Co-reporter:C. C. Beto;E. D. Holt;Y. Yang;I. Ghiviriga;K. S. Schanze;A. S. Veige
Chemical Communications 2017 vol. 53(Issue 71) pp:9934-9937
Publication Date(Web):2017/08/31
DOI:10.1039/C7CC06289C
The first example of an in-chain metallo-poly(triazolate) synthesized by CuAAC is reported. Azido–platinum–acetylide (A–M–B) monomers are catalytically polymerized with copper(I) acetate to yield 1,2,3-triazolate linked Pt(II) units. The metallopolymers are characterized by multinuclear NMR, IR, UV/Vis, GPC, and MS.
Co-reporter:Matthew E. O’Reilly, Saikat Dutta, and Adam S. Veige
Chemical Reviews 2016 Volume 116(Issue 14) pp:8105-8145
Publication Date(Web):July 1, 2016
DOI:10.1021/acs.chemrev.6b00054
This review describes organometallic compounds and materials that are capable of mediating a rarely encountered but fundamentally important reaction: β-alkyl elimination at the metal–Cα–Cβ–R moiety, in which an alkyl group attached to the Cβ atom is transferred to the metal or to a coordinated substrate. The objectives of this review are to provide a cohesive fundamental understanding of β-alkyl-elimination reactions and to highlight its applications in olefin polymerization, alkane hydrogenolysis, depolymerization of branched polymers, ring-opening polymerization of cycloalkanes, and other useful organic reactions. To provide a coherent understanding of the β-alkyl elimination reaction, special attention is given to conditions and strategies used to facilitate β-alkyl-elimination/transfer events in metal-catalyzed olefin polymerization, which provide the well-studied examples.
Co-reporter:Stella A. Gonsales; Tomohiro Kubo; Madison K. Flint; Khalil A. Abboud; Brent S. Sumerlin
Journal of the American Chemical Society 2016 Volume 138(Issue 15) pp:4996-4999
Publication Date(Web):April 4, 2016
DOI:10.1021/jacs.6b00014
The tungsten alkylidyne [tBuOCO]W≡C(tBu) (THF)2 (1) reacts with CO2, leading to complete cleavage of one C═O bond, followed by migratory insertion to generate the tungsten-oxo alkylidene 2. Complex 2 is the first catalyst to polymerize norbornene via ring expansion metathesis polymerization to yield highly cis-syndiotactic cyclic polynorbornene.
Co-reporter:Soufiane S. Nadif; Tomohiro Kubo; Stella A. Gonsales; Sudarsan VenkatRamani; Ion Ghiviriga; Brent S. Sumerlin
Journal of the American Chemical Society 2016 Volume 138(Issue 20) pp:6408-6411
Publication Date(Web):May 12, 2016
DOI:10.1021/jacs.6b03247
Tungsten alkylidynes [CF3–ONO]W≡CC(CH3)3(THF)2 (1) and [tBuOCO]W≡CC(CH3)3(THF)2 (3) react with ethylene. Complex 1 reacts reversibly with ethylene to give the metallacyclobutene (2). Complex 3 reacts with ethylene to form the tethered alkylidene (4) featuring a tetraanionic pincer ligand. Complexes 1 and 3 initiate the polymerization of norbornene at room temperature. The polymerization of norbornene by 1 is not stereoselective, whereas 3 generates a highly cis and syndiotactic cyclic polynorbornene. Comparison of the intrinsic viscosity, radius of gyration, and elution time of the synthesized cyclic polynorbornene with those of linear analogues provides conclusive evidence for a cyclic topology.
Co-reporter:Matias E. Pascualini, Sebastian A. Stoian, Andrew Ozarowski, Khalil A. Abboud, and Adam S. Veige
Inorganic Chemistry 2016 Volume 55(Issue 11) pp:5191-5200
Publication Date(Web):May 16, 2016
DOI:10.1021/acs.inorgchem.6b00075
Square-planar high-spin Fe(II) molecular compounds are rare, and until recently, the only four examples of non-macrocyclic or sterically driven molecular compounds of this kind shared a common FeO4 core. The trianionic pincer-type ligand [CF3-ONO]H3 (1) supports the high-spin square-planar Fe(II) complex {[CF3-ONO]FeCl}{Li(Sv)2}2 (2). In the solid state, 2 forms the dimer complex {[CF3-ONO]Fe}2{(μ-Cl)2(μ-LiTHF)4} (3) in 96% yield by simply applying a vacuum or stirring it with pentane for 2 h. A detailed high-frequency electron paramagnetic resonance and field-dependent 57Fe Mössbauer investigation of 3 revealed a weak antiferromagnetic exchange interaction between the local iron spins which exhibit a zero-field splitting tensor characterized by negative D parameter. In solution, 2 is in equilibrium with the solvento complex {[CF3-ONO]FeCl(THF)}{Li2(Sv)4} (2·Sv) and the dimer 3. A combination of frozen solution 57Fe Mössbauer spectroscopy and single crystal X-ray crystallography helped elucidate the solvent dependent equilibrium between these three species. The oxidation chemistry of 2·Sv was investigated. Complex 2 reacts readily with the one-electron oxidizing agent CuCl2 to give the Fe(III) complex {[CF3-ONO]FeCl2}{Li(THF)2}2 (4). Also, 2·Sv reacts with 2 equiv of TlPF6 to form the Fe(III) complex [CF3-ONO]Fe(THF)3 (5).
Co-reporter:Christopher C. Beto, Xi Yang, Andrew R. Powers, Ion Ghiviriga, Khalil A. Abboud, Adam S. Veige
Polyhedron 2016 Volume 108() pp:87-92
Publication Date(Web):29 March 2016
DOI:10.1016/j.poly.2015.08.032
The iClick (inorganic click) reactions between gold-acetylides and group 9 transition metal-azide complexes are presented. Complexes [Rh(CO)(PPh3)2][PPh3Au](μ-N3C2C6H4NO2) (3), {[Rh(CO)(PPh3)][PPh3Au](μ-N3C2C6H4NO2)}2 (4), and [(CO)(PPh3)2IrAuPPh3](μ-N3C2C6H4NO2) (6) have been synthesized via M-azide/M-acetylide cycloaddition reactions between PPh3Au(CCC6H4NO2) (2) and either Rh(CO)(PPh3)2N3 (1), or Ir(CO)(PPh3)2N3 (5). Complexes 3, 4, and 6 have been characterized by a combination of NMR spectroscopies, crystallography and combustion analysis.Gold-azides undergo cycloaddition (iClick) to group 9 azido complexes to give triazolate bridged heterobimetallic complexes.
Co-reporter:Sudarsan VenkatRamani, Christopher D. Roland, James G. Zhang, Ion Ghiviriga, Khalil A. Abboud, and Adam S. Veige
Organometallics 2016 Volume 35(Issue 16) pp:2675-2682
Publication Date(Web):July 19, 2016
DOI:10.1021/acs.organomet.6b00421
This report details the synthesis and characterization of a series of Nb complexes and one tantalum complex supported by the [CF3–ONO]3– trianionic pincer-type ligand. Access to the trianionic Nb dichloride complex, [CF3–ONO]NbCl2(OEt2) (3-Et2O), allows for the synthesis of Nb-dialkyl complexes, [CF3–ONO]NbR2 (where R = benzyl (5), neopentylsilyl (6), neophyl (7)). The sterically encumbered Nb-neophyl complex (7) is thermally stable and fails to convert to the alkylidene even in the presence of donor ligands. Complex 7, however, promotes catalytic ring-opening metathesis polymerization (ROMP) of norbornene, suggesting the plausible intermediacy of a Nb-alkylidene. The corresponding Ta analogue, [CF3–ONO]Ta(CH2C(CH3)2(C6H5))2 (9), requires dramatically higher temperatures to initiate ROMP and provides poor yields of polymer. Complexes 5 and 6 also promote ROMP of norbornene. Characterization of all new complexes includes multinuclear NMR spectroscopy and combustion analysis. For complex 7, characterization also includes solid-state structure elucidation via a single crystal X-ray diffraction experiment.
