Co-reporter:Xinde Chen; Kai Wang; Wenwei Xu; Quanxin Ma; Minli Chen; Lili Du; Mingguang Mo; Yiping Wang
Journal of Medicinal Chemistry 2016 Volume 59(Issue 6) pp:2674-2687
Publication Date(Web):February 29, 2016
DOI:10.1021/acs.jmedchem.5b01930
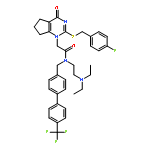
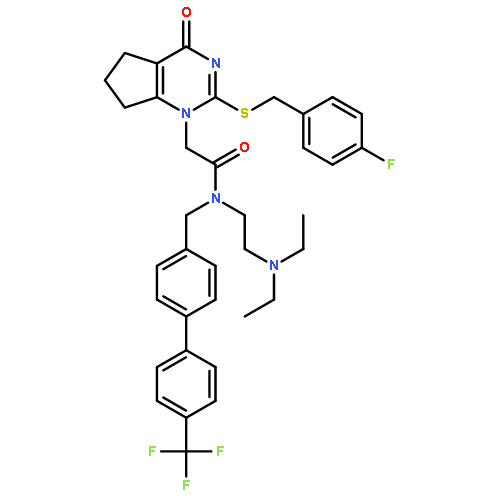
Lipoprotein-associated phospholipase A2 (Lp-PLA2) is considered to be a promising therapeutic target for several inflammation-associated diseases. Herein, we describe the discovery of a series of pyrimidone derivatives as Lp-PLA2 inhibitors. Systematic structural modifications led to the identification of several pyrimidone compounds with promising in vitro inhibitory potency and pharmacokinetic properties. Compound 14c, selected for in vivo evaluation, demonstrated decent pharmacokinetic profiles and robust inhibitory potency against Lp-PLA2 in Sprague–Dawley (SD) rats. Furthermore, 14c significantly inhibited retinal thickening in STZ-induced diabetic SD rats as a model of diabetic macular edema (DME) after oral dosing for 4 weeks. Taken together, these results suggested that 14c can serve as a valuable lead in the search for new Lp-PLA2 inhibitors for prevention and/or treatment of DME.
Co-reporter:Xinde Chen; Wenwei Xu; Kai Wang; Mingguang Mo; Wei Zhang; Lili Du; Xiaojing Yuan; Yechun Xu; Yiping Wang
Journal of Medicinal Chemistry 2015 Volume 58(Issue 21) pp:8529-8541
Publication Date(Web):October 19, 2015
DOI:10.1021/acs.jmedchem.5b01024
Inhibition of lipoprotein-associated phospholipase A2 (Lp-PLA2) has been suggested to be a promising therapeutic strategy for several inflammation-associated diseases, including atherosclerosis, Alzheimer’s disease, and diabetic macular edema. Herein, we report the discovery of a novel series of Lp-PLA2 inhibitors constructed on an imidazo[1,2-a]pyrimidine scaffold through a conformational restriction strategy. Structure–activity relationship (SAR) analysis resulted in the identification of several compounds with high potency in vitro and good metabolic stability in liver S9 fractions. Compounds 7c and 14b selected for further exploration in vivo demonstrated excellent pharmacokinetic profiles and exhibited significant inhibitory efficacy in SD rats upon oral dosing.
Co-reporter:Hongliang Duan; Mengmeng Ning; Qingan Zou; Yangliang Ye; Ying Feng; Lina Zhang; Ying Leng
Journal of Medicinal Chemistry 2015 Volume 58(Issue 8) pp:3315-3328
Publication Date(Web):February 24, 2015
DOI:10.1021/jm500829b
Activation of TGR5 stimulates intestinal glucagon-like peptide-1 (GLP-1) release, but activation of the receptors in gallbladder and heart has been shown to cause severe on-target side effects. A series of low-absorbed TGR5 agonists was prepared by modifying compound 2 with polar functional groups to limit systemic exposure and specifically activate TGR5 in the intestine. Compound 15c, with a molecular weight of 1401, a PSA value of 223 Å2, and low permeability on Caco-2 cells, exhibited satisfactory potency both in vitro and in vivo. Low levels of 15c were detected in blood, bile, and gallbladder tissue, and gallbladder-related side effects were substantially decreased compared to the absorbed small-molecule TGR5 agonist 2.
