Co-reporter:Aurélie Mallinger; Kai Schiemann; Christian Rink; Frank Stieber; Michel Calderini; Simon Crumpler; Mark Stubbs; Olajumoke Adeniji-Popoola; Oliver Poeschke; Michael Busch; Paul Czodrowski; Djordje Musil; Daniel Schwarz; Maria-Jesus Ortiz-Ruiz; Richard Schneider; Ching Thai; Melanie Valenti; Alexis de Haven Brandon; Rosemary Burke; Paul Workman; Trevor Dale; Dirk Wienke; Paul A. Clarke; Christina Esdar; Florence I. Raynaud; Suzanne A. Eccles; Felix Rohdich
Journal of Medicinal Chemistry 2016 Volume 59(Issue 3) pp:1078-1101
Publication Date(Web):January 21, 2016
DOI:10.1021/acs.jmedchem.5b01685
The Mediator complex-associated cyclin-dependent kinase CDK8 has been implicated in human disease, particularly in colorectal cancer where it has been reported as a putative oncogene. Here we report the discovery of 109 (CCT251921), a potent, selective, and orally bioavailable inhibitor of CDK8 with equipotent affinity for CDK19. We describe a structure-based design approach leading to the discovery of a 3,4,5-trisubstituted-2-aminopyridine series and present the application of physicochemical property analyses to successfully reduce in vivo metabolic clearance, minimize transporter-mediated biliary elimination while maintaining acceptable aqueous solubility. Compound 109 affords the optimal compromise of in vitro biochemical, pharmacokinetic, and physicochemical properties and is suitable for progression to animal models of cancer.
Co-reporter:Vassilios Bavetsias; Rachel M. Lanigan; Gian Filippo Ruda; Butrus Atrash; Mark G. McLaughlin; Anthony Tumber; N. Yi Mok; Yann-Vaï Le Bihan; Sally Dempster; Katherine J. Boxall; Fiona Jeganathan; Stephanie B. Hatch; Pavel Savitsky; Srikannathasan Velupillai; Tobias Krojer; Katherine S. England; Jimmy Sejberg; Ching Thai; Adam Donovan; Akos Pal; Giuseppe Scozzafava; James M. Bennett; Akane Kawamura; Catrine Johansson; Aleksandra Szykowska; Carina Gileadi; Nicola A. Burgess-Brown; Frank von Delft; Udo Oppermann; Zoe Walters; Janet Shipley; Florence I. Raynaud; Susan M. Westaway◆; Rab K. Prinjha◆; Oleg Fedorov; Rosemary Burke; Christopher J. Schofield; Isaac M. Westwood; Chas Bountra; Susanne Müller; Rob L. M. van Montfort; Paul E. Brennan
Journal of Medicinal Chemistry 2016 Volume 59(Issue 4) pp:1388-1409
Publication Date(Web):January 7, 2016
DOI:10.1021/acs.jmedchem.5b01635
We report the discovery of N-substituted 4-(pyridin-2-yl)thiazole-2-amine derivatives and their subsequent optimization, guided by structure-based design, to give 8-(1H-pyrazol-3-yl)pyrido[3,4-d]pyrimidin-4(3H)-ones, a series of potent JmjC histone N-methyl lysine demethylase (KDM) inhibitors which bind to Fe(II) in the active site. Substitution from C4 of the pyrazole moiety allows access to the histone peptide substrate binding site; incorporation of a conformationally constrained 4-phenylpiperidine linker gives derivatives such as 54j and 54k which demonstrate equipotent activity versus the KDM4 (JMJD2) and KDM5 (JARID1) subfamily demethylases, selectivity over representative exemplars of the KDM2, KDM3, and KDM6 subfamilies, cellular permeability in the Caco-2 assay, and, for 54k, inhibition of H3K9Me3 and H3K4Me3 demethylation in a cell-based assay.
