au(QPQ)PF6 exhibit two and three AuIII/AuII processes, respectively. These unusual multistep sequential AuIII/AuII processes were monitored by thin-layer spectroelectrochemistry and a reduction/oxidation mechanism for Au(PQ)PF6 and Au(QPQ)PF6 in acidic media is proposed.
Co-reporter:Karl M. Kadish, Ping Chen, Yulia Yu. Enakieva, Sergey E. Nefedov, Yulia G. Gorbunova, Aslan Yu. Tsivadze, Alla Bessmertnykh-Lemeune, Christine Stern, Roger Guilard
Journal of Electroanalytical Chemistry 2011 Volume 656(1–2) pp:61-71
Publication Date(Web):15 June 2011
DOI:10.1016/j.jelechem.2011.01.011
The synthesis and electrochemical characterization of two related series of porphyrins bearing diethoxyphosphoryl groups are reported. One group of compounds is represented as (T(p-R)PP)M where R = phos = P(O)(OEt)2 and M = Zn(II) or H2 while the other is represented as (di(p-R)Pdi(phos)P)M where R = P(O)(OEt)2, H or CH3 and M = Zn(II) or H2. Each porphyrin was investigated by electrochemistry and thin-layer spectroelectrochemistry in CH2Cl2, CDCl3, CHCl3 or PhCN containing tetra-n-butylammonium perchlorate (TBAP) as supporting electrolyte. The highly electron-withdrawing P(O)(OEt)2 groups lead to easier reductions and harder oxidations than the two comparison compounds, (TPP)Zn and (TPP)H2 where TPP = the dianion of the tetraphenylporphyrin. The P(O)(OEt)2 groups located on the two meso-positions of the porphyrin macrocycle in (di(p-R)Pdi(phos)P)M or on the para-positions of the substituted phenyl groups in (T(p-R)PP)M can also bind to the Zn(II) ion of another porphyrin in solution, leading to the formation of aggregates which was both concentration and solvent dependent as determined by UV–visible spectroscopy and electrochemistry. Binding constants for addition of triphenylphosphine oxide (PPh3O) to the zinc porphyrins were also measured in CDCl3 and ranged from log K = 1.7 to 3.1 for formation of the five-coordinate complex. The comparison of X-ray diffraction data for the investigated diethoxyphosphoryl-substituted porphyrin (diPdi(phos)P)H2 and its Zn complex showed that the metallation induces almost no change in the geometry of the porphyrin macrocycle, but plays the key role in the process of supramolecular assembling.Research highlights► Cross-coupling methodology towards poly(phosphoryl)porphyrins. ► Phosphorylporphyrins – new structural building blocks for supramolecular networks. ► Influence of position of phosphoryl groups in porphyrin on redox and self-assembling properties.
Co-reporter:Giuseppe Pomarico ; Xiao Xiao ; Sara Nardis ; Roberto Paolesse ; Frank R. Fronczek ; Kevin M. Smith ; Yuanyuan Fang ; Zhongping Ou
Inorganic Chemistry 2010 Volume 49(Issue 12) pp:5766-5774
Publication Date(Web):May 25, 2010
DOI:10.1021/ic100730j
A series of free-base and metalated isocorroles represented as (TT-n-iso-Cor)H2 and (TT-n-iso-Cor)MII, where n = 5 or 10 and M = Ni or Cu, were synthesized and characterized by electrochemistry and spectroelectrochemistry in CH2Cl2 containing 0.1 M tetra-n-butylammonium perchlorate. The metalation of the free-base macrocycles with CoII, MnIII, or ZnII was also attempted but was unsuccessful. Six isocorroles were isolated and shown to undergo two stepwise oxidations to give π-cation radicals and dications in CH2Cl2, with the most stable products being obtained in the case of the 10-substituted derivatives. The same isocorroles could also be reduced by one or two electrons, but the initial one-electron addition products are unstable and undergo a rapid chemical reaction giving a reduced corrole or corrole-like product, which could be reoxidized to the corresponding (TTCor)M at a controlled positive potential. This series of reactions effectively illustrates an isocorrole to corrole conversion upon reduction and reoxidation and was monitored by both electrochemistry and thin-layer spectroelectrochemistry.
Co-reporter:Weihua Zhu ; Maxine Sintic ; Zhongping Ou ; Paul J. Sintic ; James A. McDonald ; Peter R. Brotherhood ; Maxwell J. Crossley
Inorganic Chemistry 2010 Volume 49(Issue 3) pp:1027-1038
Publication Date(Web):December 22, 2009
DOI:10.1021/ic901851u
A series of cobalt(II) and cobalt(III) porphyrins with fused quinoxaline rings at one or more β,β′-pyrrolic units of the macrocycle were synthesized and characterized as to their electrochemical properties in nonaqueous media. Their UV−visible spectra were also measured before and during oxidation or reduction in a thin-layer cell. The investigated quinoxalinoporphyrins are represented as (PQ)Co, (QPQ)CoCl, (PQ2)CoCl, Co(P)-TA-(P)Co, and Co(PQ)-(QP)Co, where PQ = the dianion of 5,10,15,20-tetrakis(3,5-di-tert-butylphenyl)-quinoxalino[2,3-b′]porphyrin, QPQ = the dianion of the corresponding linear bisquinoxalino[2,3-b′:12,13-b′′]porphyrin, PQ2 = the dianion of the corresponding corner bisquinoxalino[2,3-b′:7,8-b′′]porphyrin, and (P)-TA-(P) = the tetraanion of the bis-porphyrin with 5,10,15,20-tetrakis(3,5-di-tert-butylphenyl)porphyrins fused at opposite ends of tetraazaanthracene. (P)Co and (P)CoCl were also characterized where P = the dianion of 5,10,15,20-tetrakis(3,5-di-tert-butylphenyl)porphyrin. Each compound could be cycled between their Co(III), Co(II), and Co(I) forms under the application of a given oxidizing or reducing potential, although a one-electron reduction of the Co(II) quinoxalinoporphyrins led to a product with mixed Co(I) and porphyrin π-anion radical character followed by generation of a pure Co(I) π-anion radical species at more negative potentials. The effect of the position and number of quinoxaline groups on the redox potentials and mechanisms of each electron transfer were elucidated, and comparisons made to structurally similar compounds containing both redox active and redox inactive central metal ions. Surprisingly, the position and number of quinoxaline groups on the macrocycle has little or no effect on the redox potentials for the Co(II) → Co(III) or Co(III) → Co(II) processes, but this is not the case for other electron transfer reactions where significant differences are seen between the examined compounds. Significant interactions are also observed between the two porphyrin macrocycles of the laterally bridged dicobalt(II) bis-porphyrin dyad Co(P)-TA-(P)Co in its singly and doubly reduced form, but only weak interactions are seen between the two Co(PQ) units of the single bond biquinolalinyl-bridged dicobalt(II) bis-porphyrin dyad Co(PQ)-(QP)Co.