Co-reporter:Yang Liu, Lin Guo, Hongliang Duan, Liming Zhang, Neng Jiang, Xuechu Zhen and Jianhua Shen
RSC Advances 2015 vol. 5(Issue 51) pp:40964-40977
Publication Date(Web):30 Apr 2015
DOI:10.1039/C5RA04714E
Regulation of glycine transporter 1 (GlyT1) activity is a currently investigated strategy in drug discovery for schizophrenia. This study developed a series of new 4-benzoylpiperidine derivatives as GlyT1 inhibitors by bioisosteric replacement and mimicking of the pyridine ring of RG1678. Among the 4-benzoylpiperidine derivatives, 23q showed an IC50 of 30 nM. Preliminary optimization of the blood–brain barrier penetration led to the discovery of 3-(piperidin-4-yl)benzo[d]isoxazole derivatives. Both series showed good selectivity over GlyT2, D1, D2, D3, 5-HT1A and 5-HT2A receptors. Moreover, behavioral testing showed 23q (40 mg kg−1, intragastric) can inhibit the hyperlocomotion induced by acute treatment of phencyclidine, and improve the impaired negative and cognitive symptoms in chronic phencyclidine-induced C57BL/6J mice. An interesting finding showed that 3-(piperidin-4-yl)benzo[d]isoxazole was a privileged scaffold of atypical antipsychotic agents but exhibited high selectivity and potency as a GlyT1 inhibitor.
Co-reporter:Qingan Zou, Hongliang Duan, Mengmeng Ning, Jia Liu, Ying Feng, Liming Zhang, Junjie Zhu, Ying Leng, Jianhua Shen
European Journal of Medicinal Chemistry 2014 Volume 82() pp:1-15
Publication Date(Web):23 July 2014
DOI:10.1016/j.ejmech.2014.05.031
•4-Benzofuranyloxynicotinamide derivatives were novel potent TGR5 agonists.•The hTGR5 and mTGR5 EC50 for 9r was 0.28 nM and 0.92 nM, respectively.•19 displayed low permeability (Papp = 0.21 × 10−6 cm/s) and moderate potency.•Both 9r and 19 could effectively lower the blood glucose in ICR mice.•19 still increased the gallbladder volume of mice.A series of 4-benzofuranyloxynicotinamide derivatives were identified to be novel, potent, and orally available TGR5 agonists. Among them, compound 9r had the highest potency in vitro (hTGR5 EC50 = 0.28 nM, mTGR5 EC50 = 0.92 nM). Further in vivo studies disclosed that 9r could effectively lower the blood glucose, but meantime caused an increase in the gallbladder volume of mice. Subsequent research toward eliminating the gallbladder toxicity resulted in compound 19 with low permeability. Although the EC50 of mTGR5 of 19 was larger one order than that of 9r, it still had good glucose-lowing activity. Nevertheless, 19 also caused the adverse effects to the gallbladder. The drug levels detection disclosed that the concentration of 19 was only lower than that of 9r in plasma but was higher in bile and gallbladder tissue. This result indicated that low exposure in plasma could not guarantee low exposure in bile and gallbladder tissue, and thus resulting in the gallbladder toxicity of 19.
