Co-reporter:Takafumi Ide, Shuya Masuda, Yuji Kawato, Hiromichi Egami, and Yoshitaka Hamashima
Organic Letters September 1, 2017 Volume 19(Issue 17) pp:
Publication Date(Web):August 11, 2017
DOI:10.1021/acs.orglett.7b01971
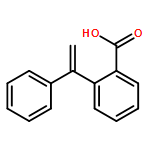
Photoenols generated in situ from ortho-methyl-substituted phenylketones such as benzophenones and acetophenones were trifluoromethylated with Togni reagent without any additive or catalyst. This trifluoromethylation reaction proceeded smoothly under photoirradiation conditions (365 nm). Various functional groups were tolerant of the reaction conditions. Interestingly, the trifluoromethyl group was exclusively introduced at the ortho-benzylic position. Mechanistic studies suggested that this reaction proceeds via formation of a photoenol, not via a radical pathway.
Co-reporter:Dr. Yuji Kawato;Hiromi Ono;Akino Kubota;Yoshihiro Nagao;Naoki Morita;Dr. Hiromichi Egami ;Dr. Yoshitaka Hamashima
Chemistry - A European Journal 2016 Volume 22( Issue 6) pp:2127-2133
Publication Date(Web):
DOI:10.1002/chem.201503153
Abstract
The enantioselective bromocyclization of allylic amides catalyzed by phosphorus-containing Lewis bases was examined in detail. A series of control experiments and NMR studies showed that a partially oxidized bis-phosphine generated in situ serves as the actual enantioselective catalyst. The reaction mechanism involves distinct roles of two Lewis basic sites, P and P=O, with P+Br serving as a fine-tuning element for substrate fixation in the chiral environment, and P+OBr as the Br+ transfer agent to the olefin. Catalyst loading could be reduced to as little as 1 mol %, and the reaction affords enantioenriched oxazolines with up to >99.5 % ee.
Co-reporter:Hiromichi Egami, Takahiro Yoneda, Minako Uku, Takafumi Ide, Yuji Kawato, and Yoshitaka Hamashima
The Journal of Organic Chemistry 2016 Volume 81(Issue 10) pp:4020-4030
Publication Date(Web):April 21, 2016
DOI:10.1021/acs.joc.6b00295
Difunctionalization of alkenes with 1-chloro-1,2-benziodoxol-3-(1H)-one (1) was investigated. Various additional nucleophiles were tested, and oxychlorination, dichlorination, azidochlorination, chlorothiocyanation, and iodoesterfication were demonstrated. The oxychlorination product was obtained efficiently when the reaction was operated in water. Dichlorination occurred in the presence of a Lewis basic promoter, such as 4-phenylpyridine N-oxide, as an additive. The reaction with in situ-generated azido anion afforded azidochlorinated compounds with a chlorine atom at the terminal position, while the reaction with trimethylsilyl isothiocyanate produced chlorothiocyanation adducts with a chlorine atom at the benzylic position. On the other hand, when 1 was treated with tetra-n-butylammonium iodide prior to the addition of alkenes, only iodoesterification occurred selectively. These mild reactions enable convenient site-selective difunctionalizations of substrates having two alkene moieties. NMR experiments suggested that the electrophilic reactive species in each reaction varied depending on the nature of the added nucleophile.
Co-reporter:Hiromichi Egami; Junshi Asada; Kentaro Sato; Daisuke Hashizume; Yuji Kawato
