Co-reporter:Shobanbabu Bommagani, Jessica Ponder, Narsimha R. Penthala, Venumadhav Janganati, Craig T. Jordan, Michael J. Borrelli, Peter A. Crooks
European Journal of Medicinal Chemistry 2017 Volume 136(Volume 136) pp:
Publication Date(Web):18 August 2017
DOI:10.1016/j.ejmech.2017.05.031
•Design and synthesis of novel MMB-indole carboxylic acid ester analogs.•Screening of analogs for anticancer activity against NCI-60 cancer cell panel.•Potent growth inhibitory effects of analogs on human cancer cell lines.•Potent anti-leukemic activity of analogs against cell specimens from AML patients.•Potent antiproliferative effects against rat 9L-SF gliosarcoma cells.A series of novel, heteroaryl carboxylic acid conjugates of the sesquiterpene melampomagnolide-B (MMB, 3) has been evaluated as antitumor agents against an NCI panel of 64 human hematopoetic and solid tumor cell lines. The indole-3-acrylic acid conjugate 7j and the indole-3-carboxylic acid conjugate 7k were found to be the most potent analogs in the series. Compounds 7j and 7k exhibited remarkable growth inhibition, with GI50 values in the range 0.03–0.30 μM and 0.04–0.28 μM, respectively, against the cell lines in the leukemia sub-panel, and GI50 values of 0.05–0.40 μM and 0.04–0.61 μM, respectively, against 90% of the solid tumor cell lines in the NCI panel. Compound 7a was particularly effective against the sub-panel of breast cancer cell lines with GI50 values in the range <0.01–0.30 μM. Compounds 7j, 7a and its water soluble analog 7p also exhibited potent anticancer activity against rat 9L-SF gliosarcoma cells in culture. Compound 7j was the most potent compound in the series in the M9-ENL1 AML cell assay with a lethal dose concentration EC50 value of 720 nM, and exhibited the greatest cytotoxicity against a collection of primary AML stem cell specimens, which included a specimen that was unresponsive to PTL, affording EC50 values in the range 0.33–1.0 μM in three out of four specimens. The results from this study provide further evidence that analogs of the sesquiterpene MMB can be designed to afford molecules with significantly improved anticancer activity. Thus, both 7j and 7k are considered potential lead molecules in the search for new anticancer agents that can be used as treatments for both hematopoetic and solid tumors.Download high-res image (312KB)Download full-size image
Co-reporter:Venumadhav Janganati, Jessica Ponder, Shraddha Thakkar, Craig T. Jordan, Peter A. Crooks
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 14(Issue 14) pp:
Publication Date(Web):15 July 2017
DOI:10.1016/j.bmc.2017.05.008
A series of succinamide derivatives of melampomagnolide B have been synthesized by coupling MMB monosuccinate (2) with various heterocyclic amines to afford compounds 3a–3l. MMB monosuccinate was also reacted with terminal diaminoalkanes to afford dimeric succinamido analogs of MMB (4a–4h). These succinamide analogs of MMB were evaluated for their anti-cancer activity against a panel of sixty human cancer cell lines. Analogs 3d–3i and dimers 4f–4g exhibited promising anti-cancer activity with GI50 values ranging from 0.28 to 33.5 µM against most of the cell lines in the panel. The dimeric analogs 4f and 4g were identified as lead compounds with GI50 values in the nanomolar range (GI50 = 280–980 nM) against several cell lines in the panel; i.e. leukemia cell lines CCRF-CEM, HL-60(TB), K-562, MOLT-4, RPMI-8226 and SR; and solid tumor cell lines NCI-H522 (non-small cell lung cancer), SW-620 and HCT-116 (colon cancer), LOX IMVI (melanoma), RXF 393 (renal cancer), and MCF7, BT-549 and MDA-MB-468 (breast cancer). Succinamide analogs 3a, 3c–3l and 4b–4h were also evaluated for their apoptotic activity against M9-ENL1 acute myelogenous leukemia cells; compounds 3h–3j and 4g were equipotent with parthenolide, exhibiting LC50 values in the range 4.1–8.1 μM. Molecular docking studies indicate that these molecules interact covalently with the highly conserved Cys-46 residue of the N-terminal lobe (1–109) of human IKKβ to inhibit the NFκB transcription factor complex, resulting in down-regulation of anti-apoptotic genes under NFκB control.Download high-res image (207KB)Download full-size image
Co-reporter:Emily R. Hankosky, Shyam R. Joolakanti, Justin R. Nickell, Venumadhav Janganati, Linda P. Dwoskin, Peter A. Crooks
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 24(Issue 24) pp:
Publication Date(Web):15 December 2017
DOI:10.1016/j.bmcl.2017.10.039
A small library of fluoroethoxy-1,4-diphenethyl piperidine and fluoroethoxy-1,4-diphenethyl piperazine derivatives were designed, synthesized and evaluated for their ability to inhibit [3H]dopamine (DA) uptake at the vesicular monoamine transporter-2 (VMAT2) and dopamine transporter (DAT), [3H]serotonin (5-HT) uptake at the serotonin transporter (SERT), and [3H]dofetilide binding at the human-ether-a-go-go-related gene (hERG) channel. The majority of the compounds exhibited potent inhibition of [3H]DA uptake at VMAT2, Ki changes in the nanomolar range (Ki = 0.014–0.073 µM). Compound 15d exhibited the highest affinity (Ki = 0.014 µM) at VMAT2, and had 160-, 5-, and 60-fold greater selectivity for VMAT2 vs. DAT, SERT and hERG, respectively. Compound 15b exhibited the greatest selectivity (>60-fold) for VMAT2 relative to all the other targets evaluated, and 15b had high affinity for VMAT2 (Ki = 0.073 µM). Compound 15b was considered the lead compound from this analog series due to its high affinity and selectivity for VMAT2.Download high-res image (148KB)Download full-size image
Co-reporter:Benjamin M. Ford, Lirit N. Franks, Sherrica Tai, William E. Fantegrossi, Edward L. Stahl, Michael D. Berquist, Christian V. Cabanlong, Catheryn D. Wilson, Narsimha R. Penthala, Peter A. Crooks, Paul L. Prather
Pharmacological Research 2017 Volume 125, Part B(Volume 125, Part B) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.phrs.2017.08.008
The human cannabinoid subtype 1 receptor (hCB1R) is highly expressed in the CNS and serves as a therapeutic target for endogenous ligands as well as plant-derived and synthetic cannabinoids. Unfortunately, acute use of hCB1R agonists produces unwanted psychotropic effects and chronic administration results in development of tolerance and dependence, limiting the potential clinical use of these ligands. Studies in β-arrestin knockout mice suggest that interaction of certain GPCRs, including μ-, δ-, κ-opioid and hCB1Rs, with β-arrestins might be responsible for several adverse effects produced by agonists acting at these receptors. Indeed, agonists that bias opioid receptor activation toward G-protein, relative to β-arrestin signaling, produce less severe adverse effects. These observations indicate that therapeutic utility of agonists acting at hCB1Rs might be improved by development of G-protein biased hCB1R agonists. Our laboratory recently reported a novel class of indole quinulidinone (IQD) compounds that bind cannabinoid receptors with relatively high affinity and act with varying efficacy. The purpose of this study was to determine whether agonists in this novel cannabinoid class exhibit ligand bias at hCB1 receptors. Our studies found that a novel IQD-derived hCB1 receptor agonist PNR-4-20 elicits robust G protein-dependent signaling, with transduction ratios similar to the non-biased hCB1R agonist CP-55,940. In marked contrast to CP-55,940, PNR-4-20 produces little to no β-arrestin 2 recruitment. Quantitative calculation of bias factors indicates that PNR-4-20 exhibits from 5.4-fold to 29.5-fold bias for G protein, relative to β-arrestin 2 signaling (when compared to G protein activation or inhibition of forskolin-stimulated cAMP accumulation, respectively). Importantly, as expected due to reduced β-arrestin 2 recruitment, chronic exposure of cells to PNR-4-20 results in significantly less desensitization and down-regulation of hCB1Rs compared to similar treatment with CP-55,940. PNR-4-20 (i.p.) is active in the cannabinoid tetrad in mice and chronic treatment results in development of less persistent tolerance and no significant withdrawal signs when compared to animals repeatedly exposed to the non-biased full agoinst JWH-018 or Δ9-THC. Finally, studies of a structurally similar analog PNR- 4-02 show that it is also a G protein biased hCB1R agonist. It is predicted that cannabinoid agonists that bias hCB1R activation toward G protein, relative to β-arrestin 2 signaling, will produce fewer and less severe adverse effects both acutely and chronically.Download high-res image (205KB)Download full-size image