Co-reporter:Aurélie Mallinger, Kai Schiemann, Christian Rink, Jimmy Sejberg, Mark A. Honey, Paul Czodrowski, Mark Stubbs, Oliver Poeschke, Michael Busch, Richard Schneider, Daniel Schwarz, Djordje Musil, Rosemary Burke, Klaus Urbahns, Paul Workman, Dirk Wienke, Paul A. Clarke, Florence I. Raynaud, Suzanne A. Eccles, Christina Esdar, Felix Rohdich, and Julian Blagg
ACS Medicinal Chemistry Letters 2016 Volume 7(Issue 6) pp:573
Publication Date(Web):March 28, 2016
DOI:10.1021/acsmedchemlett.6b00022
We demonstrate a designed scaffold-hop approach to the discovery of 2,8-disubstituted-1,6-naphthyridine- and 4,6-disubstituted-isoquinoline-based dual CDK8/19 ligands. Optimized compounds in both series exhibited rapid aldehyde oxidase-mediated metabolism, which could be abrogated by introduction of an amino substituent at C5 of the 1,6-naphthyridine scaffold or at C1 of the isoquinoline scaffold. Compounds 51 and 59 were progressed to in vivo pharmacokinetic studies, and 51 also demonstrated sustained inhibition of STAT1SER727 phosphorylation, a biomarker of CDK8 inhibition, in an SW620 colorectal carcinoma human tumor xenograft model following oral dosing.Keywords: aldehyde oxidase; CDK19; CDK8; kinase inhibitor; mediator complex
Co-reporter:Aurélie Mallinger; Simon Crumpler; Mark Pichowicz; Dennis Waalboer; Mark Stubbs; Olajumoke Adeniji-Popoola; Bozena Wood; Elizabeth Smith; Ching Thai; Alan T. Henley; Katrin Georgi; William Court; Steve Hobbs; Gary Box; Maria-Jesus Ortiz-Ruiz; Melanie Valenti; Alexis De Haven Brandon; Robert TePoele; Birgitta Leuthner; Paul Workman; Wynne Aherne; Oliver Poeschke; Trevor Dale; Dirk Wienke; Christina Esdar; Felix Rohdich; Florence Raynaud; Paul A. Clarke; Suzanne A. Eccles; Frank Stieber; Kai Schiemann
Journal of Medicinal Chemistry 2015 Volume 58(Issue 4) pp:1717-1735
Publication Date(Web):February 13, 2015
DOI:10.1021/jm501436m
WNT signaling is frequently deregulated in malignancy, particularly in colon cancer, and plays a key role in the generation and maintenance of cancer stem cells. We report the discovery and optimization of a 3,4,5-trisubstituted pyridine 9 using a high-throughput cell-based reporter assay of WNT pathway activity. We demonstrate a twisted conformation about the pyridine–piperidine bond of 9 by small-molecule X-ray crystallography. Medicinal chemistry optimization to maintain this twisted conformation, cognisant of physicochemical properties likely to maintain good cell permeability, led to 74 (CCT251545), a potent small-molecule inhibitor of WNT signaling with good oral pharmacokinetics. We demonstrate inhibition of WNT pathway activity in a solid human tumor xenograft model with evidence for tumor growth inhibition following oral dosing. This work provides a successful example of hypothesis-driven medicinal chemistry optimization from a singleton hit against a cell-based pathway assay without knowledge of the biochemical target.
Co-reporter:Spiros Linardopoulos
Journal of Medicinal Chemistry 2015 Volume 58(Issue 13) pp:5186-5188
Publication Date(Web):June 25, 2015
DOI:10.1021/acs.jmedchem.5b00918
The quest for potent and selective small molecule inhibitors of the Aurora kinases has been long and resource intensive with multiple agents progressed to the clinic. To definitively explore the potential for clinical efficacy at well-tolerated dosing schedules requires a well-characterized, selective inhibitor with pharmacokinetic properties, flexible dosing regimen, and suite of target engagement biomarkers suitable for clinical use. AMG900 is a promising opportunity to definitively test the clinical benefit of dual Aurora kinase A and B inhibition.
Co-reporter:Nicholas C. Firth; Butrus Atrash; Nathan Brown
Journal of Chemical Information and Modeling 2015 Volume 55(Issue 6) pp:1169-1180
Publication Date(Web):June 9, 2015
DOI:10.1021/acs.jcim.5b00073
We describe the development and application of an integrated, multiobjective optimization workflow (MOARF) for directed medicinal chemistry design. This workflow couples a rule-based molecular fragmentation scheme (SynDiR) with a pharmacophore fingerprint-based fragment replacement algorithm (RATS) to broaden the scope of reconnection options considered in the generation of potential solution structures. Solutions are ranked by a multiobjective scoring algorithm comprising ligand-based (shape similarity) biochemical activity predictions as well as physicochemical property calculations. Application of this iterative workflow to optimization of the CDK2 inhibitor Seliciclib (CYC202, R-roscovitine) generated solution molecules in desired physicochemical property space. Synthesis and experimental evaluation of optimal solution molecules demonstrates CDK2 biochemical activity and improved human metabolic stability.
