Co-reporter:Guojun Pan, Xuehui Li, Long Zhao, Meng Wu, Chao Su, Xuzhe Li, Yongmin Zhang, Peng Yu, Yuou Teng, Kui Lu
European Journal of Medicinal Chemistry 2017 Volume 138(Volume 138) pp:
Publication Date(Web):29 September 2017
DOI:10.1016/j.ejmech.2017.06.054
•Anastatins A and B as well as their analogs were synthesized.•Aurone derivatives showed better antioxidant activity than flavone counterparts.•Compound 24c was identified as the most potent antioxidant activity.•Compound 24c decreased apoptosis in H2O2-treated PC12 cells.Two novel flavonoids (±)-Anastatins A and B as well as 14 analogs, which containing a benzofuran moiety, were synthesized by using halogenation, Suzuki coupling reaction and an oxidation/Oxa-Michael reaction cascade as the key steps. The structures of the new flavonoids were confirmed by 1H NMR, 13C NMR and HRMS. The antioxidant activities of them as well as the key intermediates were evaluated by ferric reducing antioxidant power (FRAP) assay and the active compounds were evaluated in the PC12 cell model of hydrogen peroxide (H2O2)-induced oxidative damage. SAR studies suggested that, for in vitro antioxidant activity, aurone derivatives showed better bioactivity than flavone counterparts. However, cyclization to benzofuran and connecting the two conjugated parts as a whole conjugated system by a double bond diminished the in vitro antioxidant activity. Among them, the most potent compound 24c was significantly decreased H2O2-caused cell injury. The apoptotic rate (Annexin V+) of H2O2-damaged PC12 cells was 60.7% while that of the compound 24c-treated cells decreased to 5.9% and 4.1% at 10 μM and 100 μM respectively.Two novel flavonoids Anastatins A and B as well as their analogs were synthesized. The antioxidant activities of these flavonoids and the key intermediates were evaluated by ferric reducing antioxidant power (FRAP) assay and PC12 cell-based antioxidant assay.Download high-res image (167KB)Download full-size image
Co-reporter:Hao-Meng Wang, Li Zhang, Jiang Liu, Zhao-Liang Yang, Hong-Ye Zhao, Yao Yang, Di Shen, Kui Lu, Zhen-Chuan Fan, Qing-Wei Yao, Yong-Min Zhang, Yu-Ou Teng, Yu Peng
European Journal of Medicinal Chemistry 2015 Volume 92() pp:439-448
Publication Date(Web):6 March 2015
DOI:10.1016/j.ejmech.2015.01.007
•Four natural chalcones bearing prenyl or geranyl groups were synthesized.•Eleven 3′ or/and 5′-prenylated/geranylated chalcones analogs were prepared.•5′-prenylation/geranylation of the chalcones enhanced the cytotoxicy 7–10 folds.•These chalcone derivatives inhibited K562' proliferation by inducing apoptosis.Four natural chalcones bearing prenyl or geranyl groups, i.e., bavachalcone (1a), xanthoangelol (1b), isobavachalcone (1c), and isoxanthoangelol (1d) were synthesized by using a regio-selective iodination and the Suzuki coupling reaction as key steps. The first total synthesis of isoxanthoangelol (1d) was achieved in 36% overall yield. A series of diprenylated and digeranylated chalcone analogs were also synthesized by alkylation, regio-selective iodination, aldol condensation, Suzuki coupling and [1,3]-sigmatropic rearrangement. The structures of the 11 new derivatives were confirmed by 1H NMR, 13C NMR and HRMS. The anticancer activity of these new chalcone derivatives against human tumor cell line K562 were evaluated by MTT assay in vitro. SAR studies suggested that the 5′-prenylation/geranylation of the chalcones significantly enhance their cytotoxic activity. Among them, Bavachalcone (1a) displayed the most potent cytotoxic activity against K562 with IC50 value of 2.7 μM. The morphology changes and annexin-V/PI staining studies suggested that those chalcone derivatives inhibited the proliferation of K562 cells by inducing apoptosis.A series of prenyl and geranyl substituted chalcone natural products and their derivatives were synthesized. The anti-cancer activity evaluation results revealed these chalcone derivatives inhibit K562 proliferation by inducing apoptosis.
Co-reporter:Kailin Han, Yao Zhou, Fengxi Liu, Qiannan Guo, Pengfei Wang, Yao Yang, Binbin Song, Wei Liu, Qingwei Yao, Yuou Teng, Peng Yu
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 2) pp:591-594
Publication Date(Web):15 January 2014
DOI:10.1016/j.bmcl.2013.12.001
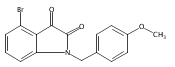
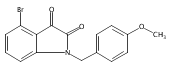
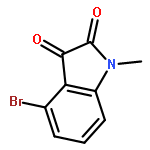
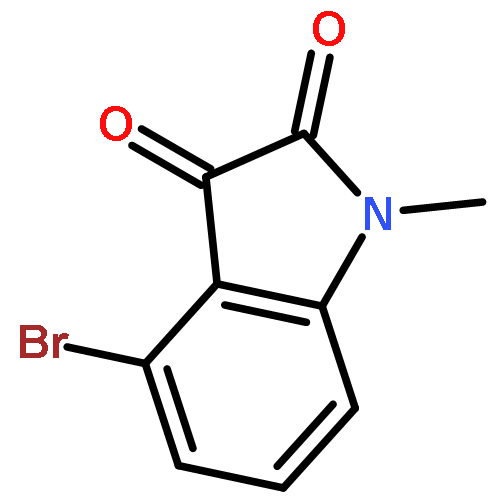
Forty four di- or trisubstituted novel isatin derivatives were designed and synthesized in 5–6 steps in 25–45% overall yields. Their structures were confirmed by 1H NMR and 13C NMR as well as LC–MS. The anticancer activity of these new isatin derivatives against three human tumor cell lines, K562, HepG2 and HT-29, were evaluated by MTT assay in vitro. SAR studies suggested that the combination of 1-benzyl and 5-[trans-2-(methoxycarbonyl)ethen-1-yl] substitution greatly enhance their cytotoxic activity, whereas an intact carbonyl functionality on C-3 as present in the parent ring is required to such a potency. This study leads to the identification of two highly active molecules, compounds 2h (IC50 = 3 nM) and 2k (IC50 = 6 nM), against human leukemia K562 cells.