Journal of the American Chemical Society 2015 Volume 137(Issue 32) pp:10132-10135
Publication Date(Web):August 7, 2015
DOI:10.1021/jacs.5b06546
We report the first successful example of a highly enantioselective fluorolactonization with an electrophilic fluorinating reagent, Selectfluor®, in the presence of a novel bifunctional organocatalyst. The catalyst design includes a carboxylate anion functioning as a phase-transfer agent and a benzyl alcohol unit to capture the substrate through hydrogen bonding. Fluorinated isobenzofuranones were obtained in good yields with up to 94% ee (97:3 er). On the basis of mechanistic studies, we propose a unique reaction mechanism with potential for further applications.
Co-reporter:Hiromichi Egami, Takafumi Ide, Yuji Kawato and Yoshitaka Hamashima
Chemical Communications 2015 vol. 51(Issue 93) pp:16675-16678
Publication Date(Web):25 Sep 2015
DOI:10.1039/C5CC07011B
Phenol derivatives were trifluoromethylated using copper/Togni reagent. In dimethylformamide, the benzylic C–H bond at the para position of the hydroxyl group was selectively substituted with a CF3 group. In contrast, aromatic C–H trifluoromethylation occurred in alcoholic solvents. Practical utility of the reactions was demonstrated by application to the synthesis of a potent enoyl-acyl carrier protein reductase (FabI) inhibitor.
Co-reporter:Yuji Kawato, Akino Kubota, Hiromi Ono, Hiromichi Egami, and Yoshitaka Hamashima
Organic Letters 2015 Volume 17(Issue 5) pp:1244-1247
Publication Date(Web):February 20, 2015
DOI:10.1021/acs.orglett.5b00220
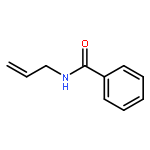
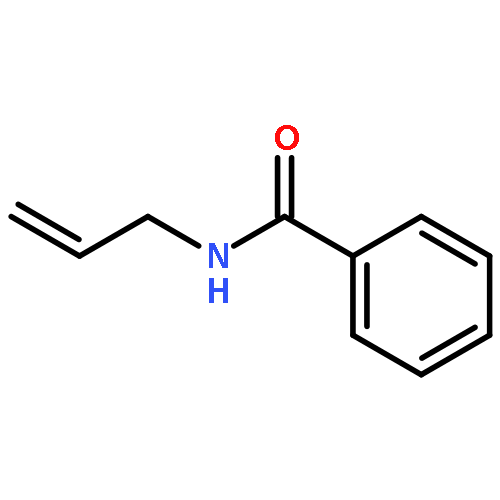
A highly enantioselective bromocyclization of allylic amides with N-bromosuccinimide (NBS) was developed with DTBM-BINAP as a catalyst, affording chiral oxazolines with a tetrasubstituted carbon center in high yield with up to 99% ee. By utilizing the bromo substituent as a handle, the obtained compounds were converted to synthetically useful chiral building blocks.
Co-reporter:Hiromichi Egami, Kentaro Sato, Junshi Asada, Yuji Kawato, Yoshitaka Hamashima
Tetrahedron 2015 Volume 71(Issue 37) pp:6384-6388
Publication Date(Web):16 September 2015
DOI:10.1016/j.tet.2015.05.041
3,3′-Disubstituted 1,1′-binaphthyl-2,2′-dicarboxylic acids (1) were synthesized in three or four steps from commercially available BINOL via carbon dioxide fixation with organolithium to incorporate the carboxylic acid moieties, followed by either carboxylate-directed ortho-C–H arylation or Suzuki cross-coupling. This method provides easy access to various types of axiially chiral dicarboxylic acids, which should be useful for studies of chiral Brønsted acid-catalyzed asymmetric reactions.
Co-reporter:Hitoshi Ouchi, Aya Asahina, Tomohiro Asakawa, Makoto Inai, Yoshitaka Hamashima, and Toshiyuki Kan
Organic Letters 2014 Volume 16(Issue 7) pp:1980-1983
Publication Date(Web):March 24, 2014
DOI:10.1021/ol500529w
Practical total syntheses of acromelic acids A (1) and B (2), which have potent neuro-excitatory activity, were accomplished in 13 (36% total yield) and 17 steps (6.9% total yield), respectively, from 2,6-dichloropyridine (8). Regioselective transformation of symmetric 8 provided nitroalkenes 15 and 16. The pyrrolidine ring was efficiently constructed by Ni-catalyzed asymmetric conjugate addition followed by intramolecular reductive amination.