Co-reporter:Vassilios Bavetsias, Yolanda Pérez-Fuertes, Patrick J. McIntyre, Butrus Atrash, Magda Kosmopoulou, Lisa O’Fee, Rosemary Burke, Chongbo Sun, Amir Faisal, Katherine Bush, Sian Avery, Alan Henley, Florence I. Raynaud, Spiros Linardopoulos, Richard Bayliss, Julian Blagg
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 19) pp:4203-4209
Publication Date(Web):1 October 2015
DOI:10.1016/j.bmcl.2015.08.003
Introduction of a 1-benzyl-1H-pyrazol-4-yl moiety at C7 of the imidazo[4,5-b]pyridine scaffold provided 7a which inhibited a range of kinases including Aurora-A. Modification of the benzyl group in 7a, and subsequent co-crystallisation of the resulting analogues with Aurora-A indicated distinct differences in binding mode dependent upon the pyrazole N-substituent. Compounds 7a and 14d interact with the P-loop whereas 14a and 14b engage with Thr217 in the post-hinge region. These crystallographic insights provide options for the design of compounds interacting with the DFG motif or with Thr217.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Vassilios Bavetsias ; Amir Faisal ; Simon Crumpler ; Nathan Brown ; Magda Kosmopoulou ; Amar Joshi ; Butrus Atrash ; Yolanda Pérez-Fuertes ; Jessica A. Schmitt ; Katherine J. Boxall ; Rosemary Burke ; Chongbo Sun ; Sian Avery ; Katherine Bush ; Alan Henley ; Florence I. Raynaud ; Paul Workman ; Richard Bayliss ; Spiros Linardopoulos
Journal of Medicinal Chemistry 2013 Volume 56(Issue 22) pp:9122-9135
Publication Date(Web):November 6, 2013
DOI:10.1021/jm401115g
Aurora-A differs from Aurora-B/C at three positions in the ATP-binding pocket (L215, T217, and R220). Exploiting these differences, crystal structures of ligand–Aurora protein interactions formed the basis of a design principle for imidazo[4,5-b]pyridine-derived Aurora-A-selective inhibitors. Guided by a computational modeling approach, appropriate C7-imidazo[4,5-b]pyridine derivatization led to the discovery of highly selective inhibitors, such as compound 28c, of Aurora-A over Aurora-B. In HCT116 human colon carcinoma cells, 28c and 40f inhibited the Aurora-A L215R and R220K mutants with IC50 values similar to those seen for the Aurora-A wild type. However, the Aurora-A T217E mutant was significantly less sensitive to inhibition by 28c and 40f compared to the Aurora-A wild type, suggesting that the T217 residue plays a critical role in governing the observed isoform selectivity for Aurora-A inhibition. These compounds are useful small-molecule chemical tools to further explore the function of Aurora-A in cells.
Co-reporter:Sébastien Naud ; Isaac M. Westwood ; Amir Faisal ; Peter Sheldrake ; Vassilios Bavetsias ; Butrus Atrash ; Kwai-Ming J. Cheung ; Manjuan Liu ; Angela Hayes ; Jessica Schmitt ; Amy Wood ; Vanessa Choi ; Kathy Boxall ; Grace Mak ; Mark Gurden ; Melanie Valenti ; Alexis de Haven Brandon ; Alan Henley ; Ross Baker ; Craig McAndrew ; Berry Matijssen ; Rosemary Burke ; Swen Hoelder ; Suzanne A. Eccles ; Florence I. Raynaud ; Spiros Linardopoulos ; Rob L. M. van Montfort
Journal of Medicinal Chemistry 2013 Volume 56(Issue 24) pp:10045-10065
Publication Date(Web):November 20, 2013
DOI:10.1021/jm401395s
The protein kinase MPS1 is a crucial component of the spindle assembly checkpoint signal and is aberrantly overexpressed in many human cancers. MPS1 is one of the top 25 genes overexpressed in tumors with chromosomal instability and aneuploidy. PTEN-deficient breast tumor cells are particularly dependent upon MPS1 for their survival, making it a target of significant interest in oncology. We report the discovery and optimization of potent and selective MPS1 inhibitors based on the 1H-pyrrolo[3,2-c]pyridine scaffold, guided by structure-based design and cellular characterization of MPS1 inhibition, leading to 65 (CCT251455). This potent and selective chemical tool stabilizes an inactive conformation of MPS1 with the activation loop ordered in a manner incompatible with ATP and substrate-peptide binding; it displays a favorable oral pharmacokinetic profile, shows dose-dependent inhibition of MPS1 in an HCT116 human tumor xenograft model, and is an attractive tool compound to elucidate further the therapeutic potential of MPS1 inhibition.
Co-reporter:Fiona C. Rowan, Meirion Richards, Rachel A. Bibby, Andrew Thompson, Richard Bayliss, and Julian Blagg
ACS Chemical Biology 2013 Volume 8(Issue 10) pp:2184
Publication Date(Web):August 7, 2013
DOI:10.1021/cb400425t
Most protein kinases are regulated through activation loop phosphorylation, but the contributions of individual sites are largely unresolved due to insufficient control over sample phosphorylation. Aurora-A is a mitotic Ser/Thr protein kinase that has two regulatory phosphorylation sites on its activation loop, T287 and T288. While phosphorylation of T288 is known to activate the kinase, the function of T287 phosphorylation is unclear. We applied site-directed mutagenesis and selective chemical modification to specifically introduce bioisosteres for phospho-threonine and other unnatural amino acids at these positions. Modified Aurora-A proteins were characterized using a biochemical assay measuring substrate phosphorylation. Replacement of T288 with glutamate and aspartate weakly stimulated activity. Phospho-cysteine, installed by chemical synthesis from a corresponding cysteine residue introduced at position 288, showed catalytic activity approaching that of the comparable phospho-serine protein. Unnatural amino acid residues, with longer side chains, inserted at position 288 were autophosphorylated and supported substrate phosphorylation. Aurora-A activity is enhanced by phosphorylation at position 287 alone but is suppressed when position 288 is also phosphorylated. This is rationalized by competition between phosphorylated T287 and T288 for a binding site composed of arginines, based on a structure of Aurora-A in which phospho-T287 occupies this site. This is, to our knowledge, the first example of a Ser/Thr kinase whose activity is controlled by the phosphorylation state of adjacent residues in its activation loop. Overall we demonstrate an approach that combines mutagenesis and selective chemical modification of selected cysteine residues to investigate otherwise impenetrable aspects of kinase regulation.
