Co-reporter:Qin Yin, Yashar Soltani, Rebecca L. Melen, and Martin Oestreich
Organometallics July 10, 2017 Volume 36(Issue 13) pp:2381-2381
Publication Date(Web):June 19, 2017
DOI:10.1021/acs.organomet.7b00381
The rarely used boron Lewis acid tris[3,5-bis(trifluoromethyl)phenyl]borane (BArF3) is found to be an excellent catalyst for metal-free hydroboration of imines. In the presence of 1.0 mol % of BArF3, several ketimines and aldimines undergo hydroboration with pinacolborane (HBpin) at room temperature without the aid of an external Lewis base. BArF3 is more reactive than other Lewis acidic boranes, including the often-used tris(pentafluorophenyl)borane (B(C6F5)3). The steric hindrance imparted by the six fluorine atoms ortho to the boron center in B(C6F5)3 accounts for this. Mechanistic control experiments indicate conventional Lewis acid catalysis involving imine activation and hydride transfer from HBpin.
Co-reporter:Jonas Scharfbier, Hamideh Hazrati, Elisabeth Irran, and Martin Oestreich
Organic Letters December 15, 2017 Volume 19(Issue 24) pp:6562-6562
Publication Date(Web):December 1, 2017
DOI:10.1021/acs.orglett.7b03279
A copper-catalyzed nucleophilic displacement of α-triflyloxy nitriles and esters with silicon nucleophiles allows for the stereospecific generation of highly enantioenriched α-silylated carboxyl compounds. The enantioselective synthesis of α-silylated nitriles is unprecedented. The catalytic system exhibits good functional group tolerance. The stereochemical course of the substitution is shown to proceed with inversion of the configuration. The new reaction is an addition to the still limited number of methods for catalytic C(sp3)–Si cross-coupling.
Co-reporter:Julien Fuchs, Hendrik F. T. Klare, and Martin Oestreich
ACS Catalysis December 1, 2017 Volume 7(Issue 12) pp:8338-8338
Publication Date(Web):October 26, 2017
DOI:10.1021/acscatal.7b03336
An experimental analysis proves that Nikonov’s carbonyl hydrosilylation proceeds through a two-silicon cycle rather than the originally proposed one-silicon cycle. The intermediate ruthenium(II) monohydride is not sufficiently hydridic to transfer its hydride onto the silylcarboxonium ion. However, that hydricity is enhanced by oxidative addition of another hydrosilane molecule to afford the corresponding ruthenium(IV) silyl dihydride as the actual hydride donor. The present study also demonstrates that the acetonitrile ligands in Nikonov’s ruthenium(II) catalyst are not innocent. That complex is able to hydrosilylate its own ligand(s), and the resulting N,N-disilylated amine base accounts for competing dehydrogenative silylation of enolizable carbonyl compounds, explaining the formation of a silyl enol ether in substantial quantities next to the expected silyl ether. Both findings lead to a revised mechanistic picture that provides the basis for the development of more efficient and chemoselective catalysts.Keywords: homogeneous catalysis; hydrosilanes; ionic hydrosilylation; Lewis-acid catalysis; reaction mechanism; ruthenium; Si−H bond activation;
Co-reporter:Sebastian Keess and Martin Oestreich
Organic Letters April 7, 2017 Volume 19(Issue 7) pp:
Publication Date(Web):March 30, 2017
DOI:10.1021/acs.orglett.7b00672
Various cyclohexa-2,5-dien-1-yl-substituted germanes are shown to serve as easy-to-handle surrogates of hydrogermanes, including gaseous MeGeH3 and Me2GeH2. The Ge–H functional group is liberated by treatment with catalytic amounts of B(C6F5)3 and participates in situ in the B(C6F5)3-catalyzed hydrogermylation of alkenes. The range of suitable alkenes is broad, and the overall procedure provides a convenient access to tetraalkyl-substituted germanes at room temperature. Transfer hydrogermylation of internal alkynes works equally well and selectively forms the trans or cis diastereomer depending on the electronic bias of the C≡C bond.
Co-reporter:Lukas OmannMartin Oestreich
Organometallics 2017 Volume 36(Issue 4) pp:
Publication Date(Web):December 16, 2016
DOI:10.1021/acs.organomet.6b00801
A protocol for the catalytic synthesis of indole-fused benzosiloles starting from 2-aryl-substituted indoles and dihydrosilanes is reported. Compared to known procedures, this method does not require prefunctionalized starting materials and, hence, allows for a rapid access to those siloles. The net reaction is a 2-fold electrophilic C–H silylation catalyzed by cationic Ru–S complexes. Both reaction steps were separately investigated, and these results eventually led to the development of a two-step procedure. By preparing new Ru–S complexes with different weakly coordinating anions (WCAs), it is also shown that the latter can have a dramatic influence on the outcome of these reactions. Furthermore, the substrate scope of the new method is discussed.
Co-reporter:Dr. Qin Yin; Dr. Martin Oestreich
Angewandte Chemie International Edition 2017 Volume 56(Issue 27) pp:7716-7718
Publication Date(Web):2017/06/26
DOI:10.1002/anie.201703536
Sunburned: Visible light facilitates the acceptorless dehydrogenation of saturated hetero- and carbocycles with an annulated benzene ring (see scheme; X=N and CH2). These aromatizations occur at room temperature. The new methods are not only useful for the synthesis of bicyclic heteroarenes and arenes with diverse substitution patterns but also potentially attractive for hydrogen-storage materials.
Co-reporter:Dr. Qin Yin; Dr. Martin Oestreich
Angewandte Chemie 2017 Volume 129(Issue 27) pp:7824-7826
Publication Date(Web):2017/06/26
DOI:10.1002/ange.201703536
Sonnenverbrannt: Sichtbares Licht ermöglicht die akzeptorfreie Dehydrierung von gesättigten Hetero- und Carbocyclen mit anelliertem Benzolring bei Raumtemperatur (siehe Schema, X=N und CH2). Die neuen Reaktionsvorschriften sind nicht nur nützlich für die Synthese bicyclischer (Hetero)Arene mit vielfältigen Substitutionsmustern, sondern unter Umständen auch für die Wasserstoffspeicherung attraktiv.
Co-reporter:Sebastian Keess
Chemical Science (2010-Present) 2017 vol. 8(Issue 7) pp:4688-4695
Publication Date(Web):2017/06/26
DOI:10.1039/C7SC01657C
Safe- and convenient-to-handle surrogates of hazardous chemicals are always in demand. Recently introduced cyclohexa-1,4-dienes with adequate substitution fulfil this role as El+/H− equivalents in B(C6F5)3-catalysed transfer reactions of El–H to π- and σ-donors (CC/CC and CO/CN). Surrogates of Si–H/Ge–H, H–H and even C–H bonds have been designed and successfully applied to ionic transfer hydrosilylation/hydrogermylation, hydrogenation and hydro-tert-butylation, respectively. These processes and their basic principles are summarised in this Minireview. The similarities and differences between these transfer reactions as well as the challenges associated with these transformations are discussed.
Co-reporter:Weiming Yuan;Patrizio Orecchia
Chemical Communications 2017 vol. 53(Issue 75) pp:10390-10393
Publication Date(Web):2017/09/19
DOI:10.1039/C7CC06195A
The cyclohexa-1,3-diene motif is introduced as an equally effective alternative to the cyclohexa-1,4-diene platform in B(C6F5)3-catalysed transfer processes. The transfer hydrogenation of alkenes is realised with α-terpinene and the related transfer hydrosilylation is achieved with 5-trimethylsilyl-substituted cyclohexa-1,3-diene. Both yields and substrate scope are comparable with the prior systems.
Co-reporter:Weichao Xue; Dr. Martin Oestreich
Angewandte Chemie 2017 Volume 129(Issue 38) pp:11808-11811
Publication Date(Web):2017/09/11
DOI:10.1002/ange.201706611
AbstractEs wird über eine decarboxylierende Silylierung von aliphatischen Estern des N-Hydroxyphthalimids (NHPI) mit Si-B-Reagenzien als Siliciumpronukleophile berichtet. Diese C(sp3)-Si-Kreuzkupplung wird durch Kupfer(I) katalysiert und folgt selbst unter Lichtausschluss einem Radikalmechanismus. Primäre und sekundäre Alkylgruppen gehen die Kupplung gleichermaßen effektiv ein, während tertiäre Gruppen vermutlich sterisch zu gehindert sind. Die Toleranz gegenüber funktionellen Gruppen ist insgesamt hervorragend, und α-heteroatomsubstituierte Substrate reagieren ebenfalls gut. Das macht beispielsweise die Darstellung von α-silylierten Aminen ausgehend von NHPI-Estern abgeleitet von α-Aminosäuren möglich. Die neue Methode ergänzt die noch immer überschaubare Zahl an C(sp3)-Si-Kreuzkupplungen nichtaktivierter Alkylelektrophile.
Co-reporter:Susanne BährMartin Oestreich
Organometallics 2017 Volume 36(Issue 4) pp:
Publication Date(Web):February 13, 2017
DOI:10.1021/acs.organomet.7b00030
Ruthenium thiolate complexes with one chiral monodentate phosphine ligand are applied to enantioselective hydrosilylation of enolizable imines and ketones. The structural features of the catalyst exclude the presence of more than one phosphine ligand at the ruthenium center in the enantioselectivity-determining step. The enantiomeric excesses obtained in these reduction reactions are moderate (up to 66% ee), but the stereochemical outcome enables an experimental analysis of the reaction pathways operative in this catalysis. A two-step sequence consisting of successive N–Si/O–Si dehydrogenative coupling and enamine/enol ether hydrogenation is the prevailing mechanism of action. Both steps involve cooperative bond activation at the Ru–S bond of the coordinatively unsaturated ruthenium complex: Si–H bond activation in the dehydrogenative coupling and heterolytic H–H splitting in the hydrogenation. Previously documented side reactions such as deprotonation/protonation equilibria as well as competing direct C═N or C═O hydrogenation have been excluded.
Co-reporter:Weichao Xue, Zheng-Wang Qu, Stefan Grimme, and Martin Oestreich
Journal of the American Chemical Society 2016 Volume 138(Issue 43) pp:14222-14225
Publication Date(Web):October 16, 2016
DOI:10.1021/jacs.6b09596
A copper-catalyzed C(sp3)–Si cross-coupling of aliphatic C(sp3)–I electrophiles using a Si–B reagent as the silicon pronucleophile is reported. The reaction involves an alkyl radical intermediate that also engages in 5-exo-trig ring closures onto pendant alkenes prior to the terminating C(sp3)–Si bond formation. Several Ueno–Stork-type precursors cyclized with excellent diastereocontrol in good yields. The base-mediated release of the silicon nucleophile and the copper-catalyzed radical process are analyzed by quantum-chemical calculations, leading to a full mechanistic picture.
Co-reporter:Lars Süsse; Julia Hermeke
Journal of the American Chemical Society 2016 Volume 138(Issue 22) pp:6940-6943
Publication Date(Web):May 23, 2016
DOI:10.1021/jacs.6b03443
An axially chiral, cyclic borane decorated with just one C6F5 group at the boron atom promotes the highly enantioselective hydrosilylation of acetophenone derivatives without assistance of an additional Lewis base (up to 99% ee). The reaction is an unprecedented asymmetric variant of Piers’ B(C6F5)3-catalyzed carbonyl hydrosilylation. The steric congestion imparted by the 3,3′-disubstituted binaphthyl backbone of the borane catalyst as well as the use of reactive trihydrosilanes as reducing agents are keys to success.
Co-reporter:Qing-An Chen; Hendrik F. T. Klare
Journal of the American Chemical Society 2016 Volume 138(Issue 25) pp:7868-7871
Publication Date(Web):June 15, 2016
DOI:10.1021/jacs.6b04878