Co-reporter:Yusuke Kawabe, Ryo Ishikawa, Yusuke Akao, Atsushi Yoshida, Makoto Inai, Tomohiro Asakawa, Yoshitaka Hamashima, and Toshiyuki Kan
Organic Letters 2014 Volume 16(Issue 7) pp:1976-1979
Publication Date(Web):March 24, 2014
DOI:10.1021/ol500524y
The total synthesis of hedyotol A (1), a natural product isolated from Hedyotis lawsoniae (DC.) Wight et Arn. (Rubiaceae), was accomplished in a highly stereocontrolled manner. Key steps include an l-proline-catalyzed cross-aldol reaction and the biomimetic construction of a furofuran lignan skeleton through a quinomethide intermediate.
Co-reporter:Shingo Sasaki, Hiroto Suzuki, Hitoshi Ouchi, Tomohiro Asakawa, Makoto Inai, Ryuichi Sakai, Keiko Shimamoto, Yoshitaka Hamashima, and Toshiyuki Kan
Organic Letters 2014 Volume 16(Issue 2) pp:564-567
Publication Date(Web):January 7, 2014
DOI:10.1021/ol403434e
A practical total synthesis of kainoid MFPA (5) was achieved in only six steps, via a novel Ni-catalyst-mediated asymmetric conjugate addition reaction. Furthermore, a fluorescein-based fluorescent ionotropic glutamate receptor probe 28 was efficiently synthesized from a precursor derived from a synthetic intermediate of 5.
Co-reporter:Dr. Hiromichi Egami;Takafumi Ide;Masashi Fujita;Toshifumi Tojo;Dr. Yoshitaka Hamashima;Dr. Mikiko Sodeoka
Chemistry - A European Journal 2014 Volume 20( Issue 38) pp:12061-12065
Publication Date(Web):
DOI:10.1002/chem.201403447
Abstract
Trifluoromethylation of propargylic alcohols to provide (Z)-α-trifluoromethylated enones and β-unsubstituted α-trifluoromethylated enones proceeded with high yield and selectivity in the presence of CuI/Re2O7. The Z isomer was formed under kinetic control, though it is less stable than the E isomer in terms of steric repulsion.
Co-reporter:Kazutada Ikeuchi, Shunsuke Ido, Satoshi Yoshimura, Tomohiro Asakawa, Makoto Inai, Yoshitaka Hamashima, and Toshiyuki Kan
Organic Letters 2012 Volume 14(Issue 23) pp:6016-6019
Publication Date(Web):November 13, 2012
DOI:10.1021/ol302908a
Asymmetric bromolactonization of prochiral cyclohexadiene derivatives with N-bromosuccimide proceeded in the presence of (DHQD)2PHAL as a chiral catalyst to afford the corresponding bromolactones with up to 93% ee. This reaction was also applicable to the kinetic resolution of a racemic cyclic ene-carboxylic acid, where the starting material was recovered with high enantioselectivity.
Co-reporter:Hiromichi Egami, Takafumi Ide, Yuji Kawato and Yoshitaka Hamashima
Chemical Communications 2015 - vol. 51(Issue 93) pp:NaN16678-16678
Publication Date(Web):2015/09/25
DOI:10.1039/C5CC07011B
Phenol derivatives were trifluoromethylated using copper/Togni reagent. In dimethylformamide, the benzylic C–H bond at the para position of the hydroxyl group was selectively substituted with a CF3 group. In contrast, aromatic C–H trifluoromethylation occurred in alcoholic solvents. Practical utility of the reactions was demonstrated by application to the synthesis of a potent enoyl-acyl carrier protein reductase (FabI) inhibitor.