Co-reporter:Sarah R. Langdon, Isaac M. Westwood, Rob L. M. van Montfort, Nathan Brown, and Julian Blagg
Journal of Chemical Information and Modeling 2013 Volume 53(Issue 5) pp:1100-1112
Publication Date(Web):May 14, 2013
DOI:10.1021/ci400100c
We describe and apply a scaffold-focused virtual screen based upon scaffold trees to the mitotic kinase TTK (MPS1). Using level 1 of the scaffold tree, we perform both 2D and 3D similarity searches between a query scaffold and a level 1 scaffold library derived from a 2 million compound library; 98 compounds from 27 unique top-ranked level 1 scaffolds are selected for biochemical screening. We show that this scaffold-focused virtual screen prospectively identifies eight confirmed active compounds that are structurally differentiated from the query compound. In comparison, 100 compounds were selected for biochemical screening using a virtual screen based upon whole molecule similarity resulting in 12 confirmed active compounds that are structurally similar to the query compound. We elucidated the binding mode for four of the eight confirmed scaffold hops to TTK by determining their protein–ligand crystal structures; each represents a ligand-efficient scaffold for inhibitor design.
Co-reporter:Jonathan Macdonald, Victoria Oldfield, Vassilios Bavetsias and Julian Blagg
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 14) pp:2335-2347
Publication Date(Web):21 Feb 2013
DOI:10.1039/C3OB27477B
We show that N3-MEM-protected imidazo[4,5-b]pyridines undergo efficient C2-functionalisation via direct C–H arylation. Twenty-two substituted imidazo[4,5-b]pyridines are prepared and iterative, selective elaboration of functionalised imidazo[4,5-b]pyridines gives 2,7- and 2,6-disubstituted derivatives in good yields from common intermediates. Mechanistic observations are consistent with a concerted-metallation-deprotonation mechanism facilitated by coordination of copper(I)iodide to the imidazo[4,5-b]pyridine.
Co-reporter:Vassilios Bavetsias ; Simon Crumpler ; Chongbo Sun ; Sian Avery ; Butrus Atrash ; Amir Faisal ; Andrew S. Moore ; Magda Kosmopoulou ; Nathan Brown ; Peter W. Sheldrake ; Katherine Bush ; Alan Henley ; Gary Box ; Melanie Valenti ; Alexis de Haven Brandon ; Florence I. Raynaud ; Paul Workman ; Suzanne A. Eccles ; Richard Bayliss ; Spiros Linardopoulos
Journal of Medicinal Chemistry 2012 Volume 55(Issue 20) pp:8721-8734
Publication Date(Web):October 8, 2012
DOI:10.1021/jm300952s
Optimization of the imidazo[4,5-b]pyridine-based series of Aurora kinase inhibitors led to the identification of 6-chloro-7-(4-(4-chlorobenzyl)piperazin-1-yl)-2-(1,3-dimethyl-1H-pyrazol-4-yl)-3H-imidazo[4,5-b]pyridine (27e), a potent inhibitor of Aurora kinases (Aurora-A Kd = 7.5 nM, Aurora-B Kd = 48 nM), FLT3 kinase (Kd = 6.2 nM), and FLT3 mutants including FLT3-ITD (Kd = 38 nM) and FLT3(D835Y) (Kd = 14 nM). FLT3-ITD causes constitutive FLT3 kinase activation and is detected in 20–35% of adults and 15% of children with acute myeloid leukemia (AML), conferring a poor prognosis in both age groups. In an in vivo setting, 27e strongly inhibited the growth of a FLT3-ITD-positive AML human tumor xenograft (MV4–11) following oral administration, with in vivo biomarker modulation and plasma free drug exposures consistent with dual FLT3 and Aurora kinase inhibition. Compound 27e, an orally bioavailable dual FLT3 and Aurora kinase inhibitor, was selected as a preclinical development candidate for the treatment of human malignancies, in particular AML, in adults and children.