Co-reporter:Stella A. Gonsales; Matias E. Pascualini; Ion Ghiviriga; Khalil A. Abboud
Journal of the American Chemical Society 2015 Volume 137(Issue 14) pp:4840-4845
Publication Date(Web):March 20, 2015
DOI:10.1021/jacs.5b01599
The tungsten alkylidyne [CF3–ONO]W≡CC(CH3)3(THF)2 (3) {where CF3–ONO = (MeC6H3[C(CF3)2O])2N3–} supported by a trianionic pincer-type ligand demonstrates enhanced nucleophilicity in unusually fast “Wittig-like” reactions. Experiments are designed to provide support for an inorganic enamine effect that is the origin of the enhanced nucleophilicity. Treating complex 3 with various carbonyl-containing substrates provides tungsten-oxo-vinyl complexes upon oxygen atom transfer. The rates of reactivity of 3 are compared with the known alkylidyne (DIPP)3W≡CC(CH3)3 (DIPP = 2,6-diisopropylphenoxide). In all cases (except acetone), complex 3 exhibits significantly faster overall rates than (DIPP)3W≡CC(CH3)3. New oxo-vinyl complexes are characterized by NMR, combustion analysis and single crystal X-ray diffraction. Treating 3 with acid chlorides provides the tungsten oxo chloride species [CF3–ONO]W(O)Cl (4) and disubstituted alkynes. In the case of acetone the oxo-vinyl complex results in two rotational isomers 10syn and 10anti. The rate of isomerization was determined for the forward and reverse directions and was complimented with DFT calculations.
Co-reporter:M. E. Pascualini, N. V. Di Russo, A. E. Thuijs, A. Ozarowski, S. A. Stoian, K. A. Abboud, G. Christou and A. S. Veige
Chemical Science 2015 vol. 6(Issue 1) pp:608-612
Publication Date(Web):15 Oct 2014
DOI:10.1039/C4SC02634A
Square-planar high-spin Fe(II) molecular compounds are rare and the only three non-macrocyclic or sterically-driven examples reported share a common FeO4 core. Using an easily modifiable pincer-type ligand, the successful synthesis of the first compound of this type that breaks the FeO4 motif was achieved. In addition, we present the first evidence that geometry and spin state persist in solution. Extensive characterization includes the first high-field EPR and variable field/temperature Mössbauer spectra for this class of compounds. Analysis of the spectroscopic data indicates this complex exhibits a large and positive zero-field splitting tensor. Furthermore, the unusually small ΔEQ value determined for this compound is rationalized on the basis of DFT calculations.
Co-reporter:S. A. Gonsales, M. E. Pascualini, I. Ghiviriga and A. S. Veige
Chemical Communications 2015 vol. 51(Issue 69) pp:13404-13407
Publication Date(Web):21 Jul 2015
DOI:10.1039/C5CC04851F
The trianionic pincer supported tungsten–vinyl complex [CF3-ONO]W(O){(CH3)3CCC(CH3)2} (3syn) undergoes facile double bond rotation at ambient temperature. The degenerate methyl exchange rates were measured via selective inversion-recovery experiments. DFT computations in conjunction with experimentally determined rate constants support a double bond rotation that proceeds via a Zwiterionic transition state.
Co-reporter:Mary E. Garner, Weijia Niu, Xigao Chen, Ion Ghiviriga, Khalil A. Abboud, Weihong Tan and Adam S. Veige
Dalton Transactions 2015 vol. 44(Issue 4) pp:1914-1923
Publication Date(Web):14 Nov 2014
DOI:10.1039/C4DT02850C
This work describes several synthetic approaches to append organic functional groups to gold and silver N-heterocyclic carbene (NHC) complexes suitable for applications in biomolecule conjugation. Carboxylate appended NHC ligands (3) lead to unstable AuI complexes that convert into bis-NHC species (4). A benzyl protected carboxylate NHC–AuI complex 2 was synthesized but deprotection to produce the carboxylic acid functionality could not be achieved. A small library of new alkyne functionalized NHC proligands were synthesized and used for subsequent silver and gold metalation reactions. The alkyne appended NHC gold complex 13 readily reacts with benzyl azide in a copper catalyzed azide–alkyne cycloaddition reaction to form the triazole appended NHC gold complex 14. Cell cytotoxicity studies were performed on DLD-1 (colorectal adenocarcinoma), Hep-G2 (hepatocellular carcinoma), MCF-7 (breast adenocarcinoma), CCRF-CEM (human T-Cell leukemia), and HEK (human embryonic kidney). Complete spectroscopic characterization of the ligands and complexes was achieved using 1H and 13C NMR, gHMBC, ESI-MS, and combustion analysis.
Co-reporter:M. E. Pascualini, S. A. Stoian, A. Ozarowski, N. V. Di Russo, A. E. Thuijs, K. A. Abboud, G. Christou and A. S. Veige
Dalton Transactions 2015 vol. 44(Issue 46) pp:20207-20215
Publication Date(Web):29 Oct 2015
DOI:10.1039/C5DT03960F
High-spin square-planar molecular compounds are rare. In an effort to access this unique combination of geometry and spin state, we report the synthesis of a series of M(II) compounds stabilized by a trianionic pincer-type ligand, highlighting the formation of a high-spin square-planar Co(II) complex. Low-temperature, variable-frequency EPR measurements reveal that the ground electronic state of the Co(II) analogue is a highly anisotropic Kramers doublet (effective g values 7.35, 2.51, 1.48). This doublet can be identified with the lowest doublet of a quartet, S = 3/2 spin state that exhibits a very large ZFS, D ≥ 50 cm−1. The observation of an effective g value considerably greater than the largest spin-only value 6, demonstrates that the orbital angular moment is essentially unquenched along one spatial direction. Density Functional Theory (DFT) and time-dependent DFT calculations reveal the electronic configurations of the ground and excited orbital states. A qualitative crystal field description of the geff tensor shows that it originates from the spin–orbit coupling acting on states obtained through the transfer of a β electron from the doubly occupied xy to the singly-occupied {xz/yz} orbitals.
Co-reporter:Sudarsan VenkatRamani, Ion Ghiviriga, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2015 vol. 44(Issue 42) pp:18475-18486
Publication Date(Web):04 Sep 2015
DOI:10.1039/C5DT02911B
This report details the synthesis and characterization of the semi-flexible [ONCH2O]H3 (1) ligand and its W(VI)-alkylidene and alkylidyne complexes. The alkylidyne complex [ONHCH2O]WCtBu(OtBu) (2) forms as a result of alcoholysis of 1 with (tBuO)3WCtBu. Complex 2 evolves to [ONCH2O]WCHtBu(OtBu) (3) through proton migration from the N atom of the pincer ligand to the WCα bond. Deprotonation of 2 or 3 with Ph3PCH2 affords the anionic alkylidyne {CH3PPh3}{[ONCH2O]WCtBu(OtBu)} (4). Complex 4 exhibits pincer-ligand-centered reactivity with electrophiles (H+, Me+, and TMS+), in spite of its enhanced inorganic enamine interaction. Addition of 2 equiv. of HCl to 4 yields the W(VI)-neopentyl complex [ONCH2O]W(CH2tBu)(OtBu)(Cl) (5). MeOTf or TMSOTf addition to 4 generates the dianionic pincer ligated alkylidynes [ONRCH2O]WCtBu(OtBu) (R = Me (6-Me); TMS (6-TMS)). Complexes 2–5 were characterized by multinuclear NMR spectroscopy, and combustion analysis. Complexes 4 and 5 were also characterized by single crystal X-ray diffraction. This work bridges the gap in the series involving W(VI)-alkylidynes ligated to the rigid [CF3-ONO]3−, and the flexible [OCH2NCH2O]3− ligands. DFT computations permit comparison of the inorganic enamine effect within alkylidynes supported by all three trianionic-pincer type ONO ligands.
Co-reporter:Xi Yang, Shanshan Wang, Ion Ghiviriga, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2015 vol. 44(Issue 25) pp:11437-11443
Publication Date(Web):19 May 2015
DOI:10.1039/C5DT00282F
A novel synthetic method to create gold based metallo–oligomers/polymers via the combination of inorganic click (iClick) with intermolecular aurophilic interactions is demonstrated. Complexes [PEt3Au]4(μ-N3C2C6H5) (1) and [PPhMe2Au]4(μ-N3C2C6H5) (2) and {[PEt3Au]4[(μ-N3C2)2-9,9-dihexyl-9H-fluorene]}n (8) have been synthesized via iClick. The tetranuclear structures of 1 and 2, induced by aurophilic bonding, are confirmed in the solid state through single crystal X-ray diffraction experiments and in solution via variable temperature NMR spectroscopy. The extended 1D structure of 8 is constructed by aurophilic induced self-assembly. 1H DOSY NMR analysis reveals that the aurophilic bonds in 1, 2, and 8 are retained in the solution phase. The degree of polymerization within complex 8 is temperature and concentration dependent, as determined by 1H DOSY NMR. Complex 8 is a rare example of a solution stable higher ordered structure linked by aurophilic interactions.