A counterintuitive approach to electrophilic aromatic substitution with silicon electrophiles is disclosed. A strong Brønsted acid that would usually promote the reverse reaction, i.e., protodesilylation, was found to initiate the C–H silylation of electron-rich (hetero)arenes with hydrosilanes. Protonation of the hydrosilane followed by liberation of dihydrogen is key to success, fulfilling two purposes: to generate the stabilized silylium ion and to remove the proton released from the Wheland intermediate.
Co-reporter:Carolin Fopp, Elise Romain, Kevin Isaac, Fabrice Chemla, Franck Ferreira, Olivier Jackowski, Martin Oestreich, and Alejandro Perez-Luna
Organic Letters 2016 Volume 18(Issue 9) pp:2054-2057
Publication Date(Web):April 19, 2016
DOI:10.1021/acs.orglett.6b00680
Zinc reagents (Me2PhSi)2Zn and [(Me3Si)3Si]2Zn undergo highly regio- and stereoselective addition across the carbon–carbon triple bond of nitrogen-, sulfur-, oxygen-, and phosphorus-substituted terminal alkynes in the absence of copper or any other catalyst. Both reagents yield exclusively β-isomers, and the stereoselectivity is determined by the silyl group: Me2PhSi for cis or (Me3Si)3Si for trans. These stereodivergent silylzincation protocols offer an efficient access to heteroatom-substituted vinylsilanes with either double bond geometry, including trisubstituted vinylsilanes by one-pot electrophilic substitution of the intermediate C(sp2)–Zn bond by copper(I)-mediated carbon–carbon bond formation.
Co-reporter:Indranil Chatterjee and Martin Oestreich
Organic Letters 2016 Volume 18(Issue 10) pp:2463-2466
Publication Date(Web):May 11, 2016
DOI:10.1021/acs.orglett.6b01016
Cyclohexa-1,4-dienes are introduced to Brønsted acid-catalyzed transfer hydrogenation as an alternative to the widely used Hantzsch dihydropyridines. While these hydrocarbon-based dihydrogen surrogates do offer little advantage over established protocols in imine reduction as well as reductive amination, their use enables the previously unprecedented transfer hydrogenation of structurally and electronically unbiased 1,1-di- and trisubstituted alkenes. The mild procedure requires 5.0 mol % of Tf2NH, but the less acidic sulfonic acids TfOH and TsOH work equally well.
Co-reporter:Lin-Yu Jiao;André V. Ferreira;Dr. Martin Oestreich
Chemistry – An Asian Journal 2016 Volume 11( Issue 3) pp:367-370
Publication Date(Web):
DOI:10.1002/asia.201500829
Abstract
A phosphinic amide is introduced as a directing group for the ortho C−H alkenylation of anilines. The new donor group distinguishes itself from existing ones by assisting the C−H bond activation of anilides without (NH group) and with alkylation (NMe group) at the amide nitrogen atom. The reactivity is even reversed with the methyl-substituted anilide being more reactive than its unsubstituted counterpart. Electron-donating substituents at the arene ring enhance their reactivity while halogenation is not tolerated. The phosphinic amide also enables the C-7-selective C−H alkenylation of indoline.
Co-reporter:Dr. Martin Oestreich
Angewandte Chemie International Edition 2016 Volume 55( Issue 2) pp:494-499
Publication Date(Web):
DOI:10.1002/anie.201508879
Abstract
Transfer hydrogenation is without question a common technology in industry and academia. Unlike its countless varieties, conceptually related transfer hydrosilylations had essentially been unreported until the recent development of a radical and an ionic variant. The new methods are both based on a silicon-substituted cyclohexa-1,4-diene and hinge on the aromatization of the corresponding cyclohexadienyl radical and cation intermediates, respectively, concomitant with homo- or heterolytic fission of the SiC bond. Both the radical and ionic transfer hydrosilylation are brought into context with one other in this Minireview, and early insight into the possibility of transfer hydrosilylation is included. Although the current state-of-the-art is certainly still limited, the recent advances have already revealed the promising potential of transfer hydrosilylation.
Co-reporter:Dr. Qin Yin;Dr. Hendrik F. T. Klare ;Dr. Martin Oestreich
Angewandte Chemie International Edition 2016 Volume 55( Issue 9) pp:3204-3207
Publication Date(Web):
DOI:10.1002/anie.201510469
Abstract
An electrophilic aromatic substitution (SEAr) with a catalytically generated silicon electrophile is reported. Essentially any commercially available base-metal salt acts as an initiator/catalyst when activated with NaBArF4 . The thus-generated Lewis acid then promotes the SEAr of electron-rich arenes with hydrosilanes but not halosilanes. This new C−H silylation was optimized for FeCl2 /NaBArF4 , affording good yields at catalyst loadings as low as 0.5 mol %. The procedure is exceedingly straightforward and comes close to typical Friedel–Crafts methods, where no added base is needed to absorb the released protons.
Co-reporter:Dr. Martin Oestreich
Angewandte Chemie 2016 Volume 128( Issue 2) pp:504-509
Publication Date(Web):
DOI:10.1002/ange.201508879
Abstract
Es steht außer Frage, dass Transferhydrierung ein gängiges Verfahren in der Industrie und der akademischen Welt ist. Trotz ihrer Vielfältigkeit war die konzeptionell verwandte Transferhydrosilylierung bis zur jüngsten Entwicklung einer radikalischen und einer ionischen Variante im Grunde unbekannt gewesen. Die neuen Methoden basieren beide auf dem Motiv eines siliciumsubstituierten Cyclohexa-1,4-diens und hängen von der Aromatisierung der entsprechenden radikalischen bzw. kationischen Cyclohexadienylzwischenstufen begleitet von homo- oder heterolytischer Spaltung der Si-C-Bindung ab. Die radikalische und ionische Transferhydrosilylierung werden in diesem Kurzaufsatz miteinander in Bezug gesetzt, und frühe Hinweise auf die Möglichkeit einer Transferhydrosilylierung werden auch berücksichtigt. Der derzeitige Stand der Forschung befindet sich zweifellos noch in den Anfängen, aber die jüngsten Fortschritte lassen das vielversprechende Potenzial von Transferhydrosilylierungen bereits erkennen.
Co-reporter:Dr. Qin Yin;Dr. Hendrik F. T. Klare ;Dr. Martin Oestreich
Angewandte Chemie 2016 Volume 128( Issue 9) pp:3256-3260
Publication Date(Web):
DOI:10.1002/ange.201510469
Abstract
Es wird über eine elektrophile aromatische Substitution (SEAr) mit einem katalytisch erzeugten Siliciumelektrophil berichtet. Bei Aktivierung mit NaBArF4 wirkt im Grunde jedes käufliche Nichtedelmetallsalz als Initiator/Katalysator. Die auf diese Weise gebildete Lewis-Säure vermittelt dann die SEAr von elektronenreichen Arenen mit Hydrosilanen, nicht aber mit Halogensilanen. Diese neue C-H-Silylierung ist für FeCl2/NaBArF4 optimiert und erbringt gute Ausbeuten bei Katalysatorbeladungen von lediglich 0.5 Mol-%. Die Vorgehensweise ist denkbar einfach und kommt typischen Friedel-Crafts-Reaktionsvorschriften nahe, bei denen keine zusätzliche Base zum Abfangen der freigesetzten Protonen vonnöten ist.
Co-reporter:Meera Mehta, Isaac Garcia de la Arada, Manuel Perez, Digvijay Porwal, Martin Oestreich, and Douglas W. Stephan
Organometallics 2016 Volume 35(Issue 7) pp:1030-1035
Publication Date(Web):March 29, 2016
DOI:10.1021/acs.organomet.6b00158
The hydrosilylation/reduction of tertiary and secondary phosphine oxides to phosphines is catalyzed by B(C6F5)3 or electrophilic fluorophosphonium cations (EPCs). B(C6F5)3 is an effective catalyst for phosphine oxide reduction using (EtO)3SiH, PhSiH3, and Ph2SiH2 at elevated temperature (105 °C), while EPCs effect the same reduction at significantly lower temperature with PhSiH3 as reducing agent, allowing for good functional-group tolerance.
Co-reporter:Polina Shaykhutdinova and Martin Oestreich
Organometallics 2016 Volume 35(Issue 16) pp:2768-2771
Publication Date(Web):August 12, 2016
DOI:10.1021/acs.organomet.6b00548
The stereoselective preparation of diastereomeric dihydrosilepine-derived silicon cations decorated with another binaphthyl unit at the silicon atom is described. A sulfide donor attached to that additional binaphthyl substituent forms an intramolecular Lewis pair with the electron-deficient silicon atom, as verified by 29Si NMR spectroscopy. Both chiral sulfur-stabilized silicon cations act as catalysts in the difficult Diels–Alder reaction of cyclohexa-1,3-diene and chalcone derivatives. Both Lewis acids induce enantioselectivity, but the S,S relative configuration is superior to the S,R configuration. With the former diastereomer, enantiomeric excesses of close to 60% are obtained. These values are the highest achieved to date in this seemingly trivial cycloaddition.
Co-reporter:Dr. Ruth K. Schmidt;Dr. Hendrik F. T. Klare;Dr. Rol Fröhlich;Dr. Martin Oestreich
Chemistry - A European Journal 2016 Volume 22( Issue 15) pp:5376-5383
Publication Date(Web):
DOI:10.1002/chem.201504777
Abstract
The preparation of a series of planar chiral, ferrocenyl-substituted hydrosilanes as precursors of ferrocene-stabilized silicon cations is described. These molecules also feature stereogenicity at the silicon atom. The generation and 29Si NMR spectroscopic characterization of the corresponding silicon cations is reported, and problems arising from interactions of the electron-deficient silicon atom and adjacent C(sp3)−H bonds or aromatic π donors are discussed. These issues are overcome by tethering another substituent at the silicon atom to the ferrocene backbone. The resulting annulation also imparts conformational rigidity and steric hindrance in such a way that the central chirality at the silicon atom is set with complete diastereocontrol. These chiral Lewis acid catalysts were then tested in difficult Diels–Alder reactions, but no enantioinduction was seen.
Co-reporter:Polina Smirnov and Martin Oestreich
Organometallics 2016 Volume 35(Issue 15) pp:2433-2434
Publication Date(Web):July 27, 2016
DOI:10.1021/acs.organomet.6b00505
The monosilane (SiH4) surrogate di(cyclohexa-2,5-dien-1-yl)silane is shown to be compatible with platinum-catalyzed hydrosilylation of α-olefins. The cyclohexa-2,5-dien-1-yl substituents in the monohydrosilylation adducts serve as protecting groups, and treatment with catalytic amounts of B(C6F5)3 liberates the Si–H bonds along with benzene. By this, trihydrosilanes become accessible in two steps without the formation of salt waste.
Co-reporter:Susanne Bähr, Antoine Simonneau, Elisabeth Irran, and Martin Oestreich
Organometallics 2016 Volume 35(Issue 7) pp:925-928
Publication Date(Web):March 24, 2016
DOI:10.1021/acs.organomet.6b00110
The preparation of a coordinatively unsaturated NHC ruthenium complex with a tethered 2,6-dimesitylphenyl thiolate is described. Unlike related mononuclear phosphine complexes, the NHC complex forms an air-stable dimer with bridging sulfur ligands in both the solid state and solution. The dinuclear complex is a precatalyst and activates Si–H bonds upon heat- or donor-assisted dissociation into the catalytically active monomer. Its usefulness is illustrated with representative dehydrogenative Si–X coupling and hydrosilylation reactions.
Co-reporter:Martin Oestreich, Julia Hermeke and Jens Mohr
Chemical Society Reviews 2015 vol. 44(Issue 8) pp:2202-2220
Publication Date(Web):13 Feb 2015
DOI:10.1039/C4CS00451E
