Co-reporter:Francisco J. Sarabia and Eric M. Ferreira
Organic Letters June 2, 2017 Volume 19(Issue 11) pp:
Publication Date(Web):May 12, 2017
DOI:10.1021/acs.orglett.7b01095
The chromium photocatalyzed cyclopropanation of diazo reagents with electron-rich alkenes is described. The transformation occurs under mild conditions and features specific distinctions from traditional diazo-based cyclopropanations (e.g., avoiding β-hydride elimination, chemoselectivity considerations, etc.). The reaction appears to work most effectively using chromium catalysis, and a number of decorated cyclopropanes can be accessed in generally good yields.
Co-reporter:Khoi Q. Huynh, Curtis A. Seizert, Tarik J. Ozumerzifon, Paul A. Allegretti, and Eric M. Ferreira
Organic Letters 2017 Volume 19(Issue 1) pp:294-297
Publication Date(Web):December 20, 2016
DOI:10.1021/acs.orglett.6b03682
Formal syntheses of tetracyclic terpenoids frondosin B and liphagal are described. Both synthetic routes rely on the use of platinum-catalyzed α,β-unsaturated carbene formation for the key C–C bond forming transformations. The successful route toward frondosin B utilizes a formal (4 + 3) cycloaddition, while the liphagal synthesis features the vinylogous addition of an enol nucleophile as a key step. Both synthetic routes are discussed, revealing insights into structural requirements in the catalytic α,β-unsaturated carbene reaction manifold.
Co-reporter:Susan M. Stevenson;Robert F. Higgins;Matthew P. Shores
Chemical Science (2010-Present) 2017 vol. 8(Issue 1) pp:654-660
Publication Date(Web):2016/12/19
DOI:10.1039/C6SC03303B
A chromium-catalyzed, visible light-activated net [4 + 2] cycloaddition between dienes and electron-deficient alkenes is described. Gathered evidence, via control experiments, isolated intermediates, and measured redox potentials, points to several converging reaction pathways that afford the cyclohexene adducts, including a photochemical [2 + 2] cycloaddition/vinylcyclobutane rearrangement cascade and a substrate excitation/oxidation sequence to a radical cation intermediate. Notably, the accompanying mechanistic stipulations result in a process that yields regioisomeric compounds from those generated by traditional Diels–Alder cycloadditions.
Co-reporter:Curtis A. Seizert, Eric M. Ferreira
Tetrahedron 2017 Volume 73, Issue 29(Issue 29) pp:
Publication Date(Web):20 July 2017
DOI:10.1016/j.tet.2016.11.030
A synthetic approach to pordamacrine A that features two key transformations is discussed. The first transformation applies an Ireland-Claisen rearrangement to establish sterically congested vicinal carbon centers. Although a hard enolization technique for accessing the silyl ketene acetal failed, a soft enolization, boron-based reaction was highly successful. The second step involves a proposed cascade palladium-catalyzed biscyclization to construct two carbocycles of the natural product. The overall strategy is presented, demonstrating the challenges of the cyclization events in this complex setting.Download high-res image (138KB)Download full-size image
Co-reporter:Brian J. Knight, Jacob O. Rothbaum and Eric M. Ferreira
Chemical Science 2016 vol. 7(Issue 3) pp:1982-1987
Publication Date(Web):14 Dec 2015
DOI:10.1039/C5SC03948G
We describe herein the design of a novel molecular scaffold that can induce facile oxidative olefinations when attached to alcohols. Benzylic, homo-, and bishomobenzylic alcohols are utilized. The scaffold can act as a protecting group for the alcohol in other transformations, and it is recoverable in excellent yield. The overall sequence can also be telescoped without purifications of intermediates, representing a net alcohol-based directed ortho-alkenylation.
Co-reporter:Paul A. Allegretti, Khoi Huynh, Tarik J. Ozumerzifon, and Eric M. Ferreira
Organic Letters 2016 Volume 18(Issue 1) pp:64-67
Publication Date(Web):December 14, 2015
DOI:10.1021/acs.orglett.5b03246
A variety of substituted indoles and benzofurans are accessed via a platinum catalyzed annulation and vinylogous addition of enol nucleophiles. Several β-dicarbonyl compounds participate in the reaction, as do α-nitro and α-cyano carbonyl species. Subjecting the indole products to acidic conditions results in the formation of fused heterocycles.