Co-reporter:Benjamin M. Ford, Lirit N. Franks, Sherrica Tai, William E. Fantegrossi, Edward L. Stahl, Michael D. Berquist, Christian V. Cabanlong, Catheryn D. Wilson, Narsimha R. Penthala, Peter A. Crooks, Paul L. Prather
Pharmacological Research 2017 Volume 125, Part B(Volume 125, Part B) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.phrs.2017.08.008
The human cannabinoid subtype 1 receptor (hCB1R) is highly expressed in the CNS and serves as a therapeutic target for endogenous ligands as well as plant-derived and synthetic cannabinoids. Unfortunately, acute use of hCB1R agonists produces unwanted psychotropic effects and chronic administration results in development of tolerance and dependence, limiting the potential clinical use of these ligands. Studies in β-arrestin knockout mice suggest that interaction of certain GPCRs, including μ-, δ-, κ-opioid and hCB1Rs, with β-arrestins might be responsible for several adverse effects produced by agonists acting at these receptors. Indeed, agonists that bias opioid receptor activation toward G-protein, relative to β-arrestin signaling, produce less severe adverse effects. These observations indicate that therapeutic utility of agonists acting at hCB1Rs might be improved by development of G-protein biased hCB1R agonists. Our laboratory recently reported a novel class of indole quinulidinone (IQD) compounds that bind cannabinoid receptors with relatively high affinity and act with varying efficacy. The purpose of this study was to determine whether agonists in this novel cannabinoid class exhibit ligand bias at hCB1 receptors. Our studies found that a novel IQD-derived hCB1 receptor agonist PNR-4-20 elicits robust G protein-dependent signaling, with transduction ratios similar to the non-biased hCB1R agonist CP-55,940. In marked contrast to CP-55,940, PNR-4-20 produces little to no β-arrestin 2 recruitment. Quantitative calculation of bias factors indicates that PNR-4-20 exhibits from 5.4-fold to 29.5-fold bias for G protein, relative to β-arrestin 2 signaling (when compared to G protein activation or inhibition of forskolin-stimulated cAMP accumulation, respectively). Importantly, as expected due to reduced β-arrestin 2 recruitment, chronic exposure of cells to PNR-4-20 results in significantly less desensitization and down-regulation of hCB1Rs compared to similar treatment with CP-55,940. PNR-4-20 (i.p.) is active in the cannabinoid tetrad in mice and chronic treatment results in development of less persistent tolerance and no significant withdrawal signs when compared to animals repeatedly exposed to the non-biased full agoinst JWH-018 or Δ9-THC. Finally, studies of a structurally similar analog PNR- 4-02 show that it is also a G protein biased hCB1R agonist. It is predicted that cannabinoid agonists that bias hCB1R activation toward G protein, relative to β-arrestin 2 signaling, will produce fewer and less severe adverse effects both acutely and chronically.Download high-res image (205KB)Download full-size image
Co-reporter:Justin R. Nickell, John P. Culver, Venumadhav Janganati, Guangrong Zheng, Linda P. Dwoskin, Peter A. Crooks
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 18) pp:4441-4445
Publication Date(Web):15 September 2016
DOI:10.1016/j.bmcl.2016.08.001
A small library of 1,4-diphenethylpiperazine analogs was synthesized and evaluated for inhibition of [3H]dihydrotetrabenazine binding and [3H]dopamine uptake at the vesicular monoamine transporter-2 (VMAT2). Results from these studies identified three novel molecules, 6b, 6e and 9a (Ki = 35 nM, 48 nM and 37 nM, respectively) that exhibit similar potency for inhibition of VMAT2 function compared with lobelane (Ki = 45 nM), and importantly, have enhanced water-solubility when compared to the previously reported 1,4-diphenethylpiperidine analogs. These 1,4-diphenethylpiperazine analogs constitute promising new leads in the discovery of potential pharmacotherapeutics for treatment of methamphetamine use disorders.
Co-reporter:Narsimha Reddy Penthala, Shobanbabu Bommagani, Jaishankar Yadlapalli, Peter A. Crooks
Tetrahedron Letters 2016 Volume 57(Issue 16) pp:1807-1810
Publication Date(Web):20 April 2016
DOI:10.1016/j.tetlet.2016.03.040
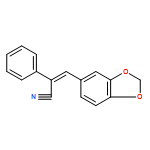
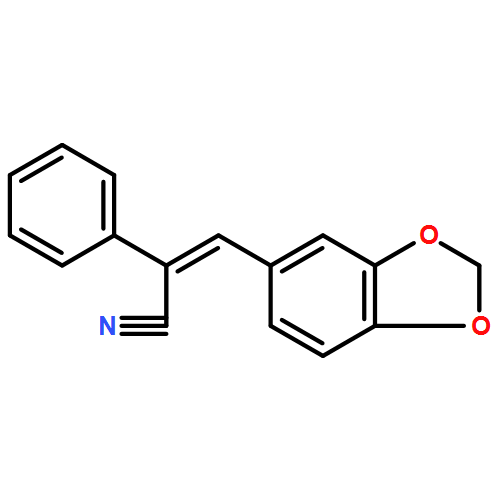
•Synthesis of a series of 1H-tetrazolylstilbenes from cyanostilbenes.•Novel and efficient tributyltin azide-mediated synthesis.•Cyanostilbenes containing benzo[b]thiophene, benzo[b]furan, and indole moieties.•An efficient, simple, and cost-effective procedure for the synthesis of novel tetrazolylstilbenes.A novel and efficient route was established for the synthesis of a series of (Z)-5-(2-(heteroaryl-2-yl) and (Z)-5-(3-(heteroaryl-3-yl)-1-(phenyl)vinyl)-1H-tetrazoles (3a–g and 6a–f, respectively) from cyanostilbene analogs containing benzo[b]thiophene, benzo[b]furan, and indole moieties utilizing tributyltin azide as a Lewis acid. This 1,3-dipolar [3+2]cycloaddition of azide to the cyano group of the cyanostilbene precursor molecule affords good yields of the corresponding tetrazolylstilbene analog and constitutes a simple and cost-effective procedure.
Co-reporter:Nikhil R. Madadi, Amit Ketkar, Narsimha R. Penthala, April C.L. Bostian, Robert L. Eoff, Peter A. Crooks
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 9) pp:2164-2169
Publication Date(Web):1 May 2016
DOI:10.1016/j.bmcl.2016.03.068
•Synthesized a variety of novel dioxol and dihydrodioxin analogs of 2- and 3-phenylacetonitriles.•Evaluated the novel analogs for their anti-cancer activity against the NCI 60 human cancer cell panel.•Carried out in silico docking studies at the colchicine binding site on tubulin to rationalize the anti-cancer properties of the synthesized compounds.•Tubulin polymerization assays were conducted to establish further the mechanism of action.•Analogue 3j showed promising anticancer activity against most of the cancer cells with GI50 of <100 nM.A small library of (Z)-2-(benzo[d][1,3]dioxol-5-yl) and (Z)-2,3-dihydrobenzo[b][1,4]dioxin-6-yl analogs of 2- and 3-phenylacetonitriles has been synthesized and evaluated for their anti-cancer activities against a panel of 60 human cancer cell lines. The dihydrodioxin analog 3j and dioxol analogs 5e and 7e exhibited the most potent anti-cancer activity of all the analogs synthesized in this study, with GI50 values of <100 nM against almost all of the cell lines in the human cancer cell panel. Of these three, only compound 3j inhibited tubulin polymerization to any degree in vitro. The binding modes of 3j and the structurally related tubulin-inhibitor DMU-212 were determined by virtual docking studies with tubulin dimer. Compound 3j docked at the colchicine-binding site at the dimer interface of tubulin. The Full-Fitness (FF) score of 3j was observed to be substantially higher than DMU-212, which agrees well with the observed anti-cancer potency (GI50 values). The mechanism by which dioxol analogs 5e and 7e exert their cytotoxic effects remains unknown at this stage, but it is unlikely that they affect tubulin dynamics. Nevertheless, these findings suggest that both dioxol and dihydrodioxin analogs of phenylacrylonitrile may have potential for development as clinical candidates to treat a variety of human cancers.