The bond activation chemistry of B(C6F5)3 and related electron-deficient boranes is currently experiencing a renaissance due to the fascinating development of frustrated Lewis pairs (FLPs). B(C6F5)3's ability to catalytically activate Si–H bonds through η1 coordination opened the door to several unique reduction processes. The ground-breaking finding that the same family of fully or partially fluorinated boron Lewis acids allows for the related H–H bond activation, either alone or as a component of an FLP, brought considerable momentum into the area of transition-metal-free hydrogenation and, likewise, hydrosilylation. This review comprehensively summarises synthetic methods involving borane-catalysed Si–H and H–H bond activation. Systems corresponding to an FLP-type situation are not covered. Aside from the broad manifold of CX bond reductions and CX/C–X defunctionalisations, dehydrogenative (oxidative) Si–H couplings are also included.
Co-reporter:Toni T. Metsänen, Daniel Gallego, Tibor Szilvási, Matthias Driess and Martin Oestreich
Chemical Science 2015 vol. 6(Issue 12) pp:7143-7149
Publication Date(Web):14 Sep 2015
DOI:10.1039/C5SC02855H
Combined experimental and theoretical analysis of the carbonyl hydrosilylation catalysed by an iron(0) pincer complex reveals an unprecedented mechanism of action. The iron(0) complex is in fact a precatalyst that is converted into an iron(II) catalyst through oxidative addition of a hydrosilane. Neither the hydrogen atom nor the silicon atom bound to the iron(II) centre are subsequently transferred onto the carbonyl acceptor, instead remaining at the sterically inaccessible iron(II) atom throughout the catalytic cycle. A series of labelling, crossover and competition experiments as well as the use of a silicon-stereogenic hydrosilane as a stereochemical probe suggest that the iron(II) site is not directly involved in the hydrosilylation. Strikingly, it is the silyl ligand attached to the iron(II) atom that acts as a Lewis acid for carbonyl activation in this catalysis. The whole catalytic process occurs on the periphery of the transition metal. Computation of the new peripheral as well as plausible alternative inner and outer sphere mechanisms support the experimental findings.
Co-reporter:Timo Stahl, Peter Hrobárik, C. David F. Königs, Yasuhiro Ohki, Kazuyuki Tatsumi, Sebastian Kemper, Martin Kaupp, Hendrik F. T. Klare and Martin Oestreich
Chemical Science 2015 vol. 6(Issue 7) pp:4324-4334
Publication Date(Web):18 May 2015
DOI:10.1039/C5SC01035G
The nature of the hydrosilane activation mediated by ruthenium(II) thiolate complexes of type [(R3P)Ru(SDmp)]+[BArF4]− is elucidated by an in-depth experimental and theoretical study. The combination of various ruthenium(II) thiolate complexes and tertiary hydrosilanes under variation of the phosphine ligand and the substitution pattern at the silicon atom is investigated, providing detailed insight into the activation mode. The mechanism of action involves reversible heterolytic splitting of the Si–H bond across the polar Ru–S bond without changing the oxidation state of the metal, generating a ruthenium(II) hydride and sulfur-stabilized silicon cations, i.e. metallasilylsulfonium ions. These stable yet highly reactive adducts, which serve as potent silicon electrophiles in various catalytic transformations, are fully characterized by systematic multinuclear NMR spectroscopy. The structural assignment is further verified by successful isolation and crystallographic characterization of these key intermediates. Quantum-chemical analyses of diverse bonding scenarios are in excellent agreement with the experimental findings. Moreover, the calculations reveal that formation of the hydrosilane adducts proceeds via barrierless electrophilic activation of the hydrosilane by sterically controlled η1 (end-on) or η2 (side-on) coordination of the Si–H bond to the Lewis acidic metal center, followed by heterolytic cleavage of the Si–H bond through a concerted four-membered transition state. The Ru–S bond remains virtually intact during the Si–H bond activation event and also preserves appreciable bonding character in the hydrosilane adducts. The overall Si–H bond activation process is exergonic with ΔG0r ranging from −20 to −40 kJ mol−1, proceeding instantly already at low temperatures.
Co-reporter:Manish Pareek, Thomas Fallon, and Martin Oestreich
Organic Letters 2015 Volume 17(Issue 9) pp:2082-2085
Publication Date(Web):April 16, 2015
DOI:10.1021/acs.orglett.5b00604
Indolynes are converted into previously unprecedented indole building blocks by platinum(0)-catalyzed insertion into a symmetrically substituted boron–boron bond. The two boron sites in these indoles must be differentiated in a subsequent step, and the 6,7-bis[(pinacolato)boryl]indole was shown to undergo site-selective Suzuki–Miyaura cross-coupling with perfect C7 selectivity. The net reaction is the regioselective installation of two different substituents in the C6 and C7 positions of a 6,7-indolyne precursor.
Co-reporter:Toni T. Metsänen and Martin Oestreich
Organometallics 2015 Volume 34(Issue 3) pp:543-546
Publication Date(Web):December 19, 2014
DOI:10.1021/om501279a
Tethered Ru–S complexes were found to catalyze the hydrosilylation of carbon dioxide with a variety of simple monohydrosilanes selectively to bis(silyl)acetals or silylated methanol, respectively. The chemoselective conversion to either the formaldehyde or methanol oxidation state was solely dependent on the reaction temperature. This is the first example of a catalyst system that gives access to both of the intermediate oxidation states in a controlled way.
Co-reporter:Sebastian Keess, Antoine Simonneau, and Martin Oestreich
Organometallics 2015 Volume 34(Issue 4) pp:790-799
Publication Date(Web):January 27, 2015
DOI:10.1021/om501284a
The present survey serves several purposes. Selected electron-deficient boron Lewis acids catalyze the release of hydrosilanes from cyclohexa-2,5-dien-1-yl-substituted silanes. The two-step process consists of a hydride abstraction to generate a silicon-stabilized Wheland complex and capture of the arene-stabilized silicon cation by the borohydride formed in the previous step. The same boron catalyst will then activate the Si–H bond for the reaction with representative π- and σ-donating substrates, alkenes/alkynes and ketones/ketimines, respectively. The net transformation is a transfer hydrosilylation, and the effect that the substitution pattern of the cyclohexa-1,4-diene core and the subsituents at the silicon atom exert on these hydrosilane surrogates is systematically investigated. The results are compared with those obtained employing the hydrosilane directly. Another part of this comprehensive analysis is dedicated to the comparison of literature-known fully or partially fluorinated triarylboranes in both the direct and the transfer hydrosilylation of the aforementioned substrates. The data are tabulated and color-coded, finally providing an overview of promising substrate/reductant/borane combinations. The often different reactivities of π- and σ-basic substrates are explained, and it is shown that the Lewis acidity of the boron atom, estimated by the Gutmann–Beckett method, is not the only decisive feature of these boron Lewis acids. Practical mechanistic models are presented to rationalize the interplay between the Lewis acidity and steric situation at the boron and, likewise, the silicon atom as well as the need for fluorination ortho to the boron atom in certain cases.
Co-reporter:Antoine Simonneau, Tobias Biberger, and Martin Oestreich
Organometallics 2015 Volume 34(Issue 16) pp:3927-3929
Publication Date(Web):August 5, 2015
DOI:10.1021/acs.organomet.5b00609
The cyclohexa-2,5-dien-1-yl group is established as a leaving group at silicon as an alternative to the Bartlett–Condon–Schneider silicon-to-carbon hydride transfer and the allyl-leaving-group approach to generate silylium ions. Hydride abstraction from the skipped diene unit employing trityl tetrakis(pentafluorophenyl)borate (1, [Ph3C]+[B(C6F5)4]−) yields the silicon cation along with benzene. Our investigations reveal that the presence of an internal or external donor group is mandatory to allow for the formation of intra- or intermolecularly stabilized silylium ions. If not, degradation of the precursor is observed as a result of the reaction of the allylic silane units with the released silylium ion. It is also shown that such allylic silanes do form remarkably stable alkyl-substituted carbenium ions when reacted stoichiometrically with benzene-stabilized silylium ions.
Co-reporter:Volker H. G. Rohde, Maria F. Müller, and Martin Oestreich
Organometallics 2015 Volume 34(Issue 13) pp:3358-3373
Publication Date(Web):June 12, 2015
DOI:10.1021/acs.organomet.5b00351
The formation and 29Si NMR spectroscopic characterization of silicon cations that are intramolecularly stabilized by a dialkyl thioether are described. The chemical stability of the silicon–sulfur Lewis pair and, hence, the viability of the approach, were probed with a 2-[(alkylthio)methyl]phenyl-substituted hydrosilane as a proxy before three different motifs with chiral binaphthyl backbones were prepared in multistep sequences. The degree of shielding of the silicon atom in these cations was found to depend on the substitution pattern at the silicon atom and the ring size generated by the silicon–sulfur interaction. These sulfur-stabilized silicon cations are sufficiently reactive to promote Diels–Alder reactions of cyclohexa-1,3-diene with various dienophiles; the same set of reactions with cyclopentadiene is also reported. One of the three chiral Lewis acids induces low, but promising, enantioselectivity, and 24% ee is the highest value so far obtained with a cationic tetracoordinate silicon catalyst.
Co-reporter:Alexer Hensel ;Dr. Martin Oestreich
Chemistry - A European Journal 2015 Volume 21( Issue 25) pp:9062-9065
Publication Date(Web):
DOI:10.1002/chem.201501371
Abstract
An enantio- and regioselective allylic silylation of linear allylic phosphates that makes use of catalytically generated cuprate-type silicon nucleophiles is reported. The method relies on soft bis(triorganosilyl) zincs as silicon pronucleophiles that are prepared in situ from the corresponding hard lithium reagents by transmetalation with ZnCl2. With a preformed chiral N-heterocyclic carbene–copper(I) complex as catalyst, exceedingly high enantiomeric excesses are achieved. The new method is superior to existing ones using a silicon–boron reagent as the source of the silicon nucleophile.
Co-reporter:Jens Mohr;Digvijay Porwal;Dr. Indranil Chatterjee ;Dr. Martin Oestreich
Chemistry - A European Journal 2015 Volume 21( Issue 49) pp:17583-17586
Publication Date(Web):
DOI:10.1002/chem.201503509
Abstract
The B(C6F5)3-catalyzed hydrogenation is applied to aldoxime triisopropylsilyl ethers and hydrazones bearing an easily removable phthaloyl protective group. The CN reduction of aldehyde-derived substrates (oxime ethers and hydrazones) is enabled by using 1,4-dioxane as the solvent known to participate as the Lewis-basic component in FLP-type heterolytic dihydrogen splitting. More basic ketone-derived hydrazones act as Lewis bases themselves in the FLP-type dihydrogen activation and are therefore successfully hydrogenated in nondonating toluene. The difference in reactivity between aldehyde- and ketone-derived substrates is also reflected in the required catalyst loading and dihydrogen pressure.
Co-reporter:Lukas Omann ;Dr. Martin Oestreich
Angewandte Chemie International Edition 2015 Volume 54( Issue 35) pp:10276-10279
Publication Date(Web):
DOI:10.1002/anie.201504066
Abstract
