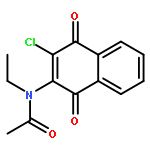
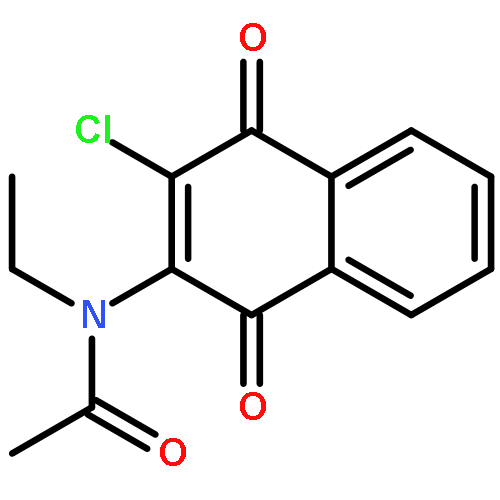
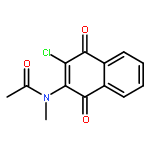
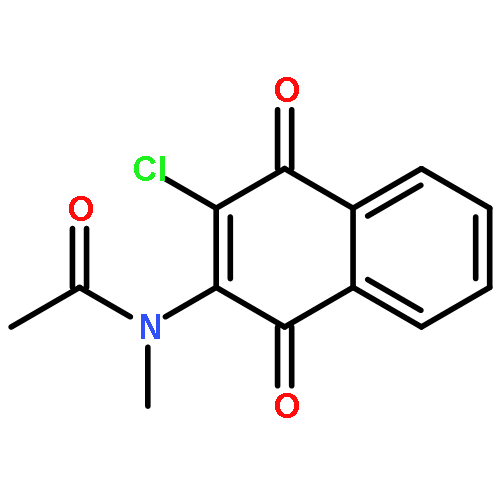
Co-reporter:Ming Li, Samuel A. Nyantakyi, Pooja Gopal, Dinah binte Aziz, Thomas Dick, and Mei-Lin Go
ACS Medicinal Chemistry Letters November 9, 2017 Volume 8(Issue 11) pp:1165-1165
Publication Date(Web):October 9, 2017
DOI:10.1021/acsmedchemlett.7b00287
Agents that selectively target the mycobacterial membrane could potentially shorten treatment time for tuberculosis, reduce relapse, and curtail emergence of resistant strains. The lipophilicity and extensive charge-delocalized state of the triphenylphosphonium cation strongly favor accumulation within bacterial membranes. Here, we explored the antimycobacterial activities and membrane-targeting properties of indolylalkyltriphenylphosphonium analogues. The most active analogues preferentially inhibited growth of Mycobacterium tuberculosis H37Rv (MIC50 2–4 μM) and were bactericidal against Mycobacterium bovis BCG (MBC99 3 μM). In spite of their propensity to accumulate within membranes, we found no evidence that these compounds permeabilized mycobacterial membranes or induced cell-envelope stress. Our investigations indicated that their bacterical effects stem from sustained depolarization of mycobacterial membranes and ensuing disruptive effects on electron transfer and cell division.Keywords: Antimycobacterial activity; Cationic amphiphilic indoles; Membrane depolarization; Triphenylphosphonium cations;
Co-reporter:Tianming Yang, Wilfried Moreira, Samuel Agyei Nyantakyi, Huan Chen, Dinah binte Aziz, Mei-Lin Go, and Thomas Dick
Journal of Medicinal Chemistry April 13, 2017 Volume 60(Issue 7) pp:2745-2745
Publication Date(Web):March 14, 2017
DOI:10.1021/acs.jmedchem.6b01530
Antibacterials that disrupt cell membrane function have the potential to eradicate “persister” organisms and delay the emergence of resistance. Here we report the antimycobacterial activities of 4-fluoro and 6-methoxyindoles bearing a cationic amphiphilic motif represented by a lipophilic n-octyl side chain at position 1 and a positively charged azepanyl or 1,4-dioxa-8-azaspiro[4.5]decane moiety at position 3. These analogues exhibited balanced profiles of potency (Mycobacterium bovis BCG, M tuberculosis H37Rv), selective activity, solubility, and metabolic stability. Bacteriological mechanism of action investigations on a representative analogue revealed cell membrane permeabilization and depolarization in M bovis BCG. These membrane-related changes preceded cell death indicating that the loss in membrane integrity was not an epiphenomenon. Bactericidal activity was observed against both growing and nongrowing mycobacterial cultures. The analogue also upregulated cell envelope stress-inducible promoters piniBAC and pclgR, implicating the involvement of envelope-related targets in its mode of action.
Co-reporter:Tianming Yang, Xiao Chen, Hai-xiao Jin, Gautam Sethi, Mei-Lin Go
European Journal of Medicinal Chemistry 2015 Volume 92() pp:145-155
Publication Date(Web):6 March 2015
DOI:10.1016/j.ejmech.2014.12.027
•Sirtuins are protein deacylases that regulate metabolism and stress responses.•Tetrahydropyridoindoles were identified as preferential Sirt2 inhibitors.•A well defined SAR was deduced from in vitro inhibitory activities.•A potent member 18 induced hyperacetylation of p53 and α-tubulin in cancer cells.•Molecular docking pointed to occupancy of the NAD+ and substrate sites of Sirt2.Sirtuins are protein deacylases with regulatory roles in metabolism and stress response. Functionalized tetrahydro-1H-pyrido[4,3-b]indoles were identified as preferential sirtuin 2 inhibitors, with in vitro inhibitory potencies in the low micromolar concentrations (IC50 3–4 μM) for the more promising candidates. The functional relevance of sirtuin inhibition was corroborated in western blots that showed hyperacetylation of p53 and α-tubulin in treated HepG2 and MDA-MB-231 cells. Molecular docking showed that the tetrahydropyridoindole scaffold was positioned in the NAD + pocket and the acetylated substrate channel of the sirtuin 2 protein by van der Waals/hydrophobic, H bonding and stacking interactions. Functionalized tetrahydropyridoindoles represent a novel class of sirtuin 2 inhibitors that could be further explored for its therapeutic potential.