Co-reporter:Eric T. Newcomb; Phil C. Knutson; Blaine A. Pedersen
Journal of the American Chemical Society 2015 Volume 138(Issue 1) pp:108-111
Publication Date(Web):December 30, 2015
DOI:10.1021/jacs.5b12263
The first total synthesis of (±)-gelsenicine is reported. The synthetic route is highly efficient (13 steps), featuring (1) a pivotal metal-catalyzed isomerization/rearrangement process that forges the central core of the molecule and (2) two facile C–N bond-forming steps that establish the flanking heterocycles.
Co-reporter:Brian J. Knight, Erin E. Stache, Eric M. Ferreira
Tetrahedron 2015 Volume 71(Issue 35) pp:5814-5823
Publication Date(Web):2 September 2015
DOI:10.1016/j.tet.2015.05.010
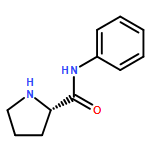
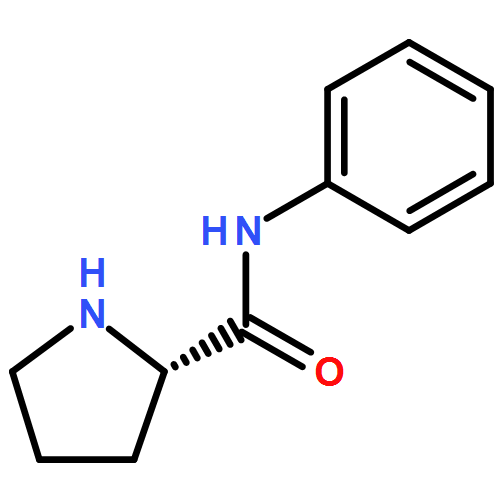
Alkylations of proline-based imidazolidinones are described based on the principle of self-regeneration of stereocenters (SRS), affording high levels of either the cis or trans configured products. Stereoselectivity is dictated solely on the nature of the ‘temporary’ group, where isobutyraldehyde-derived imidazolidinones provide the cis configured products and 1-naphthaldehyde-derived imidazolidinones afford the complementary trans configured products. These stereodivergent products can be readily cleaved to afford both α-alkylated proline enantiomers from readily available l-proline. A series of imidazolidinones were alkylated to investigate the origin of the anti-selectivity. Potential contributions toward the observed anti-selectivity are discussed on the basis of these experiments, suggesting a refined hypothesis for selectivity may be in order.
Co-reporter:Susan M. Stevenson, Eric T. Newcomb and Eric M. Ferreira
Inorganic Chemistry Frontiers 2016 - vol. 3(Issue 10) pp:NaN1235-1235
Publication Date(Web):2016/06/30
DOI:10.1039/C6QO00224B
A full account of our investigation of C–C bond migration in the cycloisomerization of oxygen-tethered 1,6-enynes is described. Under Pt(II) and/or Ir(I) catalysis, cyclic and acylic alkyl groups were found to undergo 1,2-shifts into metal carbenoid intermediates. Interestingly, this process does not appear to be driven by the release of ring strain, and thus provides access to large carbocyclic frameworks. The beneficial effect of CO on the Pt(II) and Ir(I) catalytic systems is also evaluated.
Co-reporter:Susan M. Stevenson, Robert F. Higgins, Matthew P. Shores and Eric M. Ferreira
Chemical Science (2010-Present) 2017 - vol. 8(Issue 1) pp:NaN660-660
Publication Date(Web):2016/09/12
DOI:10.1039/C6SC03303B
A chromium-catalyzed, visible light-activated net [4 + 2] cycloaddition between dienes and electron-deficient alkenes is described. Gathered evidence, via control experiments, isolated intermediates, and measured redox potentials, points to several converging reaction pathways that afford the cyclohexene adducts, including a photochemical [2 + 2] cycloaddition/vinylcyclobutane rearrangement cascade and a substrate excitation/oxidation sequence to a radical cation intermediate. Notably, the accompanying mechanistic stipulations result in a process that yields regioisomeric compounds from those generated by traditional Diels–Alder cycloadditions.
Co-reporter:Brian J. Knight, Jacob O. Rothbaum and Eric M. Ferreira
Chemical Science (2010-Present) 2016 - vol. 7(Issue 3) pp:NaN1987-1987
Publication Date(Web):2015/12/14
DOI:10.1039/C5SC03948G
We describe herein the design of a novel molecular scaffold that can induce facile oxidative olefinations when attached to alcohols. Benzylic, homo-, and bishomobenzylic alcohols are utilized. The scaffold can act as a protecting group for the alcohol in other transformations, and it is recoverable in excellent yield. The overall sequence can also be telescoped without purifications of intermediates, representing a net alcohol-based directed ortho-alkenylation.
.jpg)
![1H-Benzo[b]benzo[6,7]cyclohepta[1,2-d]furan-9-carboxaldehyde, 2,3,4,4a,5,6,7,12c-octahydro-10,11-dihydroxy-4,4,7,12c-tetramethyl-, (4aS,7R,12cS)-](/data/chemimg/109600/874292-32-7.png)
![1H-Benzo[b]benzo[6,7]cyclohepta[1,2-d]furan-9-carboxaldehyde, 2,3,4,4a,5,6,7,12c-octahydro-10,11-dihydroxy-4,4,7,12c-tetramethyl-, (4aS,7R,12cS)-](/data/chemimg/109600/874292-32-7_b.png)


