Co-reporter:Jingheng Wu, Sixiang Wen, Yiwei Zhou, Hui Chao, and Yong Shen
Journal of Chemical Information and Modeling 2016 Volume 56(Issue 12) pp:2421-2433
Publication Date(Web):November 1, 2016
DOI:10.1021/acs.jcim.6b00216
Ferrochelatase catalyzes the insertion of ferrous iron into protoporphyrin IX, the terminal step in heme biosynthesis. Some disputes in its mechanism remain unsolved, especially for human ferrochelatase. In this paper, high-level quantum mechanical/molecular mechanics (QM/MM) and free-energy studies were performed to address these controversial issues including the iron-binding site, the optimal reaction path, the substrate porphyrin distortion, and the presence of the sitting-atop (SAT) complex. Our results reveal that the ferrous iron is probably at the binding site coordinating with Met76, and His263 plays the role of proton acceptor. The rate-determining step is either the first proton removed by His263 or the proton transition within the porphyrin with an energy barrier of 14.99 or 14.87 kcal/mol by the quantum mechanical thermodynamic cycle perturbation (QTCP) calculations, respectively. The fast deprotonation step with the conservative residues rather than porphyrin deformation found in solution provides the driving force for biochelation. The SAT complex is not a necessity for the catalysis though it induces a modest distortion on the porphyrin ring.
Co-reporter:Shuqin Liu, Yiwei Zhou, Juan Zheng, Jianqiao Xu, Ruifeng Jiang, Yong Shen, Jijun Jiang, Fang Zhu, Chengyong Su and Gangfeng Ouyang
Analyst 2015 vol. 140(Issue 13) pp:4384-4387
Publication Date(Web):07 May 2015
DOI:10.1039/C5AN00775E
Here we report the successful utilization of the stepwise ligand exchange strategy for the improvement of adsorption ability of a series of bio-MOFs. The fast extraction rate and the different adsorption performances of the three bio-MOF coatings were dominated by their pore structures.
Co-reporter:Li Qian;Si-Yan Liao;Zu-Liang Huang
Journal of Molecular Modeling 2010 Volume 16( Issue 6) pp:1139-1150
Publication Date(Web):2010 June
DOI:10.1007/s00894-009-0609-8
Theoretical studies on the three-dimensional (3D) quantitative structure-activity relationship (QSAR) and mechanisms of action of a series of pyrimidine substituent derivatives as dual inhibitors of AP-1 and NF-κB were carried out using comparative molecular field analysis (CoMFA) and docking methods. The established 3D-QSAR model exhibits a satisfying statistical quality and prediction ability. Docking results show somewhat lower average values of the flexible and rigid energy scores in the chosen binding sites. The docking analysis offers appropriate orientations and conformations of these compounds at the binding sites to both AP-1 and NF-κB in good agreement with the 3D-QSAR model from CoMFA. The combined CoMFA and docking study suggests the following substituent selections: substituent R2 should be a kind of H–N–thienyl or CH3–N–thienyl group; substituent R5 should be a kind of COO–tBu or COOEt group; and substituent R4 should be a CH2CH3 or 2-thienyl group. The docking analysis also shows that the binding sites fall just at the joint regions between AP-1 (or NF-κB) and DNA, where these compounds can effectively prevent free AP-1 and NF-κB from binding to DNA, and this may be the reason that derivatives with pyrimidine substituents have an inhibition function. In addition, a very interesting finding was that the binding sites of both AP-1 and NF-κB have a common structural characteristic, thereby providing a reasonable explanation for the dual inhibition functions of these compounds towards both AP-1 and NF-κB. These theoretical results help to deepen our understanding of the inhibition mechanism of these pyrimidine substituent derivatives, and will aid in directing further drug-molecular design.
Co-reporter:Yaxue Wang, Yong Shen, Ulf Ryde
Journal of Inorganic Biochemistry 2009 Volume 103(Issue 12) pp:1680-1686
Publication Date(Web):December 2009
DOI:10.1016/j.jinorgbio.2009.09.013
Ferrochelatase catalyzes the metallation of protoporphyrin IX in the terminal step of heme biosynthesis. Mutations in the ferrochelatase gene can lead to the disease erythropoietic porphyria. The catalyzing mechanism of ferrochelatase is still not fully understood. In this paper, we have studied the insertion of Fe2+ into the protoporphyrin IX ring by Bacillussubtilis ferrochelatase using combined quantum mechanical and molecular mechanics (QM/MM) calculations. Geometries were optimized at the BP86/6-31G∗ level and energies were calculated at the B3LYP/TZVP level. The overall process involves the stepwise displacement of Glu-264, His-183, and a water molecule from Fe2+, and the removal of two protons from the porphyrin ring. The rate-determining step is the cleavage of the bond between the oxygen atom of Glu-264 and Fe2+, concomitant with the formation of the first Fe–N bond. It has an energy barrier of 57 kJ mol−1. The porphyrin ring is only slightly distorted in the enzyme active site. The residue Tyr-13 plays a key role for the catalytic process extracting two protons from protoporphyrin IX.