Co-reporter:Junjie Zhu, Mengmeng Ning, Cen Guo, Lina Zhang, Guoyu Pan, Ying Leng, Jianhua Shen
European Journal of Medicinal Chemistry 2013 Volume 69() pp:55-68
Publication Date(Web):November 2013
DOI:10.1016/j.ejmech.2013.07.050
•A new class of TGR5 agonists based on a 4-phenyl pyridine scaffold was designed and synthesized.•Structure–activity relationships of this series were summarized.•Several compounds, like 15a, 18b and 18c, showed excellent potency in vitro.•15a displayed an unfavorable PK profile and was found to be ineffective during an OGTT in ICR mice at a dose of 50 mg/kg.TGR5, a GPCR, is involved in energy and glucose homeostasis, and as such, is a target for the treatment of diabetes, obesity and other metabolic syndromes. A new class of TGR5 agonists based on a 4-phenyl pyridine scaffold was designed, synthesized and evaluated in vitro and in vivo. Extensive structure–activity relationship studies are reported herein. The most potent compounds 15a, 18b and 18c showed comparable activity with the lead compound 2. 15a had the best potency in vitro but displayed an unfavorable pharmacokinetic profile and was found to be ineffective during an oral glucose tolerance test in imprinting control region mice at a dose of 50 mg/kg.A novel class of amides, sulfonamides and α-amino nitriles as potent TGR5 agonists was evaluated in vitro and in vivo.
Co-reporter:Kai Wang, Wenwei Xu, Wei Zhang, Mingguang Mo, Yiping Wang, Jianhua Shen
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 10) pp:2897-2901
Publication Date(Web):15 May 2013
DOI:10.1016/j.bmcl.2013.03.062
This Letter reports our efforts towards the optimization of our previously identified series of imidazole and triazole derivatives that lead to the discovery of a series of orally active Lp-PLA2 inhibitors in C57 mice. These inhibitors are characterized by the presence of a diamine side chain in the molecules, such as 2c, 2f, and 4a. The introduction of the terminal-end amine succeeded in maintaining the in vitro activities at sub-nanomolar levels. The vivo activities could be greatly affected by variations in the two amines via modulating the metabolic stability and lipophilicity of the compounds.Relative serum Lp-PLA2 Activition in C57 mice after a single dose (50mg/kg, po, n = 5).
Co-reporter:Kai Wang, Wenwei Xu, Yang Liu, Wei Zhang, Wenyi Wang, Jianhua Shen, Yiping Wang
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 5) pp:1187-1192
Publication Date(Web):1 March 2013
DOI:10.1016/j.bmcl.2013.01.029
New Lp-PLA2 inhibitors were synthesized by the bioisosteric replacement of the amide group of Darapladib with an imidazole or a triazole. Unfortunately, the inhibitory activities of these derivatives were lower than that of Darapladib. But interestingly, a series of quaternary ammonium salts that were isolated as by-products during this synthetic work were found with high potency. Of these by-products, compound 22c showed a similar profile to Darapladib both in vitro and in vivo.
Co-reporter:Junjie Zhu;Yangliang Ye;Mengmeng Ning;Attila Mándi;Ying Feng;Qingan Zou;Dr. Tibor Kurtán;Dr. Ying Leng;Dr. Jianhua Shen
ChemMedChem 2013 Volume 8( Issue 7) pp:1210-1223
Publication Date(Web):
DOI:10.1002/cmdc.201300144
Abstract
Given its role in the mediation of energy and glucose homeostasis, the G-protein-coupled bile acid receptor 1 (TGR5) is considered a potential target for the treatment of type 2 diabetes mellitus and other metabolic disorders. By thorough analysis of diverse structures of published TGR5 agonists, a hypothetical ligand-based pharmacophore model was built, and a new class of potent TGR5 agonists, based on the novel 3,4,5-trisubstituted 4,5-dihydro-1,2,4-oxadiazole core, was discovered by rational design. Three distinct synthetic methods for constructing 4,5-dihydro-1,2,4-oxadiazoles and extensive structure–activity relationship studies are reported herein. Compound (R)-54 n, the structure of which was determined by single-crystal X-ray diffraction and quantum chemical solid-state TDDFT-ECD calculations, showed the best potency, with an EC50 value of 1.4 nM toward hTGR5. Its favorable properties in vitro warrant further investigation.