Co-reporter:Maria Pia Donzello ; Elisa Viola ; Xiaohui Cai ; Luisa Mannina ; Claudio Ercolani
Inorganic Chemistry 2010 Volume 49(Issue 5) pp:2447-2456
Publication Date(Web):January 26, 2010
DOI:10.1021/ic902317h
A series of heteropentametallic porphyrazine macrocycles, represented as [(PdCl2)4LM], where L = dianion of tetrakis-2,3-[5,6-di(2-pyridyl)pyrazino]porphyrazine and M = ZnII, CuII MgII(H2O) or CdII, were prepared by reaction of the corresponding mononuclear [LM] species, and their behavior was examined by UV−visible and NMR spectroscopy, electrochemistry, and thin layer spectroelectrochemistry in nonaqueous media. The PdCl2 units in [(PdCl2)4LM] are coordinated at the pyridine N atoms of the external dipyridinopyrazine fragments (“py-py” coordination) and are displaced out of the plane of the central pyrazinoporphyrazine macrocycle as verified by 1H and 13C NMR data on [(PdCl2)4LZn]. The same arrangement is also strongly suggested by similar NMR data on the MgII and CdII analogues. The predominant component in the synthesized materials among the four predictable macrocyclic isomers has the four exocyclic N2(py)PdCl2 square planar coordination sites on the same side of the central macrocyclic framework (4:0 isomer, C4v symmetry), and this is accompanied by a minor isomeric component (2:2 cis or trans), in line with previous findings on the pentapalladated species [(PdCl2)4LPd]. IR, UV−visible, and NMR spectral data also provide evidence for transmetalation reactions of the type [(PdCl2)4LMg(H2O)] → [(PdCl2)4LPd] and [(PdCl2)4LCd] → [(PdCl2)4LPd], with the amount of [(PdCl2)4LPd] formed varying from batch to batch. Dissociation of the four exocyclic PdCl2 units from [(PdCl2)4LM] occurs in pyridine, but the compounds are stable in N,N-dimethylformamide (DMF) or dimethylsulfoxide (DMSO) and can be stepwise reduced via two one-electron reversible or quasi-reversible processes, prior to an irreversible electroreduction of the bound PdCl2 group at more negative potentials. This metal-centered reduction leads to a [LM]2− product which is then further reduced to [LM]3− and [LM]4− at the electrode surface. The first two reductions of the heteropentametallic compounds are easier than those of the monometallic [LM] species but generally more difficult than reduction of the related octacationic [L′M]8+ derivatives (L′ = the octamethylated free-base dianion) whose redox properties were previously reported. The CdII octacation [L′Cd]8+, isolated as an iodide salt, was also synthesized for the first time in the current study, and its spectroscopic and electrochemical properties are compared to that of the previously examined analogues.
Co-reporter:Jean-Michel Barbe, Benoit Habermeyer, Tony Khoury, Claude P. Gros, Philippe Richard, Ping Chen, and Karl M. Kadish
Inorganic Chemistry 2010 Volume 49(Issue 19) pp:8929-8940
Publication Date(Web):September 7, 2010
DOI:10.1021/ic101170k
The synthesis and characterization of a new type of bisporphyrin system is reported where the two macrocycles are linked in a cofacial arrangement by a substituted carbazole bridge. The three nitrogen atoms of the carbazole bridge in the compounds may complex a metal ion and thus provide a new parameter for varying the physical properties and flexibility of the dyad after formation of a three-metal system. In the present study, four bis-metalloporphyrin complexes were synthesized and examined by electrochemistry and thin-layer spectroelectrochemistry in CH2Cl2 and PhCN. Two of the examined compounds contain Cu(II) or Zn(II) porphyrins and a carbazole linker with a bound Cu(II) ion, giving a three metal system, while the other two examined compounds contained the same porphyrins but with a carbazole bridge which lacks the Cu(II) component. Since carbazoles and Cu(II) ions are both electroactive, redox properties of several unlinked carbazoles with and without bound Cu(II) ions were also examined as to their electrochemical behavior under the same solution conditions as the dyads to better understand the redox reactions which may occur at the carbazole group linking the two porphyrin macrocycles. Several mononuclear porphyrins with structures related to macrocycles in the dyads were also investigated.
Co-reporter:Zhongping Ou, Ping Chen and Karl M. Kadish
Dalton Transactions 2010 vol. 39(Issue 46) pp:11272-11276
Publication Date(Web):25 Oct 2010
DOI:10.1039/C0DT00899K
The first example for electrogeneration of a Pt(IV) porphyrin from its Pt(II) form is presented and the PtII/IV and reverse PtIV/II oxidation-reduction processes are elucidated by electrochemistry and thin-layer UV-visible spectroelectrochemistry. Three products, [(TPP˙+)PtII]+, [(TPP)PtIV]2+ and [(TPP˙+)PtIV]3+, produced by electrooxidation of the Pt(II) porphyrin have been characterized by in situ spectroelectrochemistry and ESR measurements after controlled potential bulk electrolysis. The first definitive evidence for the electrochemical conversion of a Pt(IV) porphyrin to its Pt(II) form is also presented. The potential for this electroreduction is highly dependent upon the nature of the anion, ClO4− or Cl−. A mechanism for the reversible conversion between Pt(II) and Pt(IV) tetraphenylporphyrins is proposed.
Co-reporter:Luciano Cuesta ; Elizabeth Karnas ; Vincent M. Lynch ; Ping Chen ; Jing Shen ; Karl M. Kadish ; Kei Ohkubo ; Shunichi Fukuzumi ;Jonathan L. Sessler
Journal of the American Chemical Society 2009 Volume 131(Issue 37) pp:13538-13547
Publication Date(Web):August 31, 2009
DOI:10.1021/ja905284d
Unprecedented porphycene complexes, containing a [RuCp*] (Cp*: pentamethylcyclopentadienyl) fragment accommodated in the central N4 core or directly bonded to the “π-face” of the macrocycle have been prepared and fully characterized, including via single crystal X-ray diffraction analysis. The optical and electrochemical properties of these new families of compounds were examined in detail, revealing fluorescence in the case of the “sitting-atop” complexes for which the lifetime was determined. For both metal (M = Cu, Ni) porphycene derivatives with a “fused” ruthenocene moiety, strong electronic communication was observed through efficient photoinduced electron transfer from the ruthenocene unit to the macrocycle after laser flash photolysis, affording a charge-separated state. This ruthenocene-macrocycle communication was also confirmed by observation of strong spin−spin coupling in the EPR spectra of the one-electron oxidized species; this allowed for calculation of the distance between the two metal centers.
Co-reporter:Karl M. Kadish, Laurent Frémond, Jing Shen, Ping Chen, Kei Ohkubo, Shunichi Fukuzumi, Maya El Ojaimi, Claude P. Gros, Jean-Michel Barbe and Roger Guilard
Inorganic Chemistry 2009 Volume 48(Issue 6) pp:2571-2582
Publication Date(Web):February 12, 2009
DOI:10.1021/ic802092n
A series of biscobalt cofacial porphyrin-corrole dyads bearing mesityl substituents at the meso positions of the corrole ring were investigated as to their electrochemistry, spectroelectrochemistry, and CO binding properties in nonaqueous media and then applied to the surface of a graphite electrode and tested as electrocatalysts for the reduction of dioxygen to water or hydrogen peroxide in air-saturated aqueous solutions containing 1 M HClO4. The catalytic reduction of O2 with the same dyads was also investigated in the homogeneous phase using 1,1′-dimethylferrocene as a reductant in PhCN containing HClO4. The examined compounds are represented as (PMes2CY)Co2, where P = a porphyrin dianion, Mes2C = a corrole trianion with two mesityl groups in trans meso-positions of the macrocycle, and Y is one of three bridging groups separating the two metallomacrocycles in a face-to-face arrangement, either with 9,9-dimethylxanthene, dibenzofuran, or diphenylether as linkers. Cyclic voltammetry and rotating disk electrode voltammetry revealed that the examined compounds are all catalytically active toward the electroreduction of dioxygen in acid media giving H2O2 or H2O depending upon the type of linkage (Y) and the initial site of electron transfer which, in nonaqueous media, could be switched between the corrole and the porphyrin metal center by variations of substituents on the corrole macrocycle or the gas above the solution. The homogeneous reduction of dioxygen via a two- or four-electron transfer process was also investigated using 1,1′-dimethylferrocene as reductant in PhCN containing HClO4.
Co-reporter:Manuela Stefanelli, Jing Shen, Weihua Zhu, Marco Mastroianni, Federica Mandoj, Sara Nardis, Zhongping Ou, Karl M. Kadish, Frank R. Fronczek, Kevin M. Smith and Roberto Paolesse
Inorganic Chemistry 2009 Volume 48(Issue 14) pp:6879-6887
Publication Date(Web):June 23, 2009
DOI:10.1021/ic900859a
Several procedures for the demetalation of silver(III) corrolates have been tested. Acidic conditions induce removal of the silver ion but they can also promote concomitant oxidation of the corrole nucleus to an isocorrole species, the degree of which will depend upon the specific acidic media. This oxidation cannot be completely avoided by addition of hydrazine, particularly in the case of 3-NO2 substituted complexes which are quantitatively converted into the corresponding 3-NO2, 5-hydroxy isocorroles upon silver ion removal. Several β-nitro isocorrole products were isolated, and one was structurally characterized. Electrochemical and chemical reductive methods for silver(III) corrolates demetalation were then tested with the aim to avoid the formation of isocorroles. While reaction with sodium borohydride was shown to be quite effective to demetalate unsubstituted silver corrolates this was not the case for the β-nitro derivatives where the peripheral nitro group is reduced by borohydride giving the corresponding 3-amino free base corrole species. For the β-nitro corrole silver complexes, a successful approach was obtained using DBU/THF solutions which afforded the 3-NO2 corrole free-base compound as a single reaction product in good yield. These conditions were also effective for unsubstituted corroles although longer reaction times were necessary in this case. To study in greater detail the corrole demetalation behavior, selected Ag(III) derivatives were characterized by cyclic voltammetry in pyridine, and the demetalation products spectrally characterized after controlled potential reduction in a thin-layer spectroelectrochemical cell.