A general procedure for the catalytic preparation of dibenzosiloles functionalized at one or both benzene rings starting from readily available ortho-silylated biphenyls is reported. This method provides rapid access to silole building blocks substituted with chlorine atoms at both phenylene groups, thereby allowing catalytic access to directly polymerizable dibenzosiloles. Moreover, it is shown that, despite the involvement of highly electrophilic intermediates, a considerable range of Lewis-basic, for example, oxygen- and nitrogen-containing, functional groups is tolerated. The mechanism of this intramolecular electrophilic aromatic substitution (SEAr) proceeds through a sulfur-stabilized silicon cation, generated catalytically from the hydrosilane precursor.
Co-reporter:Lukas Omann ;Dr. Martin Oestreich
Angewandte Chemie 2015 Volume 127( Issue 35) pp:10414-10418
Publication Date(Web):
DOI:10.1002/ange.201504066
Abstract
Es wird über eine allgemeine Reaktionsvorschrift zur katalytischen Synthese von an einem oder beiden Benzolringen funktionalisierten Dibenzosilolen berichtet, die von leicht verfügbaren ortho-silylierten Biphenylen ausgeht. Diese Methode ermöglicht einen schnellen Zugang zu Silolbausteinen mit Chloratomen an beiden Phenylengruppen und beschreibt somit einen katalytischen Weg zu direkt polymerisierbaren Dibenzosilolen. Es wird außerdem gezeigt, dass trotz hochgradig elektrophiler Zwischenstufen eine beachtliche Breite an Lewis-basischen, beispielsweise sauerstoff- und stickstoffhaltigen, funktionellen Gruppen toleriert wird. Der Mechanismus dieser intramolekularen elektrophilen aromatischen Substitution (SEAr) durchläuft ein schwefelstabilisiertes Siliciumkation, das katalytisch aus der Hydrosilanvorstufe generiert wird.
Co-reporter:Dr. Indranil Chatterjee ;Dr. Martin Oestreich
Angewandte Chemie 2015 Volume 127( Issue 6) pp:1988-1991
Publication Date(Web):
DOI:10.1002/ange.201409246
Abstract
Die Freisetzung von Wasserstoff aus entsprechend donorsubstituierten Cyclohexa-1,4-dienen nach Hydridabstraktion durch die starke Bor-Lewis-Säure Tris(pentafluorphenyl)boran, B(C6F5)3, wird vorgestellt. Dieses Verfahren ist dann an die FLP-artige Hydrierung (FLP=frustiertes Lewis-Paar) von Iminen und stickstoffenthaltenden Heteroaromaten, die durch dieselbe Lewis-Säure katalysiert wird, gekoppelt. Die Nettoreaktion ist eine B(C6F5)3-katalysierte, d. h. übergangsmetallfreie, Transferhydrierung mit leicht zugänglichen Cyclohexa-1,4-dienen als Reduktionsmitteln. Konkurrierende Reaktionspfade mit oder ohne Einbeziehung von freiem Wasserstoff werden diskutiert.
Co-reporter:Dr. Antoine Simonneau ;Dr. Martin Oestreich
Angewandte Chemie 2015 Volume 127( Issue 12) pp:3626-3628
Publication Date(Web):
DOI:10.1002/ange.201500557
Co-reporter:Simon Wübbolt ;Dr. Martin Oestreich
Angewandte Chemie 2015 Volume 127( Issue 52) pp:16103-16106
Publication Date(Web):
DOI:10.1002/ange.201508181
Abstract
Eine C-H-Silylierung von Pyridinen wurde entwickelt, die scheinbar dem Mechanismus einer elektrophilen aromatischen Substitution (SEAr) folgt. Reaktionen von 2- und 3-substituierten Pyridinen mit Hydrosilanen in Gegenwart eines Katalysators, welcher die Si-H-Bindung in ein Hydrid und ein Siliciumelektrophil spaltet, ergeben die entsprechenden 5-silylierten Pyridine. Diese formale Silylierung einer aromatischen C-H-Bindung ist das Ergebnis einer dreistufigen Reaktionssequenz bestehend aus einer Pyridinhydrosilylierung, einer dehydrierenden C-H-Silylierung der Enaminzwischenstufe und einer Retro-Hydrosilylierung des 1,4-Dihydropyridins. Die Schlüsselzwischenstufen wurden 1H-NMR-spektroskopisch nachgewiesen und schrittweise einzeln hergestellt. Dieses komplexe Wechselspiel elektrophiler Silylierungen, Hydridübertragungen und Protonenabstraktionen wird durch einen einzigen Katalysator vermittelt.
Co-reporter:Dr. Indranil Chatterjee ;Dr. Martin Oestreich
Angewandte Chemie International Edition 2015 Volume 54( Issue 6) pp:1965-1968
Publication Date(Web):
DOI:10.1002/anie.201409246
Abstract
The strong boron Lewis acid tris(pentafluorophenyl)borane, B(C6F5)3, is shown to abstract a hydride from suitably donor-substituted cyclohexa-1,4-dienes, eventually releasing dihydrogen. This process is coupled with the FLP-type (FLP=frustrated Lewis pair) hydrogenation of imines and nitrogen-containing heteroarenes that are catalyzed by the same Lewis acid. The net reaction is a B(C6F5)3-catalyzed, i.e., transition-metal-free, transfer hydrogenation using easy-to-access cyclohexa-1,4-dienes as reducing agents. Competing reaction pathways with or without the involvement of free dihydrogen are discussed.
Co-reporter:Dr. Antoine Simonneau ;Dr. Martin Oestreich
Angewandte Chemie International Edition 2015 Volume 54( Issue 12) pp:3556-3558
Publication Date(Web):
DOI:10.1002/anie.201500557
Co-reporter:Simon Wübbolt ;Dr. Martin Oestreich
Angewandte Chemie International Edition 2015 Volume 54( Issue 52) pp:15876-15879
Publication Date(Web):
DOI:10.1002/anie.201508181
Abstract
A CH silylation of pyridines that seemingly proceeds through electrophilic aromatic substitution (SEAr) is reported. Reactions of 2- and 3-substituted pyridines with hydrosilanes in the presence of a catalyst that splits the SiH bond into a hydride and a silicon electrophile yield the corresponding 5-silylated pyridines. This formal silylation of an aromatic CH bond is the result of a three-step sequence, consisting of a pyridine hydrosilylation, a dehydrogenative CH silylation of the intermediate enamine, and a 1,4-dihydropyridine retro-hydrosilylation. The key intermediates were detected by 1H NMR spectroscopy and prepared through the individual steps. This complex interplay of electrophilic silylation, hydride transfer, and proton abstraction is promoted by a single catalyst.
Co-reporter:Toni T. Metsänen ; Peter Hrobárik ; Hendrik F. T. Klare ; Martin Kaupp
Journal of the American Chemical Society 2014 Volume 136(Issue 19) pp:6912-6915
Publication Date(Web):May 2, 2014
DOI:10.1021/ja503254f
New experimental findings suggest partial revision of the currently accepted mechanism of the carbonyl hydrosilylation catalyzed by the iridium(III) pincer complex introduced by Brookhart. Employing silicon-stereogenic silanes as a stereochemical probe results in racemization rather than inversion of the configuration at the silicon atom. The degree of the racemization is, however, affected by the silane/carbonyl compound ratio, and inversion is seen with excess silane. Independently preparing the silylcarboxonium ion intermediate and testing its reactivity then helped to rationalize that effect. The stereochemical analysis together with these control experiments, rigorous multinuclear NMR analysis, and quantum-chemical calculations clearly prove that another silane molecule participates in the hydride transfer. The activating role of the silane is unexpected but, in fact, vital for the catalytic cycle to close.
Co-reporter:Lin-Yu Jiao, Polina Smirnov, and Martin Oestreich
Organic Letters 2014 Volume 16(Issue 22) pp:6020-6023
Publication Date(Web):November 6, 2014
DOI:10.1021/ol503035z
An improved method for the dehydrogenative C–H/C–H cross-coupling at the C-7 position of indolines containing a urea as a directing group is reported. The new protocol is a rare example of an aerobic palladium(II)-catalyzed cross dehydrogenative coupling (CDC) reaction that proceeds at low temperature. The use of either Cu(OAc)2 in an open flask or dioxygen (balloon) at 50 °C tolerates indolines not substituted at C-2 and C-3, thereby extending the scope of the previous method that suffers from indoline-to-indole oxidation.
Co-reporter:Seep R. Kukuri;Lin-Yu Jiao;Axel B. Machotta
Advanced Synthesis & Catalysis 2014 Volume 356( Issue 7) pp:1597-1609
Publication Date(Web):
DOI:10.1002/adsc.201301108
Co-reporter:Antoine Simonneau;Jonas Friebel
European Journal of Organic Chemistry 2014 Volume 2014( Issue 10) pp:2077-2083
Publication Date(Web):
DOI:10.1002/ejoc.201301840
Abstract
An unprecedented transfer silylation of alcohols catalyzed by the strong Lewis acid B(C6F5)3 is described. Gaseous Me3SiH is released in situ by B(C6F5)3-catalyzed decomposition of 3-trimethylsilylcyclohexa-1,4-diene and subsequently reacts with an alcohol in a dehydrogenative Si–O coupling promoted by the same boron catalyst. Benzene and dihydrogen are formed during the reaction, but no salt waste is. This expedient protocol is applicable to several silicon groups, and the preparation of trimethylsilyl ethers presented here is potentially useful for alcohol derivatization prior to GLC analysis.
Co-reporter:Julia Hermeke;Dr. Hendrik F. T. Klare ;Dr. Martin Oestreich
Chemistry - A European Journal 2014 Volume 20( Issue 30) pp:9250-9254
Publication Date(Web):
DOI:10.1002/chem.201402866
Abstract
A procedure for the synthesis of otherwise difficult-to-make N-silylated enamines, that is masked enamines derived from primary amines, is reported. The approach is based on formation of a silyliminium ion and subsequent abstraction of the acidified α-proton rather than α-deprotonation of the enolizable imine followed by reaction with an electrophilic silicon reagent. The silicon electrophile, stabilized by a sulfur atom, is generated by cooperative activation of an SiH bond at the RuS bond of a tethered ruthenium(II) thiolate complex. After transfer of the silicon cation onto the imine nitrogen atom, the remaining ruthenium(II) hydride fulfills the role of the base. Deprotonation and release of dihydrogen close the catalytic cycle. The net reaction is a dehydrogenative SiN coupling of enolizable imines and hydrosilanes.
Co-reporter:Kai M. Redies, Thomas Fallon, and Martin Oestreich
Organometallics 2014 Volume 33(Issue 13) pp:3235-3238
Publication Date(Web):June 16, 2014
DOI:10.1021/om500485m
The synthesis of a bis(α-spirocyclopropyl)silylene is reported and its reactivity revealed. Liberation of the silylene was accomplished by UV-light-mediated photolysis of a trisilane precursor. Insertion and addition reactions prove the existence and versatility of this new family of bis(α-spirocyclopropyl)-substituted silylenes. Substitution on the flanking cyclopropyls for improved steric shielding of the reactive center remains challenging.
Co-reporter:Julia Hermeke, Marius Mewald, Elisabeth Irran, and Martin Oestreich
Organometallics 2014 Volume 33(Issue 19) pp:5097-5100
Publication Date(Web):September 9, 2014
DOI:10.1021/om500851r
C(sp2)–Sn bonds generally react more quickly than C(sp3)–Sn bonds in tin–boron exchange processes, and discrimination between (hetero)aryl groups to be transferred from tin to boron and those to remain at the tin atom (usually methyl groups) is reliable. Conversely, transmetalation of benzylic carbon–tin bonds is disfavored, and the chemoselectivity against other C(sp3)–Sn bonds at the tin atom is poor. That inherent problem is overcome by the installation of one or more Me3SiCH2 (TMSM) groups at the tin atom as nontransferable substituents. With these groups, the tin–boron metathesis of benzylic C(sp3)–Sn bonds occurs cleanly and rapidly. The efficiency of the new method is demonstrated in the demanding synthesis of C6F5-substituted dihydroborepines with binaphthyl backbones.
Co-reporter:Alexander R. Nödling, Kristine Müther, Volker H. G. Rohde, Gerhard Hilt, and Martin Oestreich
Organometallics 2014 Volume 33(Issue 1) pp:302-308
Publication Date(Web):December 10, 2013
DOI:10.1021/om401040y