Co-reporter:Shyamsunder R. Joolakanti, Justin R. Nickell, Venumadhav Janganati, Guangrong Zheng, Linda P. Dwoskin, Peter A. Crooks
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 10) pp:2422-2427
Publication Date(Web):15 May 2016
DOI:10.1016/j.bmcl.2016.03.119
A series of lobelane and GZ-793A analogues that incorporate aromatic 4-hydroxy and 4-(2-fluoroethoxy) substituents were synthesized and evaluated for inhibition of [3H]dopamine (DA) uptake at the vesicular monoamine transporter-2 (VMAT2) and the dopamine transporter (DAT), and [3H]serotonin uptake at the serotonin transporter (SERT). Most of these compounds exhibited potent inhibition of DA uptake at VMAT2 in the nanomolar range (Ki = 30–70 nM). The two most potent analogues, 7 and 14, both exhibited a Ki value of 31 nM for inhibition of VMAT2. The lobelane analogue 14, incorporating 4-(2-fluoroethoxy) and 4-hydroxy aromatic substituents, exhibited 96- and 335-fold greater selectivity for VMAT2 versus DAT and SERT, respectively, in comparison to lobelane. Thus, lobelane analogues bearing hydroxyl and fluoroethoxy moieties retain the high affinity for VMAT2 of the parent compound, while enhancing selectivity for VMAT2 versus the plasmalemma transporters.
Co-reporter:Justin R. Nickell, John P. Culver, Venumadhav Janganati, Guangrong Zheng, Linda P. Dwoskin, Peter A. Crooks
Bioorganic & Medicinal Chemistry Letters 2016 26(13) pp: 2997-3000
Publication Date(Web):1 July 2016
DOI:10.1016/j.bmcl.2016.05.025
A series of 1,4-diphenalkylpiperidine analogs were synthesized and evaluated for their affinity and inhibitory potency at the [3H]dihydrotetrabenazine (DTBZ) binding site and [3H]dopamine (DA) uptake site on the vesicular monoamine transporter-2 (VMAT2). Results revealed that translocation of the phenethyl side chains of lobelane from C2 and C6 to C1 and C4 around the central piperidine ring slightly reduces affinity and inhibitory potency at VMAT2 with respect to lobelane. However, methoxy and fluoro-substitution of either phenyl ring of these 1,4-diphenethyl analogs afforded VMAT2 inhibition comparable or higher (5-fold) affinity at the DTBZ binding and DA uptake sites relative to lobelane, whereas replacement of the 4-phenethyl moiety in these analogs with a 4-phenmethyl moiety markedly reduced affinity for the DTBZ binding and DA uptake sites by 3- and 5-fold, respectively. Among the twenty five 1,4-diphenethylpiperidine analogs evaluated, compounds containing a 4-(2-methoxyphenethyl) moiety exhibited the most potent inhibition of DTBZ binding and vesicular DA uptake. From this subgroup, analogs 8h, 8j and 8m exhibited Ki values of 9.3 nM, 13 nM and 13 nM, respectively, for inhibition of [3H]DA uptake by VMAT2, and represent some of the most potent inhibitors of VMAT2 function reported thus far.
Co-reporter:Venumadhav Janganati; Jessica Ponder; Craig T. Jordan; Michael J. Borrelli; Narsimha Reddy Penthala;Peter A. Crooks
Journal of Medicinal Chemistry 2015 Volume 58(Issue 22) pp:8896-8906
Publication Date(Web):November 5, 2015
DOI:10.1021/acs.jmedchem.5b01187
Novel carbamate (7a–7h) and carbonate (7i, 7j, and 8) dimers of melampomagnolide B have been synthesized by reaction of the melampomagnolide-B-triazole carbamate synthon 6 with various terminal diamino- and dihydroxyalkanes. Dimeric carbamate products 7b, 7c, and 7f exhibited potent growth inhibition (GI50 = 0.16–0.99 μM) against the majority of cell lines in the NCI panel of 60 human hematological and solid tumor cell lines. Compound 7f and 8 exhibited anticancer activity that was 300-fold and 1 × 106-fold more cytotoxic than DMAPT, respectively, at a concentration of 10 μM against rat 9L-SF gliosarcoma cells. Compounds 7a–7j and 8 were also screened against M9-ENL1 and acute myelogenous leukemia (AML) primary cell lines and exhibited 2- to 10-fold more potent antileukemic activity against M9-ENL1 cells (EC50 = 0.57–2.90 μM) when compared to parthenolide (EC50 = 6.0) and showed potent antileukemic activity against five primary AML cell lines (EC50 = 0.76–7.3 μM).
Co-reporter:Nikhil R. Madadi, Narsimha R. Penthala, Kevin Howk, Amit Ketkar, Robert L. Eoff, Michael J. Borrelli, Peter A. Crooks
European Journal of Medicinal Chemistry 2015 Volume 103() pp:123-132
Publication Date(Web):20 October 2015
DOI:10.1016/j.ejmech.2015.08.041
•Novel 4,5-disubstituted 2H-1,2,3-triazoles were synthesized.•Evaluated for their anti-cancer activity against NCI 60 human cancer cell lines.•Conducted in silico docking studies.•Cell cycle redistribution assay conducted to further understand the mechanism of action of synthesized CA-4 analogues.•One analogue, 2e, showed promising anticancer activity with an LD50 value of 7.5 nM against 9LSF rat gliosarcoma cells.A series of combretastatin A-4 (CA-4) analogues have been prepared from (Z)-substituted diarylacrylonitriles (1a-1p) obtained in a two-step synthesis from appropriate arylaldehydes and acrylonitriles. The resulting 4,5-disubstituted 2H-1,2,3-triazoles were evaluated for their anti-cancer activities against a panel of 60 human cancer cell lines. The diarylacrylonitrile analogue 2l exhibited the most potent anti-cancer activity in the screening studies, with GI50 values of <10 nM against almost all the cell lines in the human cancer cell panel and TGI values of <10 nM against cancer cell lines SF-539, MDA-MB-435, OVCAR-3 and A498. Furthermore, in silico docking studies of compounds 2l, 2e and 2h within the active site of tubulin were carried out in order to rationalize the mechanism of the anti-cancer properties of these compounds. From the in silico studies, compound 2e was predicted to have better affinity for the colchicine binding site on tubulin compared to compounds 2l and 2h. Analogue 2e was also evaluated for its anti-cancer activity by colony formation assay against 9LSF rat gliosarcoma cells and afforded an LD50 of 7.5 nM. A cell cycle redistribution assay using analogue 2e was conducted to further understand the mechanism of action of these CA-4 analogues. From this study, analogues 2e and 2l were the most potent anti-cancer agents in this structural class, and were considered lead compounds for further development as anti-cancer drugs.