Co-reporter:Andrew R. Powers, Ion Ghiviriga, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2015 vol. 44(Issue 33) pp:14747-14752
Publication Date(Web):20 Jul 2015
DOI:10.1039/C5DT02405F
This report outlines the investigation of the iClick mechanism between gold(I)–azides and gold(I)–acetylides to yield digold triazolates. Isolation of digold triazolate complexes offer compelling support for the role of two copper(I) ions in CuAAC. In addition, a kinetic investigation reveals the reaction is first order in both Au(I)–N3 and Au(I)–CC–R, thus second order overall. A Hammett plot with a ρ = 1.02(5) signifies electron-withdrawing groups accelerate the cycloaddition by facilitating the coordination of the second gold ion in a π-complex. Rate inhibition by the addition of free triphenylphosphine to the reaction indicates that ligand dissociation is a prerequisite for the reaction. The mechanistic conclusions mirror those proposed for the CuAAC reaction.
Co-reporter:Soufiane S. Nadif;Dr. Matthew E. O'Reilly;Dr. Ion Ghiviriga;Dr. Khalil A. Abboud ; Adam S. Veige
Angewandte Chemie International Edition 2015 Volume 54( Issue 50) pp:15138-15142
Publication Date(Web):
DOI:10.1002/anie.201507008
Abstract
A chemically non-innocent pyrrole-based trianionic (ONO)3− pincer ligand within [(pyr-ONO)TiCl(thf)2] (2) can access the dianionic [(3H-pyr-ONO)TiCl2(thf)] (1-THF) and monoanionic [(3H,4H-pyr-ONO)TiCl2(OEt2)][B{3,5-(CF3)2C6H3}4] (3-Et2O) states through remote protonation of the pyrrole γ-C π-bonds. The homoleptic [(3H-pyr-ONO)2Zr] (4) was synthesized and characterized by X-ray diffraction and NMR spectroscopy in solution. The protonation of 4 by [H(OEt2)2][B{C6H3(CF3)2}4] yields [(3H,4H-pyr-ONO)(3H-pyr-ONO)Zr][B{3,5-(CF3)2C6H3}4] (5), thus demonstrating the storage of three protons.
Co-reporter:Soufiane S. Nadif;Dr. Matthew E. O'Reilly;Dr. Ion Ghiviriga;Dr. Khalil A. Abboud ; Adam S. Veige
Angewandte Chemie 2015 Volume 127( Issue 50) pp:15353-15357
Publication Date(Web):
DOI:10.1002/ange.201507008
Abstract
A chemically non-innocent pyrrole-based trianionic (ONO)3− pincer ligand within [(pyr-ONO)TiCl(thf)2] (2) can access the dianionic [(3H-pyr-ONO)TiCl2(thf)] (1-THF) and monoanionic [(3H,4H-pyr-ONO)TiCl2(OEt2)][B{3,5-(CF3)2C6H3}4] (3-Et2O) states through remote protonation of the pyrrole γ-C π-bonds. The homoleptic [(3H-pyr-ONO)2Zr] (4) was synthesized and characterized by X-ray diffraction and NMR spectroscopy in solution. The protonation of 4 by [H(OEt2)2][B{C6H3(CF3)2}4] yields [(3H,4H-pyr-ONO)(3H-pyr-ONO)Zr][B{3,5-(CF3)2C6H3}4] (5), thus demonstrating the storage of three protons.
Co-reporter:Soufiane S. Nadif, Jakub Pedziwiatr, Ion Ghiviriga, Khalil A. Abboud, and Adam S. Veige
Organometallics 2015 Volume 34(Issue 6) pp:1107-1117
Publication Date(Web):March 4, 2015
DOI:10.1021/acs.organomet.5b00036
This report describes the synthesis and characterization of a new series of group 4 complexes supported by a trianionic ONO3– pincer-type ligand. Treating TiCl4 with the proligand [CF3–ONO]H3 (1) and NEt3 in benzene afforded {[CF3–ONO]TiCl3}{HNEt3}2 (2). By means of a lithium transmetalation route, the neutral monochloride complex [CF3–ONO]TiCl(THF) (3) was synthesized in 91% yield. The analogous Hf(IV) derivative could not be obtained using this method. Instead, transmetalation with thallium(I) resulted in the formation of the seven-coordinate complex [CF3–ONHO]HfCl2(THF)2 (4-(THF)2), which was characterized by combustion analysis and X-ray crystallography. Applying vacuum to 4-(THF)2 liberated the THF ligands to provide the five-coordinate THF-free complex [CF3–ONHO]HfCl2 (4). Alkylation of complex 4 with alkyllithium or Grignard reagents resulted in a mixture of unidentifiable products. However, access to the neutral complex 3 enabled the subsequent preparation of organotitanium complexes [CF3–ONO]TiR(THF) (5-R; R = Me, Bn, Mes). Single-crystal X-ray analysis of 5-Me indicated that the organotitanium complexes are mononuclear. Single-crystal X-ray diffraction and NMR studies in solution confirmed that complex 5-Mes exhibits rare through-space 19F–19F coupling (5 Hz).
Co-reporter:Sudarsan VenkatRamani, Nicholas B. Huff, Muhammad Tariq Jan, Ion Ghiviriga, Khalil A. Abboud, and Adam S. Veige
Organometallics 2015 Volume 34(Issue 12) pp:2841-2848
Publication Date(Web):May 28, 2015
DOI:10.1021/acs.organomet.5b00155
Metal–carbon multiple bonds exhibit enhanced nucleophilicity at the α carbon within ONO trianionic pincer alkylidyne complexes. Defined as the inorganic enamine effect, this phenomenon is a result of the overlap of the N atom lone pair within the ONO3– ligand and a π bond from the metal–carbon multiple bond. Treating the proligand [OCH2NCH2O]H3 (2) with (tBuO)3W≡CR (where R = Et, tBu) results in the formation of the dianionic pincer complexes [OCH2NHCH2O]W≡CR(OtBu) (where R = Et (3-Et), tBu (3-tBuanti)). Deprotonation of 3-tBuanti by treatment with Ph3P═CH2 forms the anionic alkylidyne complex {[OCH2NCH2O]W≡CtBu(OtBu)}{CH3PPh3} (4-tBu). DFT calculations modeling 4-tBu reveal overlap of the N atom lone pair with a W≡C π bond. However, 4-tBu reacts with electrophiles preferentially at the pincer N atom as opposed to the W≡Cα group. Multinuclear NMR spectroscopy, combustion analysis, and single-crystal X-ray crystallography are employed to characterize complexes 3-Et, 3-tBuanti, and 4-tBu.
Co-reporter:Matthew E. O'Reilly and Adam S. Veige
Chemical Society Reviews 2014 vol. 43(Issue 17) pp:6325-6369
Publication Date(Web):13 Jun 2014
DOI:10.1039/C4CS00111G
Trianionic pincer and pincer-type ligands are the focus of this review. Metal ions from across the periodic table, from main group elements, transition metals, and the rare earths, are combined with trianionic pincer ligands to produce some of the most interesting complexes to appear in the literature over the past decade. This review provides a comprehensive examination of the synthesis, characterization, properties, and catalytic applications of trianionic pincer metal complexes. Some of the interesting applications employing trianionic pincer and pincer-type complexes include: (1) catalyzed aerobic oxidation, (2) alkene isomerization, (3) alkene and alkyne polymerization, (4) nitrene and carbene group transfer, (5) fundamental transformations such as oxygen-atom transfer, (6) nitrogen-atom transfer, (7) O2 activation, (8) C–H bond activation, (9) disulfide reduction, and (10) ligand centered storage of redox equivalents (i.e. redox active ligands). Expansion of the architecture, type of donor atoms, chelate ring size, and steric and electronic properties of trianionic pincer ligands has occurred rapidly over the past ten years. This review is structured according to the type of pincer donor atoms that bind to the metal ion. The type of donor atoms within trianionic pincer and pincer-type ligands to be discussed include: NCN3−, OCO3−, CCC3−, redox active NNN3−, NNN3−, redox active ONO3−, ONO3−, and SNS3−. Since this is the first review of trianionic pincer and pincer-type ligands, an emphasis is placed on providing the reader with in-depth discussion of synthetic methods, characterization data, and highlights of these complexes as catalysts.
Co-reporter:Matthew E. O’Reilly, Soufiane S. Nadif, Ion Ghiviriga, Khalil A. Abboud, and Adam S. Veige
Organometallics 2014 Volume 33(Issue 4) pp:836-839
Publication Date(Web):November 1, 2013
DOI:10.1021/om4009422
Synthetic protocols for a pyrrolide-centered ONO3– trianionic pincer-type ligand are presented. Treating (tBuO)3W≡CtBu with the proligand [pyr-ONO]H3 (2) results in the formation of the trianionic pincer alkylidene complex [pyr-ONO]W═CHtBu(OtBu) (3). Addition of a mild base to complex 3 provides the trianionic pincer alkylidyne complex {MePPh3}{[pyr-ONO]W≡CtBu(OtBu)} (4). All new compounds were characterized by NMR spectroscopy, combustion analysis, and, in the case of complex 4, single-crystal X-ray crystallography. DFT calculations performed on 4 provide insight into its electronic structure and indicate that the HOMO is ligand-based and localized on the pyrrolide π orbitals.