Co-reporter:Hongliang Duan ; Mengmeng Ning ; Xiaoyan Chen ; Qingan Zou ; Liming Zhang ; Ying Feng ; Lina Zhang ; Ying Leng
Journal of Medicinal Chemistry 2012 Volume 55(Issue 23) pp:10475-10489
Publication Date(Web):November 13, 2012
DOI:10.1021/jm301071h
4-Phenoxynicotinamide and 4-phenoxypyrimidine-5-carboxamide derivatives as potent and orally efficacious TGR5 agonists are reported. Several 4-phenoxynicotinamide derivatives were found to activate human and mouse TGR5 (hTGR5 and mTGR5) with EC50 values in the low nanomolar range. Compound 23g, with an EC50 value of 0.72 nM on hTGR5 and an EC50 value of 6.2 nM on mTGR5, was selected for further in vivo efficacy studies. This compound exhibited a significant dose-dependent glucagon-like peptide-1 (GLP-1) secretion effect. A single oral dose of 23g (50 mg/kg) significantly reduced blood glucose levels in db/db mice and caused a 49% reduction in the area under the blood glucose curve (AUC)0–120 min following an oral glucose tolerance test (OGTT) in imprinting control region (ICR) mice. However, 23g stimulated gallbladder filling, which might result in side effects to the gallbladder.
Co-reporter:Liming Zhang, Junhua Chen, Mengmeng Ning, Qingan Zou, Ying Leng, Jianhua Shen
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 8) pp:2748-2752
Publication Date(Web):15 April 2012
DOI:10.1016/j.bmcl.2012.02.095
11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) has attracted considerable attention as a potential target for the treatment of diabetes and metabolic syndrome. Herein we report the design, synthesis and efficacy evaluation of novel amide and urea 11β-HSD1 inhibitors. Structure–activity relationship studies led to the identification of 10c, which was efficacious in a diabetic ob/ob mouse model and reduced fasting and non-fasting blood glucose levels after ip dosing.Novel piperidine ureas were designed as potent 11β-HSD1 inhibitors. SAR studies led to the identification of 10c, which reduced fasting and non-fasting blood glucose levels after ip dosing in diabetic ob/ob mice.
Co-reporter:Xishan Xiong;Li Wang;Yangliang Ye;Lili Fu;Minli Chen
Investigational New Drugs 2010 Volume 28( Issue 4) pp:472-481
Publication Date(Web):2010 August
DOI:10.1007/s10637-009-9278-9
Numerous studies have documented that various naturally derived ligands or synthetic non-thiazolidinediones (TZD) as peroxisome proliferator-activated receptor gamma (PPARγ) agonists have shown moderate or potent antitumor activities, which is PPARγ independent or partially dependent. However, the PPARγ agonistic or glucose-lowering activity is ranked first more often than antitumor activity to determine promising novel PPARγ agonists for potential clinical use. In this study, we hypothesized that there might exist some compounds with less PPARγ agonistic activity but potent antitumor activity. Thereafter, we evaluated the PPARγ agonistic and antitumor activity of a novel series of α-aryloxy-α-methylhydrocinnamic acid derivatives synthesized with the initial aim of developing novel PPARγ agonists as hypoglycemic agents. MTT assay results revealed that several compounds were able to inhibit cell proliferation in a dose-dependent manner with IC50 12.7–29.7 μM, better than that of rosiglitazone (45.9–141 μM), although the PPARγ agonistic activity of most compounds is much lower than rosiglitazone. Some compounds induced cell cycle arrest and apoptosis tested by Flow Cytometry. Oral administration of DH9 (100 mg/kg/d) for 21 days to BALB/c nude mice bearing xenografts including MGC-803, NCI-H460, HT-29 and OS-RC-2 cells significantly retarded tumor growth. DG8 and DJ5 showed benefits in some of the above four xenografts. Our findings demonstrate that these compounds have potent antitumor activity in vitro and in vivo and pyrimidinyl-arylpropionic acid derivatives might be viable resources in the development of new antineoplastic agents
Co-reporter:Huaiyu Yang, Yu Shen, Junhua Chen, Qunfeng Jiang, Ying Leng, Jianhua Shen
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 3) pp:1167-1171
Publication Date(Web):March 2009
DOI:10.1016/j.ejmech.2008.06.005
Structure-based pharmacophore models were built by using LigandScout and used for virtual screening of the SPECS database to identify new potential 11β-HSD1 inhibitors. As a refinement of the results obtained from virtual 3D pharmacophore screening, the best fitting virtual hits were subjected to docking study. The resulting compounds were tested in an enzyme assay and revealed several compounds with novel scaffolds that show sub-micromolar activity and high selectivity for 11β-HSD1 against 11β-HSD2.