Co-reporter:Nicholas C. Firth, Nathan Brown, and Julian Blagg
Journal of Chemical Information and Modeling 2012 Volume 52(Issue 10) pp:2516-2525
Publication Date(Web):September 26, 2012
DOI:10.1021/ci300293f
We describe a computational method, plane of best fit (PBF), to quantify and characterize the 3D character of molecules. This method is rapid and amenable to analysis of large diverse data sets. We compare PBF with alternative literature methods used to assess 3D character and apply the method to diverse data sets of fragment-like, drug-like, and natural product compound libraries. We show that exemplar fragment libraries underexploit the potential of 3D character in fragment-like chemical space and that drug-like molecules in the libraries examined are predominantly 2D in character.
Co-reporter:Cornelis Matijssen, M. Cris Silva-Santisteban, Isaac M. Westwood, Samerene Siddique, Vanessa Choi, Peter Sheldrake, Rob L.M. van Montfort, Julian Blagg
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 22) pp:6630-6639
Publication Date(Web):15 November 2012
DOI:10.1016/j.bmc.2012.09.024
Two closely related binding modes have previously been proposed for the ATP-competitive benzimidazole class of checkpoint kinase 2 (CHK2) inhibitors; however, neither binding mode is entirely consistent with the reported SAR. Unconstrained rigid docking of benzimidazole ligands into representative CHK2 protein crystal structures reveals an alternative binding mode involving a water-mediated interaction with the hinge region; docking which incorporates protein side chain flexibility for selected residues in the ATP binding site resulted in a refinement of the water-mediated hinge binding mode that is consistent with observed SAR. The flexible docking results are in good agreement with the crystal structures of four exemplar benzimidazole ligands bound to CHK2 which unambiguously confirmed the binding mode of these inhibitors, including the water-mediated interaction with the hinge region, and which is significantly different from binding modes previously postulated in the literature.Unconstrained rigid docking, flexible side chain docking and protein crystal structure determinations reveal a water-mediated hinge binding mode for a series of benzimidazole ligands of the protein kinase CHK2. This binding mode is different from those previously postulated in the literature and may provide a useful approach to selective small molecule inhibitor design.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Sarah R. Langdon, Nathan Brown, and Julian Blagg
Journal of Chemical Information and Modeling 2011 Volume 51(Issue 9) pp:2174-2185
Publication Date(Web):August 31, 2011
DOI:10.1021/ci2001428
The scaffold diversity of 7 representative commercial and proprietary compound libraries is explored for the first time using both Murcko frameworks and Scaffold Trees. We show that Level 1 of the Scaffold Tree is useful for the characterization of scaffold diversity in compound libraries and offers advantages over the use of Murcko frameworks. This analysis also demonstrates that the majority of compounds in the libraries we analyzed contain only a small number of well represented scaffolds and that a high percentage of singleton scaffolds represent the remaining compounds. We use Tree Maps to clearly visualize the scaffold space of representative compound libraries, for example, to display highly populated scaffolds and clusters of structurally similar scaffolds. This study further highlights the need for diversification of compound libraries used in hit discovery by focusing library enrichment on the synthesis of compounds with novel or underrepresented scaffolds.
Co-reporter:Mark Pichowicz, Simon Crumpler, Edward McDonald, Julian Blagg
Tetrahedron 2010 66(13) pp: 2398-2403
Publication Date(Web):
DOI:10.1016/j.tet.2010.01.101
Co-reporter:Julian Blagg, Charlotte M.N. Allerton, David V.J. Batchelor, Andrew D. Baxter, Denise J. Burring, Christopher L. Carr, Andrew S. Cook, Carly L. Nichols, Joanne Phipps, Vivienne G. Sanderson, Hugh Verrier, Stephen Wong
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 24) pp:6691-6696
Publication Date(Web):15 December 2007
DOI:10.1016/j.bmcl.2007.10.059
This paper reports the synthesis and biological activity of a novel series of aryl-morpholine dopamine receptor agonists. Several compounds show high levels of functional selectivity for the D3 over the D2 dopamine receptor. Compound 26 has >1000-fold functional selectivity and has been successfully progressed in vivo using an intranasal delivery route.Synthesis and dopamine receptor agonist activity of a novel series of aryl-morpholines is disclosed. Compound 26 shows high functional in vitro selectivity for activation the dopamine D3 receptor. In vivo properties of this compound are also reported.
Co-reporter:Julian Blagg, Paul Workman
Current Opinion in Pharmacology (August 2014) Volume 17() pp:87-100
Publication Date(Web):1 August 2014
DOI:10.1016/j.coph.2014.07.007
•Target validation is especially critical in the context of drugging the cancer genome and clinical drug resistance.•We review how chemical biology approaches benefit target validation.•We illustrate how critically assessed small molecule chemical inhibitors complement genetic approaches.•We highlight recent progress, including reagents for less druggable targets.Target validation is a crucial element of drug discovery. Especially given the wealth of potential targets emerging from cancer genome sequencing and functional genetic screens, and also considering the time and cost of downstream drug discovery efforts, it is essential to build confidence in a proposed target, ideally using different technical approaches. We argue that complementary biological and chemical biology strategies are essential for robust target validation. We discuss recent progress in the discovery and application of high quality chemical tools and other chemical biology approaches to target validation in cancer. Among other topical examples, we highlight the emergence of designed irreversible chemical tools to study potential target proteins and oncogenic pathways that were hitherto regarded as poorly druggable.
