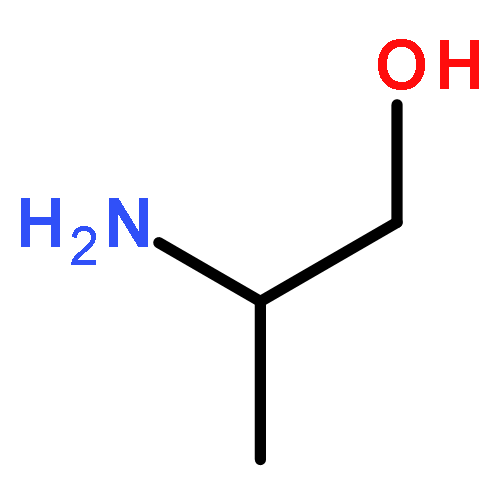
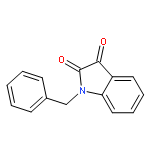
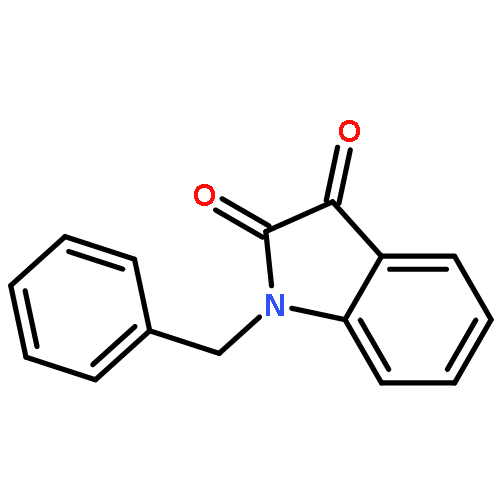
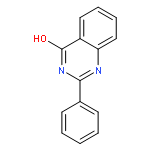
Co-reporter:Dinesh Kumar, Sandeep R. Vemula, Narayanaganesh Balasubramanian, and Gregory R. Cook
Accounts of Chemical Research 2016 Volume 49(Issue 10) pp:2169
Publication Date(Web):October 4, 2016
DOI:10.1021/acs.accounts.6b00362
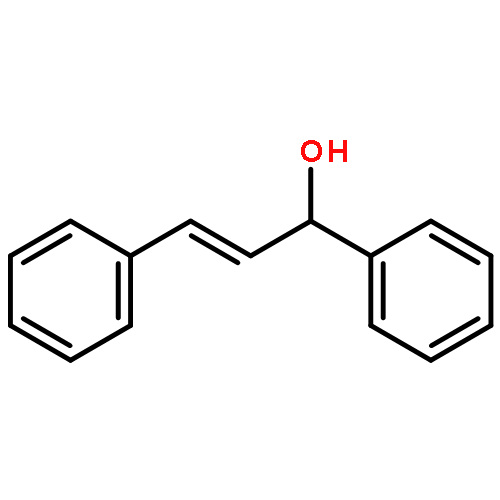
Stereoselective indium-mediated organic reactions have enjoyed tremendous growth in the last 25 years. This is in part due to the insensitivity of allylindium to moisture, affording facile and practical reaction conditions coupled with outstanding functional group tolerance and minimal side reactions. Despite the plethora of articles about allylindium, there is much yet to be discovered and exploited for efficient and sustainable synthesis.In this Account, we describe indium-mediated synthetic methods for the preparation of chiral amines with the aim to present a balance of practical method development, novel asymmetric chemistry, and mechanistic understanding that impact multiple chemical and materials science disciplines. In 2005, we demonstrated the indium-mediated allylation of chiral hydrazones with complete diastereoselectivity (>99:1) and quantitative yields. Further, we revealed the first example of enantioselective indium-mediated allylation of hydrazones using catalytic (R)-3,3′-bis(trifluoromethyl)-BINOL ligands to afford homoallylic amines with high enantioselectivity. The use of enantiopure perfluoroalkylsulfonate BINOLs greatly improved the indium-mediated allylation of N-acylhydrazones with exquisite enantiocontrol (99% yield, 99% ee). This laboratory has also investigated indium-mediated asymmetric intramolecular cyclization in the presence of amino acid additives to deliver biologically relevant chromanes with excellent diastereoselectivity (dr >99:1). The effect of amino acid additives (N-Boc-glycine) was further investigated during the indium-mediated allylation of isatins with allyl bromide to yield homoallylic alcohols in excellent yields in a short time with a wide range of functional group tolerance. Critical mechanistic insight was gained, and evidence suggests that the additive plays two roles: (1) to increase the rate of formation of allylindium from allyl bromide and In(0) and (2) to increase the nucleophilicity of the allylindium reagent, probably through disruption of aggregates and coordination to the metal. We recently reported the palladium-catalyzed umpolung allylation of hydrazones with allyl acetates in the presence of indium(I) iodide (InI) with excellent diastereoselectivity (up to 99:1). The conversion was found to be inversely proportional to the phosphine concentration, providing insight into the mechanism of the critical redox transmetalation process that has implications for other Pd-catalyzed umpolung-type allylation processes.A detailed overview of the work in our lab is presented with the intention of stimulating further research interest in organoindium chemistry and its application in organic synthesis.