Co-reporter:Si-Han Sherman Ho, Mei-Yi Sim, Wei-Loong Sherman Yee, Tianming Yang, Shyi-Peng John Yuen, Mei-Lin Go
European Journal of Medicinal Chemistry 2015 Volume 104() pp:42-56
Publication Date(Web):2 November 2015
DOI:10.1016/j.ejmech.2015.09.026
•YM155, a survivin suppressor, is implicated in DNA damage and its SAR is unknown.•53 analogs were synthesized to define the growth inhibition SAR of YM155.•AB1 and AB7 emerged as potent analogs against RCC and NSCLC cell lines.•YM155 and analogs were not potent DNA intercalators and redox cyclers.•YM155 and analogs induced apoptotic cell death that resulted in DNA digestion.The anticancer agent YM155 is widely investigated as a specific survivin suppressant. More recently, YM155 was found to induce DNA damage and this has raised doubts as to whether survivin is its primary target. In an effort to assess the contribution of DNA damage to the anticancer activity of YM155, several analogs were prepared and evaluated for antiproliferative activity on malignant cells, participation in DNA intercalation and free radical generation by redox cycling. The intact positively charged scaffold was found to be essential for antiproliferative activity and intercalation but was less critical for redox cycling where the minimal requirement was a pared down bicyclic quinone. Side chain requirements at the N1 and N3 positions of the scaffold were more alike for redox cycling and intercalation than antiproliferative activity, underscoring yet again, the limited structural overlaps for these activities. Furthermore, antiproliferative activities were poorly correlated to DNA intercalation and redox cycling. Potent antiproliferative activity (IC50 9–23 nM), exceeding that of YM155, was found for a minimally substituted methyl analog AB7. Like YM155 and other dioxonaphthoimidazoliums, AB7 was a modest DNA intercalator but with weak redox cycling activity. Thus, the capacity of this scaffold to inflict direct DNA damage leading to cell death may not be significant and YM155 should not be routinely classified as a DNA damaging agent.
Co-reporter:Dr. Xiao Chen;Dr. Tianming Yang;Amudha Deivasigamani;Dr. Muthu K. Shanmugam; Kam-Man Hui; Gautam Sethi; Mei-Lin Go
ChemMedChem 2015 Volume 10( Issue 9) pp:1548-1558
Publication Date(Web):
DOI:10.1002/cmdc.201500235
Abstract
The benzylideneindolinone 6-chloro-3-(3′-trifluoromethylbenzylidene)-1,3-dihydroindol-2-one (4) was reported to exhibit potent and selective growth inhibitory effects on hepatocellular carcinoma (HCC). Corroborative evidence supported multi-receptor tyrosine kinase (RTK) inhibition as a possible mode of action. However, the poor physicochemical properties of 4 limited its furtherance as a lead compound. In this study, the modification of 4 was investigated with the aim of improving its potency and physicochemical profile. The 6-fluorobenzylideneindolinone 3-12 bearing a 3′-N-propylaminosulfonyl substituent was found to be a promising substitute. Compound 3-12 [6-fluoro-3-(3′-N-propylaminosulfonylbenzylidene)-1,3-dihydroindol-2-one] was found to be tenfold more soluble than 4 and to have sub-micromolar growth inhibitory activities on HCC cells. It is apoptogenic and inhibits the phosphorylation of several RTKs in HuH7, of which the inhibition of FGFR4 and HER3 are prominent. Compound 3-12 decreased the tumor load in a physiologically relevant orthotopic HCC xenograft murine model. Structure–activity relationships support pivotal roles for the fluoro and N′-propylaminosulfonyl moieties in enhancing cell-based activity and moderating the physicochemical profile (solubility, permeability) of 3-12.
Co-reporter:Kheng-Lin Tan ; Azhar Ali ; Yuhong Du ; Haian Fu ; Hai-Xiao Jin ; Tan-Min Chin ; Matiullah Khan
Journal of Medicinal Chemistry 2014 Volume 57(Issue 14) pp:5904-5918
Publication Date(Web):June 24, 2014
DOI:10.1021/jm401352a
Curcumin is known to trigger ER-stress induced cell death of acute promyelocytic leukemic (APL) cells by intercepting the degradation of nuclear co-repressor (N-CoR) protein which has a key role in the pathogenesis of APL. Replacing the heptadienedione moiety of curcumin with a monocarbonyl cross-conjugated dienone embedded in a tetrahydrothiopyranone dioxide ring resulted in thiopyranone dioxides that were more resilient to hydrolysis and had greater growth inhibitory activities than curcumin on APL cells. Several members intercepted the degradation of misfolded N-CoR and triggered the signaling cascade in the unfolded protein response (UPR) which led to apoptotic cell death. Microarray analysis showed that genes involved in protein processing pathways that were germane to the activation of the UPR were preferentially up-regulated in treated APL cells, supporting the notion that the UPR was a consequential mechanistic pathway affected by thiopyranone dioxides. The Michael acceptor reactivity of the scaffold may have a role in exacerbating ER stress in APL cells.