Co-reporter:Jonathan Macdonald, Victoria Oldfield, Vassilios Bavetsias and Julian Blagg
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 14) pp:NaN2347-2347
Publication Date(Web):2013/02/21
DOI:10.1039/C3OB27477B
We show that N3-MEM-protected imidazo[4,5-b]pyridines undergo efficient C2-functionalisation via direct C–H arylation. Twenty-two substituted imidazo[4,5-b]pyridines are prepared and iterative, selective elaboration of functionalised imidazo[4,5-b]pyridines gives 2,7- and 2,6-disubstituted derivatives in good yields from common intermediates. Mechanistic observations are consistent with a concerted-metallation-deprotonation mechanism facilitated by coordination of copper(I)iodide to the imidazo[4,5-b]pyridine.

![N8-[(2S)-3,3-Dimethylbutan-2-yl]-N2-[2-methoxy-4-(1-methyl-1H-pyrazol-4-yl)phenyl]pyrido[3,4-d]pyrimidine-2,8-diamine](http://img.cochemist.com/ccimg/1578300/1578244-34-4.png)
![N8-[(2S)-3,3-Dimethylbutan-2-yl]-N2-[2-methoxy-4-(1-methyl-1H-pyrazol-4-yl)phenyl]pyrido[3,4-d]pyrimidine-2,8-diamine](http://img.cochemist.com/ccimg/1578300/1578244-34-4_b.png)
![N-[2-(2-pyridinyl)-6-(1,2,4,5-tetrahydro-3h-3-benzazepin-3-yl)-4- Pyrimidinyl]-β-alanine](http://img.cochemist.com/ccimg/1373500/1373422-53-7.png)
![N-[2-(2-pyridinyl)-6-(1,2,4,5-tetrahydro-3h-3-benzazepin-3-yl)-4- Pyrimidinyl]-β-alanine](http://img.cochemist.com/ccimg/1373500/1373422-53-7_b.png)
![8H-Purin-8-one, 9-cyclopentyl-7,9-dihydro-2-[[2-methoxy-4-[(1-methyl-4-piperidinyl)oxy]phenyl]amino]-7-methyl-](http://img.cochemist.com/ccimg/1124400/1124329-14-1.png)
![8H-Purin-8-one, 9-cyclopentyl-7,9-dihydro-2-[[2-methoxy-4-[(1-methyl-4-piperidinyl)oxy]phenyl]amino]-7-methyl-](http://img.cochemist.com/ccimg/1124400/1124329-14-1_b.png)
![8-boc-1,8-diazaspiro[4.5]decane oxalate](http://img.cochemist.com/ccimg/1408100/1408075-17-1.png)
![8-boc-1,8-diazaspiro[4.5]decane oxalate](http://img.cochemist.com/ccimg/1408100/1408075-17-1_b.png)


![tert-Butyl 6-oxo-2,7-diazaspiro[4.4]nonane-2-carboxylate](http://img.cochemist.com/ccimg/1194400/1194376-44-7.png)