![Benzenesulfonamide, N-[2-(3-methoxy-1-propyn-1-yl)phenyl]-4-methyl-](http://img.cochemist.com/ccimg/916700/916601-15-5.png)
![Benzenesulfonamide, N-[2-(3-methoxy-1-propyn-1-yl)phenyl]-4-methyl-](http://img.cochemist.com/ccimg/916700/916601-15-5_b.png)

![(Acetonitrile)[(2-biphenyl)di-tert-butylphosphine]gold(I) hexafluoroantimonate](http://img.cochemist.com/ccimg/866700/866641-66-9.png)
![(Acetonitrile)[(2-biphenyl)di-tert-butylphosphine]gold(I) hexafluoroantimonate](http://img.cochemist.com/ccimg/866700/866641-66-9_b.png)












![Hexanenitrile, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,2-dimethyl-](http://img.cochemist.com/ccimg/206300/206266-43-5.png)
![Hexanenitrile, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,2-dimethyl-](http://img.cochemist.com/ccimg/206300/206266-43-5_b.png)






![3-iodo-1-[(4-methylphenyl)sulfonyl]-1H-Indole](http://img.cochemist.com/ccimg/170500/170456-80-1.png)
![3-iodo-1-[(4-methylphenyl)sulfonyl]-1H-Indole](http://img.cochemist.com/ccimg/170500/170456-80-1_b.png)






![6,7-Dihydro-5H-cyclopenta[b]pyridin-7-yl acetate](http://img.cochemist.com/ccimg/90700/90685-59-9.png)
![6,7-Dihydro-5H-cyclopenta[b]pyridin-7-yl acetate](http://img.cochemist.com/ccimg/90700/90685-59-9_b.png)