Co-reporter:Thuy Nguyen, Tianming Yang, Mei-Lin Go
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 7) pp:1830-1838
Publication Date(Web):1 April 2014
DOI:10.1016/j.bmcl.2014.02.006
The in vitro neuronal cell death model based on the HT22 mouse hippocampal cell model is a convenient means of identifying compounds that protect against oxidative glutamate toxicity which plays a role in the development of certain neurodegenerative diseases. Functionalized acridin-9-yl-phenylamines were found to protect HT22 cells from glutamate challenge at submicromolar concentrations. The Aryl1-NH-Aryl2 scaffold that is embedded in these compounds was the minimal pharmacophore for activity. Mechanistically, protection against the endogenous oxidative stress generated by glutamate did not involve up-regulation of glutathione levels but attenuation of the late stage increases in mitochondrial ROS and intracellular calcium levels. The NH residue in the pharmacophore played a crucial role in this regard as seen from the loss of neuroprotection when it was structurally modified or replaced. That the same NH was essential for radical scavenging in cell-free and cell-based systems pointed to an antioxidant basis for the neuroprotective activities of these compounds.Pharmacophore for neuroprotection against glutamate challenge may involve generation of stabilized radical cation from NH residue.
Co-reporter:Pondy M. Ramanujulu, Tianming Yang, Siew-Qi Yap, Fui-Chung Wong, Patrick J. Casey, Mei Wang, Mei-Lin Go
European Journal of Medicinal Chemistry 2013 Volume 63() pp:378-386
Publication Date(Web):May 2013
DOI:10.1016/j.ejmech.2013.02.007
The enzyme isoprenylcysteine carboxyl methyltransferase (Icmt) plays an important role in the post-translational modification of proteins involved in the regulation of cell growth and oncogenesis. The biological consequences of Icmt inhibition strongly implicate the enzyme as a potential therapeutic target for cancer and provide a compelling rationale for developing specific Icmt inhibitors as anti-cancer agents. We report here the systematic modification of the known Icmt inhibitor cysmethynil to give an analog 15 with greatly improved solubility and PAMPA permeability which was achieved with concurrent gains in Icmt inhibitory and cell-based antiproliferative activities. The modifications involved replacing the amide side chain of cysmethynil with a tertiary amine, and introducing an aminopyrimidine ring in place of m-tolyl. The presence of the weakly basic and polar aminopyrimidine ring contributed significantly to the potency and drug-like profile of the final compound.Graphical abstractHighlights► Icmt is a viable anti-cancer target for which there are few lead inhibitors. ► The best known potent inhibitor cysmethynil is poorly soluble and too lipophilic. ► An indoleamine analog 15 inhibited Icmt at a submicromolar IC50. ► 15 shows improved solubility and PAMPA permeability.
Co-reporter:Sherman Si Han Ho, Mei Lin Go
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 22) pp:6127-6133
Publication Date(Web):15 November 2013
DOI:10.1016/j.bmcl.2013.09.014
The semi-synthetic lignan terameprocol inhibits the transcription of several inflammatory and oncogenic genes and has been evaluated for its anti-cancer properties. Here we investigated the effect of restricting the flexibility of the carbon linker connecting the terminal rings of terameprocol on its growth inhibitory activity. Conformational restriction was explored by introducing unsaturation, inserting polar entities with limited flexibility and cyclization of the connecting linker. Twenty three compounds were synthesized and evaluated on a panel of malignant human cells. The most promising compounds were those with non-polar linkers, as seen in butadiene 1a and the cyclized benzylideneindane analog 7. Both compounds were more potent than terameprocol on pancreatic BxPC-3 cells with GI50 values of 3.4 and 8.1 μM, respectively. Selected isomers of 1a (E,E) and 7 (Z) adopted low energy bent conformations that mimicked the low energy conformer of terameprocol. It is tempting to propose that conformational similarity to terameprocol may have contributed to their good activity. The scaffolds of 1a and 7 should be further investigated for their anticancer potential.
Co-reporter:Dr. Xi Kai Wee;Dr. Tianming Yang ;Dr. Mei Lin Go
ChemMedChem 2012 Volume 7( Issue 5) pp:777-791
Publication Date(Web):
DOI:10.1002/cmdc.201200018
Abstract
Meisoindigo has been used as an indirubin substitute for the treatment of chronic myeloid leukemia (CML) for several years. In view of its poor solubility and erratic absorption, several investigations have focused on developing analogues with more desirable physicochemical profiles. Here, we investigated the structure–activity relationship (SAR) of meisoindigo with respect to its antiproliferative activity on leukemic K562 cells and found that appending a phenalkyl side chain onto the lactam NH resulted in analogues that retained good activity. Furthermore, analogues in which the phenyl ring was substituted with a basic heterocycle were significantly more soluble than meisoindigo while retaining acceptable antiproliferative profiles. The most promising analogue (E)-1-(2-(4-methylpiperazin-1-yl)ethyl)-[3,3′-biindolinylidene]-2,2′-dione (5-4) is more potent than meisoindigo across a panel of malignant cells, with at least 40 times greater solubility than meisoindigo, little or no tendency to aggregate in solution and capable of significantly extending the lifespans of animals with K562 induced xenografts. Mechanistically, it induced apoptotic cell death and disrupted the progression of K562 cells from the G1 to G2 phase. Taken together, our findings highlighted the feasibility of addressing the physicochemical deficits of the isoindigo scaffold by systematic modifications which was achieved without overt loss of growth inhibitory activity.