![Benzonitrile, 4-[(1S)-1-hydroxy-3-butynyl]-](http://img.cochemist.com/ccimg/906100/906077-64-3.png)
![Benzonitrile, 4-[(1S)-1-hydroxy-3-butynyl]-](http://img.cochemist.com/ccimg/906100/906077-64-3_b.png)
![BENZONITRILE, 4-[(1S)-1-HYDROXY-3-BUTENYL]-](http://img.cochemist.com/ccimg/850800/850798-64-0.png)
![BENZONITRILE, 4-[(1S)-1-HYDROXY-3-BUTENYL]-](http://img.cochemist.com/ccimg/850800/850798-64-0_b.png)

![<br>(-)-1,2-Bis[(2R,5R)-diethylphospholano)benzene(cyclooctadiene]rhodium(I) tr iflate](http://img.cochemist.com/ccimg/136800/136705-77-6.png)
![<br>(-)-1,2-Bis[(2R,5R)-diethylphospholano)benzene(cyclooctadiene]rhodium(I) tr iflate](http://img.cochemist.com/ccimg/136800/136705-77-6_b.png)







![Urea, N-[2-(diethylamino)ethyl]-N'-phenyl-](http://img.cochemist.com/ccimg/4600/4559-89-1.png)
![Urea, N-[2-(diethylamino)ethyl]-N'-phenyl-](http://img.cochemist.com/ccimg/4600/4559-89-1_b.png)
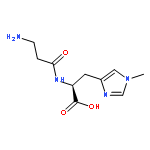
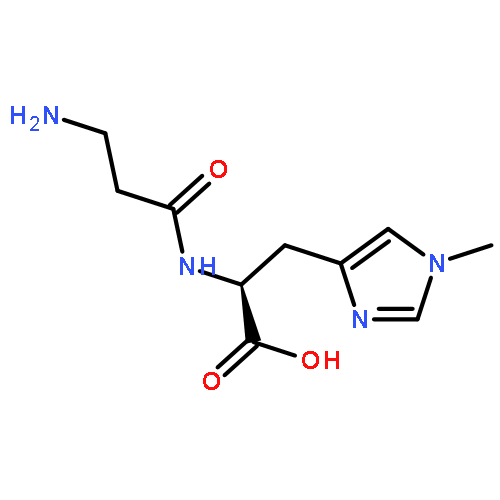

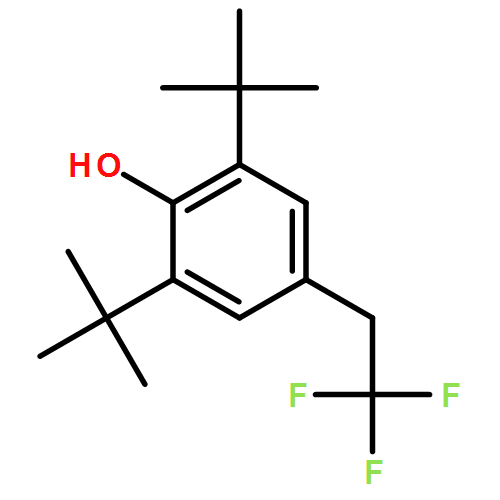
![Benzamide, N-[(3,4-dihydro-1-naphthalenyl)methyl]-](/data/chemimg/3751000/1660995-07-2.png)
![Benzamide, N-[(3,4-dihydro-1-naphthalenyl)methyl]-](/data/chemimg/3751000/1660995-07-2_b.png)





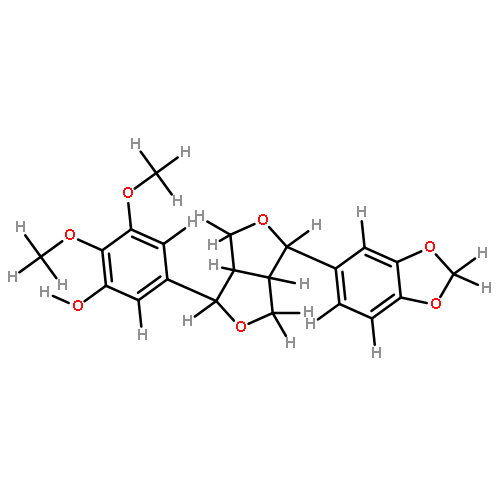






![1H-Thieno[3,4-d]imidazole-4-pentanamide, hexahydro-2-oxo-N-2-propyn-1-yl-, (3aS,4S,6aR)-](/data/chemimg/103200/773888-45-2.png)
![1H-Thieno[3,4-d]imidazole-4-pentanamide, hexahydro-2-oxo-N-2-propyn-1-yl-, (3aS,4S,6aR)-](/data/chemimg/103200/773888-45-2_b.png)




![2-[3,4-BIS(PHENYLMETHOXY)PHENYL]-3-HYDROXYCHROMEN-4-ONE](http://img.cochemist.com/ccimg/307600/307521-00-2.png)
![2-[3,4-BIS(PHENYLMETHOXY)PHENYL]-3-HYDROXYCHROMEN-4-ONE](http://img.cochemist.com/ccimg/307600/307521-00-2_b.png)