Co-reporter:JinCan Chen;Li Qian;LanMei Chen
Science China Chemistry 2008 Volume 51( Issue 2) pp:111-119
Publication Date(Web):2008 February
DOI:10.1007/s11426-007-0107-8
A quantitative structure-activity relationship (QSAR) of a series of benzothiazole derivatives showing a potent and selective cytotoxicity against a tumorigenic cell line has been studied by using the density functional theory (DFT), molecular mechanics (MM+) and statistical methods, and the QSAR equation was established via a correlation analysis and a stepwise regression analysis. A new scheme determining outliers by “leave-one-out” (LOO) cross-validation coefficient (qn-i2) was suggested and successfully used. In the established optimal equation (excluding two outliers), the steric parameter (MRR) and the net charge (QFR) of the first atom of the substituent (R), as well as the square of hydrophobic parameter (IgP)2 of the whole molecule, are the main independent factors contributing to the anticancer activities of the compounds. The fitting correlation coefficient (R2) and the cross-validation coefficient (q2) values are 0.883 and 0.797, respectively. it indicates that this model has a significantly statistical quality and an excellent prediction ability. Based on the QSAR studies, 4 new compounds with high predicted anticancer activities have been theoretically designed and they are expected to be confirmed experimentally.

![2-Propenoic acid, 3-[5'-[3,6-bis(5-hexyl-2-thienyl)-9H-carbazol-9-yl][2,2'-bithiophen]-5-yl]-2-cyano-](/data/chemimg/102000/1639059-76-9.png)
![2-Propenoic acid, 3-[5'-[3,6-bis(5-hexyl-2-thienyl)-9H-carbazol-9-yl][2,2'-bithiophen]-5-yl]-2-cyano-](/data/chemimg/102000/1639059-76-9_b.png)
![[2,2'-Bithiophene]-5-carboxaldehyde, 5'-[3,6-bis(5-hexyl-2-thienyl)-9H-carbazol-9-yl]-](/data/chemimg/102000/1639059-74-7.png)
![[2,2'-Bithiophene]-5-carboxaldehyde, 5'-[3,6-bis(5-hexyl-2-thienyl)-9H-carbazol-9-yl]-](/data/chemimg/102000/1639059-74-7_b.png)
![Thieno[3,4-b]-1,4-dioxin-5-carboxaldehyde, 7-(5'-bromo-3',4-dihexyl[2,2'-bithiophen]-5-yl)-2,3-dihydro-](/data/chemimg/101600/1519896-29-7.png)
![Thieno[3,4-b]-1,4-dioxin-5-carboxaldehyde, 7-(5'-bromo-3',4-dihexyl[2,2'-bithiophen]-5-yl)-2,3-dihydro-](/data/chemimg/101600/1519896-29-7_b.png)
![2-Propenoic acid, 3-[7-[5-[3,6-bis(5-hexyl-2-thienyl)-9H-carbazol-9-yl]-3-hexyl-2-thienyl]-2,3-dihydrothieno[3,4-b]-1,4-dioxin-5-yl]-2-cyano-](/data/chemimg/101600/1519896-22-0.png)
![2-Propenoic acid, 3-[7-[5-[3,6-bis(5-hexyl-2-thienyl)-9H-carbazol-9-yl]-3-hexyl-2-thienyl]-2,3-dihydrothieno[3,4-b]-1,4-dioxin-5-yl]-2-cyano-](/data/chemimg/101600/1519896-22-0_b.png)