Co-reporter:Narsimha Reddy Penthala, Hongliang Zong, Amit Ketkar, Nikhil Reddy Madadi, Venumadav Janganati, Robert L. Eoff, Monica L. Guzman, Peter A. Crooks
European Journal of Medicinal Chemistry 2015 Volume 92() pp:212-220
Publication Date(Web):6 March 2015
DOI:10.1016/j.ejmech.2014.12.050
•Synthesis of novel a series of heterocyclic trans cyano CA-4 analogs.•Analogs evaluated for anti-cancer activity against 60 human cancer cell lines.•Analogs evaluated against a variety of primary leukemia cell lines.•Binding modes at the colchicine binding site on tubulin have been determined.•Benzothiophen-2-yl and indol-2-yl analogs are the most potent anticancer agents.A series of heterocyclic combretastatin analogues have been synthesized and evaluated for their anticancer activity against a panel of 60 human cancer cell lines. The most potent compounds were two 3,4,5-trimethoxy phenyl analogues containing either an (Z)-indol-2-yl (8) or (Z)-benzo[b]furan-2-yl (12) moiety; these compounds exhibited GI50 values of <10 nM against 74% and 70%, respectively, of the human cancer cell lines in the 60-cell panel. Compounds 8, and 12 and two previously reported compounds in the same structural class, i.e. 29 and 31, also showed potent anti-leukemic activity against leukemia MV4-11 cell lines with LD50 values = 44 nM, 47 nM, 18 nM, and 180 nM, respectively. From the NCI anti-cancer screening results and the data from the in vitro toxicity screening on cultured AML cells, seven compounds: 8, 12, 21, 23, 25, 29 and 31 were screened for their in vitro inhibitory activity on tubulin polymerization in MV4-11 AML cells; at 50 nM, 8 and 29 inhibited polymerization of tubulin by >50%. The binding modes of the three most active compounds (8, 12 and 29) to tubulin were also investigated utilizing molecular docking studies. All three molecules were observed to bind in the same hydrophobic pocket at the interface of α- and β-tubulin that is occupied by colchicine, and were stabilized by van der Waals’ interactions with surrounding tubulin residues. The results from the tubulin polymerization and molecular docking studies indicate that compounds 8 and 29 are the most potent anti-leukemic compounds in this structural class, and are considered lead compounds for further development as anti-leukemic drugs.
Co-reporter:Narsimha Reddy Penthala, Amit Ketkar, Konjeti R. Sekhar, Michael L. Freeman, Robert L. Eoff, Ramesh Balusu, Peter A. Crooks
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 22) pp:7226-7233
Publication Date(Web):15 November 2015
DOI:10.1016/j.bmc.2015.10.019
In the present study, we have designed and synthesized a series of 1-benzyl-2-methyl-3-indolylmethylene barbituric acid analogs (7a–7h) and 1-benzyl-2-methyl-3-indolylmethylene thiobarbituric acid analogs (7i–7l) as nucleophosmin 1 (NPM1) inhibitors and have evaluated them for their anti-cancer activity against a panel of 60 different human cancer cell lines. Among these analogs 7i, 7j, and 7k demonstrated potent growth inhibitory effects in various cancer cell types with GI50 values <2 μM. Compound 7k exhibited growth inhibitory effects on a sub-panel of six leukemia cell lines with GI50 values in the range 0.22–0.35 μM. Analog 7i also exhibited GI50 values <0.35 μM against three of the leukemia cell lines in the sub-panel. Analogs 7i, 7j, 7k and 7l were also evaluated against the mutant NPM1 expressing OCI-AML3 cell line and compounds 7k and 7l were found to cause dose-dependent apoptosis (AP50 = 1.75 μM and 3.3 μM, respectively). Compound 7k also exhibited potent growth inhibition against a wide variety of solid tumor cell lines: that is, A498 renal cancer (GI50 = 0.19 μM), HOP-92 and NCI-H522 lung cancer (GI50 = 0.25 μM), COLO 205 and HCT-116 colon cancer (GI50 = 0.20 and 0.26 μM, respectively), CNS cancer SF-539 (GI50 = 0.22 μM), melanoma MDA-MB-435 (GI50 = 0.22 μM), and breast cancer HS 578T (GI50 = 0.22 μM) cell lines. Molecular docking studies suggest that compounds 7k and 7l exert their anti-leukemic activity by binding to a pocket in the central channel of the NPM1 pentameric structure. These results indicate that the small molecule inhibitors 7i, 7j, 7k, and 7l could be potentially developed into anti-NPM1 drugs for the treatment of a variety of hematologic malignancies and solid tumors.
Co-reporter:Manfred Biermann, Guangrong Zheng, Marhaba Hojahmat, Nick V. Moskalev, Peter A. Crooks
Tetrahedron Letters 2015 Volume 56(Issue 20) pp:2608-2610
Publication Date(Web):13 May 2015
DOI:10.1016/j.tetlet.2015.04.050
Asymmetric synthesis of (S)-norketamine and (R)-norketamine utilizing Sharpless asymmetric dihydroxylation followed by stereo-controlled Ritter amination and Jones oxidation is reported. This method allowed the preparation of the free base of (S)-norketamine and (R)-norketamine in pure crystalline form for the first time. The absolute configuration of both enantiomers was verified by X-ray structure and optical resolution of racemic norketamine.
Co-reporter:Narsimha Reddy Penthala, Leena Madhukuri, Shraddha Thakkar, Nikhil Reddy Madadi, Gauri Lamture, Robert L. Eoff and Peter A. Crooks
MedChemComm 2015 vol. 6(Issue 8) pp:1535-1543
Publication Date(Web):30 Jun 2015
DOI:10.1039/C5MD00219B
trans-Cyanocombretastatin A-4 (trans-CA-4) analogues have been structurally modified to afford their more stable CA-4-(2H)-1,2,3-triazole analogues. Fifteen novel, stable 4-heteroaryl-5-aryl-(2H)-1,2,3-triazole CA-4 analogues (8a–i, 9 and 11a–e) were evaluated for anti-cancer activity against a panel of 60 human cancer cell lines. These analogues displayed potent cytotoxic activity against both hematological and solid tumor cell lines with GI50 values in the low nanomolar range. The most potent compound, 8a, was a benzothiophen-2-yl analogue that incorporated a 3,4,5-trimethoxyphenyl moiety connected to the (2H)-1,2,3-triazole ring system. Compound 8a exhibited GI50 values of <10 nM against 80% of the cancer cell lines in the panel. Three triazole analogues, 8a, 8b and 8g, showed particularly potent growth inhibition against the triple negative Hs578T breast cancer cell line with GI50 values of 10.3 nM, 66.5 nM and 20.3 nM, respectively. Molecular docking studies suggest that these compounds bind to the same hydrophobic pocket at the interface of α- and β-tubulin that is occupied by colchicine and cis-CA-4, and are stabilized by Van der Waals' interactions with surrounding amino acid residues. Compound 8a was found to inhibit tubulin polymerization in vitro with an IC50 value of 1.7 μM. The potent cytotoxicity of these novel compounds and their inhibition of tubulin dynamics make these triazole analogues promising candidates for development as anti-cancer drugs.
Co-reporter:Nikhil R. Madadi, Hongliang Zong, Amit Ketkar, Chen Zheng, Narsimha R. Penthala, Venumadhav Janganati, Shobanbabu Bommagani, Robert L. Eoff, Monica L. Guzman and Peter A. Crooks
MedChemComm 2015 vol. 6(Issue 5) pp:788-794
Publication Date(Web):30 Jan 2015
DOI:10.1039/C4MD00478G
A series of novel diarylacrylonitrile and trans-stilbene analogues of resveratrol has been synthesized and evaluated for their anticancer activities against a panel of 60 human cancer cell lines. The diarylacrylonitrile analogues 3b and 4a exhibited the most potent anticancer activity of all the analogues synthesized in this study, with GI50 values of <10 nM against almost all the cell lines in the human cancer cell panel. Compounds 3b and 4a were also screened against the acute myeloid leukemia (AML) cell line, MV4-11, and were found to have potent cytotoxic properties that are likely mediated through inhibition of tubulin polymerization. Results from molecular docking studies indicate a common binding site for 4a and 3b on the α,β-tubulin heterodimer, with a slightly more favorable binding for 4a compared to 3b; this is consistent with the results from the microtubule assays, which demonstrate that 4a is more potent than 3b in inhibiting tubulin polymerization in MV4-11 cells. Taken together, these data suggest that diarylacrylonitriles 3b and 4a may have potential as antitubulin therapeutics for treatment of both solid and hematological tumors.