The 29Si NMR chemical shifts of ferrocene-stabilized silicon cations span a wide range depending on the substituents at the silicon atom. These pronounced differences in deshielding of the silicon atom do not translate into significant differences in their catalytic activity in Diels–Alder reactions. It was shown by Lewis pair formation with Lewis base probes (Et3PO and pyridine-d5) that there is hardly any difference between these silicon cations after coordination to a Lewis base. This finding not only thwarts experimental quantification of the Lewis acidity of the free Lewis acids but also demonstrates that the reactivity differences are largely due to steric effects for a given counteranion. These observations are further verified by a ReactIR kinetic analysis. The Lewis acidity of silicon cations and their performance as catalysts cannot be correlated with 29Si NMR chemical shifts as well as resonances of adducts with Lewis base probes, not even for a subset of silicon Lewis acids.
Co-reporter:Jens Mohr, Mustafa Durmaz, Elisabeth Irran, and Martin Oestreich
Organometallics 2014 Volume 33(Issue 5) pp:1108-1111
Publication Date(Web):February 24, 2014
DOI:10.1021/om500128a
Typical congeners of the boron Lewis acid tris(pentafluorophenyl)borane, B(C6F5)3, are fluorinated at the aryl groups directly attached to the boron atom. The chemistry of related electron-deficient boranes with fluorination distal to the Lewis acidic center is largely unexplored. The preparation and characterization of tris(5,6,7,8-tetrafluoronaphthalen-2-yl)borane are reported. It serves as a model system that provides sites for further substitution at C-1 and C-3 of the naphthalen-2-yl units. A Gutmann–Beckett analysis of its Lewis acidity revealed that, despite remote fluorination, it is as Lewis acidic as B(C6F5)3. The new Lewis acid performs equally well in C═O and C═N reduction as well as dehydrogenative Si–O coupling involving Si–H bond activation. Adducts with water and a phosphine oxide are crystallographically characterized.
Co-reporter:Volker H. G. Rohde, Phillip Pommerening, Hendrik F. T. Klare, and Martin Oestreich
Organometallics 2014 Volume 33(Issue 13) pp:3618-3628
Publication Date(Web):July 2, 2014
DOI:10.1021/om500570d
The synthesis and spectroscopic characterization of previously unprecedented sulfur-stabilized silicon cations are reported. Several 1,3-dithiolan-2-yl- and 1,3-dithian-2-yl-substituted silanes were prepared and successfully transformed into the corresponding silicon cations by hydride abstraction. The silicon–sulfur interaction creates three consecutive stereocenters at three different elements. It is remarkable that the present stereocenter at the silicon atom determines the stereochemical outcome at the formerly prochiral sulfur and carbon atoms with excellent diastereoselectivity. All sulfur-stabilized silicon cations are shown to be potent catalysts in a challenging Diels–Alder reaction. Moreover, structurally related oxazoline-stabilized silicon cations were generated and characterized but found to be unreactive.
Co-reporter:Dr. Chinmoy K. Hazra;Carolin Fopp;Dr. Martin Oestreich
Chemistry – An Asian Journal 2014 Volume 9( Issue 10) pp:3005-3010
Publication Date(Web):
DOI:10.1002/asia.201402643
Abstract
The copper(I) alkoxide-catalyzed release of a silicon-based cuprate reagent from a silicon–boron pronucleophile is applied to the addition across carbon–carbon triple bonds. Commercially available CuBr⋅Me2S was found to be a general precatalyst that secures high regiocontrol for both aryl- and alkyl-substituted terminal as well as internal alkynes. The solvent greatly influences the regioisomeric ratio, favoring the linear regioisomer with terminal acceptors. This facile protocol even allows for the transformation of internal acceptors with remarkable levels of regio- and diastereocontrol.
Co-reporter:Dr. Martin Oestreich
Angewandte Chemie International Edition 2014 Volume 53( Issue 9) pp:2282-2285
Publication Date(Web):
DOI:10.1002/anie.201310585
Co-reporter:Alexer Hensel;Dr. Kazuhiko Nagura;Lukas B. Delvos ;Dr. Martin Oestreich
Angewandte Chemie International Edition 2014 Volume 53( Issue 19) pp:4964-4967
Publication Date(Web):
DOI:10.1002/anie.201402086
Abstract
A remaining major challenge in the asymmetric addition of silicon nucleophiles to typical prochiral acceptors, the enantioselective 1,2-addition to aldimines, is addressed. Activation of the SiB bond in the silicon pronucleophile by a copper(I) alkoxide with McQuade’s chiral six-membered N-heterocyclic carbene as a supporting ligand releases the silicon nucleophile, which adds to various aldimines with high levels of enantioselectivity. The new method provides a catalytic asymmetric access to α-silylated amines.
Co-reporter:Elise Romain;Carolin Fopp;Dr. Fabrice Chemla;Dr. Franck Ferreira;Dr. Olivier Jackowski;Dr. Martin Oestreich;Dr. Alejro Perez-Luna
Angewandte Chemie International Edition 2014 Volume 53( Issue 42) pp:11333-11337
Publication Date(Web):
DOI:10.1002/anie.201407002
Abstract
The silylzincation of terminal ynamides is achieved through a radical-chain process involving (Me3Si)3SiH and R2Zn. A potentially competing polar mechanism is excluded on the basis of diagnostic control experiments. The unique feature of this addition across the CC bond is its trans selectivity. One-pot electrophilic substitution of the CZn bond by CuI-mediated CC bond formation and subsequent manipulation of the CSi bond provides a modular access to Z-α,β-disubstituted enamides.
Co-reporter:Jens Mohr ;Dr. Martin Oestreich
Angewandte Chemie International Edition 2014 Volume 53( Issue 48) pp:13278-13281
Publication Date(Web):
DOI:10.1002/anie.201407324
Abstract
The hydrogenation of oximes and oxime ethers is usually hampered by NO bond cleavage, hence affording amines rather than hydroxylamines. The boron Lewis acid B(C6F5)3 is found to catalyze the chemoselective hydrogenation of oxime ethers at elevated or even room temperature under 100 bar dihydrogen pressure. The use of the triisopropylsilyl group as a protecting group allows for facile liberation of the free hydroxylamines.
Co-reporter:Dr. Martin Oestreich
Angewandte Chemie 2014 Volume 126( Issue 9) pp:2314-2317
Publication Date(Web):
DOI:10.1002/ange.201310585
Co-reporter:Alexer Hensel;Dr. Kazuhiko Nagura;Lukas B. Delvos ;Dr. Martin Oestreich
Angewandte Chemie 2014 Volume 126( Issue 19) pp:5064-5067
Publication Date(Web):
DOI:10.1002/ange.201402086
Abstract
Diese Arbeit befasst sich mit einer noch verbliebenen, bedeutenden Herausforderung der asymmetrischen Addition von Siliciumnukleophilen an typische prochirale Akzeptoren, der enantioselektiven 1,2-Addition an Aldimine. Die Aktivierung der Si-B-Bindung des Siliciumpronukleophils durch ein Kupfer(I)-Alkoholat mit McQuades chiralem sechsgliedrigen N-heterocyclischen Carben als Ligand setzt das Siliciumnukleophil frei, das mit hohen Enantioselektivitäten an verschiedenartige Aldimine addiert. Die neue Methode bietet einen katalytischen asymmetrischen Zugang zu α-silylierten Aminen.
Co-reporter:Elise Romain;Carolin Fopp;Dr. Fabrice Chemla;Dr. Franck Ferreira;Dr. Olivier Jackowski;Dr. Martin Oestreich;Dr. Alejro Perez-Luna
Angewandte Chemie 2014 Volume 126( Issue 42) pp:11515-11519
Publication Date(Web):
DOI:10.1002/ange.201407002
Abstract
The silylzincation of terminal ynamides is achieved through a radical-chain process involving (Me3Si)3SiH and R2Zn. A potentially competing polar mechanism is excluded on the basis of diagnostic control experiments. The unique feature of this addition across the CC bond is its trans selectivity. One-pot electrophilic substitution of the CZn bond by CuI-mediated CC bond formation and subsequent manipulation of the CSi bond provides a modular access to Z-α,β-disubstituted enamides.
Co-reporter:Jens Mohr ;Dr. Martin Oestreich
Angewandte Chemie 2014 Volume 126( Issue 48) pp:13494-13497
Publication Date(Web):
DOI:10.1002/ange.201407324
Abstract
Die Hydrierung von Oximen und Oximethern geht normalerweise mit der Spaltung der N-O-Bindung einher und setzt daher Amine und eben nicht Hydroxylamine frei. Die Bor-Lewis-Säure B(C6F5)3 katalysiert die chemoselektive Hydrierung von Oximethern bei erhöhter oder sogar Raumtemperatur unter 100 bar Wasserstoffdruck. Die Verwendung der Triisopropylsilylgruppe als Schutzgruppe erlaubt eine mühelose Freisetzung der Hydroxylamine.
Co-reporter:Martin Oestreich, Eduard Hartmann, and Marius Mewald
Chemical Reviews 2013 Volume 113(Issue 1) pp:402
Publication Date(Web):November 20, 2012
DOI:10.1021/cr3003517
Co-reporter:Timo Stahl ; Hendrik F. T. Klare
Journal of the American Chemical Society 2013 Volume 135(Issue 4) pp:1248-1251
Publication Date(Web):January 11, 2013
DOI:10.1021/ja311398j
Heterolytic splitting of the Si–H bond mediated by a Ru–S bond forms a sulfur-stabilized silicon cation that is sufficiently electrophilic to abstract fluoride from CF3 groups attached to selected anilines. The ability of the Ru–H complex, generated in the cooperative activation step, to intramolecularly transfer its hydride to the intermediate carbenium ion (stabilized in the form of a cationic thioether complex) is markedly dependent on the electronic nature of its phosphine ligand. An electron-deficient phosphine thwarts the reduction step but, based on the Ru-S catalyst, half of an equivalent of an added alkoxide not only facilitates but also accelerates the catalysis. The intriguing effect is rationalized by the formation of a hydride-bridged Ru–S dimer that was detected by 1H NMR spectroscopy. A refined catalytic cycle is proposed.
Co-reporter:Timo Stahl ; Kristine Müther ; Yasuhiro Ohki ; Kazuyuki Tatsumi
Journal of the American Chemical Society 2013 Volume 135(Issue 30) pp:10978-10981
Publication Date(Web):July 15, 2013
DOI:10.1021/ja405925w
The B–H bond of typical boranes is heterolytically split by the polar Ru–S bond of a tethered ruthenium(II) thiolate complex, affording a ruthenium(II) hydride and borenium ions with a dative interaction with the sulfur atom. These stable adducts were spectroscopically characterized, and in one case, the B–H bond activation step was crystallographically verified, a snapshot of the σ-bond metathesis. The borenium ions derived from 9-borabicyclo[3.3.1]nonane dimer [(9-BBN)2], pinacolborane (pinBH), and catecholborane (catBH) allowed for electrophilic aromatic substitution of indoles. The unprecedented electrophilic borylation with the pinB cation was further elaborated for various nitrogen heterocycles.
Co-reporter:Julia Hermeke ; Marius Mewald
Journal of the American Chemical Society 2013 Volume 135(Issue 46) pp:17537-17546
Publication Date(Web):November 1, 2013
DOI:10.1021/ja409344w