Co-reporter:Kheng-Lin Tan;Siang-Boon Koh;Rachel Pui-Lai Ee;Dr. Matiullah Khan; Mei-Lin Go
ChemMedChem 2012 Volume 7( Issue 9) pp:1567-1579
Publication Date(Web):
DOI:10.1002/cmdc.201200293
Abstract
Curcumin arrests the proliferation of acute promyelocytic leukemia (APL) cells by stabilizing the misfolded nuclear receptor co-repressor (N-CoR) protein, thereby sensitizing APL cells to apoptosis induced by the unfolded protein response. This phenomenon was attributed to inhibition of the proteasomal and protease-induced breakdown of misfolded N-CoR by curcumin. Curcumin is, however, a modest inhibitor and affected the viability of APL cells at micromolar concentrations. Modifying curcumin at its conjugated β-diketone linker and terminal phenyl rings yielded potent congeners with sub-micromolar growth inhibitory activities which selectively kill APL cells over non-APL leukemic and nonmalignant cells. Analogues with pronounced APL-selective anti-proliferative activities, as observed in representative dibenzylidenecyclohexanones and dibenzylidenecyclopentanones, strongly promoted the accumulation of misfolded and nonfunctional N-CoR at significantly lower concentrations than their growth inhibitory IC50 values. These compounds also inhibited the human 20S proteasome in an enzyme-based assay, thus providing convincing support for the prevailing hypothesis that impeding the degradation of N-CoR is a key mechanistic event contributing to APL cell death.
Co-reporter:Wee Kiang Yeo;Kheng Lin Tan;Siang Boon Koh;Dr. Matiullah Khan;Dr. Shahul Nilar; Mei Lin Go
ChemMedChem 2012 Volume 7( Issue 6) pp:977-982
Publication Date(Web):
DOI:10.1002/cmdc.201200106
Co-reporter:Thuy Nguyen, Yuji Sakasegawa, Katsumi Doh-ura, Mei-Lin Go
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 7) pp:2917-2929
Publication Date(Web):July 2011
DOI:10.1016/j.ejmech.2011.04.016
In this paper, we report the synthesis and cell-based anti-prion activity of quinacrine analogs derived by replacing the basic alkyl side chain of quinacrine with 4-(4-methylpiperazin-I-yl)phenyl, (1-benzylpiperidin-4-yl) and their structural variants. Several promising analogs were found that have a more favorable anti-prion profile than quinacrine in terms of potency and activity across different prion-infected murine cell models. They also exhibited greater binding affinities for a human prion protein fragment (hPrP121–231) than quinacrine, and had permeabilities on the PAMPA-BBB assay that fall within the range of CNS permeant candidates. When evaluated on bidirectional assays on a Pgp overexpressing cell line, one analog was less susceptible to Pgp efflux activity compared to quinacrine. Taken together, the results point to an important role for the substituted 9-amino side chain attached to the acridine, tetrahydroacridine and quinoline scaffolds. The nature of this side chain influenced cell-based potency, PAMPA permeability and binding affinity to hPrP121–231.Highlights► Functionalized quinacrine analogs with basic phenyl residues were investigated. ► Compounds were more potent and have broad ranging cell-based anti-prion activity. ► A promising analog cleared PrPSc in 3 cell models with nanomolar EC50. ► It binds to hPrP121–231, is CNS + (PAMPA-BBB), and is a weaker Pgp substrate. ► Modifications have resulted in more potent and drug-like analogs.
Co-reporter:Wei Zhang, Mei Lin Go
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 3) pp:1032-1035
Publication Date(Web):1 February 2011
DOI:10.1016/j.bmcl.2010.12.029
A series of methoxystilbenes (E and Z isomers) related to resveratrol were investigated for their effects on NQO1 induction in murine hepatoma cells and growth inhibitory effects on human cancer cell lines. Both activities were enhanced in compounds with methoxy groups on rings A and B of resveratrol but methoxylation of the di-meta (3,5) hydroxyl groups on ring A of resveratrol was found to be more critical for improving activity. Strikingly different structure–activity trends were observed, namely the association of E isomers with potent NQO1 induction activity and Z isomers with growth inhibitory properties. The introduction of ortho-methoxy groups on ring A greatly benefited NQO1 induction activity while meta/para methoxy groups on ring A were preferred for potent growth inhibitory effects. These results serve to highlight the contrasting effects on different activities brought about by methoxylation, which is widely employed as a structural modification approach to improve potency and bioavailability of resveratrol. It serves as a timely reminder that in the course of structural modification, a balance between optimizing desired outcomes against unwanted effects is necessary and the most potent analog need not always be the most desirable.Different structural features of resveratrol are required for induction of NQO1 and growth inhibition of human cancer cell lines.