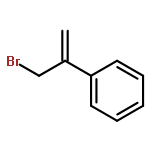
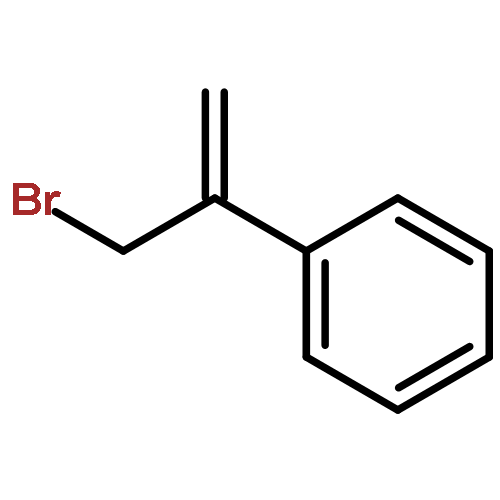
![Benzene, 1-[1-(bromomethyl)ethenyl]-3-chloro-](http://img.cochemist.com/ccimg/193600/193531-42-9.png)
![Benzene, 1-[1-(bromomethyl)ethenyl]-3-chloro-](http://img.cochemist.com/ccimg/193600/193531-42-9_b.png)


![Phosphine oxide,[(1S)-2'-(diphenylphosphino)[1,1'-binaphthalen]-2-yl]diphenyl-](/data/chemimg/1773500/183244-55-5.png)
![Phosphine oxide,[(1S)-2'-(diphenylphosphino)[1,1'-binaphthalen]-2-yl]diphenyl-](/data/chemimg/1773500/183244-55-5_b.png)






![Benzene, 1-[1-(bromomethyl)ethenyl]-4-fluoro-](http://img.cochemist.com/ccimg/133000/132927-05-0.png)
![Benzene, 1-[1-(bromomethyl)ethenyl]-4-fluoro-](http://img.cochemist.com/ccimg/133000/132927-05-0_b.png)





![3-[TERT-BUTYL(DIMETHYL)SILYL]OXY-4,5-DIMETHOXYBENZALDEHYDE](http://img.cochemist.com/ccimg/122300/122271-47-0.png)
![3-[TERT-BUTYL(DIMETHYL)SILYL]OXY-4,5-DIMETHOXYBENZALDEHYDE](http://img.cochemist.com/ccimg/122300/122271-47-0_b.png)
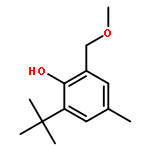
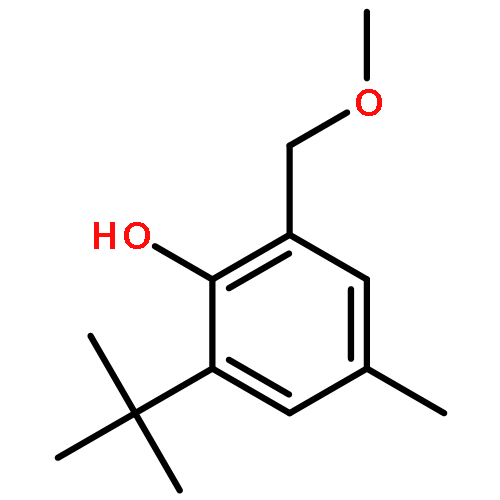
![[1,1'-Biphenyl]-2-ol, 3-(1,1-dimethylethyl)-5-methyl-](http://img.cochemist.com/ccimg/120200/120186-71-2.png)
![[1,1'-Biphenyl]-2-ol, 3-(1,1-dimethylethyl)-5-methyl-](http://img.cochemist.com/ccimg/120200/120186-71-2_b.png)







![2,3-Dihydro-2-(4-hydroxy-3-methoxyphenyl)-7-methoxy-5-[tetrahydro-4-(4-hydroxy-3-methoxyphenyl)-1H,3H-furo[3,4-c]furan-1-yl]-3-benzofuranmethanol](http://img.cochemist.com/ccimg/97500/97400-01-6.png)
![2,3-Dihydro-2-(4-hydroxy-3-methoxyphenyl)-7-methoxy-5-[tetrahydro-4-(4-hydroxy-3-methoxyphenyl)-1H,3H-furo[3,4-c]furan-1-yl]-3-benzofuranmethanol](http://img.cochemist.com/ccimg/97500/97400-01-6_b.png)