Co-reporter:Xiaohui Cai, Maria Pia Donzello, Elisa Viola, Corrado Rizzoli, Claudio Ercolani and Karl M. Kadish
Inorganic Chemistry 2009 Volume 48(Issue 15) pp:7086-7098
Publication Date(Web):July 7, 2009
DOI:10.1021/ic8023277
2,3-Dicyano-5,6-di-2-pyridylpyrazine, [(CN)2Py2Pyz], which autocyclotetramerizes to give the macrocycle tetrakis[5,6-di(2-pyridyl)-2,3-pyrazino]porphyrazine, [Py8TPyzPzH2], bearing externally four dipyridinopyrazine fragments, reacts with bis(benzonitrile)dichloroplatinum(II), [(C6H5CN)2PtCl2], in CH3CN, affording the monometalated species [(CN)2Py2PyzPtCl2]. Single-crystal X-ray work on this compound shows that PtII is bound to [(CN)2Py2Pyz] through the two pyridine N atoms (“py−py” coordination) in a way similar to that found for its monopalladium analogue, [(CN)2Py2PyzPdCl2]. Cyclic voltammetry of [(CN)2Py2PyzPtCl2] and [(CN)2Py2PyzPdCl2] in nonaqueous media (pyridine, DMSO, and DMF) indicates that the electron-withdrawing effect of the coordinated PtCl2 and PdCl2 units results in an initial one-electron reduction (E1/2 = −0.60 and −0.54 V vs SCE in DMSO, respectively), which is easier by 0.25−0.30 V than the unmetalated [(CN)2Py2Pyz] (first reduction: E1/2 = −0.87 V vs SCE). These electrochemical data are analyzed along with new results for a selected number of related pyrazine and 2,3-dicyanopyrazine molecules as well as earlier reported data on the mono- and bis-N-methylated derivatives [(CN)2Py(2-Mepy)Pyz]+ and [(CN)2(2-Mepy)2Pyz]2+, with these latter species being formed by reaction of the precursor [(CN)2Py2Pyz] with methyl iodide or p-toluensulfonate. The data in this study are also compared to electrochemical data previously reported for a triad of palladium(II) porphyrazine macrocycles obtained from the precursor [(CN)2Py2Pyz], i.e., [Py8TPyzPzPd], the corresponding pentanuclear complex [(PdCl2)4Py8TPyzPzPd] (presenting “py−py” coordination at the dipyridinopyrazine fragments), and the octacation [(2-Mepy)8TPyzPzPd]8+ (N-methylated at the pyridine rings). Thin-layer UV−visible spectra of singly reduced [(CN)2Py2Pyz]− and its metalated analogues, [(CN)2Py2PyzPtCl2]− and [(CN)2Py2PyzPdCl2]−, were measured in pyridine, DMF, and DMSO and show π−π* transitions, as well as unusually intense absorptions in the near-IR region (500−900 nm) of the spectrum.
Co-reporter:Federica Mandoj, Sara Nardis, Giuseppe Pomarico, Manuela Stefanelli, Luca Schiaffino, Gianfranco Ercolani, Luca Prodi, Damiano Genovese, Nelsi Zaccheroni, Frank R. Fronczek, Kevin M. Smith, Xiao Xiao, Jing Shen, Karl M. Kadish and Roberto Paolesse
Inorganic Chemistry 2009 Volume 48(Issue 21) pp:10346-10357
Publication Date(Web):October 1, 2009
DOI:10.1021/ic9014866
The reaction of 5,10,15-triarylcorrole with 4-amino-4H-1,2,4-triazole provides another example of corrole ring expansion to give the corresponding 6-azahemiporphycene, a novel porphyrin analogue. The facile oxidation of the corrole ring is a required step for the ring expansion and for this reason the reaction fails in the case of corroles bearing meso-phenyl groups carrying electron-withdrawing substituents. Steric requirements also limited the scope of the reaction, which is not successful in the case of 2,6-disubstituted meso-aryl corroles. The occurrence of an initial oxidation is further supported by formation of the 6-azahemiporphycene derivative when the reaction is carried out under the same conditions, using a 5- or a 10-isocorrole as starting material. 1H NMR spectra and X-ray crystal characterization of 6-azahemiporphycene evidenced the presence of an intramolecular N—H···N hydrogen bond in the inner core of the macrocycle, while photophysical characterization confirmed the aromatic character of the novel macrocycle, showing an intense Soret-like band around 410 nm in the absorption spectrum. The fluorescence emission is very modest, and 6-azahemiporphycene showed higher photostability than the corresponding corrole species. Different metal complexes of 6-azahemiporphycene were prepared following synthetic protocols usually exploited for the preparation of metalloporphyrins, demonstrating good coordination properties for the macrocycle. Both the free-base and metal derivatives were characterized by cyclic voltammetry and spectroelectrochemistry in dichloromethane and benzonitrile. To further detail the behavior of this novel macrocycle, density functional theory (DFT) calculations were carried out on the basic structure of 6-azahemiporphycene with the aim of assessing aromaticity and tautomerism, as well as calculating its stability with respect to the 5-aza isomer.
Co-reporter:Kei Ohkubo Dr.;Rachel Garcia;PaulJ. Sintic Dr.;Tony Khoury Dr.;MaxwellJ. Crossley Dr.;KarlM. Kadish Dr.;Shunichi Fukuzumi Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 40) pp:10493-10503
Publication Date(Web):
DOI:10.1002/chem.200901105
Abstract
The site of electron-transfer reduction of AuPQ+ (PQ=5,10,15,20-tetrakis(3,5-di-tert-butylphenyl)quino-xalino[2, 3−b′]porphyrin) and AuQPQ+ (QPQ=5,10,15,20-tetrakis(3,5-di-tert-butylphenyl)bisquinoxalino[2,3-b′:12,13-b′′]porphyrin) is changed from the AuIII center to the quinoxaline part of the PQ macrocycle in the presence of Sc3+ in benzonitrile because of strong binding of Sc3+ to the two nitrogen atoms of the quinoxaline moiety. Strong binding of Sc3+ to the corresponding nitrogen atoms on the quinoxaline unit of ZnPQ also occurs for the neutral form. The effects of Sc3+ on the photodynamics of an electron donor–acceptor compound containing a linked ZnII and AuIII porphyrin ([ZnPQ–AuPQ]PF6) have been examined by femto- and nanosecond laser flash photolysis measurements. The observed transient absorption bands at 630 and 670 nm after laser pulse irradiation in the absence of Sc3+ in benzonitrile are assigned to the charge-shifted (CS) state (ZnPQ.+–AuPQ). The CS state decays through back electron transfer (BET) to the ground state rather than to the triplet excited state. The BET rate was determined from the disappearance of the absorption band due to the CS state. The decay of the CS state obeys first-order kinetics. The CS lifetime was determined to be 250 ps in benzonitrile. Addition of Sc3+ to a solution of ZnPQ–AuPQ+ in benzonitrile caused a drastic lengthening of the CS lifetime that was determined to be 430 ns, a value 1700 times longer than the 250 ps lifetime measured in the absence of Sc3+. Such remarkable prolongation of the CS lifetime in the presence of Sc3+ results from a change in the site of electron transfer from the AuIII center to the quinoxaline part of the PQ macrocycle when Sc3+ binds to the quinoxaline moiety, which decelerate BET due to a large reorganization energy of electron transfer. The change in the site of electron transfer was confirmed by ESR measurements, redox potentials, and UV/Vis spectra of the singly reduced products.