![tert-Butyl 6-oxo-2,7-diazaspiro[4.4]nonane-2-carboxylate](http://img.cochemist.com/ccimg/1194400/1194376-44-7_b.png)
![1-[2-(Oxan-2-yloxy)ethyl]-4-(tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole](http://img.cochemist.com/ccimg/957000/956907-34-9.png)
![1-[2-(Oxan-2-yloxy)ethyl]-4-(tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole](http://img.cochemist.com/ccimg/957000/956907-34-9_b.png)
![3-Oxa-1,8-diazaspiro[4.5]decan-2-one](http://img.cochemist.com/ccimg/946000/945947-99-9.png)
![3-Oxa-1,8-diazaspiro[4.5]decan-2-one](http://img.cochemist.com/ccimg/946000/945947-99-9_b.png)
![2,8-Diazaspiro[4.5]decan-3-one, hydrochloride (1:1)](http://img.cochemist.com/ccimg/945900/945892-88-6.png)
![2,8-Diazaspiro[4.5]decan-3-one, hydrochloride (1:1)](http://img.cochemist.com/ccimg/945900/945892-88-6_b.png)


![2,8-Diazaspiro[4.5]decan-1-one, 2-methyl-, monohydrochloride](http://img.cochemist.com/ccimg/848600/848580-34-7.png)
![2,8-Diazaspiro[4.5]decan-1-one, 2-methyl-, monohydrochloride](http://img.cochemist.com/ccimg/848600/848580-34-7_b.png)

![Piperidine, 4-[(3,4-dichlorophenyl)methyl]-](http://img.cochemist.com/ccimg/220800/220772-32-7.png)
![Piperidine, 4-[(3,4-dichlorophenyl)methyl]-](http://img.cochemist.com/ccimg/220800/220772-32-7_b.png)
![Piperidine, 4-[[4-(trifluoromethyl)phenyl]methyl]-](http://img.cochemist.com/ccimg/193000/192990-03-7.png)
![Piperidine, 4-[[4-(trifluoromethyl)phenyl]methyl]-](http://img.cochemist.com/ccimg/193000/192990-03-7_b.png)
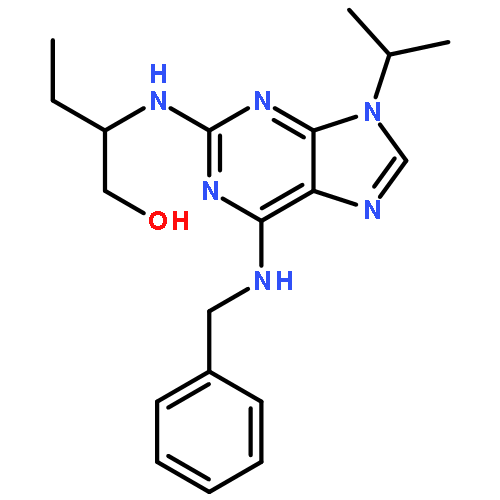
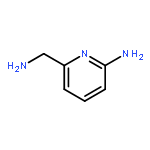
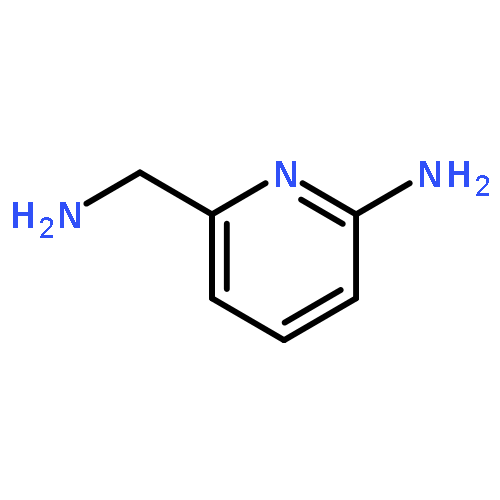
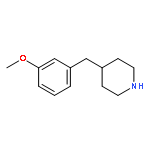
![3-PHENYL-1H-PYRAZOLO[4,3-C]PYRIDINE](http://img.cochemist.com/ccimg/113300/113277-54-6.png)
![3-PHENYL-1H-PYRAZOLO[4,3-C]PYRIDINE](http://img.cochemist.com/ccimg/113300/113277-54-6_b.png)

![PYRIDO[3,4-D]PYRIMIDIN-4(1H)-ONE, 8-CHLORO-](http://img.cochemist.com/ccimg/84400/84341-13-9.png)
![PYRIDO[3,4-D]PYRIMIDIN-4(1H)-ONE, 8-CHLORO-](http://img.cochemist.com/ccimg/84400/84341-13-9_b.png)