![2-Propen-1-one,3-(4-hydroxyphenyl)-1-[2,4,6-trihydroxy-3-(3-methyl-2-buten-1-yl)phenyl]-,(2E)-](http://img.cochemist.com/ccimg/115100/115063-39-3.png)
![2-Propen-1-one,3-(4-hydroxyphenyl)-1-[2,4,6-trihydroxy-3-(3-methyl-2-buten-1-yl)phenyl]-,(2E)-](http://img.cochemist.com/ccimg/115100/115063-39-3_b.png)
![(2E)-1-{3-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-2,4-dihydroxyphenyl}-3-(4-hydroxyphenyl)prop-2-en-1-one](http://img.cochemist.com/ccimg/63000/62949-76-2.png)
![(2E)-1-{3-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-2,4-dihydroxyphenyl}-3-(4-hydroxyphenyl)prop-2-en-1-one](http://img.cochemist.com/ccimg/63000/62949-76-2_b.png)
![ETHANONE, 1-[2,6-DIHYDROXY-4-(METHOXYMETHOXY)PHENYL]-](http://img.cochemist.com/ccimg/257700/257621-57-1.png)
![ETHANONE, 1-[2,6-DIHYDROXY-4-(METHOXYMETHOXY)PHENYL]-](http://img.cochemist.com/ccimg/257700/257621-57-1_b.png)