Co-reporter:John L. Bear, Baocheng Han, Yulan Li, Siyabonga Ngubane, Eric Van Caemelbecke, Karl M. Kadish
Polyhedron 2009 28(8) pp: 1551-1555
Publication Date(Web):
DOI:10.1016/j.poly.2009.03.012
Co-reporter:Minh Nguyen, Tuan Phan, Eric Van Caemelbecke, Xin Wei, John L. Bear and Karl M. Kadish
Inorganic Chemistry 2008 Volume 47(Issue 10) pp:4392-4400
Publication Date(Web):April 17, 2008
DOI:10.1021/ic8000703
Two isothiocyanate diruthenium complexes, (3,1) Ru2(F3ap)4(NCS) 1 and (3,1) Ru2(F3ap)3(F2Oap)(NCS) 2 (where F3ap = 2,4,6-trifluoroanilinopyridinate anion), were synthesized from (3,1) Ru2(F3ap)4Cl and SCN− under different experimental conditions. Each compound was examined as to its structural, electrochemical, spectroscopic, and magnetic properties. Compound 1 contains three unpaired electrons as its parent compound but 2 is diamagnetic. The X-ray molecular structures of 1 and 2 reveal that the NCS group is coordinated to the dimetal unit via nitrogen in both compounds with the Ru−N−C bond angle being 176.5° for 1 and 166.0° for 2. An elongation of the Ru−Ru bond distance and a shortening of both the Ru−Np (p = pyridyl) and the Ru−Na (a = anilino) bond lengths is seen upon going from (3,1) Ru2(F3ap)4Cl to 2, but the conversion of (3,1) Ru2(F3ap)4Cl to 1 does not affect significantly structural features of the Ru2(L)4 framework. Compound 1 undergoes one reduction and two oxidations, all three of which involve the dimetal core, whereas 2 undergoes two metal-centered reductions, one metal-centered oxidation, and one ligand-based oxidation due to the presence of the F2Oap ligand on the Ru2 complex. The reactivity of 1 with SCN− was also investigated.
Co-reporter:Karl M. Kadish ; Jing Shen ; Laurent Frémond ; Ping Chen ; Maya El Ojaimi ; Mohammed Chkounda ; Claude P. Gros ; Jean-Michel Barbe ; Kei Ohkubo ; Shunichi Fukuzumi ;Roger Guilard
Inorganic Chemistry 2008 Volume 47(Issue 15) pp:6726-6737
Publication Date(Web):June 27, 2008
DOI:10.1021/ic800458s
Co(III) corroles were investigated as efficient catalysts for the reduction of dioxygen in the presence of perchloric acid in both heterogeneous and homogeneous systems. The investigated compounds are (5,10,15-tris(pentafluorophenyl)corrole)cobalt (TPFCor)Co, (10-pentafluorophenyl-5,15-dimesitylcorrole)cobalt (F5PhMes2Cor)Co, and (5,10,15-trismesitylcorrole)cobalt (Mes3Cor)Co, all of which contain bulky substituents at the three meso positions of the corrole macrocycle. Cyclic voltammetry and rotating ring-disk electrode voltammetry were used to examine the catalytic activity of the compounds when adsorbed on the surface of a graphite electrode in the presence of 1.0 M perchloric acid, and this data is compared to results for the homogeneous catalytic reduction of O2 in benzonitrile containing 10−2 M HClO4. The corroles were also investigated as to their redox properties in nonaqueous media. A reversible one-electron oxidation occurs at E1/2 values between 0.42 and 0.89 V versus SCE depending upon the solvent and number of fluorine substituents on the compounds, and this is followed by a second reversible one-electron abstraction at E1/2 = 0.86 to 1.18 V in CH2Cl2, THF, or PhCN. Two reductions of each corrole are also observed in the three solvents. A linear relationship is observed between E1/2 for oxidation or reduction and the number of electron-withdrawing fluorine groups on the compounds, and the magnitude of the substituent effect is compared to what is observed in the case of tetraphenylporphyrins containing meso-substituted C6F5 substituents. The electrochemically generated forms of the corrole can exist with Co(I), Co(II), or Co(IV) central metal ions, and the site of the electron-transfer in each oxidation or reduction of the initial Co(III) complex was examined by UV−vis spectroelectrochemistry. ESR characterization was also used to characterize singly oxidized (F5PhMes2Cor)Co, which is unambiguously assigned as a Co(III) radical cation rather than the expected Co(IV) corrole with an unoxidized macrocyclic ring.
Co-reporter:Minh Nguyen ; Tuan Phan ; Eric Van Caemelbecke ; Wiroaj Kajonkijya ; John L. Bear
Inorganic Chemistry 2008 Volume 47(Issue 17) pp:7775-7783
Publication Date(Web):August 1, 2008
DOI:10.1021/ic800787p
A reaction between the (4,0) isomer of Ru2(ap)4Cl and LiC≡CC5H4N leads to a (3,1) isomer of Ru2(ap)4(C≡CC5H4N)2 1 (ap = anilinopyridinate anion), whereas a reaction involving the (3,1) isomer of Ru2(F3ap)4Cl and TBACl·H2O leads to (4,0) Ru2(F3ap)4Cl 2 (F3ap = 2-(2,4,6-trifluoroanilino)pyridinate anion). To our knowledge, these are the first documented examples for isomeric conversion involving diruthenium compounds with tetracarboxylate-type structures. The structural, electrochemical, and spectroscopic properties of 1 and 2 were examined. The reversible Ru25+/6+ process of (3,1) [Ru2(F3ap)4Cl]+ is located at 0.62 V in CH2Cl2, 0.1 M TBAP but shifts to 0.29 V upon formation of (3,1) Ru2(F3ap)4Cl2 in CH2Cl2 containing chloride from added TBACl·H2O and shifts even further to E1/2 = 0.10 V after generation of (4,0) Ru2(F3ap)4Cl2 in solution. The 190 mV potential difference between the Ru26+/5+ redox couples of (3,1) Ru2(F3ap)4Cl2 and (4,0) Ru2(F3ap)4Cl2 in chloride-containing media can be compared to a smaller potential difference of only 60 mV between the Ru26+/5+ redox couples of (3,1) Ru2(F3ap)4Cl and (4,0) Ru2(F3ap)4Cl in CH2Cl2 containing 0.1 M tetrabutylammonium perchlorate (TBAP) as supporting electrolyte. The larger ΔE1/2 in the case of the bis-chloride complexes in solutions containing 0.1 M TBACl·H2O can be accounted for in large part by structural differences that manifest themselves in different strengths of axial coordination to the Ru25+ form of the compounds.
Co-reporter:Jing Shen ; Maya El Ojaimi ; Mohammed Chkounda ; Claude P. Gros ; Jean-Michel Barbe ; Jianguo Shao ; Roger Guilard
Inorganic Chemistry 2008 Volume 47(Issue 17) pp:7717-7727
Publication Date(Web):July 31, 2008
DOI:10.1021/ic8007415
A series of manganese(III) corroles were investigated as to their electrochemistry and spectroelectrochemistry in nonaqueous solvents. Up to three oxidations and one reduction were obtained for each complex depending on the solvents. The main compound discussed in this paper is the meso-substituted manganese corrole, (Mes2PhCor)Mn, and the main points are how changes in axially coordinated anion and solvent will affect the redox potentials and UV−vis spectra of each electrogenerated species in oxidation states of Mn(III), Mn(IV), or Mn(II). The anions OAc−, Cl−, CN−, and SCN− were found to form five-coordinate complexes with the neutral Mn(III) corrole while two OH− or F− anions were shown to bind axially in a stepwise addition to give the five- and six-coordinate complexes in nonaqueous media. In each case, complexation with one or two anionic axial ligands led to an easier oxidation and a harder reduction as compared to the uncomplexed four-coordinate species.