Co-reporter:Kevin P. McGowan, Matthew E. O'Reilly, Ion Ghiviriga, Khalil A. Abboud and Adam S. Veige
Chemical Science 2013 vol. 4(Issue 3) pp:1145-1155
Publication Date(Web):03 Dec 2012
DOI:10.1039/C2SC21750C
Complex [tBuOCO]WC(tBu)(THF)2 (1) {where tBuOCO = [2,6-(tBuC6H3O)2C6H3]3−, THF = tetrahydrofuran} polymerizes acetylenes (R-phenylacetylene (R = H, p-OMe, p-F, 3,5-diCF3), 1-decyne, 3,3-dimethyl-1-butyne, and trimethylsilylacetylene) to form π-conjugating polymers. Upon treating 1 with 2 equiv. of phenylacetylene in toluene-d8 at −35 °C, two isolable products form. These two products are [O2C(tBuC)W(η2-HCCPh)] (2-tttBu) and [O2C(PhC)W(η2-HCCtBu)] (2-Ph) {where OC(tBuC)O = [2,6-(tBuC6H3O)2C6H3(tBuC)]4−, OC(PhC)O = [2,6-(tBuC6H3O)2C6H3(PhC)]4−} and derived from an apparent reductive alkylidyne migratory insertion into a metal–arene bond. Complexes 2-tttBu and 2-Ph polymerize acetylene and a wide variety of monosubstituted acetylenes including phenylacetylene derivatives, 1-decyne, 3,3-dimethyl-1-butyne and trimethylsilylacetylene. With a substrate to catalyst loading ratio of 25000:1, complex 2-tttBu polymerizes phenylacetylene with a turnover number (TON) of 17233. Additionally, 2-tttBu polymerizes phenylacetylene and 1-decyne with catalytic activities up to 5.64 × 106 gPPA mol−1 h−1 and 7.98 × 106 gPA mol−1 h−1, respectively. 2-tttBu also polymerizes the disubstituted acetylene, 1-phenyl-1-propyne. NMR spectroscopic and single crystal X-ray structural studies provide compelling evidence for polymer chain growth via an insertion ring-expansion mechanism.
Co-reporter:Andrew R. Powers, Xi Yang, Trevor J. Del Castillo, Ion Ghiviriga, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2013 vol. 42(Issue 42) pp:14963-14966
Publication Date(Web):04 Sep 2013
DOI:10.1039/C3DT52105B
Metal-azide–metal-acetylide cycloaddition (iClick) reactions to synthesize heterotrimetallics and an unexpected novel tetranuclear gold(I) complex, are described. In addition, a discussion regarding the connection between traditional azide–alkyne cycloaddition reactions and iClick is presented focusing on applications towards linking multiple metal ions.
Co-reporter:Matthew E. O'Reilly, Ion Ghiviriga, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2013 vol. 42(Issue 10) pp:3326-3336
Publication Date(Web):18 Dec 2012
DOI:10.1039/C2DT32653A
This report presents the synthesis of the first neutral trianionic ONO pincer-type tungsten alkylidyne complex, [CF3–ONO]WC(tBu)(OEt2) (5) {where CF3–ONO = (MeC6H3[C(CF3)2O])2N3−}. Treating 5 with 1-phenylpropyne, 4,4-dimethyl-2-pentyne, and cyclooctyne yields the corresponding tungstenacyclobutadiene complexes [CF3–ONO]W[κ2-C(tBu)C(Me)C(Ph)] (6), [CF3–ONO]W[κ2-C(tBu)C(Me)C(tBu)] (7), and [CF3–ONO]W[κ2-C(tBu)C(CH2)6C] (8). Complexes 6, 7, and 8 do not undergo retro-[2 + 2]-cycloaddition even at 200 °C or in the presence of PMe3. DFT methods to elucidate the electronic structure of complexes 5 and 6 reveal important electronic factors that contribute to the lack of reactivity for the tungstenacyclobutadienes. An important bonding combination between the pincer N-atom lone pair and the WC bond within 5, termed an inorganic enamine, provides an explanation for the lack of retro-[2 + 2]-cycloaddition from 6, 7, and 8. 15N NMR spectroscopy was used to confirm the computational finding of an inorganic enamine bonding combination. Single crystal X-ray analysis of 5, 6, 7, and 8 provides insight into possible steric inadequacies within the CF3–ONO3− ligand to promote catalytic metathesis.
Co-reporter:Amrita B. Mullick, Yun Min Chang, Ion Ghiviriga, Khalil A. Abboud, Weihong Tan and Adam S. Veige
Dalton Transactions 2013 vol. 42(Issue 20) pp:7440-7446
Publication Date(Web):19 Feb 2013
DOI:10.1039/C3DT32844A
A novel eighteen membered chiral macrocyclic dicarbene–digold complex [(μ-diNHC)AuI]2[OTF]2(8-(+/−)) was synthesized and characterized. Starting with enantiopure diNHC imidazolium salt ligand precursors enables access to the enantiopure versions of the digold(I) metallamacrocycles, 8-(+) and 8-(−). In vitro cytotoxicity studies indicate 8-(+/−) is moderately cytotoxic to both healthy and cancerous cell-lines, with no specificity. Confocal microscopy indicates the digold metallamacrocycle penetrates the cell membrane and causes cell death via apoptosis, as evidenced by DNA electrophoresis. The complex 8-(+/−) is characterized by a combination of NMR techniques (gDQCOSY, gHSQC, gHMBC and ROESY), single crystal X-ray diffraction, and combustion analysis.
Co-reporter:Sudarsan VenkatRamani, Matias E. Pascualini, Ion Ghiviriga, Khalil A. Abboud, Adam S. Veige
Polyhedron 2013 64() pp: 377-387
Publication Date(Web):
DOI:10.1016/j.poly.2013.07.004
Co-reporter:Soumya Sarkar ; Kevin P. McGowan ; Subramaniam Kuppuswamy ; Ion Ghiviriga ; Khalil A. Abboud
Journal of the American Chemical Society 2012 Volume 134(Issue 10) pp:4509-4512
Publication Date(Web):February 21, 2012
DOI:10.1021/ja2117975
Synthesis, characterization, and catalytic alkyne polymerization results for the first trianionic pincer alkylidyne complex, [tBuOCO]W≡CC(CH3)3(THF)2 (6), are described. Complex 6 is a highly active catalyst for the polymerization of acetylenes and exhibits a high turnover number (4371), activity (1.05 × 106 gPPA molcat–1 h–1),and yield (87%) for the polymerization of 1-ethynyl-4-fluorobenzene.
Co-reporter:Matthew E. O’Reilly ; Ion Ghiviriga ; Khalil A. Abboud
Journal of the American Chemical Society 2012 Volume 134(Issue 27) pp:11185-11195
Publication Date(Web):June 8, 2012
DOI:10.1021/ja302222s
Appending an amine to a C═C double bond drastically increases the nucleophilicity of the β-carbon atom of the alkene to form an enamine. In this report, we present the synthesis and characterization of a novel CF3–ONO3- trianionic pincer-type ligand, rationally designed to mimic enamines within a metal coordination sphere. Presented is a synthetic strategy to create enhanced nucleophilic tungsten–alkylidene and −alkylidyne complexes. Specifically, we present the synthesis and characterization of the new CF3–ONO3- trianionic pincer tungsten–alkylidene [CF3–ONO]W═CH(Et)(OtBu) (2) and −alkylidyne {MePPh3}{[CF3–ONO]W≡C(Et)(OtBu)} (3) complexes. Characterization involves a combination of multinuclear NMR spectroscopy, combustion analysis, DFT computations, and single crystal X-ray analysis for complexes 2 and 3. Exhibiting unique nucleophilic reactivity, 3 reacts with MeOTf to yield [CF3–ONO]W═C(Me)(Et)(OtBu) (4), but the bulkier Me3SiOTf silylates the tert-butoxide, which subsequently undergoes isobutylene expulsion to form [CF3–ONO]W═CH(Et)(OSiMe3) (5). A DFT calculation performed on a model complex of 3, namely, [CF3–ONO]W≡C(Et)(OtBu) (3′), reveals the amide participates in an enamine-type bonding combination. For complex 2, the Lewis acids MeOTf, Me3SiOTf, and B(C6F5)3 catalyze isobutylene expulsion to yield the tungsten–oxo complex [CF3–ONO]W(O)(nPr) (6).