Co-reporter:Xishan Xiong;Yangliang Ye;Lili Fu;Bing Dai;Jieqiong Liu
Investigational New Drugs 2009 Volume 27( Issue 3) pp:223-232
Publication Date(Web):2009 June
DOI:10.1007/s10637-008-9161-0
Peroxisome proliferator-activated receptor gamma (PPARγ) agonists have shown benefit in treating diabetes mellitus, atherosclerosis and cancer. However, widespread use of thiazolidinediones (TZDs), the clinically used synthetic PPARγ agonists, has been limited by adverse cardiovascular effects. Consequently, numerous novel non-TZD compounds were synthesized and antidiabetic efficacy was evaluated to identify PPARγ agonists for potential clinical use. On the other hand, many studies have documented that the antitumor activity of PPARγ agonists is PPARγ independent. Here we hypothesized that there might exist some compounds with less PPARγ agonistic activity or antidiabetic efficacy but potent antitumor activity. In this study, we evaluated the PPARγ agonistic and antitumor activity of several newly synthesized α-aryloxy-α-methylhydrocinnamic acid derivatives as PPARγ agonists in a panel of human cancer cell lines, which showed promising antitumor activity without appreciable PPARγ agonistic activity. The results of MTT assay revealed that cell viability was inhibited in a dose dependent manner with IC50 17.1–55.1 μM for all the novel compounds and rosiglitazone (17.2–165 μM). They induced cell cycle arrest and apoptosis tested by Flow Cytometry. In conclusion, our findings demonstrate that these compounds have potent in vitro cytotoxicity, the possible mechanism of which is through induction of apoptosis and cell cycle arrest
Co-reporter:Huaiyu Yang, Wei Dou, Jing Lou, Ying Leng, Jianhua Shen
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 4) pp:1340-1345
Publication Date(Web):15 February 2008
DOI:10.1016/j.bmcl.2008.01.020
11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) is a potential target for treatment of diabetes and metabolic syndrome. Docking and pharmacophore modeling have been used to discover novel inhibitors of 11β-HSD1. Several compounds, with large structural diversity and good potency against 11β-HSD1, have been found and their potency was determined by the enzyme assay. New scaffolds of 11β-HSD1 inhibitors are also reported.Docking and pharmacophore Modeling were used to discover novel inhibitors of 11β-HSD1. Several compounds with large structural diversity and good potency were found. New scaffolds are reported.