Co-reporter:Karl M. Kadish ; Rachel Garcia ; Tuan Phan ; Julien Wellhoff ; Eric Van Caemelbecke ;John L. Bear
Inorganic Chemistry 2008 Volume 47(Issue 23) pp:11423-11428
Publication Date(Web):November 4, 2008
DOI:10.1021/ic8017369
The electrochemistry and spectroelectrochemistry of a novel series of mixed-ligand diruthenium compounds were examined. The investigated compounds having the formula Ru2(CH3CO2)x(Fap)4−xCl where x = 1−3 and Fap is 2-(2-fluoroanilino)pyridinate anion were made from the reaction of Ru2(CH3CO2)4Cl with 2-(2-fluoroanilino)pyridine (HFap) in refluxing methanol. The previously characterized Ru2(Fap)4Cl as well as the three newly isolated compounds represented as Ru2(CH3CO2)(Fap)3Cl (1), Ru2(CH3CO2)2(Fap)2Cl (2), and Ru2(CH3CO2)3(Fap)Cl (3) possess three unpaired electrons with a Ru25+ dimetal core. Complexes 1 and 2 have well-defined Ru25+/4+ and Ru25+/6+ redox couples in CH2Cl2, but 3 exhibits a more complicated electrochemical behavior due to equilibria involving association or dissociation of the anionic chloride axial ligand on the initial and oxidized or reduced forms of the compound. The E1/2 values for the Ru25+/4+ and Ru25+/6+ processes vary linearly with the number of CH3CO2− bridging ligands on Ru2(CH3CO2)x(Fap)4−xCl and plots of reversible half-wave potentials vs the number of acetate groups follow linear free energy relationships with the largest substituent effect being observed for the oxidation. The major UV−visible band of the examined compounds in their neutral Ru25+ form is located between 550 and 800 nm in CH2Cl2 and also varies linearly with the number of CH3CO2− ligands on Ru2(CH3CO2)x(Fap)4−xCl. The electronic spectra of the singly oxidized and singly reduced forms of each diruthenium species were characterized by UV−visible spectroelectrochemistry in CH2Cl2.
Co-reporter:Marco Mastroianni, Weihua Zhu, Manuela Stefanelli, Sara Nardis, Frank R. Fronczek, Kevin M. Smith, Zhongping Ou, Karl M. Kadish and Roberto Paolesse
Inorganic Chemistry 2008 Volume 47(Issue 24) pp:11680-11687
Publication Date(Web):November 8, 2008
DOI:10.1021/ic801421a
The reaction between germanium(IV) meso-triphenylcorrolates and nitrate salts affords the corresponding β-nitro substituted corroles in good yield. Chromatographic separation of the crude reaction mixtures enables isolation of a μ-oxo dimer along with the corresponding monomers bearing a hydroxy or methoxy group at an axial position of the germanium central metal ion. Depending on the reaction conditions, mono- or dinitro substituted complexes can be obtained. The substitution is highly regioselective in each case, giving only the 3-nitro or 3,17-dinitro derivative among the different possible isomers. Five of the synthesized complexes were examined by cyclic voltammetry and UV−visible spectroelectrochemistry in dichloromethane, and the dinitro μ-oxo dimer is structurally characterized.
Co-reporter:Paul J. Sintic, Wenbo E, Zhongping Ou, Jianguo Shao, James A. McDonald, Zheng-Li Cai, Karl M. Kadish, Maxwell J. Crossley and Jeffrey R. Reimers
Physical Chemistry Chemical Physics 2008 vol. 10(Issue 2) pp:268-280
Publication Date(Web):14 Nov 2007
DOI:10.1039/B711320J
Quinoxalino[2,3-b′]porphyrins are π-expanded porphyrins, having a quinoxaline fused to a β,β′-pyrrolic position of the porphyrin. They are used as components in systems proposed as ‘molecular wires’. Knowledge of their redox properties is of value in the design of electron- or hole-conduction systems. In particular, the location of the charge density in the radical anions of quinoxalinoporphyrins can be modulated by peripheral functionalization. New theoretical treatments of electrochemical potentials are developed that identify the site of reduction in both the anions and the dianions of 33 quinoxalinoporphyrins. These molecules include free-base and metallated macrocycles substituted on the quinoxaline with electron-withdrawing groups (NO2, Cl, Br) and/or electron-donating groups (NH2, OCH3). Spectroelectrochemistry, density-functional theory calculations, and substituent-parameter models are used to verify the analysis. Five distinct patterns are observed for the locations of the first and second reductions; some of these patterns involve delocalized charges. Nitroquinoxalinoporphyrins with the nitro groups at the 5- and 6-quinoxaline positions are found to have quite different properties owing to distortions caused by peri interactions that force the nitro group of the 5-nitro regioisomer out of conjugation. Charge localization on the nitroquinoxaline fragment is found for some molecules, and this is attributed to ion-pairing with the 0.1 M tetrabutylammonium perchlorate electrolyte used, leading to the verified prediction that electron-paramagnetic resonance spectra of these molecules taken without the electrolyte yield delocalized anions. These properties enable the control of conduction through molecular wires synthesised from quinoxalinoporphyrins.
Co-reporter:Karl M. Kadish, Laurent Frémond, Fabien Burdet, Jean-Michel Barbe, Claude P. Gros, Roger Guilard
Journal of Inorganic Biochemistry 2006 Volume 100(Issue 4) pp:858-868
Publication Date(Web):April 2006
DOI:10.1016/j.jinorgbio.2006.01.010
A series of heterobinuclear cofacial porphyrin–corrole dyads containing a Co(IV) corrole linked by one of four different spacers in a face-to-face arrangement with an Fe(III) or Mn(III) porphyrin have been examined as catalysts for the electroreduction of O2 to H2O and/or H2O2 when adsorbed on the surface of a graphite electrode in air-saturated aqueous solutions containing 1 M HClO4. The examined compounds are represented as (PCY)MIIIClCoIVCl where P is a porphyrin dianion, C is a corrole trianion and Y is a biphenylene (B), 9,9-dimethylxanthene (X), dibenzofuran (O) or anthracene (A) spacer. The catalytic behavior of the seven investigated dyads in the two heterobimetallic (PCY)MClCoCl series of catalysts is compared on one hand to what was previously reported for related dyads with a single Co(III) corrole macrocycle linked to a free-base porphyrin with the same set of linking bridges, (PCY)H2Co, and on the other hand to dicobalt porphyrin–corrole dyads of the form (PCY)Co2 which were shown to efficiently electrocatalyze the four electron reduction of O2 at a graphite electrode in acid media. Comparisons between the four series of porphyrin–corrole dyads, (PCY)Co2, (PCY)H2Co, (PCY)FeClCoCl and (PCY)MnClCoCl, show that in all cases the biscobalt dyads catalyze O2 electroreduction at potentials more positive by an average 110 mV as compared to the related series of compounds containing a Co(III) or Co(IV) corrole macrocycle linked to a free-base metalloporphyrin or a metalloporphyrin with an Fe(III) or Mn(III) central metal ion. The data indicates that the E1/2 values where electrocatalysis is initiated is related to the initial site of electron transfer, which is the Co(III)/Co(II) porphyrin reduction process in the case of (PCY)Co2 and the Co(IV)/Co(III) corrole reduction in the case of (PCY)MnClCoCl, (PCY)FeClCoCl and (PCY)H2Co. The overall data also suggests that the catalytically active form of the biscobalt dyad in (PCY)Co2 contains a Co(II) porphyrin and a Co(IV) corrole.
Co-reporter:Thomas Köhler Dr.;Zhongping Ou Dr.;Jeong Tae Lee;Daniel Seidel Dr.;Vincent Lynch Dr. ;Jonathan L. Sessler
Angewandte Chemie International Edition 2005 Volume 44(Issue 1) pp:
Publication Date(Web):15 DEC 2004
DOI:10.1002/anie.200461801
Four up, four down: Reductive alkylation of cyclo[8]pyrrole and its derivatives gives rise to the corresponding reduced per-N-alkylated products, a reaction unknown in the case of porphyrins. These bowl-like molecules are arranged with four substituents up and four substituents down in a well-defined alternating fashion, as observed from their crystal structures (see picture; blue N).
Co-reporter:Thomas Köhler Dr.;Zhongping Ou Dr.;Jeong Tae Lee;Daniel Seidel Dr.;Vincent Lynch Dr. ;Jonathan L. Sessler
Angewandte Chemie 2005 Volume 117(Issue 1) pp:
Publication Date(Web):15 DEC 2004
DOI:10.1002/ange.200461801
Vier rauf, vier runter: Die reduktive Alkylierung von Cyclo[8]pyrrol und seinen Derivaten liefert die entsprechenden reduzierten per-N-alkylierten Produkte, eine Reaktion, die von Porphyrinen nicht bekannt ist. In diesen schalenförmigen Molekülen sind die acht Substituenten abwechselnd nach oben und nach unten gerichtet, wie die Kristallstrukturanalyse ergab (siehe Bild).