Co-reporter:Matthew E. O'Reilly, Trevor J. Del Castillo, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2012 vol. 41(Issue 8) pp:2237-2246
Publication Date(Web):18 Nov 2011
DOI:10.1039/C1DT11104C
The oxygen-atom-transfer (OAT) from [tBuOCO]CrV(O)(THF) (2) (where tBuOCO = [2,6-C6H3(6-tBuC6H3O)2]3−, THF = tetrahydrofuran) to triphenylphosphine (PPh3) in THF produces [tBuOCO]CrIII(THF)3 (1) and triphenylphosphine oxide (OPPh3) at a rate of 69.5(±1.9) M−1s−1 (22 °C). Identical rate constants were attained when acetonitrile (MeCN) and dichloromethane/THF (CH2Cl2/THF) were used as solvents. Electron paramagnetic resonance (EPR) data shows that the six-coordinate complex, [tBuOCO]CrV(O)(THF)2 (2a) forms upon addition of THF to 2, suggesting 2a as the active OAT species in THF. Similarly, addition of OPPh3 has no influence on the rate of OAT, but the addition of triphenylphosphorus ylide (CH2PPh3) to form [tBuOCO]CrV(O)(CH2PPh3) (4) prevents OAT to PPh3. In CH2Cl2, a [CrIV]2(μ-O) intermediate forms during the OAT from 2 to PPh3. The OAT from {[tBuOCO]CrIV(THF)}2(μ-O) (3) to PPh3 reveals a zero-order dependence in PPh3 indicating the dimer must first dissociate prior to OAT. The decay of 3versus time does not follow first-order kinetics due to the formation of a [tBuOCO]CrIII(THF) species (5) that inhibits the dissociation of 3. The change in concentration of 3versus time during OAT was simulated to obtain approximate rate constants.
Co-reporter:Kevin P. McGowan, Adam S. Veige
Journal of Organometallic Chemistry 2012 711() pp: 10-14
Publication Date(Web):
DOI:10.1016/j.jorganchem.2012.03.007
Co-reporter:Matthew E. O’Reilly ; Trevor J. Del Castillo ; Joseph M. Falkowski ; Vasanth Ramachandran ; Mekhala Pati ; Marie C. Correia ; Khalil A. Abboud ; Naresh S. Dalal ; David E. Richardson
Journal of the American Chemical Society 2011 Volume 133(Issue 34) pp:13661-13673
Publication Date(Web):July 22, 2011
DOI:10.1021/ja2050474
Synthetic and kinetic experiments designed to probe the mechanism of O2 activation by the trianionic pincer chromium(III) complex [tBuOCO]CrIII(THF)3 (1) (where tBuOCO = [2,6-(tBuC6H3O)2C6H3]3–, THF = tetrahydrofuran) are described. Whereas analogous porphyrin and corrole oxidation catalysts can become inactive toward O2 activation upon dimerization (forming a μ-oxo species) or product inhibition, complex 1 becomes more active toward O2 activation when dimerized. The product from O2 activation, [tBuOCO]CrV(O)(THF) (2), catalyzes the oxidation of 1 via formation of the μ-O dimer {[tBuOCO]CrIV(THF)}2(μ-O) (3). Complex 3 exists in equilibrium with 1 and 2 and thus could not be isolated in pure form. However, single crystals of 3 and 1 co-deposit, and the molecular stucture of 3 was determined using single-crystal X-ray crystallography methods. Variable (9.5, 35, and 240 GHz) frequency electron paramagnetic resonance spectroscopy supports the assignment of complex 3 as a CrIV–O–CrIV dimer, with a high (S = 2) spin ground state, based on detailed computer simulations. Complex 3 is the first conclusively assigned example of a complex containing a Cr(IV) dimer; its spin Hamiltonian parameters are giso = 1.976, D = 2400 G, and E = 750 G. The reaction of 1 with O2 was monitored by UV–visible spectrophotometry, and the kinetic orders of the reagents were determined. The reaction does not exhibit first-order behavior with respect to the concentrations of complex 1 and O2. Altering the THF concentration reveals an inverse order behavior in THF. A proposed autocatalytic mechanism, with 3 as the key intermediate, was employed in numerical simulations of concentration versus time decay plots, and the individual rate constants were calculated. The simulations agree well with the experimental observations. The acceleration is not unique to 2; for example, the presence of OPPh3 accelerates O2 activation by forming the five-coordinate complex trans-[tBuOCO]CrIII(OPPh3)2 (4).
Co-reporter:Trevor J. Del Castillo, Soumya Sarkar, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2011 vol. 40(Issue 32) pp:8140-8144
Publication Date(Web):04 Jul 2011
DOI:10.1039/C1DT10787A
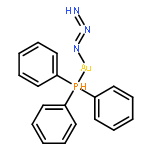
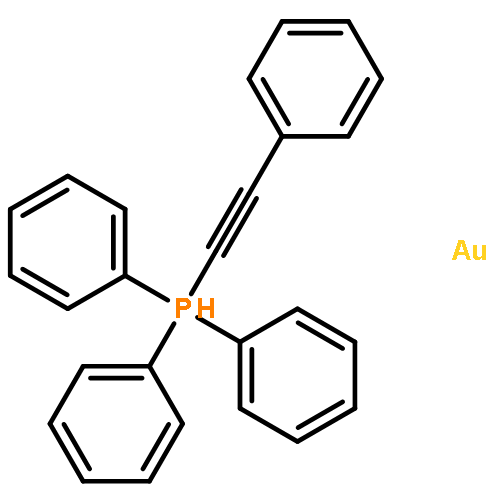
This report describes the synthesis and characterization of 1,5-bis-triphenylphosphinegold(I) 1,2,3-triazolate (3(1,5)). The synthesis of the dinuclear complex 3(1,5) is achieved via an unprecedented inorganic click (iClick) reaction between the metal–azide PPh3AuN3 (1) and the metal–acetylide PPh3Au–CCPh (2). Characterization of 3(1,5) includes multinuclear NMR spectroscopy, combustion analysis, and single crystal X-ray crystallography. Experimental characterization is complemented with density-functional-theory (DFT) calculations which indicate the 1,4-isomer 3(1,4) is less stable by 3.3 kcal mol−1. The energetic difference lies primarily in the ability of the phenyl group in the 4-position of 3(1,5) to lie coplanar with the triazolate to create a delocalized π-bonding HOMO orbital.
Co-reporter:Matthew S. Jeletic, Roxy J. Lowry, Jason M. Swails, Ion Ghiviriga, Adam S. Veige
Journal of Organometallic Chemistry 2011 696(20) pp: 3127-3134
Publication Date(Web):
DOI:10.1016/j.jorganchem.2011.05.015
Co-reporter:Matthew S. Jeletic, Claire E. Lower, Ion Ghiviriga, and Adam S. Veige
Organometallics 2011 Volume 30(Issue 21) pp:6034-6043
Publication Date(Web):October 18, 2011
DOI:10.1021/om200913c
A series of eight [(diNHC)Rh(CO)2][OTf] complexes, 1′-R, 2′-R, and 3′-R (where diNHC = DEAM, trans-9,10-dihydro-9,10-ethanoanthracene-11,12-diylmethanediyl carbene, and DEA, trans-9,10-dihydro-9,10-ethanoanthracene-11,12-diyl carbene ligands; R = R-methylphenylmethane (R-CHMePh), diphenylmethane (diPh), benzyl (Bn), methyl (Me), isopropyl (iPr), and o-methylbenzyl (o-MeBn); carbene heterocycle = imidazol-2-ylidene, and benzimidazol-2-ylidene), are synthesized and characterized. The purpose for the synthesis of these complexes was to examine the relationship between carbene N-heterocycle, R-group substitution, and metallocycle ring size (DEAM = 9, DEA = 7 atom ring) on ligand flexibility. The complexes were examined by chemical exchange saturation transfer to measure the rate of conformational isomerization. The results indicate that flexibility is not significantly altered by choice of carbene heterocycle and R group. However, the constrained DEA diNHC derivative does not undergo conformational isomerization.