![Thieno[3,4-b]-1,4-dioxin-5-carboxaldehyde, 7-[5-[3,6-bis(5-hexyl-2-thienyl)-9H-carbazol-9-yl]-3-hexyl-2-thienyl]-2,3-dihydro-](/data/chemimg/101600/1519896-19-5.png)
![Thieno[3,4-b]-1,4-dioxin-5-carboxaldehyde, 7-[5-[3,6-bis(5-hexyl-2-thienyl)-9H-carbazol-9-yl]-3-hexyl-2-thienyl]-2,3-dihydro-](/data/chemimg/101600/1519896-19-5_b.png)
![Thieno[3,4-b]-1,4-dioxin-5-carboxaldehyde, 7-(5-bromo-3-hexyl-2-thienyl)-2,3-dihydro-](/data/chemimg/101600/1519896-17-3.png)
![Thieno[3,4-b]-1,4-dioxin-5-carboxaldehyde, 7-(5-bromo-3-hexyl-2-thienyl)-2,3-dihydro-](/data/chemimg/101600/1519896-17-3_b.png)
![2-Propenoic acid, 3-[7-[5-[3,6-bis(5-hexyl-2-thienyl)-9H-carbazol-9-yl]-2-thienyl]-2,3-dihydrothieno[3,4-b]-1,4-dioxin-5-yl]-2-cyano-](/data/chemimg/101600/1519896-15-1.png)
![2-Propenoic acid, 3-[7-[5-[3,6-bis(5-hexyl-2-thienyl)-9H-carbazol-9-yl]-2-thienyl]-2,3-dihydrothieno[3,4-b]-1,4-dioxin-5-yl]-2-cyano-](/data/chemimg/101600/1519896-15-1_b.png)
![Thieno[3,4-b]-1,4-dioxin-5-carboxaldehyde, 7-[5-[3,6-bis(5-hexyl-2-thienyl)-9H-carbazol-9-yl]-2-thienyl]-2,3-dihydro-](/data/chemimg/101600/1519896-12-8.png)
![Thieno[3,4-b]-1,4-dioxin-5-carboxaldehyde, 7-[5-[3,6-bis(5-hexyl-2-thienyl)-9H-carbazol-9-yl]-2-thienyl]-2,3-dihydro-](/data/chemimg/101600/1519896-12-8_b.png)
![2-Propenoic acid, 2-cyano-3-[5'-(3,6-di-2-thienyl-9H-carbazol-9-yl)[2,2'-bithiophen]-5-yl]-](/data/chemimg/101600/1519896-03-7.png)
![2-Propenoic acid, 2-cyano-3-[5'-(3,6-di-2-thienyl-9H-carbazol-9-yl)[2,2'-bithiophen]-5-yl]-](/data/chemimg/101600/1519896-03-7_b.png)

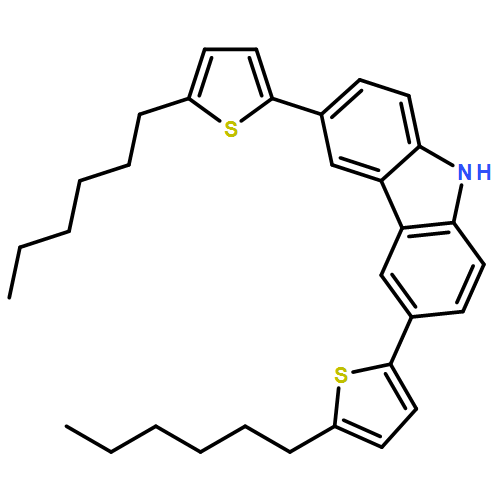
![[2,2'-Bithiophene]-5-carboxaldehyde, 5'-(3,6-di-2-thienyl-9H-carbazol-9-yl)-](/data/chemimg/101500/1519895-98-7.png)
![[2,2'-Bithiophene]-5-carboxaldehyde, 5'-(3,6-di-2-thienyl-9H-carbazol-9-yl)-](/data/chemimg/101500/1519895-98-7_b.png)
![Stannane, 1,1'-[4,8-bis[5-(2-ethylhexyl)-2-thienyl]benzo[1,2-b:4,5-b']dithiophene-2,6-diyl]bis[1,1,1-tributyl-](/data/chemimg/139800/1439862-07-3.png)
![Stannane, 1,1'-[4,8-bis[5-(2-ethylhexyl)-2-thienyl]benzo[1,2-b:4,5-b']dithiophene-2,6-diyl]bis[1,1,1-tributyl-](/data/chemimg/139800/1439862-07-3_b.png)
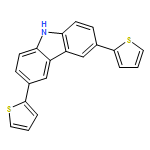

![Thieno[3,4-b]-1,4-dioxin-5-carboxaldehyde, 7-(5-bromo-2-thienyl)-2,3-dihydro-](/data/chemimg/101000/1354222-55-1.png)
![Thieno[3,4-b]-1,4-dioxin-5-carboxaldehyde, 7-(5-bromo-2-thienyl)-2,3-dihydro-](/data/chemimg/101000/1354222-55-1_b.png)
![Benzo[1,2-b:4,5-b']dithiophene, 4,8-bis[5-(2-ethylhexyl)-2-thienyl]-](/data/chemimg/139500/1352642-35-3.png)
![Benzo[1,2-b:4,5-b']dithiophene, 4,8-bis[5-(2-ethylhexyl)-2-thienyl]-](/data/chemimg/139500/1352642-35-3_b.png)