Co-reporter:Kei Ohkubo Dr.;Hiroaki Kotani;Jianguo Shao Dr.;Zhongping Ou Dr. Dr.;Guolin Li Dr.;Ravindra K. Pey Dr.;Mamoru Fujitsuka Dr.;Osamu Ito Dr.;Hiroshi Imahori Dr.;Shunichi Fukuzumi Dr.
Angewandte Chemie 2004 Volume 116(Issue 7) pp:
Publication Date(Web):22 JAN 2004
DOI:10.1002/ange.200352870
Ein sehr langlebiger Ladungstrennungs-Zustand (CS-Zustand) wird durch Photoanregung einer Zinkporphyrin-Fulleren-Dyade erzeugt. Zugrunde liegt ein einstufiger photoinduzierter Elektronentransfer, der anders als mehrstufige Elektronentransferprozesse ohne Energieverlust abläuft. Die Lebensdauer des CS-Zustands in gefrorenem Benzonitril bei −150 °C beträgt 120 s (siehe Bild).
Co-reporter:Kei Ohkubo Dr.;Hiroaki Kotani;Jianguo Shao Dr.;Zhongping Ou Dr. Dr.;Guolin Li Dr.;Ravindra K. Pey Dr.;Mamoru Fujitsuka Dr.;Osamu Ito Dr.;Hiroshi Imahori Dr.;Shunichi Fukuzumi Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 7) pp:
Publication Date(Web):22 JAN 2004
DOI:10.1002/anie.200352870
Long live the state! Photoexcitation of a zinc chlorin–fullerene dyad with a short linkage results in the formation of the ultra-long-lived charge-separated (CS) state by a one-step photoinduced electron transfer without loss of energy, which is inevitable for charge separation by multistep electron-transfer processes. The lifetime of the charge-separated state was 120 s in frozen PhCN at −150 °C (see picture).
Co-reporter:Roger Guilard, François Jérôme, Claude P Gros, Jean-Michel Barbe, Zhongping Ou, Jianguo Shao, Karl M Kadish
Comptes Rendus de l'Académie des Sciences - Series IIC - Chemistry 2001 Volume 4(Issue 3) pp:245-254
Publication Date(Web):March 2001
DOI:10.1016/S1387-1609(00)01226-3
Dicobalt or heterobimetallic cofacial bisporphyrins are up till now amongst the very few molecular electrocatalysts able to promote the direct reduction of dioxygen to water via a four-electron process in acidic medium. Numerous studies have been devoted to elucidate the key steps of this catalytic reaction and an important result has revealed an unexpected high dioxygen affinity for a mixed valence Co(II)/Co(III) cofacial porphyrin, the key intermediate complex being a μ-superoxo derivative. At the same time, the great importance assumed by ‘Pacman’ porphyrins and the recent developments in corrole chemistry have provided the stimulation to synthesise porphyrin–corrole dyads which might also transport and/or activate dioxygen. In the present paper, we report the stepwise synthesis and characterisation of a cofacial porphyrin–corrole bearing an anthracenyl bridge, (PCA)H5 where PCA is the pentaanion of 1-(13,17-diethyl-2,3,7,8,12,18-hexamethylporphyrin–5-yl)-8-(7,8,12,13-tetramethyl-2,3,17,18-tetraphenylcorrol-10-yl) anthracene. The synthesis and characterisation of the μ-superoxo Co(III)/Co(III) complex 〚(PCA)Co2Im2〛(μ-O2) is also described.Les complexes dicobalt ou hétérobimétalliques de bisporphyrines face à face sont les rares catalyseurs moléculaires capables d’initier la réduction directe du dioxygène en eau, selon un processus à quatre électrons, en milieu acide , , , , , , , , , , , , , , , , , , , , , , , , , , and . Un mécanisme a été proposé dans le cas du complexe dicobalt〚4〛. L’intermédiaire clé de ce cycle catalytique est l’espèce à valence mixte Co(II)/Co(III), qui doit cependant être électrogénérée par oxydation monoélectronique du complexe biscobalt(II,II) de départ. En effet, les porphyrines stabilisent le degré d’oxydation II de l’ion cobalt ; le complexe Co(II)/Co(II) en présence de dioxygène ne peut donner lieu à la formation du complexe oxygéné à l’origine du cycle catalytique.Ainsi, l’accès direct à un bimacrocycle capable de stabiliser l’espèce à valence mixte Co(II)/Co(III) présente un intérêt évident. Récemment, dans une communication préliminaire, nous avons décrit la synthèse d’un nouveau bimacrocycle, constitué d’une porphyrine et d’un corrole, maintenus face à face par un espaceur anthracényle, (PCA)H5〚29〛. La métallation de ce bimacrocycle conduit directement au complexe à valence mixte Co(II)/Co(III), le macrocycle corrole étant coordiné à l’ion cobalt au degré d’oxydation III.Le précurseur de cette synthèse multi-étape est le 1-formyl-8-(13,17-diéthyl-2,3,7,8,12,18-hexaméthylporphyrin–5-yl)anthracène 1 (figure 1) 〚29, 46〛. La porphyrine/corrole est obtenue avec un rendement voisin de 20 %. Tous les composés intervenant dans le schéma réactionnel ont été caractérisés par spectroscopies UV–visible, infrarouge, RMN 1H et par spectrométrie de masse. Les caractéristiques UV–visible du bimacrocycle porphyrine/corrole 7 sont proches de celles de composés bimacrocycliques face à face déjà décrits, telle la ‘Pacman’ porphyrine (DPA)H4〚18〛 (tableau I).La monométallation sélective du corrole de ce bimacrocycle est réalisée dans la pyridine à 60 °C (figure 2). L’obtention aisée du complexe monométallé, (PCA)H2Co 8 permet d’envisager la synthèse de nombreux dérivés hétérobimétalliques de type (PCA)CoM 〚53〛.Au départ de 8, par métallation par l’acétate de cobalt(II) en présence de 1-tert-butyl-5-phénylimidazole (Im) dans le mélange CHCl3/MeOH à reflux sous atmosphère de dioxygène, l’espèce oxygénée 〚(PCA)Co2Im2〛(μ-O2) 9 est obtenue quantitativement. La formation de ce complexe μ-superoxo est très aisément mise en évidence par spectrométrie de masse (voir § Partie expérimentale) et par spectroscopie RPE ( figure 3).La caractérisation de 9 a également été réalisée par voltamétrie cyclique et spectroélectrochimie (tableau II, figures 4 et 5). Les données électrochimiques relatives aux sites macrocycliques de 〚(PCA)Co2Im2〛(μ-O2) (9) sont à rapprocher de celles d’une monoporphyrine de cobalt et d’un monocorrole de cobalt présentant le même type de substitution. En revanche, la réduction électrochimique de 9 à –0,90 V/ECS (potentiel de pic dans CH2Cl2 contenant 0,1 M de perchlorate de tétrabutylammonium) implique le site métallique et correspond au départ du dioxygène (figure 5).
Co-reporter:Ikuo Nakanishi;Shunichi Fukuzumi;Jean-Michel Barbe;Roger Guilard;Karl M. Kadish
European Journal of Inorganic Chemistry 2000 Volume 2000(Issue 7) pp:
Publication Date(Web):4 JUL 2000
DOI:10.1002/1099-0682(200007)2000:7<1557::AID-EJIC1557>3.0.CO;2-9
Homogeneous electron-transfer kinetics for the reduction of four different manganese(III) porphyrins using different reductants were examined in deaerated acetonitrile, and the resulting data were evaluated in light of the Marcus theory of electron transfer to determine electron-exchange rate constants between manganese(III) and manganese(II) porphyrins. The investigated compounds are represented as (P)MnCl, where P = the dianion of dodecaphenylporphyrin (DPPX; X = H20, Cl12H8, or F20) or tetraphenylporphyrin (TPP). The electron transfer from semiquinone radical anion derivatives to (P)MnIIICl leads to formation of the corresponding MnII complex, [(P)MnIICl]−. The electron-exchange rate constants derived from the electron-transfer rate constants decrease with an increasing degree of nonplanarity of the porphyrin macrocycle and follow the order: (TPP)MnCl (3.1 × 103M−1·s−1) > (DPPH20)MnCl (1.1 × 10−2M−1·s−1) > (DPPCl12H8)MnCl (3.5 × 10−4M−1·s−1) > (DPPF20)MnCl (4.3 × 10−6M−1·s−1). The coordination of two molecules of pyridine (py) or DMSO to (DPPH20)MnCl to form [(DPPH20)Mn(py)2]+ or [(DPPH20)Mn(DMSO)2]+ enhances the rate of electron-transfer reduction. This indicates that there is a significant decrease in the reorganization energy upon axial ligand coordination of pyridine or DMSO.