Co-reporter:Hanjun Zou, Mingyue Zheng, Xiaomin Luo, Weiliang Zhu, Kaixian Chen, Jianhua Shen and Hualiang Jiang
The Journal of Physical Chemistry B 2008 Volume 112(Issue 41) pp:13070-13078
Publication Date(Web):September 24, 2008
DOI:10.1021/jp710964x
FadL is an important member of the family of fatty acid transport proteins within membranes. In this study, 11 conventional molecular dynamics (CMD) and 25 steered molecular dynamics (SMD) simulations were performed to investigate the dynamic mechanism of transport of long-chain fatty acids (LCFAs) across FadL. The CMD simulations addressed the intrinsically dynamic behavior of FadL. Both the CMD and SMD simulations revealed that a fatty acid molecule can move diffusively to a high-affinity site (HAS) from a low-affinity site (LAS). During this process, the swing motion of the L3 segment and the hydrophobic interaction between the fatty acid and FadL could play important roles. Furthermore, 22 of the SMD simulations revealed that fatty acids can pass through the gap between the hatch domain and the transmembrane domain (TMD) by different pathways. SMD simulations identified nine possible pathways for dodecanoic acid (DA) threading the barrel of FadL. The binding free energy profiles between DA and FadL along the MD trajectories indicate that all of the possible pathways are energetically favorable for the transport of fatty acids; however, one pathway (path VI) might be the most probable pathway for DA transport. The reasonability and reliability of this study were further demonstrated by correlating the MD simulation results with the available mutagenesis results. On the basis of the simulations, a mechanism for the full-length transport process of DA from the extracellular side to the periplasmic space mediated by FadL is proposed.
Co-reporter:Fei Zheng, Rongjian Sa, Jiagao Cheng, Hualiang Jiang, Jianhua Shen
Chemical Physics Letters 2007 Volume 435(1–3) pp:24-28
Publication Date(Web):12 February 2007
DOI:10.1016/j.cplett.2006.12.076