![4H-1-Benzopyran-4-one, 2-[4-(phenylmethoxy)phenyl]-](http://img.cochemist.com/ccimg/95200/95161-87-8.png)
![4H-1-Benzopyran-4-one, 2-[4-(phenylmethoxy)phenyl]-](http://img.cochemist.com/ccimg/95200/95161-87-8_b.png)



![3-[(3S,4S,5S)-5-carboxy-4-(carboxymethyl)pyrrolidin-3-yl]-6-oxo-1,6-dihydropyridine-2-carboxylic acid](http://img.cochemist.com/ccimg/86700/86630-10-6.png)
![3-[(3S,4S,5S)-5-carboxy-4-(carboxymethyl)pyrrolidin-3-yl]-6-oxo-1,6-dihydropyridine-2-carboxylic acid](http://img.cochemist.com/ccimg/86700/86630-10-6_b.png)


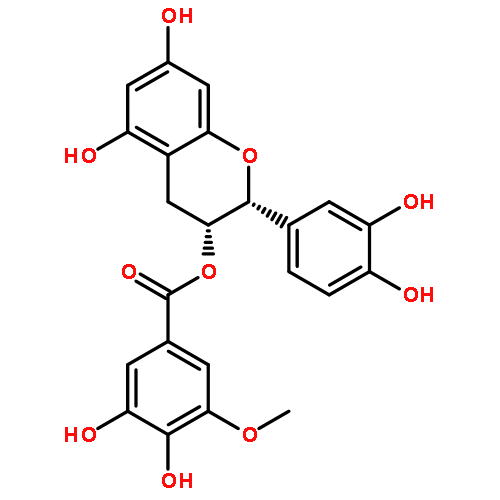


![2'-methoxy-[1,1'-binaphthalene]-2-carboxylic acid](http://img.cochemist.com/ccimg/80400/80317-69-7.png)
![2'-methoxy-[1,1'-binaphthalene]-2-carboxylic acid](http://img.cochemist.com/ccimg/80400/80317-69-7_b.png)

![Benzene, 1-[1-(bromomethyl)ethenyl]-4-methyl-](http://img.cochemist.com/ccimg/65400/65374-22-3.png)
![Benzene, 1-[1-(bromomethyl)ethenyl]-4-methyl-](http://img.cochemist.com/ccimg/65400/65374-22-3_b.png)




![Benzene, 1-[1-(bromomethyl)ethenyl]-3-methoxy-](http://img.cochemist.com/ccimg/61400/61364-70-3.png)
![Benzene, 1-[1-(bromomethyl)ethenyl]-3-methoxy-](http://img.cochemist.com/ccimg/61400/61364-70-3_b.png)
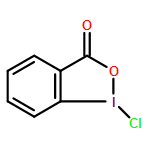
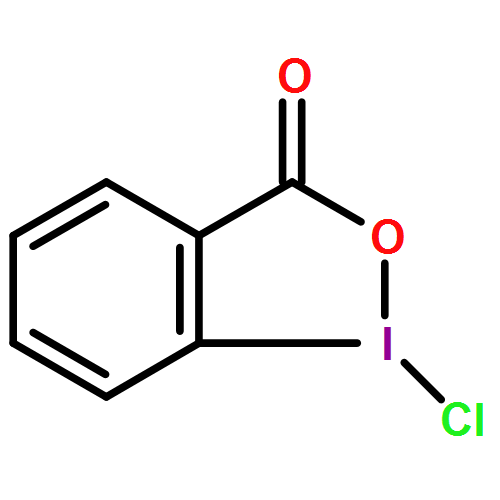




![N'-{(E)-[4-(BENZYLOXY)PHENYL]METHYLENE}-2-IODOBENZOHYDRAZIDE](http://img.cochemist.com/ccimg/39900/39887-27-9.png)
![N'-{(E)-[4-(BENZYLOXY)PHENYL]METHYLENE}-2-IODOBENZOHYDRAZIDE](http://img.cochemist.com/ccimg/39900/39887-27-9_b.png)