Co-reporter:Kevin P. McGowan, Khalil A. Abboud, and Adam S. Veige
Organometallics 2011 Volume 30(Issue 18) pp:4949-4957
Publication Date(Web):August 29, 2011
DOI:10.1021/om200547u
The synthesis and characterization of a series of four Cr complexes in the +2, +3, +4, and +5 oxidation states supported by an NCN trianionic pincer ligand are reported. Treating CrMeCl2(THF)3 with the dilithio salt pincer ligand precursor {[2,6-iPrNCHN]Li2}2 provides the CrIII complex [2,6-iPrNCN]CrIII(THF)3 (1), CrIV complex [2,6-iPrNCN]CrIVMe(THF) (2), and CrII complex [2,6-iPrNHCN]CrII(THF)2 (3). Complexes 2 and 3 are the result of disproportionation. Treating 1 with 1 equiv of styrene oxide in THF converts the CrIII complex to the CrV(O) species [2,6-iPrNCN]CrV(O)(THF) (4). Complex 2, characterized by single-crystal X-ray diffraction, is a rare CrIV methyl complex that is kinetically stable at 25 °C; at 85 °C, Cr–Me bond homolysis occurs. The homolytic cleavage results in CH4 formation and biphenyl via a radical mechanism. The metal-containing product from thermolysis is the same CrII species formed during metalation, except one of the protons is substituted for a deuterium from C6D6 (3-d). Complex 2 is a precatalyst for the selective isomerization of 1-hexene and 1-octene to the corresponding 2-alkenes. An induction period occurs during the catalytic isomerization, and the active catalyst was determined to be the CrII complex 3, not 2.
Co-reporter:Soumya Sarkar, Kevin P. McGowan, Jeffrey A. Culver, Adam R. Carlson, Jürgen Koller, Andrew J. Peloquin, Melanie K. Veige, Khalil A. Abboud and Adam S. Veige
Inorganic Chemistry 2010 Volume 49(Issue 11) pp:5143-5156
Publication Date(Web):April 29, 2010
DOI:10.1021/ic100232s
This report details the synthesis of new NCN trianionic pincer ligand precursors and metalation reactions to form group (IV) complexes. N,N′-[1,3-phenylenebis(methylene)]bis-2,6-diisopropylaniline [2,6-iPrNCN]H3 (8) was converted to the N,N′-substituted Si(IV), Sn(IV), Mg(II), and Zn(II) derivatives. [2,6-iPrNCHN](SiMe3)2 (9-Si) and [2,6-iPrNCHN](SnMe3)2 (9-Sn) form by first treating 8 with MeLi followed by Me3MCl, where M = Si or Sn. Single crystal X-ray experiments indicate 8, 9-Si, and 9-Sn have similar structural features in the solid state. [2,6-iPrNCHN](μ-MgCl·THF)2 (12) forms by treating 8 with MeMgCl, and its solid state structure revealed a bis-μ-MgCl bridging unit. The 1H NMR spectrum of 12 reveals a dynamic process occurs in solution. A variable temperature 1H NMR experiment failed to quench the dynamic process. {[2,6-iPrNCHN]Zn}2 (13) forms upon treating {[2,6-iPrNCHN]Li2}2 (10) with anhydrous ZnCl2 and is a dimer in the solid state. Again, dynamic 1H NMR behavior is observed, and a mechanism is provided to explain the apparent low symmetry of 13 in solution. Extension of the aliphatic arm of the NCN ligand provides the new NCCCN pincer ligand precursors N,N′-(2,2′-(1,3-phenylene)bis(ethane-2,1-diyl))bis(3,5-bis(trifluoromethyl)aniline) [3,5-CF3NCCCN]H3 (16) and [3,5-CF3NCCHCN](SiMe3)2 (17). A more rigid ligand architecture was accessed by synthesis of the anthracene derived pincer ligand anthracene-1,8-diylbis(N-3,5-bistrifluormethylaniline) [3,5-CF3NCCanthCN]H3 (18). Treating {Zr(NMe2)4}2 with 2 equiv of 16 provides the dimer {(μ-3,5-CF3NCCHCN)Zr(NMe2)3NHMe2}2 (19). Treating Hf(NMe2)4 with 18 provides the bimetallic complex (μ-3,5-CF3NCCHanthCN){Hf(NMe2)3NHMe2}2 (20) in which one ligand bridges two Hf(IV) ions. Salt metathesis between 10 and ZrCl2(NMe2)2(THF)2 provides the mononuclear complex [2,6-iPrNCHN]Zr(NMe2)2 (21) in which the NCN ligand is bound as a chelating diamide. Thermoysis of 21 does not lead to formation of a trianionic pincer complex. Instead, treating HfCl4 with {[2,6-iPrNCN]Li3}2 (11) followed by MeLi provides the trianionic pincerate complex [2,6-iPrNCNHfMe2][Li(DME)2] (23). In the solid state the Hf ion has distorted trigonal bipyramidal geometry.
Co-reporter:Matthew S. Jeletic, Ion Ghiviriga, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2010 vol. 39(Issue 28) pp:6392-6394
Publication Date(Web):21 Jun 2010
DOI:10.1039/C0DT00268B
This report details the synthesis and characterization of the first chiral diNHC cyclophane ligand and its palladium allyl complex. The complex was tested for 1,4-conjugate addition of phenylboronic acid to 2-cyclohexen-1-one and provides R-3-phenylcyclohexanone in 50% enantiomeric excess.
Co-reporter:Subramaniam Kuppuswamy, Ion Ghiviriga, Khalil A. Abboud, and Adam S. Veige
Organometallics 2010 Volume 29(Issue 24) pp:6711-6722
Publication Date(Web):November 24, 2010
DOI:10.1021/om100732d
This report describes the synthesis and characterization of new zirconium benzyl complexes supported by a trianionic pincer ligand. Treating Zr(CH2Ph)4 with the terphenyldiol [tBuOCO]H3 (1) in benzene affords [tBuOCHO]Zr(CH2Ph)2 (2). Thermolysis of 2 leads to formation of dinuclear {[tBuOCO]ZrCH2Ph}2 (3), which contains a five-coordinate zirconium center in a trigonal-bipyramidal (tbp) geometry. Adding 2 equiv of PMe3 to 2 affords [tBuOCO]ZrCH2Ph(PMe3)2 (4-PMe3). X-ray crystallographic analysis reveals strong agostic interactions for the benzyl ligand in both 3 and 4-PMe3. Treatment of 2 with 2 equiv of THF results in hydrocarbon-soluble [tBuOCO]ZrCH2Ph(THF)2 (5). NMR spectroscopic measurements indicate a C2v-symmetric molecule that is isostructural with 4-PMe3. Addition of the larger phosphine PMe2Ph to 2 provides the corresponding mono-PMe2Ph complex in solution, but a small amount of the bis-diphenolate [tBuOCHO]2Zr (6) also forms. Complex 6 was independently synthesized by treating Zr(CH2Ph)4 with 2 equiv of 1. When 2 equiv of pyridine (py) is added to 2, the intermediate [tBuOCHO]ZrCH2Ph(η2−C5H4N)py (7) results from pyridine o-C−H bond activation. After 48 h, complex 7 releases another 1 equiv of toluene by activation of the pincer Cipso−H bond to produce the trianionic pincer complex [tBuOCO]Zr(η2-C5H4N)(py)2 (8). Multinuclear and 2D NMR spectroscopic experiments and combustion analysis support the molecular assignment of 8. Addition of α-picoline to 2 provides different results compared to py. Initially, the α-picoline adduct [tBuOCHO]Zr(CH2Ph)2(α-picoline) (9) forms. Addition of another 1 equiv of α-picoline provides the mixed η2(N,C)-6-Me-pyridyl)/η2(N,C-CH2)-pyrid-2-yl complex [tBuOCHO]Zr(η2(N,C)-6-Me-pyridyl)(η2(N,C-CH2)-pyrid-2-yl) (10).
Co-reporter:Roxy J. Lowry, Muhammad T. Jan, Khalil A. Abboud, Ion Ghiviriga, Adam S. Veige
Polyhedron 2010 29(1) pp: 553-563
Publication Date(Web):
DOI:10.1016/j.poly.2009.06.075
Co-reporter:Subramaniam Kuppuswamy, Andrew J. Peloquin, Ion Ghiviriga, Khalil A. Abboud, and Adam S. Veige
Organometallics 2010 Volume 29(Issue 19) pp:4227-4233
Publication Date(Web):September 2, 2010
DOI:10.1021/om100189b
The synthesis and characterization of trianionic [tBuOCO]3− pincer-supported tungsten alkylidene and alkylidyne complexes are described. The reaction of an equimolar ratio of (tBuO)3W≡CC(CH3)3 (where tBuO = tert-butoxide) with [tBuOCO]H3 (9) and 2,6-diisopropylphenol affords the alkylidene [tBuOCO]W═CHC(CH3)3(O-2,6-C6H3-iPr2) (10), with a five-coordinate tungsten center that adopts a distorted square-pyramidal geometry. Treatment of (Np)3W≡CC(CH3)3 (11) with 9 provides an equilibrium mixture of the two isomeric alkylidenes with the general formula [(tBuOCO)W═CHC(CH3)3(μ-tBuOCHO)W═CHC(CH3)3(tBuOCO)] (12kin and 12therm). Single crystals of the two isomers cocrystallize and were amenable to X-ray diffraction studies, which revealed subtle differences in their molecular structures, most notably the orientation of the bridging ligand. The complexes are each comprised of two square-pyramidal tungsten ions linked by the diphenolate form of the OCO ligand. Isomer 12kin could not be isolated independently; however, adding PMe2Ph to the metalation between 11 and 9 provided 12therm exclusively, thus enabling a full set of characterization techniques including 2-D NMR spectroscopy. Salt metathesis between [tBuOCHO]K2(THF)2 (15) and (DME)Cl3W≡CC(CH3)3 (16) in diethyl ether produces the alkylidyne (tBuOCHO)W≡CC(CH3)3Cl (17) as the major product along with 12kin and other unidentified decomposition products. As a consequence, characterization of 17 was limited to 1H NMR spectroscopy and mass spectrometry.