Cation–π interaction plays an important role as a general noncovalent binding force in a wide range of systems. To examine the more stable state of carbocation–π complex, we have carried out Car–Parrinello molecular dynamics (CPMD) simulations of six models. These results are proved to be reliable from the comparison with those of investigations by ab initio calculations. Charge transfer and molecular orbital interaction are discussed to depict the interaction modes of different models. From our CPMD studies without restraints, the methyl group moves toward the side of the benzene forming a stable σ-complex. However, if the methyl group is maintained above benzene, the inter-conversions among different models of C6H6CH3+ complex, which are determined from ab initio optimization, are observed; and distances between the carbon of methyl group and the center of benzene range from 2.67 Å to 3.61 Å. Our results provide a dynamic process concerning the motion of methyl group above the benzene ring.To examine more stable state of carbocation–π complex, six models of benzene-methyl cation system are studied by Car–Parrinello molecular dynamics simulation, showing that inter-conversions among different models of C6H6CH3+ complex are possible for the carbocation–π interaction. Thus, a dynamic relationship among the different models is first constructed in our studies.

![Methanone, [4-[3-fluoro-5-(trifluoromethyl)-2-pyridinyl]-1-piperazinyl][5-(methylsulfonyl)-](http://img.cochemist.com/ccimg/845700/845614-11-1.png)
![Methanone, [4-[3-fluoro-5-(trifluoromethyl)-2-pyridinyl]-1-piperazinyl][5-(methylsulfonyl)-](http://img.cochemist.com/ccimg/845700/845614-11-1_b.png)
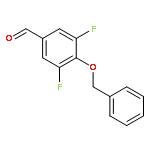
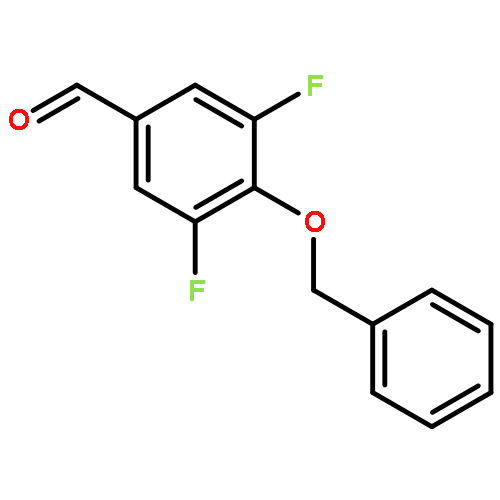
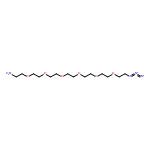

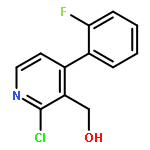


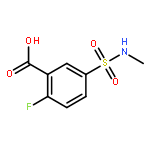
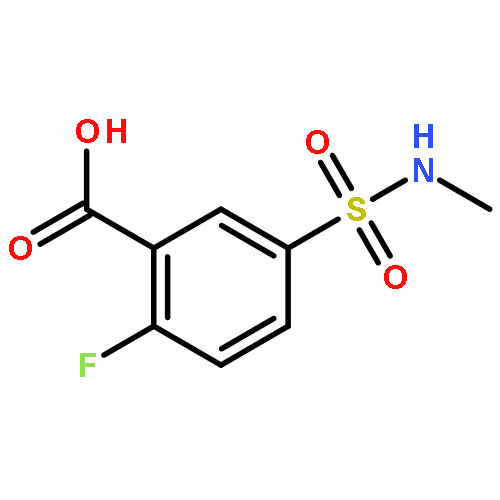
![Benzoic acid, 5-[(methylamino)sulfonyl]-2-(4-morpholinyl)-](http://img.cochemist.com/ccimg/848000/847972-01-4.png)
![Benzoic acid, 5-[(methylamino)sulfonyl]-2-(4-morpholinyl)-](http://img.cochemist.com/ccimg/848000/847972-01-4_b.png)