![Benzene, [[(2-methoxyethoxy)methoxy]methyl]-](http://img.cochemist.com/ccimg/88800/88738-41-4.png)
![Benzene, [[(2-methoxyethoxy)methoxy]methyl]-](http://img.cochemist.com/ccimg/88800/88738-41-4_b.png)
![BENZENE, [(1E)-3-BROMO-2-METHYL-1-PROPENYL]-](http://img.cochemist.com/ccimg/83200/83126-01-6.png)
![BENZENE, [(1E)-3-BROMO-2-METHYL-1-PROPENYL]-](http://img.cochemist.com/ccimg/83200/83126-01-6_b.png)


![3-Buten-2-one, 4-[4-(phenylmethoxy)phenyl]-, (E)-](http://img.cochemist.com/ccimg/75700/75676-91-4.png)
![3-Buten-2-one, 4-[4-(phenylmethoxy)phenyl]-, (E)-](http://img.cochemist.com/ccimg/75700/75676-91-4_b.png)
![SILANE, (1,1-DIMETHYLETHYL)DIMETHYL[(2E)-2,4-PENTADIENYLOXY]-](http://img.cochemist.com/ccimg/74300/74213-12-0.png)
![SILANE, (1,1-DIMETHYLETHYL)DIMETHYL[(2E)-2,4-PENTADIENYLOXY]-](http://img.cochemist.com/ccimg/74300/74213-12-0_b.png)













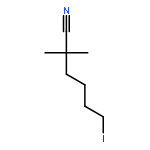



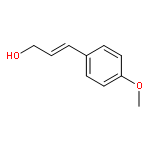
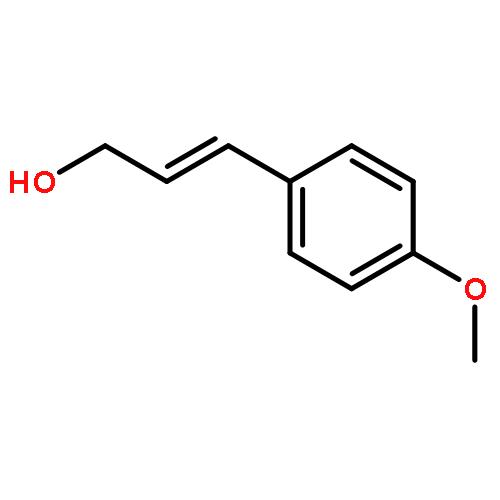
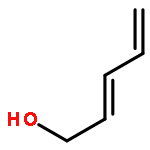


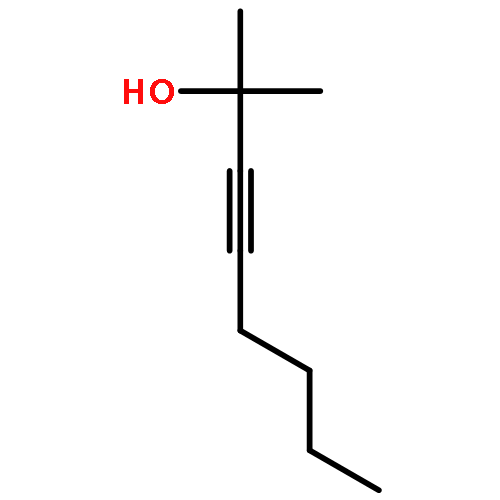
![Benzene, [(methoxymethoxy)methyl]-](http://img.cochemist.com/ccimg/31700/31600-55-2.png)
![Benzene, [(methoxymethoxy)methyl]-](http://img.cochemist.com/ccimg/31700/31600-55-2_b.png)