Co-reporter:Dr. Hong May Sim;Ker Yun Loh;Wee Kiang Yeo;Dr. Chong Yew Lee; Mei Lin Go
ChemMedChem 2011 Volume 6( Issue 4) pp:713-724
Publication Date(Web):
DOI:10.1002/cmdc.201000520
Abstract
The ability of aurones to modulate the efflux activities of ABCG2 and ABCB1 was investigated by quantifying their effects on the accumulation of pheophorbide A (PhA) in ABCG2-overexpressing MDA-MB-231/R cells and calcein AM in ABCB1-overexpressing MDCKII/MDR1 cells. Key structural features for interactions at both ABCG2 and ABCB1 are a methoxylated ring A, an intact exocyclic double bond, and the location of the carbonyl bond on ring C. Modifications on rings B and C were less critical and served primarily to moderate activity and selectivity for one or both transporters. These SAR trends were quantified by Free–Wilson analyses and are reflected in a pharmacophore model for PhA accumulation. Several compounds were found to be equipotent with fumitremorgin C (FTC) in promoting PhA accumulation, and they also demonstrated strong affinities for ABCB1. These compounds were disubstituted on ring B with methoxy or a combination of methoxy and hydroxy groups. Taken together, our findings highlight the versatility of the aurone template as a lead scaffold for the design of dual-targeting ABCG2 and ABCB1 modulators.
Co-reporter:Mei-Lin Go ; Jo Lene Leow ; Suresh Kumar Gorla ; Andreas Peter Schüller ; Mei Wang ;Patrick J. Casey
Journal of Medicinal Chemistry 2010 Volume 53(Issue 19) pp:6838-6850
Publication Date(Web):September 1, 2010
DOI:10.1021/jm1002843
The enzyme isoprenylcysteine carboxyl methyltransferase (Icmt) plays an important role in the post-translational modification of proteins that are involved in the regulation of cell growth. The indole acetamide cysmethynil is by far the most potent and widely investigated Icmt inhibitor, but it has modest antiproliferative activity and may have pharmacokinetic limitations due to its lipophilic character. We report here that cysmethynil can be structurally modified to give analogues that are as potent in inhibiting Icmt but with significantly greater antiproliferative activity. Key modifications were the replacement of the acetamide side chain by tertiary amino groups, the n-octyl side chain by isoprenyl and the 5-m-tolyl ring by fluorine. Moreover, these analogues have lower lipophilicities that could lead to improved pharmacokinetic profiles.
Co-reporter:Chong-Yew Lee, Eng-Hui Chew, Mei-Lin Go
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 7) pp:2957-2971
Publication Date(Web):July 2010
DOI:10.1016/j.ejmech.2010.03.023
The chemopreventive potential of functionalized aurones and related compounds as inducers of NAD(P)H:quinone oxidoreductase 1 (NQO1, EC 1.6.99.2) are described. Several 4,6-dimethoxy and 5-hydroxyaurones induced NQO1 activity of Hepa1c1c7 cells by 2-fold at submicromolar concentrations, making these the most potent inducers to be identified from this class. Mechanistically, induction of NQO1 was mediated by the activation of AhR/XRE and Nrf2/ARE pathways, indicating that aurones may be mixed activators of NQO1 induction or agents capable of exploiting the proposed cross-talk between the AhR and Nrf2 gene batteries. QSAR analysis by partial least squares projection to latent structures (PLS) identified size parameters, in particular those associated with non-polar surface areas, as an important determinant of induction activity. These were largely determined by the substitution on rings A and B. A stereoelectronic role for the exocyclic double bond as reflected in the ELUMO term was also identified. The electrophilicity of the double bond or its effect on the conformation of the target compound are possible key features for induction activity.The design, synthesis and biological evaluation of a series of functionalized aurones as potential chemopreventive agents is described. Several active compounds were identified.
Co-reporter:Xi Kai Wee, Wee Kiang Yeo, Bing Zhang, Vincent B.C. Tan, Kian Meng Lim, Tong Earn Tay, Mei-Lin Go
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 21) pp:7562-7571
Publication Date(Web):1 November 2009
DOI:10.1016/j.bmc.2009.09.008
A series of functionalized isoindigos structurally related to meisoindigo (1-methylisoindigo), a therapeutic agent used for the treatment of a form of leukemia, were synthesized and evaluated for antiproliferative activities on a panel of human cancer cells. Two promising compounds (1-phenpropylisoindigo and 1-(p-methoxy-phenethyl)-isoindigo) that were more potent than meisoindigo and comparable to 6-bromoindirubin-3′-oxime on leukemic K562 and liver HuH7 cells were identified. Structure–activity relationships showed the importance of keeping one of the lactam NH in an unsubstituted state. Substitution of the other lactam NH with aryl or arylalkyl side chains retained or improved activity in most instances. An intact exocyclic double bond was also essential, possibly to maintain planarity and rigidity of the isoindigo scaffold. None of the compounds were found to inhibit CDK2 in an in vitro assay, in spite of reports linking the antiproliferative activities of meisoindigo and other isoindigos to CDK2 inhibition. Hence, these functionalized isoindigos disrupted cell growth and proliferation by other mechanistic pathways that did not involve CDK2 inhibition.Lead modification of meisoindigo at position 1 of the isoindigo scaffold yield more potent analogs of meisoindigo that have low micromolar antiproliferative activities against K562 and HL60 leukemic cell lines.