![BENZOIC ACID, 5-NITRO-2-[[(TRIFLUOROMETHYL)SULFONYL]OXY]-, METHYL ESTER](http://img.cochemist.com/ccimg/474600/474521-55-6.png)
![BENZOIC ACID, 5-NITRO-2-[[(TRIFLUOROMETHYL)SULFONYL]OXY]-, METHYL ESTER](http://img.cochemist.com/ccimg/474600/474521-55-6_b.png)
![Benzoic acid, 5-[(methylamino)sulfonyl]-2-(1-piperidinyl)-](http://img.cochemist.com/ccimg/151200/151104-35-7.png)
![Benzoic acid, 5-[(methylamino)sulfonyl]-2-(1-piperidinyl)-](http://img.cochemist.com/ccimg/151200/151104-35-7_b.png)
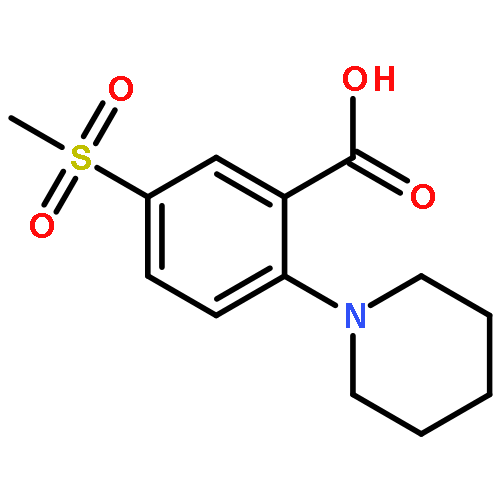
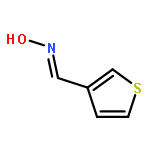
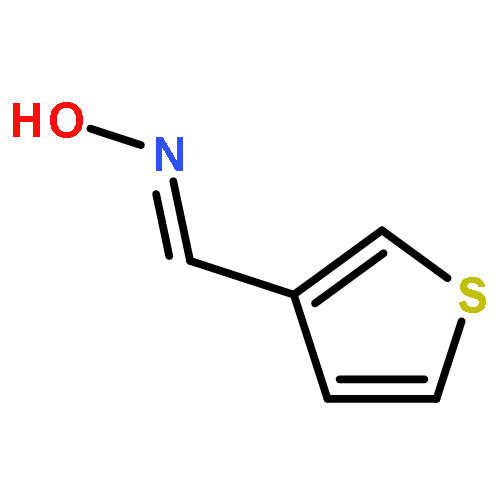
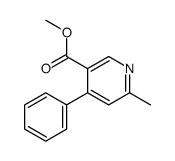
![Ethanamine, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/114300/134179-38-7.png)
![Ethanamine, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/114300/134179-38-7_b.png)


![Methyl [3,5-bis(trifluoromethyl)phenyl]acetate](http://img.cochemist.com/ccimg/95300/95299-16-4.png)
![Methyl [3,5-bis(trifluoromethyl)phenyl]acetate](http://img.cochemist.com/ccimg/95300/95299-16-4_b.png)

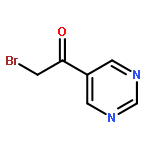
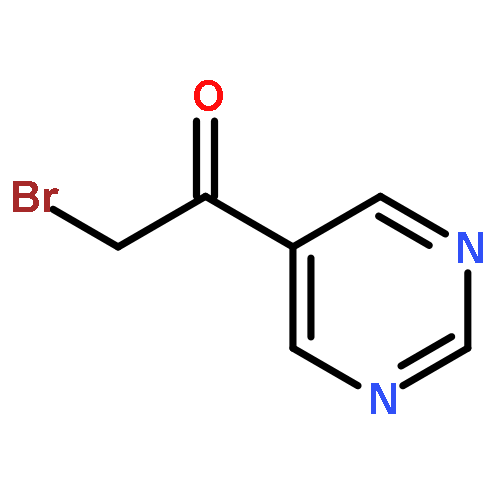
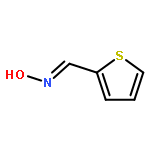
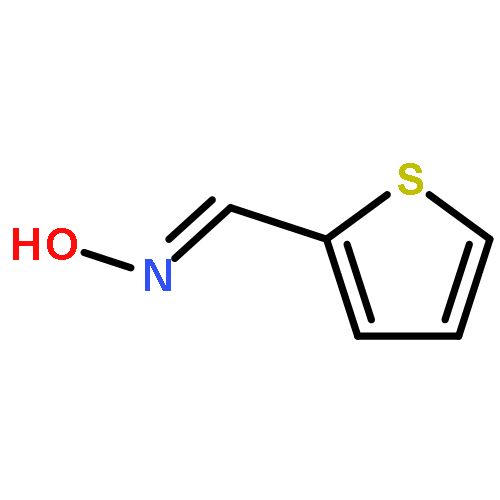





![Ethanone,1-[4-(4-fluorobenzoyl)-1-piperidinyl]-](http://img.cochemist.com/ccimg/25600/25519-77-1.png)
![Ethanone,1-[4-(4-fluorobenzoyl)-1-piperidinyl]-](http://img.cochemist.com/ccimg/25600/25519-77-1_b.png)
![Benzo[b]thiophen-5-ol](http://img.cochemist.com/ccimg/19400/19301-35-0.png)
![Benzo[b]thiophen-5-ol](http://img.cochemist.com/ccimg/19400/19301-35-0_b.png)








![[C(Z)]-2-Furancarboxaldehyde oxime](http://img.cochemist.com/ccimg/1500/1450-58-4.png)
![[C(Z)]-2-Furancarboxaldehyde oxime](http://img.cochemist.com/ccimg/1500/1450-58-4_b.png)

![5-TERT-BUTYL-2-[(4-FLUOROPHENYL)METHYL]PYRAZOLE-3-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/700/620-03-1.png)
![5-TERT-BUTYL-2-[(4-FLUOROPHENYL)METHYL]PYRAZOLE-3-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/700/620-03-1_b.png)