Co-reporter:Soumya Sarkar;Jeffrey A. Culver;Andrew J. Peloquin;Ion Ghiviriga;Dr. Khalil A. Abboud ; Adam S. Veige
Angewandte Chemie International Edition 2010 Volume 49( Issue 50) pp:9711-9714
Publication Date(Web):
DOI:10.1002/anie.201004233
Co-reporter:Soumya Sarkar;Jeffrey A. Culver;Andrew J. Peloquin;Ion Ghiviriga;Dr. Khalil A. Abboud ; Adam S. Veige
Angewandte Chemie 2010 Volume 122( Issue 50) pp:9905-9908
Publication Date(Web):
DOI:10.1002/ange.201004233
Co-reporter:Matthew O’Reilly ; Joseph M. Falkowski ; Vasanth Ramachandran ; Mekhala Pati ; Khalil A. Abboud ; Naresh S. Dalal ; Thomas G. Gray
Inorganic Chemistry 2009 Volume 48(Issue 23) pp:10901-10903
Publication Date(Web):November 6, 2009
DOI:10.1021/ic9019469
Aerobic oxidation that incorporates both O atoms into a substrate (PPh3) is achieved by employing a CrIII/CrV≡O catalytic couple. A terphenyl trianionic pincer ligand stabilizes a high oxidation state CrV≡O complex, and both the reduced (CrIII, IR/X-ray) and oxidized (CrV≡O, electron paramagnetic resonance/IR/X-ray) participants in the catalytic cycle are characterized.
Co-reporter:Matthew S. Jeletic, Muhammad T. Jan, Ion Ghiviriga, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2009 (Issue 15) pp:2764-2776
Publication Date(Web):23 Feb 2009
DOI:10.1039/B819524B
New iridium and rhodium complexes prepared from C2-symmetric trans-9,10-dihydro-9,10-ethanoanthracene-11,12-bis(1-R)-benzimidazolidine-2-ylidene ligands (R = Me, iPr, and diPh) have been synthesized and characterized. Their catalytic activities have been tested in enantioselective hydrogenation and hydroformylation reactions. The ee’s for the reactions are low. Evidence indicates that even chelating di-N-heterocyclic carbene ligands are susceptible to reductive elimination.
Co-reporter:Roxy J. Lowry, Melanie K. Veige, Olivier Clément, Khalil A. Abboud, Ion Ghiviriga and Adam S. Veige
Organometallics 2008 Volume 27(Issue 20) pp:5184-5195
Publication Date(Web):September 19, 2008
DOI:10.1021/om800471m
A new C2-symmetric di-N-heterocyclic carbene (di-NHC) ligand is synthesized, and its coordination behavior with Rh(I) salts is examined. A comprehensive list is presented describing the synthesis of all chiral di-NHC ligands reported to date, including pertinent catalytic and structural features. The historical perspective indicates few structural archetypes have been investigated and more structural alternatives are needed. Accordingly, the synthesis of the diimidazolium salts [DEA-MI](I)2 (4) and [DEA-MBI](I)2 (8) (where DEA = 9,10-dihydro-11,12-ethanoanthracene; MI = methylimidazolium, and MBI = methylbenzimidazolium) are presented. When 8 is treated with 2 equiv of KN(SiMe3)2, the strained enetetramine [DEA-MBY] (9) is obtained and is characterized by NMR spectroscopy, elemental analysis, and X-ray crystallography. The highly strained mononuclear complex [(DEA-MBY)Rh(COD)]I (10-COD) is obtained when 8 is treated with KN(SiMe3)2 followed by half an equivalent of [Rh(COD)Cl]2. A 1H NMR spectrum of 10-COD revealed an unusual downfield signal at 9.26 ppm, assigned to an aliphatic bridge proton, and provides a clue as to the relative orientation of the anthracene ligand to the metal center. Ultimately, a single-crystal X-ray diffraction experiment assisted in determining the absolute solid-state structure of 10-COD. Although an X-ray crystal structure was not obtained for the imidazole derivative [DEA-MY][Rh(COD)]I (11-COD), a similar structure is implied by the occurrence of a downfield signal for the bridge proton at 8.24 ppm. In addition, the dinuclear complexes [μ-DEA-MY][Rh(NBD)I]2 (12-NBD) and [μ-DEA-MBY][Rh(COD)Cl]2 (13-COD) are obtained. A structural comparison of the ligand precursors 2 and 7·(HCl)2 and the dinuclear species 12-NBD and 13-COD is presented.
Co-reporter:Sudarsan VenkatRamani, Matias E. Pascualini, Ion Ghiviriga, Khalil A. Abboud, Adam S. Veige
Polyhedron (12 November 2013) Volume 64() pp:377-387
Publication Date(Web):12 November 2013
DOI:10.1016/j.poly.2013.07.004
Tanatalum(V) complexes ligated with a trianionic ONO3− pincer-type ligand were synthesized and characterized. Using the ONO trianionic pincer ligand precursor 2,2′-(azanediylbis(3-methyl-6,1-phenylene))bis(1,1,1,3,3,3-hexafluoropropan-2-ol) [CF3-ONO]H3 (1) and various sources of Ta(V), the following pincer complexes were synthesized: (I) anionic trichloride complex {[CF3-ONO]TaCl3}{HNEt3}·(HNEt3Cl)x (2), (II) diamido-amine complex [CF3-ONO]Ta(NMe2)2(HNMe2) (3-NHMe2) and its amine free version [CF3-ONO]Ta(NMe2)2 (3), (III) the neutral trichloride [CF3-ONHO]TaCl3 (4), (IV) the neutral dichloride [CF3-ONO]TaCl2(THF) (5), and (V) the dibenzyl complex [CF3-ONO]TaBn2 (6). Characterization methods include the complete assignment (where possible) for 1H, 13C, 19F, and 15N NMR spectra for complexes 2–6, and single crystal X-ray diffraction characterization for complexes 2, 3, and 4. Interesting aspects regarding the interplay of the ligand in its dianionic form versus its trianionic form, and an unusual H⋯F hydrogen bonding interaction within complex 4 were discovered from these synthetic pursuits.A novel tantalum ONO pincer-type complex exhibits an N–H—F hydrogen bonding interaction in the solid and solution state.Download full-size image
Co-reporter:Stella A. Gonsales, Ion Ghiviriga, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2016 - vol. 45(Issue 40) pp:NaN15785-15785
Publication Date(Web):2016/05/23
DOI:10.1039/C6DT01049K
The cleavage of carbon dioxide by the tungsten alkylidyne [CF3-ONO]WCCtBu(THF)2 (1) {where CF3-ONO = (MeC6H3[C-(CF3)2O])2N3−}, is reported. Splitting of CO2 yields the tungsten oxo ketene complex [CF3-ONO]W(O){(CH3)3CCCO} (6). The proposed pathway occurs through initial cycloaddition of WC and CO bonds to generate a heterometallacyclobutene, which then rearranges to yield WO and CC bonds. Complex 6 was characterized by multinuclear NMR, IR, and single crystal X-ray diffraction.
Co-reporter:Matthew E. O'Reilly, Trevor J. Del Castillo, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2012 - vol. 41(Issue 8) pp:NaN2246-2246
Publication Date(Web):2011/11/18
DOI:10.1039/C1DT11104C
The oxygen-atom-transfer (OAT) from [tBuOCO]CrV(O)(THF) (2) (where tBuOCO = [2,6-C6H3(6-tBuC6H3O)2]3−, THF = tetrahydrofuran) to triphenylphosphine (PPh3) in THF produces [tBuOCO]CrIII(THF)3 (1) and triphenylphosphine oxide (OPPh3) at a rate of 69.5(±1.9) M−1s−1 (22 °C). Identical rate constants were attained when acetonitrile (MeCN) and dichloromethane/THF (CH2Cl2/THF) were used as solvents. Electron paramagnetic resonance (EPR) data shows that the six-coordinate complex, [tBuOCO]CrV(O)(THF)2 (2a) forms upon addition of THF to 2, suggesting 2a as the active OAT species in THF. Similarly, addition of OPPh3 has no influence on the rate of OAT, but the addition of triphenylphosphorus ylide (CH2PPh3) to form [tBuOCO]CrV(O)(CH2PPh3) (4) prevents OAT to PPh3. In CH2Cl2, a [CrIV]2(μ-O) intermediate forms during the OAT from 2 to PPh3. The OAT from {[tBuOCO]CrIV(THF)}2(μ-O) (3) to PPh3 reveals a zero-order dependence in PPh3 indicating the dimer must first dissociate prior to OAT. The decay of 3versus time does not follow first-order kinetics due to the formation of a [tBuOCO]CrIII(THF) species (5) that inhibits the dissociation of 3. The change in concentration of 3versus time during OAT was simulated to obtain approximate rate constants.