![4H-Imidazo[4,5-d]-1,2,3-triazin-4-one,1,7-dihydro-7-(2,3,5-tri-O-acetyl-b-D-ribofuranosyl)- (9CI)](http://img.cochemist.com/ccimg/36600/36519-17-2.png)
![4H-Imidazo[4,5-d]-1,2,3-triazin-4-one,1,7-dihydro-7-(2,3,5-tri-O-acetyl-b-D-ribofuranosyl)- (9CI)](http://img.cochemist.com/ccimg/36600/36519-17-2_b.png)
![7-[3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1h-imidazo[4,5-d]triazin-4-one](http://img.cochemist.com/ccimg/36600/36519-16-1.png)
![7-[3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1h-imidazo[4,5-d]triazin-4-one](http://img.cochemist.com/ccimg/36600/36519-16-1_b.png)
![5-[(1S,3aR,4S,6aR)-4-(3,4-dimethoxyphenyl)tetrahydro-1H,3H-furo[3,4-c]furan-1-yl]-1,3-benzodioxole](http://img.cochemist.com/ccimg/36200/36150-23-9.png)
![5-[(1S,3aR,4S,6aR)-4-(3,4-dimethoxyphenyl)tetrahydro-1H,3H-furo[3,4-c]furan-1-yl]-1,3-benzodioxole](http://img.cochemist.com/ccimg/36200/36150-23-9_b.png)

![5H-Benzocyclohepten-5-one,8-[(2R,3S)-3,4-dihydro-3,5,7-trihydroxy-2H-1-benzopyran-2-yl]-3,4,6-trihydroxy-](http://img.cochemist.com/ccimg/31800/31701-93-6.png)
![5H-Benzocyclohepten-5-one,8-[(2R,3S)-3,4-dihydro-3,5,7-trihydroxy-2H-1-benzopyran-2-yl]-3,4,6-trihydroxy-](http://img.cochemist.com/ccimg/31800/31701-93-6_b.png)


![Benzene, 1-methoxy-2-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/21300/21298-69-1.png)
![Benzene, 1-methoxy-2-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/21300/21298-69-1_b.png)

![(R)-[1,1'-Binaphthalene]-2,2'-dicarboxylic acid](http://img.cochemist.com/ccimg/18600/18531-96-9.png)
![(R)-[1,1'-Binaphthalene]-2,2'-dicarboxylic acid](http://img.cochemist.com/ccimg/18600/18531-96-9_b.png)






![1,3-Benzodioxole,5-[tetrahydro-4-(3,4,5-trimethoxyphenyl)-1H,3H-furo[3,4-c]furan-1-yl]-,(1S,3aR,4S,6aR)-](http://img.cochemist.com/ccimg/13100/13060-15-6.png)
![1,3-Benzodioxole,5-[tetrahydro-4-(3,4,5-trimethoxyphenyl)-1H,3H-furo[3,4-c]furan-1-yl]-,(1S,3aR,4S,6aR)-](http://img.cochemist.com/ccimg/13100/13060-15-6_b.png)
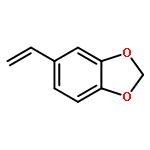
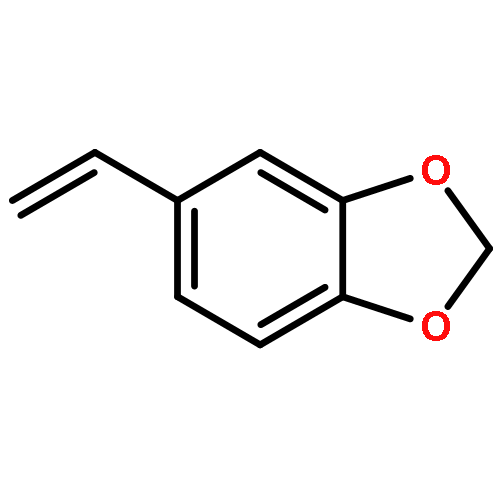