Co-reporter:Dinesh Kumar
ACS Catalysis 2016 6(8) pp: 4920-4945
Publication Date(Web):June 17, 2016
DOI:10.1021/acscatal.6b01116
α-Ketoamides and their derivatives are key constituents of natural products, biologically relevant molecules, drug and drug candidates, and functional materials. Further, they are versatile and valuable intermediates and synthons in a number of functional group transformations and total syntheses. In recent years tremendous growth has been realized in the development of synthetic methods for α-ketoamide preparation and their applications in synthetic and medicinal chemistry. Among the various catalytic methods of α-ketoamide formation, two approaches, namely double aminocarbonylation and oxidative amidation, have received much more attention and have been greatly studied because of the ready availability of the starting materials, use of carbon monoxide (CO) as a direct source of carbonyl functionalities, and use of molecular oxygen (O2) or air as a green terminal oxidant and/or reactants. Catalyzed α-ketoamide formation can be roughly classified into metal- and nonmetal-catalyzed processes. In the context of metal catalysis, most reactions involving metals are performed using palladium (Pd) and copper (Cu); however, other metals such as gold (Au), silver (Ag), and iron (Fe) based catalysts have also been investigated to some extent. On the other hand, nonmetal-catalyzed α-ketoamide syntheses are mainly restricted to iodine-based catalysts in the presence or absence of other promoters. Our objective in this review is to highlight the important research endeavors related to catalytic α-ketoamide synthesis, which include the trends in the catalytic synthesis of α-ketoamides, new breakthroughs, and recent advances up to March 2016.Keywords: amino dicarbonylation; catalysis; chemoselectivity; oxidative amidation; α-ketoamides
Co-reporter:Dinesh Kumar, Sandeep R. Vemula, and Gregory R. Cook
ACS Catalysis 2016 Volume 6(Issue 6) pp:3531
Publication Date(Web):April 22, 2016
DOI:10.1021/acscatal.6b00728
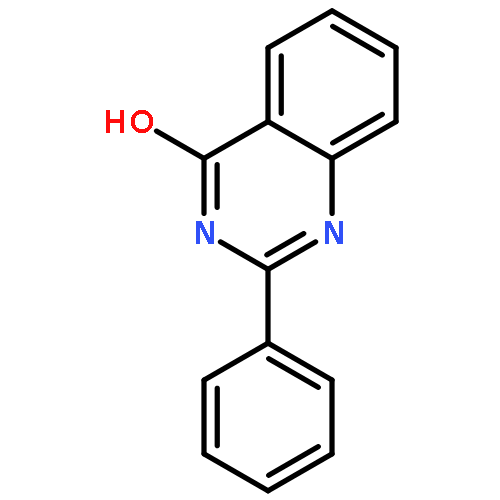
A new route for the synthesis of 2-aminopyridines has been developed that merges C–H functionalization with amide alcoholysis. The key component of this method is the ability of a quinazolinone to template the chemo- and regioselective construction of a latent pyridine ring via site-selective olefinic C–H bond functionalization under Ru(II) catalysis. Thus, highly substituted 2-aminopyridines were prepared in good yield. Mechanistic studies provide insight into the mechanism of the key oxidative C–H activation/annulation process.Keywords: 2-aminopyridine; amide alcoholysis; quinazolinone; ruthenium catalysis; site-selective C−H activation