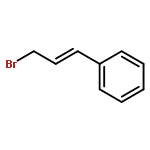
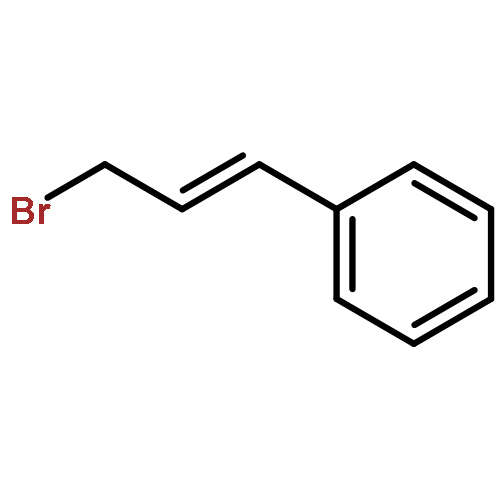







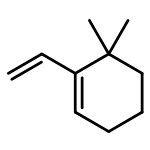

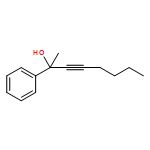







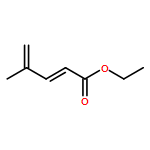
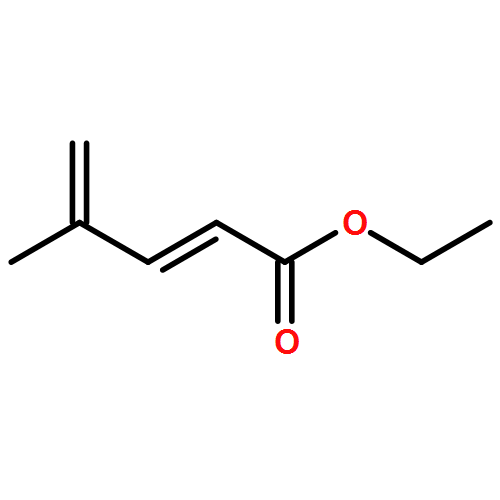


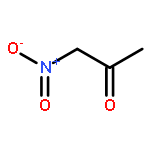
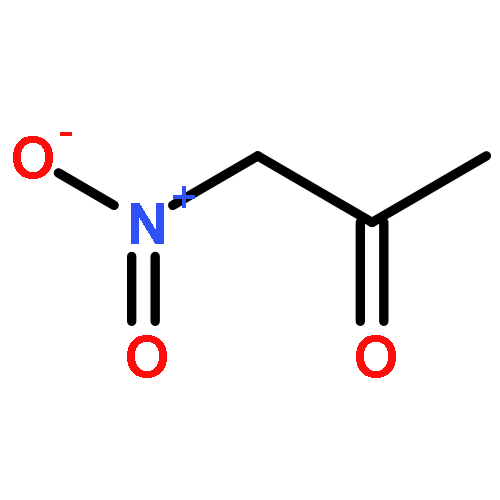








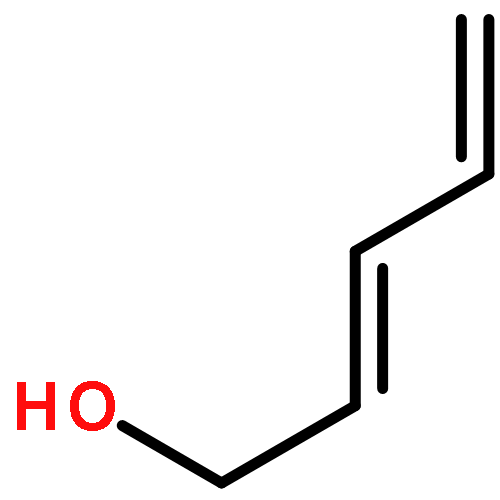
![furo[3,4-b]pyridin-5(7H)-one](http://img.cochemist.com/ccimg/4800/4733-69-1.png)
![furo[3,4-b]pyridin-5(7H)-one](http://img.cochemist.com/ccimg/4800/4733-69-1_b.png)