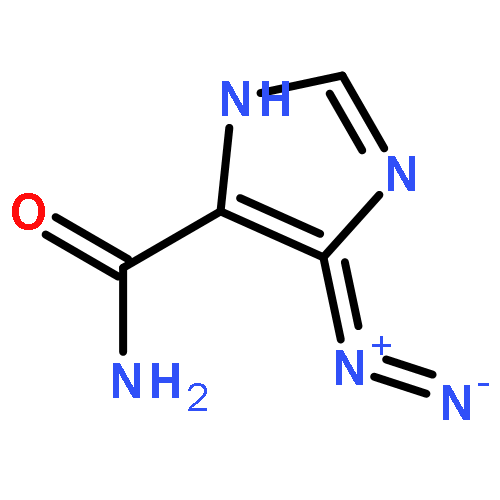
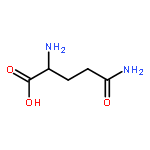
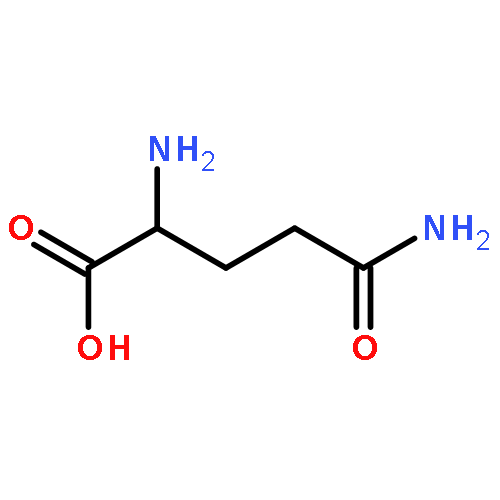


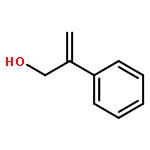
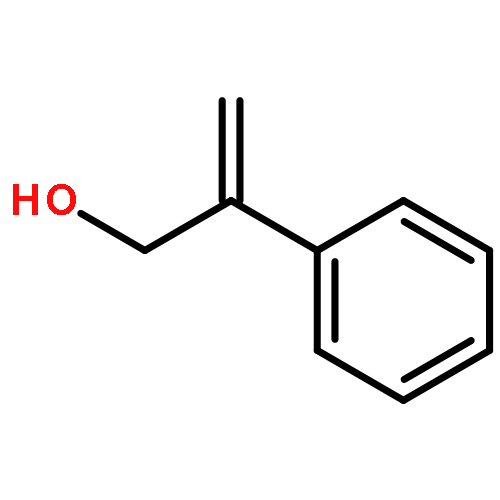


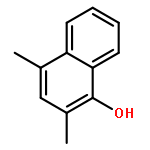

![5H-Benzocyclohepten-5-one, 1,8-bis[(2R,3R)-3,4-dihydro-3,5,7-trihydroxy-2H-1-benzopyran-2-yl]-3,4,6-trihydroxy-](http://img.cochemist.com/ccimg/4700/4670-05-7.png)
![5H-Benzocyclohepten-5-one, 1,8-bis[(2R,3R)-3,4-dihydro-3,5,7-trihydroxy-2H-1-benzopyran-2-yl]-3,4,6-trihydroxy-](http://img.cochemist.com/ccimg/4700/4670-05-7_b.png)




















![Benzamide, N-[2-[3-(trifluoromethyl)phenyl]-2-propen-1-yl]-](http://img.cochemist.com/ccimg/919400/919349-75-0.png)
![Benzamide, N-[2-[3-(trifluoromethyl)phenyl]-2-propen-1-yl]-](http://img.cochemist.com/ccimg/919400/919349-75-0_b.png)




![1,3-Benzodioxol-5-ol,6-[(1S,3aR,4S,6aR)-4-(1,3-benzodioxol-5-yl)tetrahydro-1H,3H-furo[3,4-c]furan-1-yl]-](http://img.cochemist.com/ccimg/74100/74061-79-3.png)
![1,3-Benzodioxol-5-ol,6-[(1S,3aR,4S,6aR)-4-(1,3-benzodioxol-5-yl)tetrahydro-1H,3H-furo[3,4-c]furan-1-yl]-](http://img.cochemist.com/ccimg/74100/74061-79-3_b.png)



![4H-Imidazo[4,5-d]-1,2,3-triazin-4-one,3,7-dihydro-](http://img.cochemist.com/ccimg/4700/4656-86-4.png)
![4H-Imidazo[4,5-d]-1,2,3-triazin-4-one,3,7-dihydro-](http://img.cochemist.com/ccimg/4700/4656-86-4_b.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)