The discovery of intermediates that had not been seen before in imine reduction involving borane-mediated Si–H bond activation provided new insight into the mechanism, eventually leading to a refined catalytic cycle that also bears relevance to asymmetric variants. The catalysis proceeds through an ion pair composed of a silyliminium ion and a borohydride that subsequently reacts to yield an N-silylated amine and the borane catalyst. The latter step is enantioselectivity-determining when using a chiral borane. It was now found that there are additional intermediates that profoundly influence the outcome of such enantioselective transformations. Significant amounts of the corresponding free amine and N-silylated enamine are present in equimolar ratio during the catalysis. The free amine emerges from a borohydride reduction of an iminium ion (protonated imine) that is, in turn, generated in the enamine formation step. The unexpected alternative pathway adds another enantioselectivity-determining hydride transfer to reactions employing chiral boranes. The experiments were done with an axially chiral borane that was introduced by us a few years ago, and the refined mechanistic picture helps to understand previously observed inconsistencies in the level of enantioinduction in reductions catalyzed by this borane. Our findings are general because the archetypical electron-deficient borane B(C6F5)3 shows the same reaction pattern. This must have been overlooked in the past because B(C6F5)3 is substantially more reactive than our chiral borane with just one C6F5 group. Reactions with B(C6F5)3 must be performed at low catalyst loading to allow for detection of these fundamental intermediates by NMR spectroscopy.
Co-reporter:Timo Stahl, Hendrik F. T. Klare, and Martin Oestreich
ACS Catalysis 2013 Volume 3(Issue 7) pp:1578
Publication Date(Web):June 3, 2013
DOI:10.1021/cs4003244
The significant benefits of fluorinated compounds have inspired the development of diverse techniques for the activation and subsequent (de)functionalization of rather inert C–F bonds. Although substantial progress has been made in the selective activation of C(sp2)–F bonds employing transition metal complexes, protocols that address nonactivated C(sp3)–F bonds are much less established. In this regard, the use of strong main-group Lewis acids has emerged as a powerful tool to selectively activate C(sp3)–F bonds in saturated fluorocarbons. This Perspective provides a concise overview of various cationic and neutral silicon-, boron-, and aluminum-based Lewis acids that have been identified to facilitate the heterolytic fluoride abstraction from aliphatic fluorides. The potential of these Lewis acids in hydrodefluorination as well as defluorinative C–F bond functionalization reactions is highlighted. Emphasis is placed on the underlying mechanistic principles to provide a systematic classification of the individual reactions. Finally, brief insight into the related C–F bond activation chemistry using carbocations or Brønsted acids is presented.Keywords: C−F bond activation; C−F bond functionalization; fluorine; homogeneous catalysis; hydrodefluorination; main-group Lewis acids
Co-reporter:C. David F. Königs, Maria F. Müller, Nuria Aiguabella, Hendrik F. T. Klare and Martin Oestreich
Chemical Communications 2013 vol. 49(Issue 15) pp:1506-1508
Publication Date(Web):17 Jan 2013
DOI:10.1039/C3CC38900F
A base-free, catalytic protocol for the dehydrogenative Si–N coupling of weakly nucleophilic N–H groups of heteroarenes or aryl-substituted amines with equimolar amounts of hydrosilanes is reported. Cooperative Si–H bond activation at a Ru–S bond generates a silicon electrophile that forms a Si–N bond prior to the N–H deprotonation by an intermediate Ru–H complex, only releasing H2.
Co-reporter:Lin-Yu Jiao and Martin Oestreich
Organic Letters 2013 Volume 15(Issue 20) pp:5374-5377
Publication Date(Web):October 8, 2013
DOI:10.1021/ol402687t
A mild procedure for C-7-selective C–H alkenylation of various indolines under oxidative palladium(II) catalysis is reported. A fully substituted urea, formed by carbamoylation of the indoline nitrogen atom, functions as a directing group. Both α,β-unsaturated acceptors and styrenes participate in this direct C–H functionalization. With a free NH group at the urea terminus, the nitrogen atom subsequently cyclizes in a 1,4-fashion to yield a six-membered ring.
Co-reporter:Chinmoy K. Hazra;Elisabeth Irran
European Journal of Organic Chemistry 2013 Volume 2013( Issue 22) pp:4903-4908
Publication Date(Web):
DOI:10.1002/ejoc.201300493
Abstract
A perfectly γ-selective copper(I)-catalyzed allylic substitution of protected δ-hydroxy allylic chlorides with a silicon nucleophile generated by Si–B bond activation provides diastereoselective access to β-hydroxy α-chiral allylic silanes with an anti relative configuration. The high levels of diastereocontrol of this rare allylic displacement are interpreted according to a model suggested by Nakamura where diastereofacial selectivity originates from steric control rather than an oxygen-directing effect in the Felkin–Anh transition state.
Co-reporter:Dr. Tobias Robert ;Dr. Martin Oestreich
Angewandte Chemie International Edition 2013 Volume 52( Issue 20) pp:5216-5218
Publication Date(Web):
DOI:10.1002/anie.201301205
Co-reporter:Kristine Müther;Dr. Peter Hrobárik;Dr. Veronika Hrobáriková;Dr. Martin Kaupp;Dr. Martin Oestreich
Chemistry - A European Journal 2013 Volume 19( Issue 49) pp:16579-16594
Publication Date(Web):
DOI:10.1002/chem.201302885
Abstract
The purpose of this systematic experimental and theoretical study is to deeply understand the unique bonding situation in ferrocene-stabilized silylium ions as a function of the substituents at the silicon atom and to learn about the structure parameters that determine the 29Si NMR chemical shift and electrophilicity of these strong Lewis acids. For this, ten new members of the family of ferrocene-stabilized silicon cations were prepared by a hydride abstraction reaction from silanes with the trityl cation and characterized by multinuclear 1H and 29Si NMR spectroscopy. A closer look at the NMR spectra revealed that additional minor sets of signals were not impurities but silylium ions with substitution patterns different from that of the initially formed cation. Careful assignment of these signals furnished experimental proof that sterically less hindered silylium ions are capable of exchanging substituents with unreacted silane precursors. Density functional theory calculations provided mechanistic insight into that substituent transfer in which the migrating group is exchanged between two silicon fragments in a concerted process involving a ferrocene-bridged intermediate. Moreover, the quantum-chemical analysis of the 29Si NMR chemical shifts revealed a linear relationship between δ(29Si) values and the Fe⋅⋅⋅Si distance for subsets of silicon cations. An electron localization function and electron localizability indicator analysis shows a three-center two-electron bonding attractor between the iron, silicon, and C′ipso atoms, clearly distinguishing the silicon cations from the corresponding carbenium ions and boranes. Correlations between 29Si NMR chemical shifts and Lewis acidity, evaluated in terms of fluoride ion affinities, are seen only for subsets of silylium ions, sometimes with non-intuitive trends, indicating a complicated interplay of steric and electronic effects on the degree of the Fe⋅⋅⋅Si interaction.
Co-reporter:Lin-Yu Jiao ;Dr. Martin Oestreich
Chemistry - A European Journal 2013 Volume 19( Issue 33) pp:10845-10848
Publication Date(Web):
DOI:10.1002/chem.201302140
Co-reporter:Kristine Müther, Jens Mohr, and Martin Oestreich
Organometallics 2013 Volume 32(Issue 22) pp:6643-6646
Publication Date(Web):May 9, 2013
DOI:10.1021/om4002796
The silylium ion promoted reduction of imines yielding the corresponding amines is reported. Both tert-butylferrocenylmethylsilane and triethylsilane are efficient hydride donors for the reduction of intermediate silyliminium ions, thereby regenerating the catalytically active silylium ion and closing the catalytic cycle.
Co-reporter:Lukas B. Delvos;Dr. Devendra J. Vyas;Dr. Martin Oestreich
Angewandte Chemie 2013 Volume 125( Issue 17) pp:4748-4751
Publication Date(Web):
DOI:10.1002/ange.201300648
Co-reporter:Dr. Tobias Robert ;Dr. Martin Oestreich
Angewandte Chemie 2013 Volume 125( Issue 20) pp:5324-5326
Publication Date(Web):
DOI:10.1002/ange.201301205
Co-reporter:Dipl.-Chem. C. David F. Königs;Dr. Hendrik F. T. Klare ;Dr. Martin Oestreich
Angewandte Chemie 2013 Volume 125( Issue 38) pp:10260-10263
Publication Date(Web):
DOI:10.1002/ange.201305028
Co-reporter:Dr. Antoine Simonneau ;Dr. Martin Oestreich
Angewandte Chemie 2013 Volume 125( Issue 45) pp:12121-12124
Publication Date(Web):
DOI:10.1002/ange.201305584
Co-reporter:Lukas B. Delvos;Dr. Devendra J. Vyas;Dr. Martin Oestreich
Angewandte Chemie International Edition 2013 Volume 52( Issue 17) pp:4650-4653
Publication Date(Web):
DOI:10.1002/anie.201300648
Co-reporter:Dipl.-Chem. C. David F. Königs;Dr. Hendrik F. T. Klare ;Dr. Martin Oestreich
Angewandte Chemie International Edition 2013 Volume 52( Issue 38) pp:10076-10079
Publication Date(Web):
DOI:10.1002/anie.201305028
Co-reporter:Dr. Antoine Simonneau ;Dr. Martin Oestreich
Angewandte Chemie International Edition 2013 Volume 52( Issue 45) pp:11905-11907
Publication Date(Web):
DOI:10.1002/anie.201305584
Co-reporter:Ruth K. Schmidt ; Kristine Müther ; Christian Mück-Lichtenfeld ; Stefan Grimme
Journal of the American Chemical Society 2012 Volume 134(Issue 9) pp:4421-4428
Publication Date(Web):February 6, 2012
DOI:10.1021/ja211856m
The pronounced Lewis acidity of tricoordinate silicon cations brings about unusual reactivity in Lewis acid catalysis. The downside of catalysis with strong Lewis acids is, though, that these do have the potential to mediate the formation of protons by various mechanisms, and the thus released Brønsted acid might even outcompete the Lewis acid as the true catalyst. That is an often ignored point. One way of eliminating a hidden proton-catalyzed pathway is to add a proton scavenger. The low-temperature Diels–Alder reactions catalyzed by our ferrocene-stabilized silicon cation are such a case where the possibility of proton catalysis must be meticulously examined. Addition of the common hindered base 2,6-di-tert-butylpyridine resulted, however, in slow decomposition along with formation of the corresponding pyridinium ion. Quantitative deprotonation of the silicon cation was observed with more basic (Mes)3P to yield the phosphonium ion. A deuterium-labeling experiment verified that the proton is abstracted from the ferrocene backbone. A reasonable mechanism of the proton formation is proposed on the basis of quantum-chemical calculations. This is, admittedly, a particular case but suggests that the use of proton scavengers must be carefully scrutinized, as proton formation might be provoked rather than prevented. Proton-catalyzed Diels–Alder reactions are not well-documented in the literature, and a representative survey employing TfOH is included here. The outcome of these catalyses is compared with our silylium ion-catalyzed Diels–Alder reactions, thereby clearly corroborating that hidden Brønsted acid catalysis is not operating with our Lewis acid. Several simple-looking but challenging Diels–Alder reactions with exceptionally rare dienophile/enophile combinations are reported. Another indication is obtained from the chemoselectivity of the catalyses. The silylium ion-catalyzed Diels–Alder reaction is general with regard to the oxidation level of the α,β-unsaturated dienophile (carbonyl and carboxyl), whereas proton catalysis is limited to carbonyl compounds.
Co-reporter:Eduard Hartmann and Martin Oestreich
Organic Letters 2012 Volume 14(Issue 9) pp:2406-2409
Publication Date(Web):April 16, 2012
DOI:10.1021/ol300832f
The two-directional desymmetrization of prochiral precursors with α,β-unsaturated branches by catalyst-controlled 1,4-addition of silicon and likewise boron nucleophiles allows for a general enantioselective access to syn,anti-triols with 1,n + 1,2n + 1 (n = 2 and 3) substitution patterns. The utility is demonstrated in the synthesis of the C17–C25 fragment of dermostatin A.