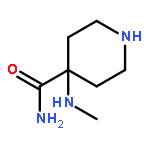
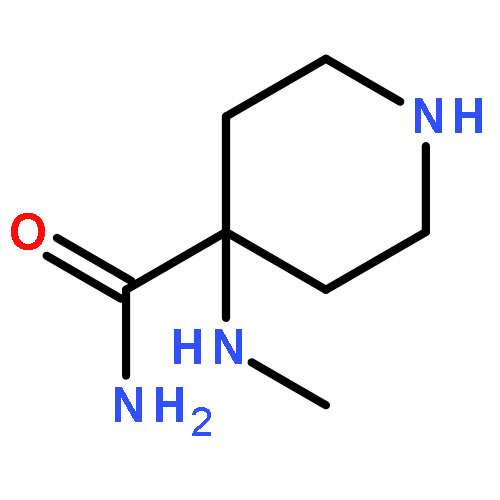
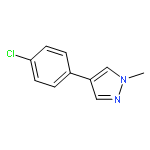
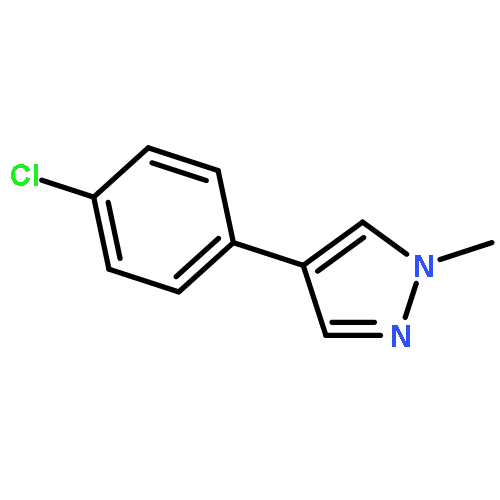

![4-BENZO[1,3]DIOXOL-5-YLMETHYL-PIPERIDINE](http://img.cochemist.com/ccimg/76700/76672-65-6.png)
![4-BENZO[1,3]DIOXOL-5-YLMETHYL-PIPERIDINE](http://img.cochemist.com/ccimg/76700/76672-65-6_b.png)



![4-[3-(TRIFLUOROMETHYL)PHENYL]PIPERIDINE](http://img.cochemist.com/ccimg/32900/32860-17-6.png)
![4-[3-(TRIFLUOROMETHYL)PHENYL]PIPERIDINE](http://img.cochemist.com/ccimg/32900/32860-17-6_b.png)




![1,3,8-Triaza-spiro[4.5]decane-2,4-dione hydrochloride](http://img.cochemist.com/ccimg/13700/13625-48-4.png)
![1,3,8-Triaza-spiro[4.5]decane-2,4-dione hydrochloride](http://img.cochemist.com/ccimg/13700/13625-48-4_b.png)







![8-Benzyl-1-oxa-3,8-diazaspiro[4.5]decan-2-one](http://img.cochemist.com/ccimg/5100/5053-14-5.png)
![8-Benzyl-1-oxa-3,8-diazaspiro[4.5]decan-2-one](http://img.cochemist.com/ccimg/5100/5053-14-5_b.png)
![1-Oxa-3,8-diazaspiro[4.5]decan-2-one](http://img.cochemist.com/ccimg/5100/5052-95-9.png)
![1-Oxa-3,8-diazaspiro[4.5]decan-2-one](http://img.cochemist.com/ccimg/5100/5052-95-9_b.png)




![2,8-Diazaspiro[4.5]decane-1,3-dione](http://img.cochemist.com/ccimg/2100/2079-25-6.png)
![2,8-Diazaspiro[4.5]decane-1,3-dione](http://img.cochemist.com/ccimg/2100/2079-25-6_b.png)


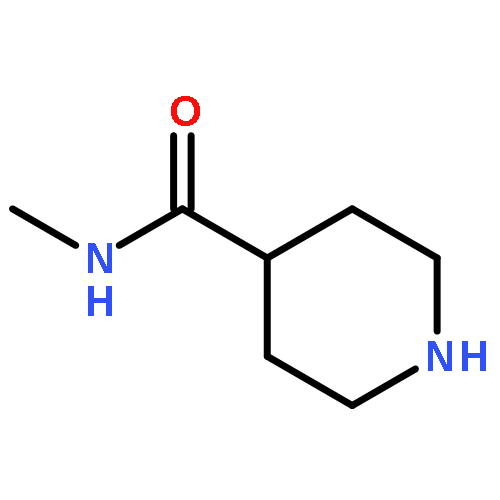
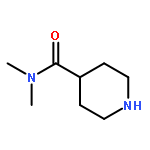
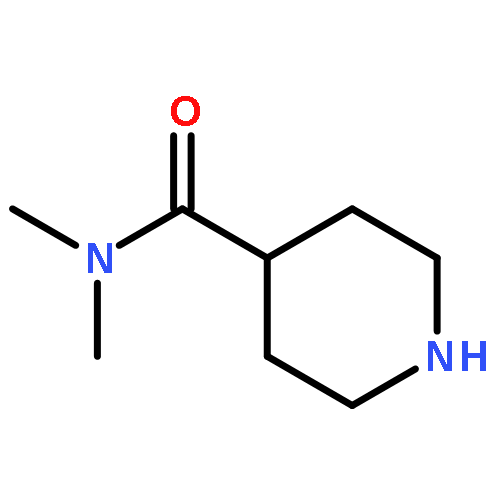
![8-Benzyl-2,8-diazaspiro[4.5]decane-1,3-dione](http://img.cochemist.com/ccimg/1500/1463-48-5.png)
![8-Benzyl-2,8-diazaspiro[4.5]decane-1,3-dione](http://img.cochemist.com/ccimg/1500/1463-48-5_b.png)


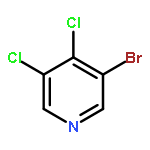
![1H-PYRAZOLE, 4-[4-(4,4,5,5-TETRAMETHYL-1,3,2-DIOXABOROLAN-2-YL)PHENYL]-](http://img.cochemist.com/ccimg/756600/756520-75-9.png)
![1H-PYRAZOLE, 4-[4-(4,4,5,5-TETRAMETHYL-1,3,2-DIOXABOROLAN-2-YL)PHENYL]-](http://img.cochemist.com/ccimg/756600/756520-75-9_b.png)