Co-reporter:Derong Ding, Justin R. Nickell, Linda P. Dwoskin, Peter A. Crooks
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 13) pp:2613-2616
Publication Date(Web):1 July 2015
DOI:10.1016/j.bmcl.2015.04.105
We have previously shown that quinolyl moieties are attractive structural replacements for the phenyl groups in lobelane. These quinolyl analogues had improved water-solubility over lobelane and retained the potent vesicular monoamine transporter-2 (VMAT-2) inhibitory properties of the parent compound, with quinlobelane (4) exhibiting potent inhibition of uptake at VMAT-2 (Ki = 51 nM). However, the VMAT-2 inhibitory properties of quinolyl analogues of norlobelane, which is equipotent with lobeline as an inhibitor of [3H]dopamine (DA) uptake at VMAT-2, have not been reported. In the current communication, we describe the synthesis of some novel des-methyl quinolyl analogues of lobelane that exhibit greater affinity (Ki = 178–647 nM) for the dihydrotetrabenazine binding site located on VMAT-2 compared with lobelane (Ki = 970 nM), norlobelane (Ki = 2310 nM) and quinlobelane (Ki = 2640 nM). The most potent compounds, 14 and 15, also exhibited inhibition of [3H]DA uptake at VMAT-2 (Ki = 42 nM) which was comparable to both lobelane (Ki = 45 nM) and norlobelane (Ki = 43 nM). Results reveal that binding affinity at VMAT-2 serves as an accurate predictor of inhibition of the function of VMAT-2 for the majority of these analogues. These novel analogues are under consideration for further development as treatments for methamphetamine abuse.
Co-reporter:Narsimha R. Penthala, Shobanbabu Bommagani, Venumadhav Janganati, Kenzie B. MacNicol, Chad E. Cragle, Nikhil R. Madadi, Linda L. Hardy, Angus M. MacNicol, Peter A. Crooks
European Journal of Medicinal Chemistry 2014 Volume 85() pp:517-525
Publication Date(Web):6 October 2014
DOI:10.1016/j.ejmech.2014.08.022
•Developed a method for the synthesis of E-olefinic coupling products of PTL and MMB.•Compounds screened for anticancer activity and effects on cell cycle progression.•Two compounds showed good growth inhibitory effects on human cancer cell lines.•The two compounds caused significant effects in oocyte maturation assays.•Screening for effects on cell cycle progression complements cancer cell screening.(E)-13-(Aryl/heteroaryl)parthenolides (5a–i and 6a–i) were synthesized and evaluated for their ability to modify cell cycle progression during progesterone-stimulated Xenopus oocyte maturation and screened for their anticancer activity against a panel of 60 human cancer cell lines. (E)-13-(4-aminophenyl) parthenolide (5b) caused a significant inhibition of progesterone-stimulated oocyte maturation, and was determined to function downstream of MAP kinase signaling, but upstream of the activation of the universal G2/M regulator, M-phase promoting factor (MPF), cyclin B/Cyclin-dependent kinase (CDK). The compound (E)-13-(2-bromo-phenyl)parthenolide (5c) activates oocyte maturation independently of progesterone stimulation. Compounds 5b and 5c displayed modest growth inhibition on select cancer cell lines at 10 μM dose when tested on the panel of 60 cancer cell lines. By contrast, compounds (5f and 7) did not modulate oocyte maturation but did exhibit micromolar level growth inhibition against most of the human cancer cell lines over a range of doses. Together, our findings indicate that screening of compounds in the oocyte maturation assay may identify additional effective cell cycle regulatory compounds that do not necessarily exert overt cytotoxicity as assessed in traditional drug screening assays.
Co-reporter:Venumadhav Janganati, Narsimha Reddy Penthala, Chad E. Cragle, Angus M. MacNicol, Peter A. Crooks
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 8) pp:1963-1967
Publication Date(Web):15 April 2014
DOI:10.1016/j.bmcl.2014.02.067
Aminoparthenolide derivatives have been prepared by reaction of parthenolide with various heterocyclic amines to afford corresponding Michael addition products. These novel compounds were evaluated for their modulatory effects on Xenopus oocyte maturation. Two compounds, 6e and 6f, were identified that promote G2-M cell cycle progression.
Co-reporter:Joshua A. Eldridge, Mikolaj Milewski, Audra L. Stinchcomb, Peter A. Crooks
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 22) pp:5212-5215
Publication Date(Web):15 November 2014
DOI:10.1016/j.bmcl.2014.09.072
A small library of amino acid ester prodrugs of 6-β-naltrexol (NTXOL, 1) was prepared in order to investigate the candidacy of these prodrugs for microneedle-enhanced transdermal delivery. Six amino acid ester prodrugs were synthesized (6a–f). 6b, 6d, and 6e were stable enough at skin pH (pH 5.0) to move forward to studies in 50% human plasma. The lead compound (6e) exhibited the most rapid bioconversion to NTXOL in human plasma (t1/2 = 2.2 ± 0.1 h).
Co-reporter:Venumadhav Janganati, Narsimha Reddy Penthala, Nikhil Reddy Madadi, Zheng Chen, Peter A. Crooks
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 15) pp:3499-3502
Publication Date(Web):1 August 2014
DOI:10.1016/j.bmcl.2014.05.059
Melampomagnolide B (MMB) is a natural sesquiterpene structurally related to parthenolide (PTL). We have shown that MMB exhibits anti-leukemic properties similar to PTL. Unlike PTL, the presence of a primary hydroxyl group in the MMB molecule allows the opportunity for examining the biological activity of a variety of conjugated analogs of MMB. We have now synthesized a series of carbamate analogs of MMB and evaluated these derivatives for anti-cancer activity against a panel of sixty human cancer cell lines. Analogs 6a and 6e exhibited promising anti-leukemic activity against human leukemia cell line CCRF-CEM with GI50 values of 680 and 620 nM, respectively. Analog 6a also showed GI50 values of 1.98 and 1.38 μM respectively, against RPMI-8226 and SR leukemia cell lines and GI50 values of 460 and 570 nM against MDA-MB-435 melanoma and MDA-MB-468 breast cancer cell lines, respectively. Analog 6e had GI50 values of 650 and 900 nM against HOP-92 non-small cell lung and RXF 393 renal cancer cell lines.
Co-reporter:Nikhil Reddy Madadi, Narsimha Reddy Penthala, Venumadhav Janganati, Peter A. Crooks
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 2) pp:601-603
Publication Date(Web):15 January 2014
DOI:10.1016/j.bmcl.2013.12.013
Based on previous SAR studies on N-benzylindole and barbituric acid hybrid molecules, we have synthesized a series of aromatic substituted 5-((1-benzyl-1H-indol-3-yl)methylene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione analogs (3a–i) and evaluated them for their in vitro growth inhibition and cytotoxicity against a panel of 60 human tumor cell lines. Compounds 3c, 3d, 3f and 3g were identified as highly potent anti-proliferative compounds against ovarian, renal and breast cancer cell lines with GI50 values in low the nanomolar range. The 4-methoxy-N-benzyl analog (3d) was the most active compound with GI50 values of 20 nM and 40 nM against OVCAR-5 ovarian cancer cells and MDA-MB-468 breast cancer cells, respectively. Two other analogs, 3c (the 4-methyl-N-benzyl analog) and 3g (the 4-fluoro-N-benzyl analog) exhibited equimolar potency against MDA-MB-468 cells GI50 = 30 nM). Analog 3f (the 4-chloro-N-benzyl analog) exhibited a GI50 value of 40 nM against renal cancer cell line A498. These results suggest that aromatic substituted N-benzylindole dimethylbarbituric acid hybrids may have potential for development as clinical candidates to treat a variety of solid tumors.