Co-reporter:C. David F. Königs, Hendrik F. T. Klare, Yasuhiro Ohki, Kazuyuki Tatsumi, and Martin Oestreich
Organic Letters 2012 Volume 14(Issue 11) pp:2842-2845
Publication Date(Web):May 23, 2012
DOI:10.1021/ol301089r
A dehydrogenative coupling between enolizable carbonyl compounds and equimolar amounts of triorganosilanes catalyzed by a tethered ruthenium complex with a Ru–S bond is reported. The complex is assumed to fulfill a dual role by activating the Si–H bond to release a silicon electrophile and by abstracting an α-proton from the intermediate silylcarboxonium ion, only liberating dihydrogen as the sole byproduct. Reaction rates are exceedingly high at room temperature with very low loadings of the ruthenium catalyst.
Co-reporter:Chinmoy K. Hazra and Martin Oestreich
Organic Letters 2012 Volume 14(Issue 15) pp:4010-4013
Publication Date(Web):July 25, 2012
DOI:10.1021/ol301827t
Copper(I)-catalyzed propargylic substitution of linear precursors with (Me2PhSi)2Zn predominantly yields the γ isomer independent of the propargylic leaving group. The thus formed allenylic silane reacts regioselectively with another equivalent of (Me2PhSi)2Zn, yielding a bifunctional building block with allylic and vinylic silicon groups. The reaction rates of both steps are well-balanced for chloride (γ:α ≥ 99:1) where the propargylic displacement occurs quantitatively prior to the addition step. Substitutions of α-branched propargylic phosphates are also reported.
Co-reporter:Thorsten H. Wöste ;Dr. Martin Oestreich
ChemCatChem 2012 Volume 4( Issue 12) pp:2096-2101
Publication Date(Web):
DOI:10.1002/cctc.201200300
Abstract
BINAP(O), rarely used as a chiral ligand, was found to induce significantly higher levels of enantioselection in representative desymmetrizing Mizoroki–Heck cyclizations where conventional BINAP produces essentially racemic material. BINAP(O) is a ligand with a stronger and weaker donor atom, and that hemilabile nature lends itself the ability to act as either a bi- or monodentate ligand. On that basis, we introduce mechanistic models where the weak donor, in one case, mediates the equilibration of diastereomeric alkene–palladium(II) complexes and, in another case, dissociates from the palladium(II) atom, thereby rendering BINAP(O) as a monodentate ligand. These new findings, along with the recently reported effects in intermolecular Mizoroki–Heck reactions, suggest that BINAP(O) ought to be included into ligand screenings.
Co-reporter:Seep R. Kukuri;Dr. Julia A. Schiffner;Dr. Martin Oestreich
Angewandte Chemie International Edition 2012 Volume 51( Issue 5) pp:1265-1269
Publication Date(Web):
DOI:10.1002/anie.201106927
Co-reporter:Marius Mewald;Dr. Julia A. Schiffner ;Dr. Martin Oestreich
Angewandte Chemie International Edition 2012 Volume 51( Issue 8) pp:1763-1765
Publication Date(Web):
DOI:10.1002/anie.201107859
Co-reporter:Seep R. Kukuri;Dr. Julia A. Schiffner;Dr. Martin Oestreich
Angewandte Chemie 2012 Volume 124( Issue 5) pp:1291-1295
Publication Date(Web):
DOI:10.1002/ange.201106927
Co-reporter:Marius Mewald ;Dr. Martin Oestreich
Chemistry - A European Journal 2012 Volume 18( Issue 44) pp:14079-14084
Publication Date(Web):
DOI:10.1002/chem.201202693
Abstract
The reduction of CO groups with silanes catalyzed by electron-deficient boranes follows a counterintuitive mechanism in which the SiH bond is activated by the boron Lewis acid prior to nucleophilic attack of the carbonyl oxygen atom at the silicon atom. The borohydride thus formed is the actual reductant. These steps were elucidated by using a silicon-stereogenic silane, but applying the same technique to the related reduction of CN groups was inconclusive due to racemization of the silicon atom. The present investigation now proves by the deliberate combination of our axially chiral borane catalyst and axially chiral silane reagents (in both enantiomeric forms) that the mechanisms of these hydrosilylations are essentially identical. Unmistakable stereochemical outcomes for the borane/silane pairs show that both participate in the enantioselectivity-determining hydride-transfer step. These experiments became possible after the discovery that our axially chiral C6F5-substituted borane induces appreciable levels of enantioinduction in the imine hydrosilylation.
Co-reporter:Sandeep R. Kandukuri and Martin Oestreich
The Journal of Organic Chemistry 2012 Volume 77(Issue 19) pp:8750-8755
Publication Date(Web):September 5, 2012
DOI:10.1021/jo301088f
A palladium(II)-catalyzed oxidative dehydrogenation of cyclohexene-1-carbonyl indole amides yielding the corresponding benzoylindoles is reported. The new aromatization is also applied to functionalized indoles such as tryptamine and tryptophan. The tethered indole is likely acting as a directing group for allylic C–H bond activation, and there is evidence for a mechanism proceeding through 1,3-diene formation followed by aromatization.
Co-reporter:Marius Mewald;Dr. Julia A. Schiffner ;Dr. Martin Oestreich
Angewandte Chemie 2012 Volume 124( Issue 8) pp:1797-1799
Publication Date(Web):
DOI:10.1002/ange.201107859
Co-reporter:Dr. Andreas Weickgenannt;Dr. Martin Oestreich
ChemCatChem 2011 Volume 3( Issue 10) pp:1527-1529
Publication Date(Web):
DOI:10.1002/cctc.201100210
Co-reporter:Toni T. Metsänen, Daniel Gallego, Tibor Szilvási, Matthias Driess and Martin Oestreich
Chemical Science (2010-Present) 2015 - vol. 6(Issue 12) pp:NaN7149-7149
Publication Date(Web):2015/09/14
DOI:10.1039/C5SC02855H
Combined experimental and theoretical analysis of the carbonyl hydrosilylation catalysed by an iron(0) pincer complex reveals an unprecedented mechanism of action. The iron(0) complex is in fact a precatalyst that is converted into an iron(II) catalyst through oxidative addition of a hydrosilane. Neither the hydrogen atom nor the silicon atom bound to the iron(II) centre are subsequently transferred onto the carbonyl acceptor, instead remaining at the sterically inaccessible iron(II) atom throughout the catalytic cycle. A series of labelling, crossover and competition experiments as well as the use of a silicon-stereogenic hydrosilane as a stereochemical probe suggest that the iron(II) site is not directly involved in the hydrosilylation. Strikingly, it is the silyl ligand attached to the iron(II) atom that acts as a Lewis acid for carbonyl activation in this catalysis. The whole catalytic process occurs on the periphery of the transition metal. Computation of the new peripheral as well as plausible alternative inner and outer sphere mechanisms support the experimental findings.
Co-reporter:Sebastian Keess and Martin Oestreich
Chemical Science (2010-Present) 2017 - vol. 8(Issue 7) pp:NaN4695-4695
Publication Date(Web):2017/05/24
DOI:10.1039/C7SC01657C
Safe- and convenient-to-handle surrogates of hazardous chemicals are always in demand. Recently introduced cyclohexa-1,4-dienes with adequate substitution fulfil this role as El+/H− equivalents in B(C6F5)3-catalysed transfer reactions of El–H to π- and σ-donors (CC/CC and CO/CN). Surrogates of Si–H/Ge–H, H–H and even C–H bonds have been designed and successfully applied to ionic transfer hydrosilylation/hydrogermylation, hydrogenation and hydro-tert-butylation, respectively. These processes and their basic principles are summarised in this Minireview. The similarities and differences between these transfer reactions as well as the challenges associated with these transformations are discussed.
Co-reporter:Martin Oestreich, Julia Hermeke and Jens Mohr
Chemical Society Reviews 2015 - vol. 44(Issue 8) pp:NaN2220-2220
Publication Date(Web):2015/02/13
DOI:10.1039/C4CS00451E
The bond activation chemistry of B(C6F5)3 and related electron-deficient boranes is currently experiencing a renaissance due to the fascinating development of frustrated Lewis pairs (FLPs). B(C6F5)3's ability to catalytically activate Si–H bonds through η1 coordination opened the door to several unique reduction processes. The ground-breaking finding that the same family of fully or partially fluorinated boron Lewis acids allows for the related H–H bond activation, either alone or as a component of an FLP, brought considerable momentum into the area of transition-metal-free hydrogenation and, likewise, hydrosilylation. This review comprehensively summarises synthetic methods involving borane-catalysed Si–H and H–H bond activation. Systems corresponding to an FLP-type situation are not covered. Aside from the broad manifold of CX bond reductions and CX/C–X defunctionalisations, dehydrogenative (oxidative) Si–H couplings are also included.
Co-reporter:Timo Stahl, Peter Hrobárik, C. David F. Königs, Yasuhiro Ohki, Kazuyuki Tatsumi, Sebastian Kemper, Martin Kaupp, Hendrik F. T. Klare and Martin Oestreich
Chemical Science (2010-Present) 2015 - vol. 6(Issue 7) pp:NaN4334-4334
Publication Date(Web):2015/05/18
DOI:10.1039/C5SC01035G
The nature of the hydrosilane activation mediated by ruthenium(II) thiolate complexes of type [(R3P)Ru(SDmp)]+[BArF4]− is elucidated by an in-depth experimental and theoretical study. The combination of various ruthenium(II) thiolate complexes and tertiary hydrosilanes under variation of the phosphine ligand and the substitution pattern at the silicon atom is investigated, providing detailed insight into the activation mode. The mechanism of action involves reversible heterolytic splitting of the Si–H bond across the polar Ru–S bond without changing the oxidation state of the metal, generating a ruthenium(II) hydride and sulfur-stabilized silicon cations, i.e. metallasilylsulfonium ions. These stable yet highly reactive adducts, which serve as potent silicon electrophiles in various catalytic transformations, are fully characterized by systematic multinuclear NMR spectroscopy. The structural assignment is further verified by successful isolation and crystallographic characterization of these key intermediates. Quantum-chemical analyses of diverse bonding scenarios are in excellent agreement with the experimental findings. Moreover, the calculations reveal that formation of the hydrosilane adducts proceeds via barrierless electrophilic activation of the hydrosilane by sterically controlled η1 (end-on) or η2 (side-on) coordination of the Si–H bond to the Lewis acidic metal center, followed by heterolytic cleavage of the Si–H bond through a concerted four-membered transition state. The Ru–S bond remains virtually intact during the Si–H bond activation event and also preserves appreciable bonding character in the hydrosilane adducts. The overall Si–H bond activation process is exergonic with ΔG0r ranging from −20 to −40 kJ mol−1, proceeding instantly already at low temperatures.
Co-reporter:C. David F. Königs, Maria F. Müller, Nuria Aiguabella, Hendrik F. T. Klare and Martin Oestreich
Chemical Communications 2013 - vol. 49(Issue 15) pp:NaN1508-1508
Publication Date(Web):2013/01/17
DOI:10.1039/C3CC38900F
A base-free, catalytic protocol for the dehydrogenative Si–N coupling of weakly nucleophilic N–H groups of heteroarenes or aryl-substituted amines with equimolar amounts of hydrosilanes is reported. Cooperative Si–H bond activation at a Ru–S bond generates a silicon electrophile that forms a Si–N bond prior to the N–H deprotonation by an intermediate Ru–H complex, only releasing H2.