Co-reporter:Wei Zhang, Mei-Lin Go
Bioorganic & Medicinal Chemistry 2009 17(5) pp: 2077-2090
Publication Date(Web):
DOI:10.1016/j.bmc.2008.12.052
Co-reporter:X.L. Liu, Y.J. Xu, M.L. Go
European Journal of Medicinal Chemistry 2008 Volume 43(Issue 8) pp:1681-1687
Publication Date(Web):August 2008
DOI:10.1016/j.ejmech.2007.10.007
A library of chalcones with basic functionalities were evaluated for antibacterial activity against drug sensitive strains of Staphylococcus aureus and Escherichia coli. The most active compounds were 2-52 and 2-57 (MIC 6.3 μM S. aureus). These compounds had no activity against E. coli (MIC > 100 μM). Both compounds were characterized by a ring A that was substituted with 2-hydroxy-4,6-dimethoxy-3-(1-methylpiperidin-4-yl) groups. The phenolic OH and 1-methylpiperidinyl groups were required for activity but the phenolic OH may play a more critical role. While the compounds were comparable to licochalcone A in terms of antibacterial activity, they caused less hemolysis of sheep erythrocytes at high concentrations (100 μM). It was noted that the structural requirements for limiting hemolytic activity were less stringent than those required for antibacterial activity. The present findings suggest that the chalcone framework is an attractive template for optimization to achieve better potency, lower toxicity and a wider spectrum of antibacterial activity.Chalcones with phenolic OH and basic heterocyclic rings had antibacterial activities comparable to licochalcone A (MIC 6.3 μM S. aureus) and protected erythrocytes from hemolysis at 100 μM.
Co-reporter:Hanh Thuy Nguyen Thi, Chong-Yew Lee, Kenta Teruya, Wei-Yi Ong, Katsumi Doh-ura, Mei-Lin Go
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 14) pp:6737-6746
Publication Date(Web):15 July 2008
DOI:10.1016/j.bmc.2008.05.060
A library of functionalized 6-chloro-2-methoxy-(N9-substituted)acridin-9-amines structurally related to quinacrine were synthesized and evaluated for antiprion activity on four different cell models persistently infected with scrapie prion strains (ScN2a, N167, Ch2) or a human disease prion strain (F3). Most of the compounds were distinguished by the side chain attached to 9-amino of the acridine ring. These were dialkylaminoalkyl and phenyl with basic groups on the phenyl ring. The most promising compound was 6-chloro-2-methoxy-N-(4-(4-methylpiperazin-1-yl)phenyl)acridin-9-amine (15) which had submicromolar EC50 values (0.1–0.7 μM) on all cell models, was able to clear PrPSc at non-toxic concentrations of 1.2–2.5 μM, and was more active than quinacrine in terms of EC50 values. Other promising compounds were 14 (a regioisomer of 15) and 17 which had a 1-benzylpiperidin-4-yl substituent attached to the 9-amino function. Activity was strongly dependent on the presence of a substituted acridine ring, which in this library comprised 6-chloro-2-methoxy substituents on the acridine ring. The side chains of 14, 15, and 17 have not been previously associated with antiprion activity and are interesting leads for further optimization of antiprion activity.Structural modification of the 9-substituted amino side chain of quinacrine resulted in compounds with better potencies and selectivities than quinacrine.
Co-reporter:Jo-Lene Leow, Rudi Baron, Patrick J. Casey, Mei-Lin Go
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 4) pp:1025-1032
Publication Date(Web):15 February 2007
DOI:10.1016/j.bmcl.2006.11.030
A QSAR is developed for the isoprenylcysteine carboxyl methyltransferase (ICMT) inhibitory activities of a series of indoloacetamides (n = 72) that are structurally related to cysmethynil, a selective ICMT inhibitor. Multivariate analytical tools (principal component analysis (PCA) and projection to latent structures (PLS)), multi-linear regression (MLR) and comparative molecular field analysis (CoMFA) are used to develop a suitably predictive model for the purpose of optimizing and identifying members with more potent inhibitory activity. The resulting model shows that good activity is determined largely by the characteristics of the substituent attached to the indole nitrogen, which should be a lipophilic residue with fairly wide dimensions. In contrast, the substituted phenyl ring attached to the indole ring must be of limited dimensions and lipophilicity.QSAR of the ICMT inhibitory activities of indoloacetamides showed contrasting steric and lipophilic preferences for the N-substituent and the substituted phenyl ring.