Co-reporter:Nikhil Reddy Madadi, Narsimha Reddy Penthala, Lin Song, Howard P. Hendrickson, Peter A. Crooks
Tetrahedron Letters 2014 Volume 55(Issue 30) pp:4207-4211
Publication Date(Web):23 July 2014
DOI:10.1016/j.tetlet.2014.05.045
2H-1,2,3-Triazoles (2) were synthesized by [3+2] cycloaddition of (Z)-2,3-diaryl substituted acrylonitriles (1) with sodium azide and ammonium chloride in DMF/water. This method represents a facile and efficient reaction procedure for the synthesis of 4,5-diaryl-2H-1,2,3-triazoles in modest to good yields.
Co-reporter:Narsimha Reddy Penthala, Nikhil Reddy Madadi, Venumadhav Janganati, Peter A. Crooks
Tetrahedron Letters 2014 Volume 55(Issue 40) pp:5562-5565
Publication Date(Web):1 October 2014
DOI:10.1016/j.tetlet.2014.08.027
•l-Proline-catalyzed one-step [3+2]cycloaddition of azide with cyanostilbenes.•A novel method for the synthesis of 2H-1,2,3-triazoles from cyanostilbenes.•Method provides a facile, efficient, and environmentally benign procedure.•Method affords good yields and relatively short reaction times.•Affords 2H-1,2,3-triazoles that cannot undergo cis–trans isomerization.Use of a novel reagent has been established for the synthesis of a series of 4,5-diaryl-2H-1,2,3-triazoles (6a–i and 9a–e) from cyanostilbene analogs of benzo[b]thiophene, benzo[b]furan, and indole, catalyzed by l-proline via Lewis base-catalyzed one-step [3+2]cycloaddition of azide. This method provides an efficient, simple, and environmentally benign procedure that affords good yields and relatively short reaction times.
Co-reporter:Narsimha Reddy Penthala, Venumadhav Janganati, Shobanbabu Bommagani and Peter A. Crooks
MedChemComm 2014 vol. 5(Issue 7) pp:886-890
Publication Date(Web):06 May 2014
DOI:10.1039/C4MD00115J
A series of novel trans-2-quinolyl-, 3-quinolyl- and 4-quinolyl cyanostilbene derivatives were synthesized as analogs of combretastatin A-4 (CA-4), and evaluated for anticancer activity against a panel of 60 human cancer cell lines. The quinolin-2-yl and quinolin-3-yl analogs containing trimethoxyphenyl (10 and 16, respectively) or dimethoxyphenyl (12 and 18, respectively) moieties showed growth inhibition against all the cancer cell lines in the panel, with GI50 values generally <1 μM. Quinolin-2-yl-analog 10 exhibited potent growth inhibition against MDA-MB-435 melanoma and NCI-H522 non-small cell lung cancer line with GI50 values of 33 nM and 37 nM respectively. Quinolin-2-yl-analog 12 showed potent growth inhibition against NCI-H522 non-small cell lung cancer lines with a GI50 value of 94 nM. Quinolin-3-yl-analog 18 exhibited potent growth inhibition against MDA-MB-435 melanoma and NCI-H522 non-small cell lung cancer lines with GI50 values of 53 nM and 69 nM, respectively. Thus, structural modification of the CA-4 molecule has afforded compounds with potential clinical utility in the treatment a variety of different solid tumors.
Co-reporter:Derong Ding, Justin R. Nickell, Agripina G. Deaciuc, Narsimha Reddy Penthala, Linda P. Dwoskin, Peter A. Crooks
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 21) pp:6771-6777
Publication Date(Web):1 November 2013
DOI:10.1016/j.bmc.2013.08.001
Lobelane analogs that incorporate a central piperidine or pyrrolidine moiety have previously been reported by our group as potent inhibitors of VMAT2 function. Further central ring size reduction of the piperidine moiety in lobelane to a four-membered heterocyclic ring has been carried out in the current study to afford novel cis-and trans-azetidine analogs. These azetidine analogs (15a–15c and 22a–22c) potently inhibited [3H]dopamine (DA) uptake into isolated synaptic vesicles (Ki ⩽ 66 nM). The cis-4-methoxy analog 22b was the most potent inhibitor (Ki = 24 nM), and was twofold more potent that either lobelane (2a, Ki = 45 nM) or norlobelane (2b, Ki = 43 nM). The trans-methylenedioxy analog, 15c (Ki = 31 nM), was equipotent with the cis-analog, 22b, in this assay. Thus, cis- and trans-azetidine analogs 22b and 15c represent potential leads in the discovery of new clinical candidates for the treatment of methamphetamine abuse.Novel cis and trans-azetidine lobelane analogs (15a–c and 22a–c) as inhibitors of vesicular [3H]DA uptake into vesicles.
Co-reporter:Joshua R. Ring, Fang Zheng, Aaron J. Haubner, John M. Littleton, Peter A. Crooks
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 7) pp:1764-1774
Publication Date(Web):1 April 2013
DOI:10.1016/j.bmc.2013.01.051
Using a combination of both the partial least squares (PLS) and back-propagation artificial neural network (ANN) pattern recognition methods, several models have been developed to predict the activity of a series of arylidenaminoguanidine analogs as inhibitory modulators of the N-methyl-d-aspartate receptor complex. This was done by correlating structural and physicochemical descriptors obtained from computation software with the experimentally observed [3H]MK-801 displacement ability of a small library of synthesized and in vitro screened arylidenaminoguanidines. Results for the generated PLS model were r2 = 0.814, rmsd = 0.208, rCV2 = 0.714, loormsd = 0.261. The ANN model was created utilizing the eleven descriptors from the PLS model for comparison. The quality of the ANN model (r2=0.828, rmsd = 0.200, rCV2 = 0.721, loormsd = 0.257) is similar to the PLS model, and indicates that the feature between the inputs and the output is majorly linear. These computational models were able to predict inhibition of the NMDA receptor complex by this series of compounds in silico, affording a predictive structure-based ‘pre-screening’ paradigm for the arylideneaminoguanidine analogs.
Co-reporter:Nikhil Reddy Madadi, Narsimha Reddy Penthala, Lisa K. Brents, Benjamin M. Ford, Paul L. Prather, Peter A. Crooks
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 7) pp:2019-2021
Publication Date(Web):1 April 2013
DOI:10.1016/j.bmcl.2013.02.025
A small library of N-benzyl indolequinuclidinone (IQD) analogs has been identified as a novel class of cannabinoid ligands. The affinity and selectivity of these IQDs for the two established cannabinoid receptor subtypes, CB1 and CB2, was evaluated. Compounds 8 (R = R2 = H, R1 = F) and 13 (R = COOCH3, R1 = R2 = H) exhibited high affinity for CB2 receptors with Ki values of 1.33 and 2.50 nM, respectively, and had lower affinities for the CB1 receptor (Ki values of 9.23 and 85.7 nM, respectively). Compound 13 had the highest selectivity of all the compounds examined, and represents a potent cannabinoid ligand with 34-times greater selectivity for CB2R over CB1R. These findings are significant for future drug development, given recent reports demonstrating beneficial use of cannabinoid ligands in a wide variety of human disease states including drug abuse, depression, schizophrenia, inflammation, chronic pain, obesity, osteoporosis and cancer.N-Benzyl indolequinuclidinone (IQD) analogs have been identified as a novel class of cannabinoid ligands. The affinity and selectivity of these IQDs for the two established cannabinoid receptor subtypes, CB1 and CB2, was evaluated. Compounds 8 (R = R2 = H, R1 = F) and 13 (R = COOCH3, R1 = R2 = H) exhibited high affinity for CB2 receptors with Ki values of 1.33 and 2.50 nM, respectively, and had lower affinity for the CB1 receptor (Ki values of 9.23 and 85.7 nM, respectively). Thus compound 13 represents a potent cannabinoid ligand with 34-times greater selectivity for CB2R over CB1R. These findings are significant for future drug development, given recent reports demonstrating beneficial use of cannabinoid ligands in a wide variety of human disease states including drug abuse, depression, schizophrenia, inflammation, chronic pain, obesity, osteoporosis and cancer.