Co-reporter:Shunichi Fukuzumi;Ikuo Nakanishi;Jean-Michel Barbe;Roger Guilard;Eric Van Caemelbecke;Ning Guo
Angewandte Chemie International Edition 1999 Volume 38(Issue 7) pp:
Publication Date(Web):26 MAR 1999
DOI:10.1002/(SICI)1521-3773(19990401)38:7<964::AID-ANIE964>3.0.CO;2-O
Slow electron transfer to manganese(iii) porphyrins results when the macrocycle deviates from planarity. This was demonstrated by measuring the kinetics of homogeneous electron transfer from a series of semiquinone radical anions to synthetic manganese porphyrins (shown schematically; R1=H, Cl, F; R2=H, F). Three of the four porphyrins studied have nonplanar macrocycles. These results could have implications for the role of manganese in biological electron transfer processes.
Co-reporter:Shunichi Fukuzumi;Ikuo Nakanishi;Jean-Michel Barbe;Roger Guilard;Eric Van Caemelbecke;Ning Guo
Angewandte Chemie 1999 Volume 111(Issue 7) pp:
Publication Date(Web):26 MAR 1999
DOI:10.1002/(SICI)1521-3757(19990401)111:7<1017::AID-ANGE1017>3.0.CO;2-C
Ein langsamer Elektronentransfer auf Mangan(III)-Porphyrine ist die Folge, wenn der Porphyrinmakrocyclus nicht planar ist. Dies konnte anhand kinetischer Messungen des homogenen Elektronentransfers von einer Reihe von Semichinon-Radikalanionen auf synthetische Manganporphyrine (siehe Formel, R1 = H, Cl, F; R2 = H, F) gezeigt werden. In drei der vier untersuchten Porphyrine ist der Makrocyclus nicht planar. Dieses Ergebnis könnte für das Verständnis der Rolle von Mangan in biologischen Elektronentransferprozessen wichtig sein.
Co-reporter:Christina M. Davis, Kei Ohkubo, I-Ting Ho, Zhan Zhang, Masatoshi Ishida, Yuanyuan Fang, Vincent M. Lynch, Karl M. Kadish, Jonathan L. Sessler and Shunichi Fukuzumi
Chemical Communications 2015 - vol. 51(Issue 31) pp:NaN6760-6760
Publication Date(Web):2015/03/12
DOI:10.1039/C5CC00903K
Photoexcitation of dichloromethane solutions of an uranyl macrocyclic complex with cyclo[1]furan[1]pyridine[4]-pyrrole (1) at the near-infrared (NIR) band (1177 nm) in the presence of electron donors and acceptors resulted in NIR-induced electron transfer without producing singlet oxygen via energy transfer.
Co-reporter:Guifen Lu, Sen Yan, Mengying Shi, Wenhan Yu, Jing Li, Weihua Zhu, Zhongping Ou and Karl M. Kadish
Chemical Communications 2015 - vol. 51(Issue 12) pp:NaN2413-2413
Publication Date(Web):2014/12/22
DOI:10.1039/C4CC09755F
The first europium triple-decker tetrapyrrole with mixed corrole and phthalocyanine macrocycles was synthesized and characterized by spectroscopic and electrochemical methods. The molecular structure was characterized by single-crystal X-ray diffraction and showed the corrole to be in the middle of the sandwich with phthalocyanine macrocycles at each extreme.
Co-reporter:Bin Sun, Zhongping Ou, Shuibo Yang, Deying Meng, Guifen Lu, Yuanyuan Fang and Karl M. Kadish
Dalton Transactions 2014 - vol. 43(Issue 28) pp:NaN10815-10815
Publication Date(Web):2014/05/02
DOI:10.1039/C4DT01072H
Four cobalt(II) porphyrins, two of which contain a β-pyrrole nitro substituent, were synthesized and characterized by electrochemistry and spectroelectrochemistry. The investigated compounds are represented as (TRPP)Co and (NO2TRPP)Co, where TRPP is the dianion of a substituted tetraphenylporphyrin and R is a CH3 or OCH3 substituent on the four phenyl rings of the macrocycle. Two reductions and three oxidations are observed for each compound in CH2Cl2 containing 0.10 M tetra-n-butylammonium perchlorate. The first reduction of the compounds without a nitro substituent is metal-centered and leads to formation of a Co(I) porphyrin which then reacts with the CH2Cl2 solvent to generate a carbon σ-bonded CoIII–R complex. A further reduction then occurs at more negative potentials to generate an unstable Co(II) σ-bonded compound. In contrast to these reactions, the first reduction of the nitro-substituted porphyrins is macrocycle-centered under the same solution conditions and gives a Co(II) porphyrin π-anion radical product. This reversible electron transfer is then followed at more negative potentials by a second reversible one-electron addition to give a Co(II) dianion. Three reversible one-electron oxidations are also seen for each compound. The first is metal-centered and the next two involve the conjugated π-system of the macrocycle. Each neutral Co(II) porphyrin was also examined as to its catalytic activity for electroreduction of molecular oxygen when coated on an edge-plane pyrolytic graphite electrode in 1.0 M HClO4. The β-pyrrole nitro-substituted derivatives were shown to be better catalysts than the non-nitro substituted compounds under the utilized experimental conditions.
Co-reporter:Pinky Yadav, Muniappan Sankar, Xiangyi Ke, Lei Cong and Karl M. Kadish
Dalton Transactions 2017 - vol. 46(Issue 30) pp:NaN10022-10022
Publication Date(Web):2017/07/13
DOI:10.1039/C7DT01814B
Di- and octa-phenylethynyl (PE) substituted π-extended copper corroles were synthesized and characterized as to their structural, electrochemical and spectroscopic properties. The addition of two or eight PE groups to the β-pyrrole positions of the corrole results in dramatic red shifts in the electronic absorption spectra and new reductions which are not seen for the parent compound lacking PE substituents. CuCor(PE)8 is reduced in four reversible one-electron transfer steps to give derivatives of [CuCor(PE)8]n− where n = 1, 2, 3 or 4. Variable temperature 1H NMR and EPR measurements were carried out and suggest that the octa- and di-PE substituted Cu-corroles can both be described as an antiferromagnetically coupled CuII corrole cation radical which is in equilibrium with a triplet state, possibly due to a lower singlet–triplet energy gap as compared to 1 and 2 at room temperature. The EPR spectra of one-electron oxidized and one electron reduced species exhibited the characteristics of Cu(II) corroles. The products generated in the first two reductions of each π-extended corrole were characterized by thin-layer spectroelectrochemistry, thus providing new insights into how UV-vis spectra of highly reduced corroles vary as a function of the number of PE groups and overall charge on the molecule. The singly reduced and singly oxidized copper corroles were also chemically generated in CH3CN and shown to have UV-visible spectra almost identical to the spectra obtained by electroreduction or electrooxidation in PhCN or THF containing 0.1 M tetrabutylammonium perchlorate.