Co-reporter:Mei Liu, Prapon Wilairat, Simon L Croft, Agnes Lay-Choo Tan, Mei-Lin Go
Bioorganic & Medicinal Chemistry 2004 Volume 12(Issue 6) pp:1569
Publication Date(Web):15 March 2004
DOI:10.1016/j.bmc.2004.01.023
Co-reporter:Mei Liu, Prapon Wilairat, Simon L Croft, Agnes Lay-Choo Tan, Mei-Lin Go
Bioorganic & Medicinal Chemistry 2003 Volume 11(Issue 13) pp:2729-2738
Publication Date(Web):3 July 2003
DOI:10.1016/S0968-0896(03)00233-5
A series of oxygenated chalcones which have been evaluated earlier for antimalarial activity (Plasmodium falciparum K1) were tested for antileishmanial activity against Leishmania donovani amastigotes. A comparison of structure–activity relationships reveal that different physicochemical and structural requirements exist for these two activities. Antileishmanial activity is associated with less lipophilic chalcones, in particular those with 4′-hydroxyl-substituted B rings and hetero/polyaromatic A rings. In contrast, chalcones with good antimalarial activity have alkoxylated B rings and electron-deficient A rings. Visualization of the steric and electrostatic fields generated from comparative molecular field analysis (CoMFA) indicate that the ring A of chalcones make a more significant contribution to antileishmanial activity while both rings A and B are important for antimalarial activity. Despite different requirements, two alkoxylated chalcones (8, 19) were identified which combined good antimalarial and antileishmanial activities.Graphic
Co-reporter:Xiang Wu, Prapon Wilairat, Mei-Lin Go
Bioorganic & Medicinal Chemistry Letters 2002 Volume 12(Issue 17) pp:2299-2302
Publication Date(Web):2 September 2002
DOI:10.1016/S0960-894X(02)00430-4
A series of ferrocenyl chalcones were synthesized and evaluated for in vitro antimalarial activity against a chloroquine-resistant strain of Plasmodium falciparum. The most active compounds were 1-(3-pyridyl)-3-ferrocenyl-2-propen-1-one (6) and 1-ferrocenyl-3-(4-nitrophenyl)-2-propen-1-one (28) with IC50 of 4.5 and 5.1 μM, respectively. Differences in activity were not readily explained by the size and lipophilicity characteristics of these compounds.Graphic





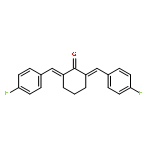
![8-Bromo-2,3,4,5-tetrahydro-1H-pyrido[4,3-b]indole](http://img.cochemist.com/ccimg/497300/497261-38-8.png)
![8-Bromo-2,3,4,5-tetrahydro-1H-pyrido[4,3-b]indole](http://img.cochemist.com/ccimg/497300/497261-38-8_b.png)
![5-[5-(2,6-dichloro-phenylmethanesulfonyl)-2-oxo-1,2-dihydro-indol-(3 Z)-ylidenemethyl]-2,4-dimethyl-1H-pyrrole-3-carboxylic Acid](http://img.cochemist.com/ccimg/477600/477574-82-6.png)
![5-[5-(2,6-dichloro-phenylmethanesulfonyl)-2-oxo-1,2-dihydro-indol-(3 Z)-ylidenemethyl]-2,4-dimethyl-1H-pyrrole-3-carboxylic Acid](http://img.cochemist.com/ccimg/477600/477574-82-6_b.png)
![2H-Indol-2-one, 5-[[(2,6-dichlorophenyl)methyl]sulfonyl]-1,3-dihydro-](http://img.cochemist.com/ccimg/477600/477573-39-0.png)