Co-reporter:Sandeep R. Vemula, Dinesh Kumar, and Gregory R. Cook
ACS Catalysis 2016 Volume 6(Issue 8) pp:5295
Publication Date(Web):July 1, 2016
DOI:10.1021/acscatal.6b01818
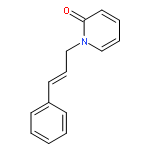
An atom-economic direct intermolecular allylic amidation of electron-deficient tautomerizable N-heterocycles is reported via allylic C–H activation of terminal olefins with a PdCl2 catalyst. The reaction did not require any activators (base or Lewis acid) or external ligands and proceeded with high chemo- (N vs O), regio- (linear vs branched), and stereoselectivity (E vs Z) for a variety of N-heterocycles and terminal olefins. Mechanistic investigation and stoichiometric studies validate the sulfoxide-ligand-assisted allylic C–H bond cleavage to form a π-allylpalladium intermediate in the reaction pathway. Excellent selectivity was observed during intermolecular competition demonstrating the differential nucleophilicity of N-heterocycles and differential susceptibility of allyl C–H bond cleavage to form π-allylpalladium complexes directly from terminal olefins.Keywords: allylic amidation; N-heterocycles; palladium catalysis; sp3 C−H activation; terminal olefins
Co-reporter:Dinesh Kumar, Sandeep R. Vemula and Gregory R. Cook
Green Chemistry 2015 vol. 17(Issue 8) pp:4300-4306
Publication Date(Web):17 Jun 2015
DOI:10.1039/C5GC01028D
Tautomerizable heteroarenes, bearing multiple interconvertible nucleophilic centers exhibit high chemo- and regioselective allylation irrespective of allylating agents used under Pd-catalysis. The achieved selectivity may be attributed to the dominant lactam form of heteroarenes and Pd-catalyzed intramolecular allylic substitution. A generalized green protocol for chemo- and regioselective allylation of biologically relevant heteroarenes with allyl alcohols in dimethyl carbonate (DMC) as solvent was developed. Excellent selectivity was observed during intermolecular competition study demonstrating the differential nucleophilicity of tautomerizable heteroarenes and differential allyl palladium forming ability of a variety of allyl alcohols.
Co-reporter:Narayanaganesh Balasubramanian, Tanmay Mandal, and Gregory R. Cook
Organic Letters 2015 Volume 17(Issue 2) pp:314-317
Publication Date(Web):January 7, 2015
DOI:10.1021/ol5034207
The general and efficient palladium-catalyzed indium-mediated allylation of chiral hydrazones was accomplished with excellent yield (72–92%) and diastereoselectivity (up to 99:1). The development of this reaction and the substrate scope are described. The conversion was found to be proportional to the phosphine concentration, which provided insight into the mechanism and competing pathways of the redox transmetalation process.