![Ethanone, 1-[2-hydroxy-4,6-bis(methoxymethoxy)phenyl]-](http://img.cochemist.com/ccimg/65500/65490-09-7.png)
![Ethanone, 1-[2-hydroxy-4,6-bis(methoxymethoxy)phenyl]-](http://img.cochemist.com/ccimg/65500/65490-09-7_b.png)






















![1-[3-(2-PYRIDINYL)-1,2,4-OXADIAZOL-5-YL]ETHANAMINE](http://img.cochemist.com/ccimg/391700/391613-98-2.png)
![1-[3-(2-PYRIDINYL)-1,2,4-OXADIAZOL-5-YL]ETHANAMINE](http://img.cochemist.com/ccimg/391700/391613-98-2_b.png)












![2,4-Imidazolidinedione, 5-[(4-fluorophenyl)methylene]-, (Z)-](http://img.cochemist.com/ccimg/140900/140894-74-2.png)
![2,4-Imidazolidinedione, 5-[(4-fluorophenyl)methylene]-, (Z)-](http://img.cochemist.com/ccimg/140900/140894-74-2_b.png)


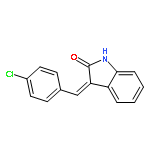
![2H-Indol-2-one, 3-[(3,4-dichlorophenyl)methylene]-1,3-dihydro-](http://img.cochemist.com/ccimg/114800/114727-43-4.png)
![2H-Indol-2-one, 3-[(3,4-dichlorophenyl)methylene]-1,3-dihydro-](http://img.cochemist.com/ccimg/114800/114727-43-4_b.png)
![L-Proline, N-[(1,1-dimethylethoxy)carbonyl]-L-tryptophyl-, methyl ester](http://img.cochemist.com/ccimg/85500/85416-63-3.png)
![L-Proline, N-[(1,1-dimethylethoxy)carbonyl]-L-tryptophyl-, methyl ester](http://img.cochemist.com/ccimg/85500/85416-63-3_b.png)


![(z)-7-[(1r,2r,3r)-3-hydroxy-2-[(e,3r)-3-hydroxy-4-phenoxybut-1-enyl]-5-oxocyclopentyl]-n-methylsulfonylhept-5-enamide](http://img.cochemist.com/ccimg/60400/60325-46-4.png)
![(z)-7-[(1r,2r,3r)-3-hydroxy-2-[(e,3r)-3-hydroxy-4-phenoxybut-1-enyl]-5-oxocyclopentyl]-n-methylsulfonylhept-5-enamide](http://img.cochemist.com/ccimg/60400/60325-46-4_b.png)


![3-[(4-METHOXYPHENYL)METHYLIDENE]-1H-INDOL-2-ONE](http://img.cochemist.com/ccimg/55200/55160-02-6.png)
![3-[(4-METHOXYPHENYL)METHYLIDENE]-1H-INDOL-2-ONE](http://img.cochemist.com/ccimg/55200/55160-02-6_b.png)





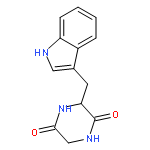
![(3S,8aS)-3-(1H-indol-3-ylmethyl)hexahydropyrrolo[1,2-a]pyrazine-1,4-dione](http://img.cochemist.com/ccimg/38200/38136-70-8.png)
![(3S,8aS)-3-(1H-indol-3-ylmethyl)hexahydropyrrolo[1,2-a]pyrazine-1,4-dione](http://img.cochemist.com/ccimg/38200/38136-70-8_b.png)
![3-[[4-(TRIFLUOROMETHYL)PHENYL]METHYLIDENE]-1H-INDOL-2-ONE](http://img.cochemist.com/ccimg/35400/35315-56-1.png)
![3-[[4-(TRIFLUOROMETHYL)PHENYL]METHYLIDENE]-1H-INDOL-2-ONE](http://img.cochemist.com/ccimg/35400/35315-56-1_b.png)