![2H-Indol-2-one, 5-[[(2,6-dichlorophenyl)methyl]sulfonyl]-1,3-dihydro-](http://img.cochemist.com/ccimg/477600/477573-39-0_b.png)


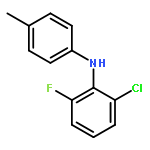
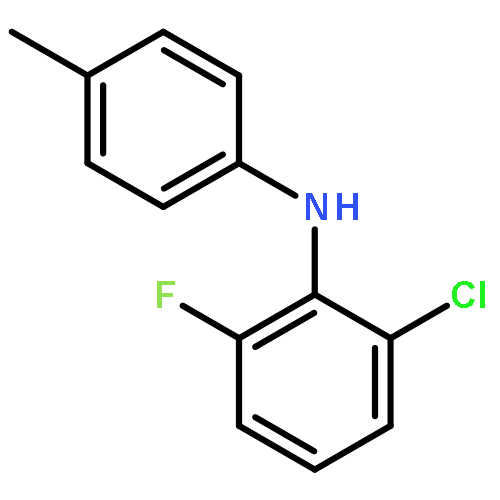
![Benzeneacetic acid, 2-[(2,6-dibromophenyl)amino]-](http://img.cochemist.com/ccimg/320800/320777-70-6.png)
![Benzeneacetic acid, 2-[(2,6-dibromophenyl)amino]-](http://img.cochemist.com/ccimg/320800/320777-70-6_b.png)
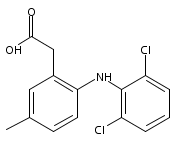
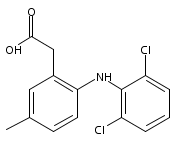




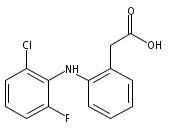
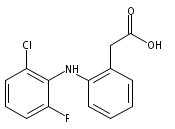
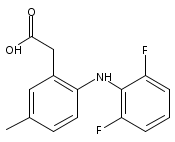
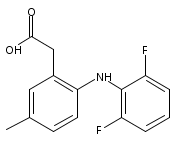
![Benzeneacetic acid, 2-[(2,6-difluorophenyl)amino]-](http://img.cochemist.com/ccimg/90300/90233-40-2.png)
![Benzeneacetic acid, 2-[(2,6-difluorophenyl)amino]-](http://img.cochemist.com/ccimg/90300/90233-40-2_b.png)


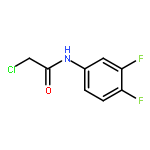
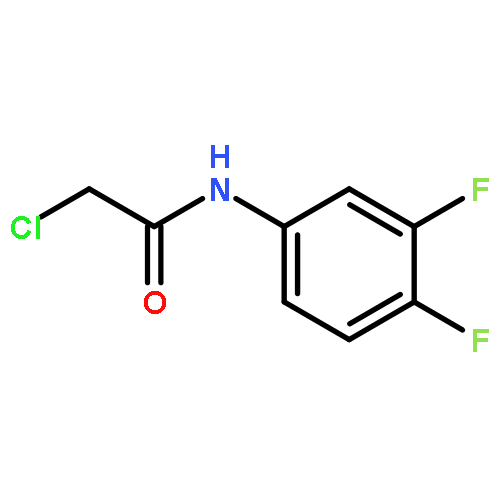
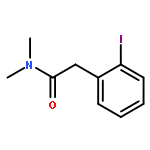
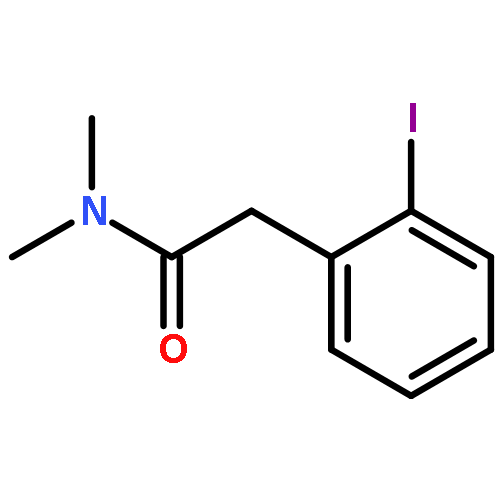
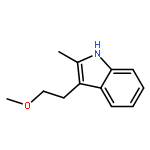
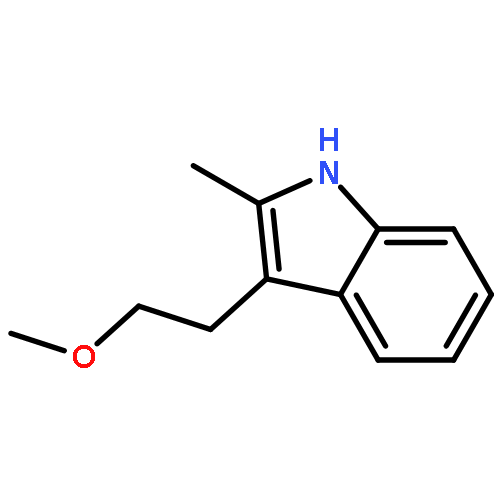
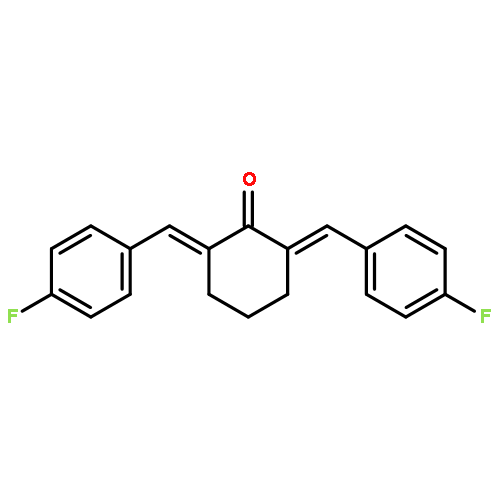
![Cyclohexanone, 2,6-bis[(3-hydroxyphenyl)methylene]-](http://img.cochemist.com/ccimg/66600/66509-01-1.png)
![Cyclohexanone, 2,6-bis[(3-hydroxyphenyl)methylene]-](http://img.cochemist.com/ccimg/66600/66509-01-1_b.png)
![2H-Pyrido[4,3-b]indole-2-carboxylic acid, 8-bromo-1,3,4,5-tetrahydro-,ethyl ester](http://img.cochemist.com/ccimg/63300/63277-57-6.png)
![2H-Pyrido[4,3-b]indole-2-carboxylic acid, 8-bromo-1,3,4,5-tetrahydro-,ethyl ester](http://img.cochemist.com/ccimg/63300/63277-57-6_b.png)

![Ethyl 8-fluoro-3,4-dihydro-1H-pyrido[4,3-b]indole-2(5H)-carboxylate](http://img.cochemist.com/ccimg/58100/58038-66-7.png)
![Ethyl 8-fluoro-3,4-dihydro-1H-pyrido[4,3-b]indole-2(5H)-carboxylate](http://img.cochemist.com/ccimg/58100/58038-66-7_b.png)
![METHYL 3-[(METHYLSULFONYL)AMINO]BENZOATE](http://img.cochemist.com/ccimg/32100/32087-05-1.png)
![METHYL 3-[(METHYLSULFONYL)AMINO]BENZOATE](http://img.cochemist.com/ccimg/32100/32087-05-1_b.png)


![2-chloro-3-[(2-methylpropyl)amino]naphthalene-1,4-dione](http://img.cochemist.com/ccimg/22300/22272-31-7.png)
![2-chloro-3-[(2-methylpropyl)amino]naphthalene-1,4-dione](http://img.cochemist.com/ccimg/22300/22272-31-7_b.png)
![1,4-Naphthalenedione, 2-chloro-3-[(2-methoxyethyl)amino]-](/data/chemimg/14900/22272-22-6.png)
![1,4-Naphthalenedione, 2-chloro-3-[(2-methoxyethyl)amino]-](/data/chemimg/14900/22272-22-6_b.png)
![1,4-Naphthalenedione,2-chloro-3-[(1-methylethyl)amino]-](http://img.cochemist.com/ccimg/15500/15455-20-6.png)
![1,4-Naphthalenedione,2-chloro-3-[(1-methylethyl)amino]-](http://img.cochemist.com/ccimg/15500/15455-20-6_b.png)




![8-bromo-2,3,4,5-tetrahydro-2-methyl-1H-Pyrido[4,3-b]indole](http://img.cochemist.com/ccimg/5100/5055-01-6.png)
![8-bromo-2,3,4,5-tetrahydro-2-methyl-1H-Pyrido[4,3-b]indole](http://img.cochemist.com/ccimg/5100/5055-01-6_b.png)