Co-reporter:Sandeep Reddy Vemula, Dinesh Kumar, Gregory R. Cook
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:3322-3325
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2015.01.036
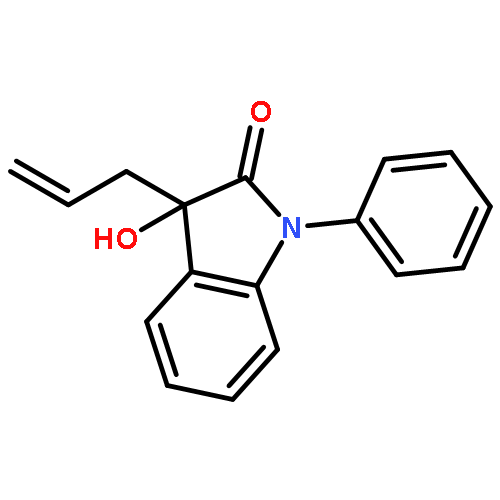
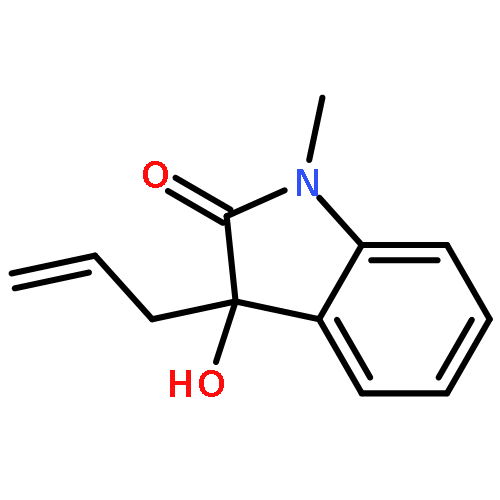
An unprecedent improvement in the reaction rate (from hours to minutes) was observed during the In-mediated allylation of isatins with allyl bromide in the presence of N-Boc-glycine. The observed phenomena were generalized as a general protocol for the allylation of differently substituted isatins with excellent yields in a short time and with a wide range of functional group tolerance. Evidence suggests the unique role of the N-Boc-glycine additive was to increase the rate of formation and increase the nucleophilicity of the allylindium organometallic reagent.
Co-reporter:Raushan K. Singh, Tanmay Mandal, Narayanaganesh Balasubramanian, Gregory Cook, D.K. Srivastava
Analytical Biochemistry 2011 408(2) pp: 309-315
Publication Date(Web):
DOI:10.1016/j.ab.2010.08.040
Co-reporter:Raushan K. Singh, Tanmay Mandal, Narayanaganesh Balsubramanian, Tajae Viaene, Travis Leedahl, Nitesh Sule, Gregory Cook, D.K. Srivastava
Bioorganic & Medicinal Chemistry Letters 2011 21(19) pp: 5920-5923
Publication Date(Web):
DOI:10.1016/j.bmcl.2011.07.080
Co-reporter:Raushan K. Singh, Tanmay Mandal, Narayanaganesh Balasubramanian, Gregory Cook, D.K. Srivastava
Analytical Biochemistry (15 January 2011) Volume 408(Issue 2) pp:309-315
Publication Date(Web):15 January 2011
DOI:10.1016/j.ab.2010.08.040
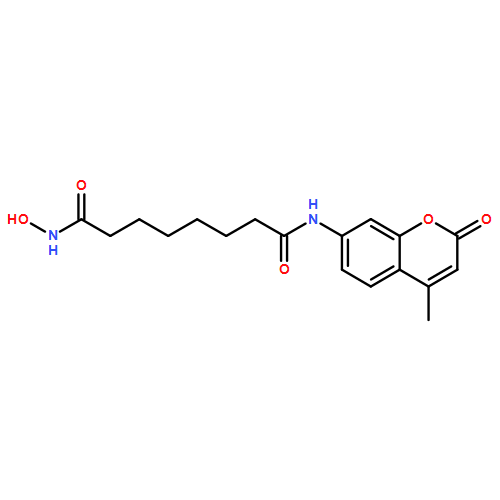
Histone deacetylases (HDACs) are intimately involved in epigenetic regulation and, thus, are one of the key therapeutic targets for cancer, and two HDAC inhibitors, namely suberoylanilide hydroxamic acid (SAHA) and romidepsin, have been recently approved for cancer treatment. Because the screening and detailed characterization of HDAC inhibitors has been time-consuming, we synthesized coumarin-SAHA (c-SAHA) as a fluorescent probe for determining the binding affinities (Kd) and the dissociation off-rates (koff) of the enzyme–inhibitor complexes. The determination of the above parameters relies on the changes in the fluorescence emission intensity (λex = 325 nm, λem = 400 nm) of c-SAHA due to its competitive binding against other HDAC inhibitors, and such determination neither requires employment of polarization accessories nor is dependent on the fluorescence energy transfer from the enzyme’s tryptophan residues to the probe. Our highly sensitive and robust analytical protocol presented here is applicable to most of the HDAC isozymes, and it can be easily adopted in a high-throughput mode for screening the HDAC inhibitors as well as for quantitatively determining their Kd and koff values.