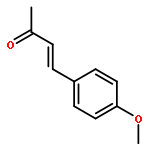
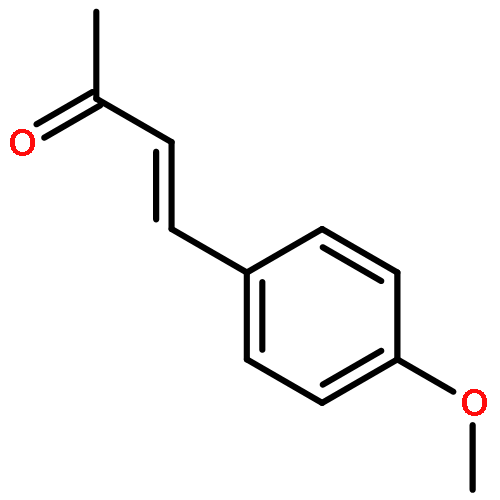
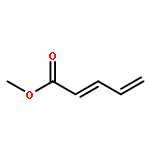
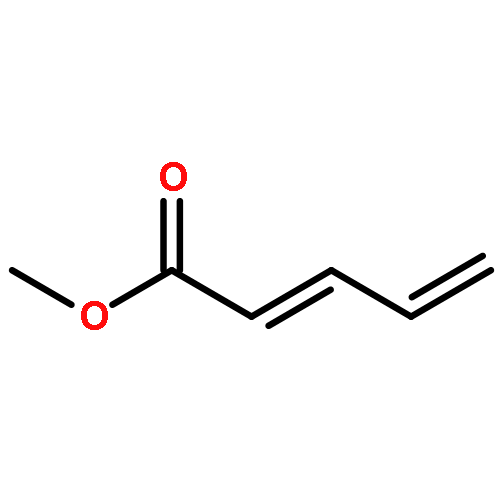

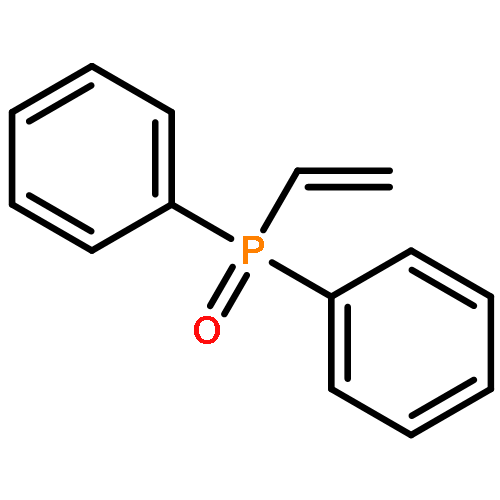




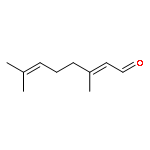


![Benzene, [[(1-methyl-2-propynyl)oxy]methyl]-](http://img.cochemist.com/ccimg/70000/69939-72-6.png)
![Benzene, [[(1-methyl-2-propynyl)oxy]methyl]-](http://img.cochemist.com/ccimg/70000/69939-72-6_b.png)



![(3R,7aS)-7a-methyl-3-(trichloromethyl)-3,5,6,7-tetrahydropyrrolo[1,2-c]oxazol-1-one](http://img.cochemist.com/ccimg/220300/220200-88-4.png)
![(3R,7aS)-7a-methyl-3-(trichloromethyl)-3,5,6,7-tetrahydropyrrolo[1,2-c]oxazol-1-one](http://img.cochemist.com/ccimg/220300/220200-88-4_b.png)
![Phosphonium, [4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]butyl]triphenyl-, iodide (1:1)](http://img.cochemist.com/data/images/190588-91-1.png)
![Phosphonium, [4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]butyl]triphenyl-, iodide (1:1)](http://img.cochemist.com/data/images/190588-91-1_b.png)






















![1-methoxy-2-[(1E)-prop-1-en-1-yl]benzene](http://img.cochemist.com/ccimg/2100/2077-36-3.png)
![1-methoxy-2-[(1E)-prop-1-en-1-yl]benzene](http://img.cochemist.com/ccimg/2100/2077-36-3_b.png)