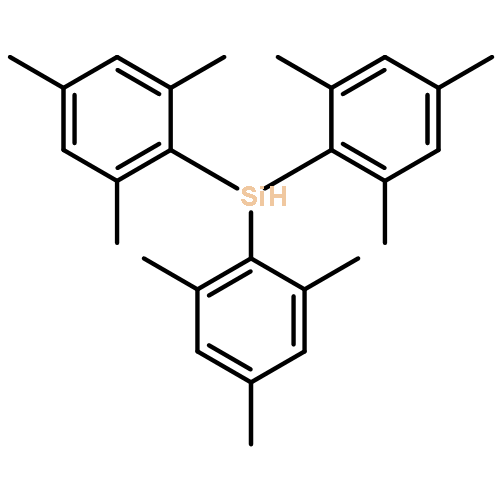


![Benzene, [(1E)-2-iodoethenyl]-](http://img.cochemist.com/ccimg/42600/42599-24-6.png)
![Benzene, [(1E)-2-iodoethenyl]-](http://img.cochemist.com/ccimg/42600/42599-24-6_b.png)




![Cyclohexane, 1-methyl-2-[(trimethylsilyl)oxy]-, (1R,2R)-rel-](/data/chemimg/1836300/39789-19-0.png)
![Cyclohexane, 1-methyl-2-[(trimethylsilyl)oxy]-, (1R,2R)-rel-](/data/chemimg/1836300/39789-19-0_b.png)
![Benzene, [(1Z)-3-chloro-1-propen-1-yl]-](/data/chemimg/1835000/39199-93-4.png)
![Benzene, [(1Z)-3-chloro-1-propen-1-yl]-](/data/chemimg/1835000/39199-93-4_b.png)

![[1,1'-Binaphthalen]-2-ol, (1R)-](/data/chemimg/1203800/35216-79-6.png)
![[1,1'-Binaphthalen]-2-ol, (1R)-](/data/chemimg/1203800/35216-79-6_b.png)
![BUTYL 4-{3-[3-(DIMETHYLAMINO)PROPOXY]-1H-INDAZOL-1-YL}BENZOATE](http://img.cochemist.com/ccimg/34600/34583-63-6.png)
![BUTYL 4-{3-[3-(DIMETHYLAMINO)PROPOXY]-1H-INDAZOL-1-YL}BENZOATE](http://img.cochemist.com/ccimg/34600/34583-63-6_b.png)


![1H-Isoindole-1,3(2H)-dione, 2-[(phenylmethylene)amino]-](http://img.cochemist.com/ccimg/32400/32386-95-1.png)
![1H-Isoindole-1,3(2H)-dione, 2-[(phenylmethylene)amino]-](http://img.cochemist.com/ccimg/32400/32386-95-1_b.png)

































![2-[(4-methoxybenzylidene)amino]-1H-isoindole-1,3(2H)-dione](http://img.cochemist.com/ccimg/19300/19279-70-0.png)
![2-[(4-methoxybenzylidene)amino]-1H-isoindole-1,3(2H)-dione](http://img.cochemist.com/ccimg/19300/19279-70-0_b.png)
























![Ethanone, 1-[4-(trifluoromethyl)phenyl]-, oxime](/data/chemimg/523900/15996-83-5.png)
![Ethanone, 1-[4-(trifluoromethyl)phenyl]-, oxime](/data/chemimg/523900/15996-83-5_b.png)











![N-[DIMETHYL(PHENYL)SILYL]ANILINE](http://img.cochemist.com/ccimg/13100/13091-06-0.png)
![N-[DIMETHYL(PHENYL)SILYL]ANILINE](http://img.cochemist.com/ccimg/13100/13091-06-0_b.png)




![Benzenesulfonamide, N-[(4-chlorophenyl)methyl]-4-methyl-](http://img.cochemist.com/ccimg/10600/10504-98-0.png)
![Benzenesulfonamide, N-[(4-chlorophenyl)methyl]-4-methyl-](http://img.cochemist.com/ccimg/10600/10504-98-0_b.png)






![[1,1'-Biphenyl]-2-amine, N,N-dimethyl-](http://img.cochemist.com/ccimg/6600/6590-81-4.png)
![[1,1'-Biphenyl]-2-amine, N,N-dimethyl-](http://img.cochemist.com/ccimg/6600/6590-81-4_b.png)






![5,5-diphenyl-5H-dibenzo[b,d]silole](http://img.cochemist.com/ccimg/5600/5550-08-3.png)
![5,5-diphenyl-5H-dibenzo[b,d]silole](http://img.cochemist.com/ccimg/5600/5550-08-3_b.png)



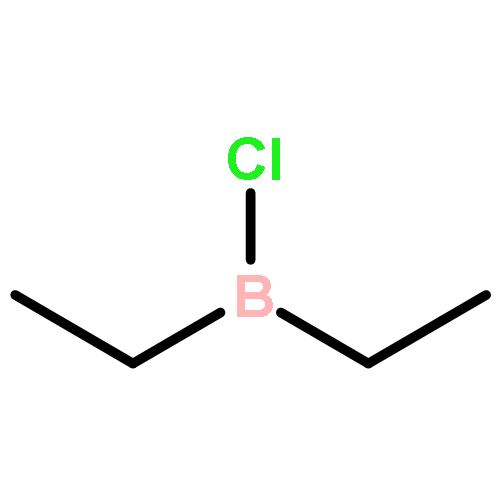


![6H-ISOINDOLO[2,1-A]INDOL-6-ONE](http://img.cochemist.com/ccimg/5000/4992-09-0.png)
![6H-ISOINDOLO[2,1-A]INDOL-6-ONE](http://img.cochemist.com/ccimg/5000/4992-09-0_b.png)





















![Bicyclo[2.2.1]hept-5-ene-2-carboxylic acid, methyl ester, (1R,2R,4R)-rel-](/data/chemimg/1178900/2903-75-5.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxylic acid, methyl ester, (1R,2R,4R)-rel-](/data/chemimg/1178900/2903-75-5_b.png)














![Cholest-5-ene,3-[(trimethylsilyl)oxy]-, (3b)-](http://img.cochemist.com/ccimg/1900/1856-05-9.png)
![Cholest-5-ene,3-[(trimethylsilyl)oxy]-, (3b)-](http://img.cochemist.com/ccimg/1900/1856-05-9_b.png)
















![N-[(2-NITROPHENYL)SULFANYL]-L-TYROSINE - N-CYCLOHEXYLCYCLOHEXANAM<WBR />INE (1:1)](http://img.cochemist.com/ccimg/400/393-56-6.png)
![N-[(2-NITROPHENYL)SULFANYL]-L-TYROSINE - N-CYCLOHEXYLCYCLOHEXANAM<WBR />INE (1:1)](http://img.cochemist.com/ccimg/400/393-56-6_b.png)


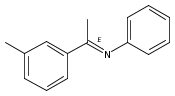
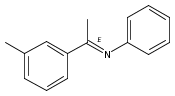







![ZINC, BIS[(1,1-DIMETHYLETHYL)DIPHENYLSILYL]-](http://img.cochemist.com/ccimg/877700/877671-99-3.png)
![ZINC, BIS[(1,1-DIMETHYLETHYL)DIPHENYLSILYL]-](http://img.cochemist.com/ccimg/877700/877671-99-3_b.png)


















![Zinc, bis[2,2,2-trimethyl-1,1-bis(trimethylsilyl)disilanyl]-](http://img.cochemist.com/ccimg/108200/108168-22-5.png)
![Zinc, bis[2,2,2-trimethyl-1,1-bis(trimethylsilyl)disilanyl]-](http://img.cochemist.com/ccimg/108200/108168-22-5_b.png)







![SILANE, DIMETHYLPHENYL[(1-PHENYLETHENYL)OXY]-](http://img.cochemist.com/ccimg/65400/65335-81-1.png)
![SILANE, DIMETHYLPHENYL[(1-PHENYLETHENYL)OXY]-](http://img.cochemist.com/ccimg/65400/65335-81-1_b.png)
![Silane, dimethylphenyl[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/64800/64788-85-8.png)
![Silane, dimethylphenyl[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/64800/64788-85-8_b.png)







![CARBAMIC ACID, [2-(1H-INDOL-3-YL)ETHYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/38800/38750-13-9.png)
![CARBAMIC ACID, [2-(1H-INDOL-3-YL)ETHYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/38800/38750-13-9_b.png)
![Silane, [(dichlorostannylene)bis(methylene)]bis[trimethyl-](http://img.cochemist.com/ccimg/38100/38047-49-3.png)
![Silane, [(dichlorostannylene)bis(methylene)]bis[trimethyl-](http://img.cochemist.com/ccimg/38100/38047-49-3_b.png)


![Bicyclo[2.2.2]oct-5-ene-2-carboxylic acid, methyl ester, (1R,2R,4R)-rel-](http://img.cochemist.com/ccimg/25600/25578-17-0.png)
![Bicyclo[2.2.2]oct-5-ene-2-carboxylic acid, methyl ester, (1R,2R,4R)-rel-](http://img.cochemist.com/ccimg/25600/25578-17-0_b.png)






![Silane, triethyl[(1-phenylethenyl)oxy]-](http://img.cochemist.com/ccimg/17800/17718-70-6.png)
![Silane, triethyl[(1-phenylethenyl)oxy]-](http://img.cochemist.com/ccimg/17800/17718-70-6_b.png)


![Benzenesulfonamide, 4-methyl-N-[(4-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/10600/10504-92-4.png)
![Benzenesulfonamide, 4-methyl-N-[(4-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/10600/10504-92-4_b.png)














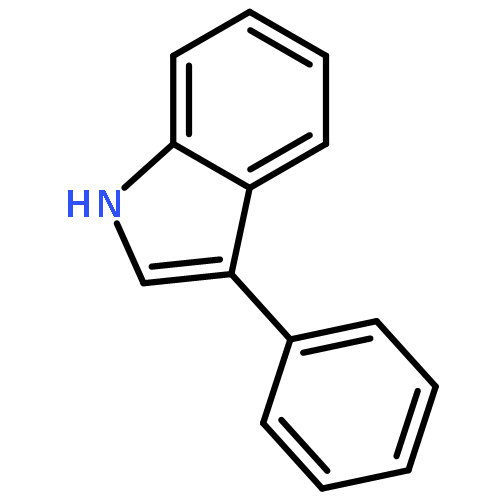




![Ethanone, 1-[(1R,2S,3S,4S)-3-phenylbicyclo[2.2.2]oct-5-en-2-yl]-, rel-](http://img.cochemist.com/ccimg/309800/309723-70-4.png)
![Ethanone, 1-[(1R,2S,3S,4S)-3-phenylbicyclo[2.2.2]oct-5-en-2-yl]-, rel-](http://img.cochemist.com/ccimg/309800/309723-70-4_b.png)