![4(1H)-Quinazolinone, 2-[(1E)-2-(4-nitrophenyl)ethenyl]-](http://img.cochemist.com/ccimg/743500/743477-76-1.png)
![4(1H)-Quinazolinone, 2-[(1E)-2-(4-nitrophenyl)ethenyl]-](http://img.cochemist.com/ccimg/743500/743477-76-1_b.png)
![4(1H)-Quinazolinone, 2-[(1E)-2-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/743500/743477-70-5.png)
![4(1H)-Quinazolinone, 2-[(1E)-2-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/743500/743477-70-5_b.png)
![4(1H)-Quinazolinone, 2-[(1E)-2-[4-(dimethylamino)phenyl]ethenyl]-](http://img.cochemist.com/ccimg/743500/743477-65-8.png)
![4(1H)-Quinazolinone, 2-[(1E)-2-[4-(dimethylamino)phenyl]ethenyl]-](http://img.cochemist.com/ccimg/743500/743477-65-8_b.png)
![4(1H)-Quinazolinone, 2-[(1E)-2-(4-chlorophenyl)ethenyl]-](http://img.cochemist.com/ccimg/743500/743477-63-6.png)
![4(1H)-Quinazolinone, 2-[(1E)-2-(4-chlorophenyl)ethenyl]-](http://img.cochemist.com/ccimg/743500/743477-63-6_b.png)
![2-OXAZOLIDINONE, 4-(1-METHYLETHYL)-3-[(E)-PROPYLIDENEAMINO]-, (4S)-](http://img.cochemist.com/ccimg/718000/717918-38-2.png)
![2-OXAZOLIDINONE, 4-(1-METHYLETHYL)-3-[(E)-PROPYLIDENEAMINO]-, (4S)-](http://img.cochemist.com/ccimg/718000/717918-38-2_b.png)










![4(1H)-Quinazolinone, 2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/83900/83800-83-3.png)
![4(1H)-Quinazolinone, 2-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/83900/83800-83-3_b.png)
![2-[4-(DIMETHYLAMINO)PHENYL]-1H-QUINAZOLIN-4-ONE](http://img.cochemist.com/ccimg/80000/79916-52-2.png)
![2-[4-(DIMETHYLAMINO)PHENYL]-1H-QUINAZOLIN-4-ONE](http://img.cochemist.com/ccimg/80000/79916-52-2_b.png)
![1,4-DIHYDRO-2H-THIENO[3,2-D][1,3]OXAZINE-2,4-DIONE](http://img.cochemist.com/ccimg/78800/78756-28-2.png)
![1,4-DIHYDRO-2H-THIENO[3,2-D][1,3]OXAZINE-2,4-DIONE](http://img.cochemist.com/ccimg/78800/78756-28-2_b.png)


![4(1H)-Quinazolinone, 2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/54000/53905-19-4.png)
![4(1H)-Quinazolinone, 2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/54000/53905-19-4_b.png)









![2-[(E)-2-phenylethenyl]quinazolin-4(1H)-one](http://img.cochemist.com/ccimg/4800/4765-58-6.png)
![2-[(E)-2-phenylethenyl]quinazolin-4(1H)-one](http://img.cochemist.com/ccimg/4800/4765-58-6_b.png)