Co-reporter:Narsimha Reddy Penthala, Purushothama Rao Ponugoti, Vinod Kasam, Peter A. Crooks
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 5) pp:1442-1446
Publication Date(Web):1 March 2013
DOI:10.1016/j.bmcl.2012.12.053
A series of novel 5-((1-aroyl-1H-indol-3-yl)methylene)-2-thioxodihydropyrimidine-4,6(1H,5H)-diones (3a–z) have been evaluated for in vitro cytotoxicity against a panel of 60 human tumor cell lines. Compound 3k exhibited the most potent growth inhibition against melanoma MDA-MB-435 cells (GI50 = 850 nM), against leukemia SR cancer cells (GI50 = 1.45 μM), and OVCAR-3 (GI50 = 1.26 μM) ovarian cancer cell lines. The structurally related compound 3s had a GI50 value of 1.77 μM against MDA-MB-435 cells. The N-naphthoyl analogue 3t had GI50 values of 1.30 and 1.91 μM against HOP-92 non-small cell lung cancer and MDA-MB-435 melanoma cell lines, respectively. The related analogue 3w had GI50 values of 1.09 μM against HOP-92 non-small cell lung cancer cell lines. Interestingly, docking of the two active molecules 3k and 3w into the active site of COX-2 indicates that these compounds are COX-2 ligands with strong hydrophobic and hydrogen bonding interactions. Thus, compounds 3k, 3t, 3s, and 3w constitute a new class of anticancer/anti-inflammatory agents that may have unique potential for cancer therapy.Compound 3k exhibited the most potent growth inhibition against melanoma MDA-MB-435 cells (GI50 = 850 nM). The structurally related compound 3s had a GI50 value of 1.77 μM against MDA-MB-435 cells. The N-naphthoyl analogue 3t had GI50 values of 1.30 μM against HOP-92 non-small cell lung cancer cell lines. The related analogue 3w had GI50 values of 1.09 μM against HOP-92 non-small cell lung cancer cell lines. Docking of the two active molecules 3k and 3w into the active site of COX-2 indicates that these compounds are COX-2 ligands with strong hydrophobic and hydrogen bonding interactions.
Co-reporter:Narsimha Reddy Penthala, Purushothama Rao Ponugoti, Justin R. Nickell, Agripina G. Deaciuc, Linda P. Dwoskin, Peter A. Crooks
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 11) pp:3342-3345
Publication Date(Web):1 June 2013
DOI:10.1016/j.bmcl.2013.03.092
Central heterocyclic ring size reduction from piperidinyl to pyrrolidinyl in the vesicular monoamine transporter-2 (VMAT2) inhibitor GZ-793A and its analogs resulted in novel N-propane-1,2(R)-diol analogs 11a–i. These compounds were evaluated for their affinity for the dihydrotetrabenazine (DTBZ) binding site on VMAT2 and for their ability to inhibit vesicular dopamine (DA) uptake. The 4-difluoromethoxyphenethyl analog 11f was the most potent inhibitor of [3H]-DTBZ binding (Ki = 560 nM), with 15-fold greater affinity for this site than GZ-793A (Ki = 8.29 μM). Analog 11f also showed similar potency of inhibition of [3H]-DA uptake into vesicles (Ki = 45 nM) compared to that for GZ-793A (Ki = 29 nM). Thus, 11f represents a new water-soluble inhibitor of VMAT function.Compound 11f has been identified as a new water-soluble inhibitor of VMAT2 function with a Ki of 45 nM in the vesicular dopamine uptake assay and a Ki of 560 nM in the [3H]DTBZ binding assay.
Co-reporter:Derong Ding, Linda P. Dwoskin, Peter A. Crooks
Tetrahedron Letters 2013 Volume 54(Issue 38) pp:5211-5213
Publication Date(Web):18 September 2013
DOI:10.1016/j.tetlet.2013.07.067
An efficient synthesis of cis-2,6-di-(2-quinolylpiperidine) has been developed. The key steps involve Wittig reaction of N-Cbz-protected cis-piperidine-2,6-dicarboxaldehyde (3) with 2-(triphenylphosphinyl-methyl)quinoline bromide (4) and sequential removal of the N-Cbz group and double bond reduction. This synthetic procedure provides an efficient preparation for this useful norlobelane analogue.
Co-reporter:Narsimha Reddy Penthala, Vijayakumar N. Sonar, Jamie Horn, Markos Leggas, Jai Shankar K. B. Yadlapalli and Peter A. Crooks
MedChemComm 2013 vol. 4(Issue 7) pp:1073-1078
Publication Date(Web):29 May 2013
DOI:10.1039/C3MD00130J
A new library of small molecules with structural features resembling combretastatin analogs was synthesized and evaluated for anticancer activity against a panel of 60 human cancer cell lines. Three novel acrylonitrile analogs (5, 6 and 13) caused a significant reduction in cell growth in almost all the cell lines examined, with GI50 values generally in the range 10–100 nM. Based on the structural characteristics of similar drugs, we hypothesized that the cytotoxic activity was likely due to interaction with tubulin. Furthermore, these compounds appeared to overcome cell-associated P-glycoprotein (P-gp)-mediated resistance, since they were equipotent in inhibiting OVCAR8 and NCI/ADR-RES cell growth. Given that antitubulin drugs are among the most effective agents for the treatment of advanced prostate cancer we sought to validate the results from the 60 cell panel by studying the representative analog 6 utilizing prostate cancer cell lines, as well as exploring the molecular mechanism of the cytotoxic action of this analog.
Co-reporter:Guangrong Zheng, David B. Horton, Narsimha Reddy Penthala, Justin R. Nickell, John P. Culver, Agripina G. Deaciuc, Linda P. Dwoskin and Peter A. Crooks
MedChemComm 2013 vol. 4(Issue 3) pp:564-568
Publication Date(Web):22 Jan 2013
DOI:10.1039/C3MD20374C
A series of N-substituted lobelane analogues was synthesized and evaluated for their [3H]dihydrotetrabenazine binding affinity at the vesicular monoamine transporter and for their inhibition of vesicular [3H]dopamine uptake. Compound 19a, which contains an N-1,2(R)-dihydroxypropyl group, had been identified as a potential clinical candidate for the treatment of methamphetamine abuse.
Co-reporter:Narsimha Reddy Penthala;Thirupathi Reddy Yerramreddy;Sean Parkin;Peter A. Crooks
Journal of Heterocyclic Chemistry 2013 Volume 50( Issue S1) pp:E156-E159
Publication Date(Web):
DOI:10.1002/jhet.1107
Novel ninhydrin–creatinine heterocyclic condensation products (3–5) were synthesized under different solvent conditions. The compound 2-(2-amino-1-methyl-4-oxo-4,5-dihydro-1H-imidazol-5-yl)-2-hydroxy-1H-ind-ene-1,3(2H)-dione (3) was formed by reacting ninhydrin (1) with creatinine (2) in the presence of sodium acetate in acetic acid. The same reactants afforded the zwitterionic compound 4 when the reaction was carried out in water, and a novel oxadiazine ring system (product 5) was generated when benzene was used as solvent.