Co-reporter:Paul J. Sintic, Wenbo E, Zhongping Ou, Jianguo Shao, James A. McDonald, Zheng-Li Cai, Karl M. Kadish, Maxwell J. Crossley and Jeffrey R. Reimers
Physical Chemistry Chemical Physics 2008 - vol. 10(Issue 2) pp:NaN280-280
Publication Date(Web):2007/11/14
DOI:10.1039/B711320J
Quinoxalino[2,3-b′]porphyrins are π-expanded porphyrins, having a quinoxaline fused to a β,β′-pyrrolic position of the porphyrin. They are used as components in systems proposed as ‘molecular wires’. Knowledge of their redox properties is of value in the design of electron- or hole-conduction systems. In particular, the location of the charge density in the radical anions of quinoxalinoporphyrins can be modulated by peripheral functionalization. New theoretical treatments of electrochemical potentials are developed that identify the site of reduction in both the anions and the dianions of 33 quinoxalinoporphyrins. These molecules include free-base and metallated macrocycles substituted on the quinoxaline with electron-withdrawing groups (NO2, Cl, Br) and/or electron-donating groups (NH2, OCH3). Spectroelectrochemistry, density-functional theory calculations, and substituent-parameter models are used to verify the analysis. Five distinct patterns are observed for the locations of the first and second reductions; some of these patterns involve delocalized charges. Nitroquinoxalinoporphyrins with the nitro groups at the 5- and 6-quinoxaline positions are found to have quite different properties owing to distortions caused by peri interactions that force the nitro group of the 5-nitro regioisomer out of conjugation. Charge localization on the nitroquinoxaline fragment is found for some molecules, and this is attributed to ion-pairing with the 0.1 M tetrabutylammonium perchlorate electrolyte used, leading to the verified prediction that electron-paramagnetic resonance spectra of these molecules taken without the electrolyte yield delocalized anions. These properties enable the control of conduction through molecular wires synthesised from quinoxalinoporphyrins.
Co-reporter:Zhongping Ou, Ping Chen and Karl M. Kadish
Dalton Transactions 2010 - vol. 39(Issue 46) pp:NaN11276-11276
Publication Date(Web):2010/10/25
DOI:10.1039/C0DT00899K
The first example for electrogeneration of a Pt(IV) porphyrin from its Pt(II) form is presented and the PtII/IV and reverse PtIV/II oxidation-reduction processes are elucidated by electrochemistry and thin-layer UV-visible spectroelectrochemistry. Three products, [(TPP˙+)PtII]+, [(TPP)PtIV]2+ and [(TPP˙+)PtIV]3+, produced by electrooxidation of the Pt(II) porphyrin have been characterized by in situ spectroelectrochemistry and ESR measurements after controlled potential bulk electrolysis. The first definitive evidence for the electrochemical conversion of a Pt(IV) porphyrin to its Pt(II) form is also presented. The potential for this electroreduction is highly dependent upon the nature of the anion, ClO4− or Cl−. A mechanism for the reversible conversion between Pt(II) and Pt(IV) tetraphenylporphyrins is proposed.
Co-reporter:Yuanyuan Fang, Dominik Koszelewski, Karl M. Kadish and Daniel T. Gryko
Chemical Communications 2014 - vol. 50(Issue 64) pp:NaN8867-8867
Publication Date(Web):2014/06/16
DOI:10.1039/C4CC02759K
The facile electrosynthesis of π-extended porphyrins is demonstrated for a series of Zn(II), In(III), Ir(III) and free-base meso-substituted derivatives containing the 4,7-dimethoxynaphthalen-1-yl substituent. Electrochemical data suggest that the overall process initially involves two stepwise one-electron oxidations, followed by an intramolecular oxidative aromatic coupling to give the electrooxidized π-extended porphyrin.
Co-reporter:Siyabonga Ngubane, Karl M. Kadish, John L. Bear, Eric Van Caemelbecke, Antoine Thuriere and Kevin P. Ramirez
Dalton Transactions 2013 - vol. 42(Issue 10) pp:NaN3580-3580
Publication Date(Web):2012/12/17
DOI:10.1039/C2DT32715E
A mixed-ligand metal–metal bonded diruthenium complex having the formula Ru2(2,4,6-(CH3)3ap)3(O2CCH3)Cl where ap is the anilinopyridinate anion was synthesized from the reaction of Ru2(O2CCH3)4Cl and H(2,4,6-(CH3)3ap), after which the isolated product was structurally, spectroscopically and electrochemically characterized. The crystal structure reveals an unusual arrangement of the bridging ligands around the dimetal unit where one ruthenium atom is coordinated to one anilino and two pyridyl nitrogen atoms while the other ruthenium atom is coordinated to one pyridyl and two anilino nitrogen atoms. To our knowledge, Ru2(2,4,6-(CH3)3ap)3(O2CCH3)Cl is the only example of a mixed-ligand diruthenium complex of the type [Ru2L3(O2CCH3)]+, where L is an unsymmetrical anionic bridging ligand that has been structurally characterized with a “(2,1)” geometric conformation of the bridging ligands, all others being “(3,0)”. The initial Ru25+ compound in CH2Cl2 or CH3CN containing 0.1 M tetra-n-butylammonium perchlorate (TBAP) undergoes up to four one-electron redox processes involving the dimetal unit. The Ru25+/4+ and Ru25+/6+ processes were characterized under N2 using thin-layer UV-visible spectroelectrochemistry and this data is compared to UV-visible spectral changes obtained during similar electrode reactions for related diruthenium compounds having the formula Ru2L4Cl or Ru2L3(O2CCH3)Cl where L is an anionic bridging ligand. Ru2(2,4,6-(CH3)3ap)3(O2CCH3)Cl was also examined by UV-visible and FTIR spectroelectrochemistry under a CO atmosphere and two singly reduced Ru24+ species, [Ru2(2,4,6-(CH3)3ap)3(O2CCH3)(CO)Cl]− and Ru2(2,4,6-(CH3)3ap)3(O2CCH3)(CO) were in situ generated for further characterization. The CO-bound complexes could be further reduced and exhibited additional reductions to their Ru23+ and Ru22+ oxidation states.
.jpg)
![Corrin, 1,2,3,7,8,12,13,17,18,19-decadehydro-5,10,15-tris[4-(1,1-dimethylethyl)phenyl]-21,22-dihydro-](/data/chemimg/115700/958259-08-0.png)
![Corrin, 1,2,3,7,8,12,13,17,18,19-decadehydro-5,10,15-tris[4-(1,1-dimethylethyl)phenyl]-21,22-dihydro-](/data/chemimg/115700/958259-08-0_b.png)



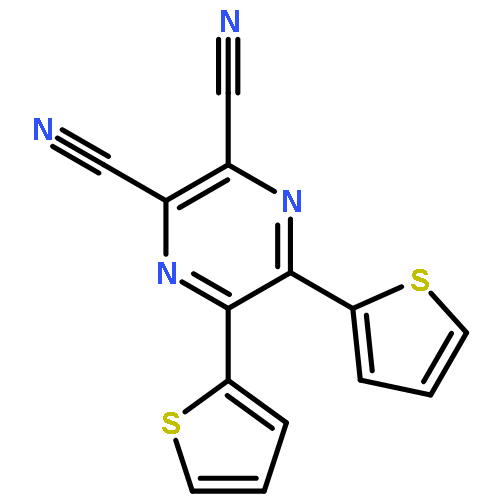
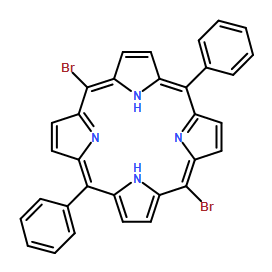








![Nickel, [2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/2500/24803-99-4.png)
![Nickel, [2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/2500/24803-99-4_b.png)
![Cobalt, [2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/1054800/17632-19-8.png)
![Cobalt, [2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/1054800/17632-19-8_b.png)
![Copper, [2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/526100/14409-63-3.png)
![Copper, [2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/526100/14409-63-3_b.png)
![Platinum, [5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/482200/14187-14-5.png)
![Platinum, [5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/482200/14187-14-5_b.png)








![Ruthenium, tetrakis[m-(acetato-kO:kO')]chlorodi-, (Ru-Ru)](http://img.cochemist.com/ccimg/38900/38833-34-0.png)
![Ruthenium, tetrakis[m-(acetato-kO:kO')]chlorodi-, (Ru-Ru)](http://img.cochemist.com/ccimg/38900/38833-34-0_b.png)
![Nickel, [5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)- (9CI)](/data/chemimg/478900/14172-92-0.png)
![Nickel, [5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)- (9CI)](/data/chemimg/478900/14172-92-0_b.png)
![Zinc, [5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/478800/14074-80-7.png)
![Zinc, [5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/478800/14074-80-7_b.png)