Co-reporter:Narsimha Reddy Penthala, Venumadhav Janganati, Terri L. Alpe, Scott M. Apana, Marc S. Berridge, Peter A. Crooks, Michael J. Borrelli
Bioorganic & Medicinal Chemistry Letters (15 December 2016) Volume 26(Issue 24) pp:5883-5886
Publication Date(Web):15 December 2016
DOI:10.1016/j.bmcl.2016.11.015
Co-reporter:Zaineb A.F. Albayati, Venumadhav Janganati, Zheng Chen, Jessica Ponder, Philip J. Breen, Craig T. Jordan, Peter A. Crooks
Bioorganic & Medicinal Chemistry (1 February 2017) Volume 25(Issue 3) pp:
Publication Date(Web):1 February 2017
DOI:10.1016/j.bmc.2016.12.036
A series of carbamate derivatives of the antileukemic sesquiterpene melampomagnolide B (MMB) has been synthesized utilizing a 1,2,4-triazole carbamate conjugate of MMB as an intermediate synthon. Five imidazole- and benzimidazole-carbamate analogs of MMB (8a–8e) were prepared and evaluated for anti-leukemic activity against cultured M9 ENL1 AML cells. All the analogs exhibited improved anti-leukemic activity (EC50 = 0.90–3.93 μM) when compared to parthenolide and the parent sesquiterpene, MMB (EC50 = 7.0 μM and 15.5 μM, respectively). The imidazole carbamate analog, 8a (EC50 = 0.9 μM), was 16 times more potent than MMB. The comparative bioavailabilities of 8a and MMB were determined in BALB/c mice following oral dosing of these compounds. It has been demonstrated that the absolute plasma bioavailabilities of MMB and 8a were 6.7 ± 0.8%, and 45.5 ± 2%, respectively. These results indicate that, compared to MMB, the PK parameters for 8a display significantly improved bioavailability and exposure after oral administration. Analog 8a is considered to be a potential clinical candidate for treatment of acute myelogenous leukemia.





![Magnesium, bromo[(3R)-3,7-dimethyl-6-octenyl]-](http://img.cochemist.com/ccimg/679900/679821-46-6.png)
![Magnesium, bromo[(3R)-3,7-dimethyl-6-octenyl]-](http://img.cochemist.com/ccimg/679900/679821-46-6_b.png)
![Benzoic acid, 4-[(3-formyl-2-methyl-1H-indol-1-yl)methyl]-, methyl ester](http://img.cochemist.com/ccimg/592600/592550-37-3.png)
![Benzoic acid, 4-[(3-formyl-2-methyl-1H-indol-1-yl)methyl]-, methyl ester](http://img.cochemist.com/ccimg/592600/592550-37-3_b.png)
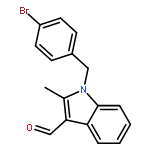


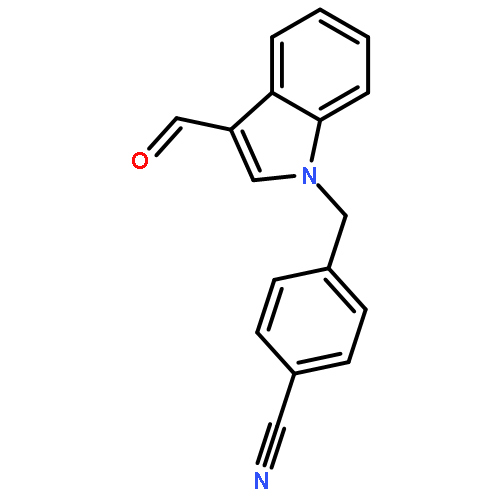


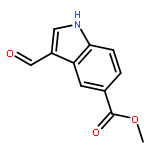
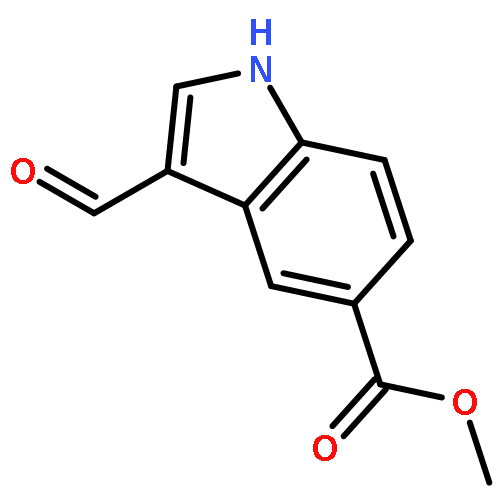

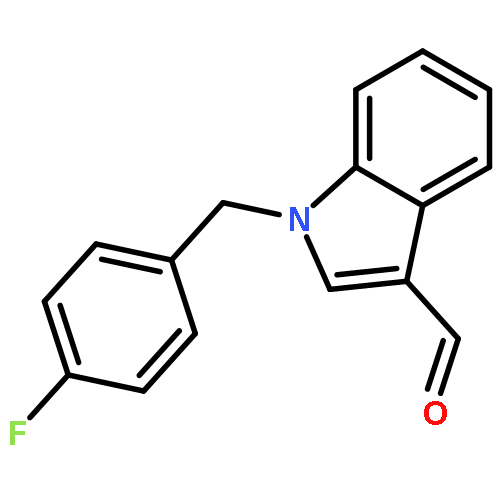
![1H-Indole-3-carboxaldehyde, 1-[(2-bromophenyl)methyl]-](http://img.cochemist.com/ccimg/171800/171734-73-9.png)
![1H-Indole-3-carboxaldehyde, 1-[(2-bromophenyl)methyl]-](http://img.cochemist.com/ccimg/171800/171734-73-9_b.png)



![CP 55,940;(-)-CIS-3-[2-HYDROXY-4-(1,1-DIMETHYLHEPTYL)PHENYL]-TRANS-4-(3-HYDROXYPROPYL)CYCLOHEXANOL](http://img.cochemist.com/ccimg/83100/83002-04-4.png)
![CP 55,940;(-)-CIS-3-[2-HYDROXY-4-(1,1-DIMETHYLHEPTYL)PHENYL]-TRANS-4-(3-HYDROXYPROPYL)CYCLOHEXANOL](http://img.cochemist.com/ccimg/83100/83002-04-4_b.png)
![ONCRASIN 1;1-[(4-CHLOROPHENYL)METHYL]-1H-INDOLE-3-CARBOXALDEHYDE](http://img.cochemist.com/ccimg/75700/75629-57-1.png)
![ONCRASIN 1;1-[(4-CHLOROPHENYL)METHYL]-1H-INDOLE-3-CARBOXALDEHYDE](http://img.cochemist.com/ccimg/75700/75629-57-1_b.png)
![2H-1-Benzopyran-6-ol,3,4-dihydro-2,8-dimethyl-2-[(3E,7E)-4,8,12-trimethyl-3,7,11-tridecatrien-1-yl]-,(2R)-](http://img.cochemist.com/ccimg/25700/25612-59-3.png)
![2H-1-Benzopyran-6-ol,3,4-dihydro-2,8-dimethyl-2-[(3E,7E)-4,8,12-trimethyl-3,7,11-tridecatrien-1-yl]-,(2R)-](http://img.cochemist.com/ccimg/25700/25612-59-3_b.png)


![2H-1-Benzopyran-6-ol,3,4-dihydro-2,7,8-trimethyl-2-[(3E,7E)-4,8,12-trimethyl-3,7,11-tridecatrien-1-yl]-,(2R)-](http://img.cochemist.com/ccimg/14200/14101-61-2.png)
![2H-1-Benzopyran-6-ol,3,4-dihydro-2,7,8-trimethyl-2-[(3E,7E)-4,8,12-trimethyl-3,7,11-tridecatrien-1-yl]-,(2R)-](http://img.cochemist.com/ccimg/14200/14101-61-2_b.png)




![2H-Benzo[a]quinolizin-2-ol,1,3,4,6,7,11b-hexahydro-9,10-dimethoxy-3-(2-methylpropyl)-](http://img.cochemist.com/ccimg/3500/3466-75-9.png)
![2H-Benzo[a]quinolizin-2-ol,1,3,4,6,7,11b-hexahydro-9,10-dimethoxy-3-(2-methylpropyl)-](http://img.cochemist.com/ccimg/3500/3466-75-9_b.png)




![Benzene,1,2,3-trimethoxy-5-[(1E)-2-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/134100/134029-62-2.png)
![Benzene,1,2,3-trimethoxy-5-[(1E)-2-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/134100/134029-62-2_b.png)