Co-reporter:Sudarsan VenkatRamani, Ion Ghiviriga, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2015 - vol. 44(Issue 42) pp:NaN18486-18486
Publication Date(Web):2015/09/04
DOI:10.1039/C5DT02911B
This report details the synthesis and characterization of the semi-flexible [ONCH2O]H3 (1) ligand and its W(VI)-alkylidene and alkylidyne complexes. The alkylidyne complex [ONHCH2O]WCtBu(OtBu) (2) forms as a result of alcoholysis of 1 with (tBuO)3WCtBu. Complex 2 evolves to [ONCH2O]WCHtBu(OtBu) (3) through proton migration from the N atom of the pincer ligand to the WCα bond. Deprotonation of 2 or 3 with Ph3PCH2 affords the anionic alkylidyne {CH3PPh3}{[ONCH2O]WCtBu(OtBu)} (4). Complex 4 exhibits pincer-ligand-centered reactivity with electrophiles (H+, Me+, and TMS+), in spite of its enhanced inorganic enamine interaction. Addition of 2 equiv. of HCl to 4 yields the W(VI)-neopentyl complex [ONCH2O]W(CH2tBu)(OtBu)(Cl) (5). MeOTf or TMSOTf addition to 4 generates the dianionic pincer ligated alkylidynes [ONRCH2O]WCtBu(OtBu) (R = Me (6-Me); TMS (6-TMS)). Complexes 2–5 were characterized by multinuclear NMR spectroscopy, and combustion analysis. Complexes 4 and 5 were also characterized by single crystal X-ray diffraction. This work bridges the gap in the series involving W(VI)-alkylidynes ligated to the rigid [CF3-ONO]3−, and the flexible [OCH2NCH2O]3− ligands. DFT computations permit comparison of the inorganic enamine effect within alkylidynes supported by all three trianionic-pincer type ONO ligands.
Co-reporter:Trevor J. Del Castillo, Soumya Sarkar, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2011 - vol. 40(Issue 32) pp:NaN8144-8144
Publication Date(Web):2011/07/04
DOI:10.1039/C1DT10787A
This report describes the synthesis and characterization of 1,5-bis-triphenylphosphinegold(I) 1,2,3-triazolate (3(1,5)). The synthesis of the dinuclear complex 3(1,5) is achieved via an unprecedented inorganic click (iClick) reaction between the metal–azide PPh3AuN3 (1) and the metal–acetylide PPh3Au–CCPh (2). Characterization of 3(1,5) includes multinuclear NMR spectroscopy, combustion analysis, and single crystal X-ray crystallography. Experimental characterization is complemented with density-functional-theory (DFT) calculations which indicate the 1,4-isomer 3(1,4) is less stable by 3.3 kcal mol−1. The energetic difference lies primarily in the ability of the phenyl group in the 4-position of 3(1,5) to lie coplanar with the triazolate to create a delocalized π-bonding HOMO orbital.
Co-reporter:Amrita B. Mullick, Yun Min Chang, Ion Ghiviriga, Khalil A. Abboud, Weihong Tan and Adam S. Veige
Dalton Transactions 2013 - vol. 42(Issue 20) pp:NaN7446-7446
Publication Date(Web):2013/02/19
DOI:10.1039/C3DT32844A
A novel eighteen membered chiral macrocyclic dicarbene–digold complex [(μ-diNHC)AuI]2[OTF]2(8-(+/−)) was synthesized and characterized. Starting with enantiopure diNHC imidazolium salt ligand precursors enables access to the enantiopure versions of the digold(I) metallamacrocycles, 8-(+) and 8-(−). In vitro cytotoxicity studies indicate 8-(+/−) is moderately cytotoxic to both healthy and cancerous cell-lines, with no specificity. Confocal microscopy indicates the digold metallamacrocycle penetrates the cell membrane and causes cell death via apoptosis, as evidenced by DNA electrophoresis. The complex 8-(+/−) is characterized by a combination of NMR techniques (gDQCOSY, gHSQC, gHMBC and ROESY), single crystal X-ray diffraction, and combustion analysis.
Co-reporter:Xi Yang, Shanshan Wang, Ion Ghiviriga, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2015 - vol. 44(Issue 25) pp:NaN11443-11443
Publication Date(Web):2015/05/19
DOI:10.1039/C5DT00282F
A novel synthetic method to create gold based metallo–oligomers/polymers via the combination of inorganic click (iClick) with intermolecular aurophilic interactions is demonstrated. Complexes [PEt3Au]4(μ-N3C2C6H5) (1) and [PPhMe2Au]4(μ-N3C2C6H5) (2) and {[PEt3Au]4[(μ-N3C2)2-9,9-dihexyl-9H-fluorene]}n (8) have been synthesized via iClick. The tetranuclear structures of 1 and 2, induced by aurophilic bonding, are confirmed in the solid state through single crystal X-ray diffraction experiments and in solution via variable temperature NMR spectroscopy. The extended 1D structure of 8 is constructed by aurophilic induced self-assembly. 1H DOSY NMR analysis reveals that the aurophilic bonds in 1, 2, and 8 are retained in the solution phase. The degree of polymerization within complex 8 is temperature and concentration dependent, as determined by 1H DOSY NMR. Complex 8 is a rare example of a solution stable higher ordered structure linked by aurophilic interactions.
Co-reporter:M. E. Pascualini, N. V. Di Russo, A. E. Thuijs, A. Ozarowski, S. A. Stoian, K. A. Abboud, G. Christou and A. S. Veige
Chemical Science (2010-Present) 2015 - vol. 6(Issue 1) pp:NaN612-612
Publication Date(Web):2014/10/15
DOI:10.1039/C4SC02634A
Square-planar high-spin Fe(II) molecular compounds are rare and the only three non-macrocyclic or sterically-driven examples reported share a common FeO4 core. Using an easily modifiable pincer-type ligand, the successful synthesis of the first compound of this type that breaks the FeO4 motif was achieved. In addition, we present the first evidence that geometry and spin state persist in solution. Extensive characterization includes the first high-field EPR and variable field/temperature Mössbauer spectra for this class of compounds. Analysis of the spectroscopic data indicates this complex exhibits a large and positive zero-field splitting tensor. Furthermore, the unusually small ΔEQ value determined for this compound is rationalized on the basis of DFT calculations.
Co-reporter:Kevin P. McGowan, Matthew E. O'Reilly, Ion Ghiviriga, Khalil A. Abboud and Adam S. Veige
Chemical Science (2010-Present) 2013 - vol. 4(Issue 3) pp:NaN1155-1155
Publication Date(Web):2012/12/03
DOI:10.1039/C2SC21750C
Complex [tBuOCO]WC(tBu)(THF)2 (1) {where tBuOCO = [2,6-(tBuC6H3O)2C6H3]3−, THF = tetrahydrofuran} polymerizes acetylenes (R-phenylacetylene (R = H, p-OMe, p-F, 3,5-diCF3), 1-decyne, 3,3-dimethyl-1-butyne, and trimethylsilylacetylene) to form π-conjugating polymers. Upon treating 1 with 2 equiv. of phenylacetylene in toluene-d8 at −35 °C, two isolable products form. These two products are [O2C(tBuC)W(η2-HCCPh)] (2-tttBu) and [O2C(PhC)W(η2-HCCtBu)] (2-Ph) {where OC(tBuC)O = [2,6-(tBuC6H3O)2C6H3(tBuC)]4−, OC(PhC)O = [2,6-(tBuC6H3O)2C6H3(PhC)]4−} and derived from an apparent reductive alkylidyne migratory insertion into a metal–arene bond. Complexes 2-tttBu and 2-Ph polymerize acetylene and a wide variety of monosubstituted acetylenes including phenylacetylene derivatives, 1-decyne, 3,3-dimethyl-1-butyne and trimethylsilylacetylene. With a substrate to catalyst loading ratio of 25000:1, complex 2-tttBu polymerizes phenylacetylene with a turnover number (TON) of 17233. Additionally, 2-tttBu polymerizes phenylacetylene and 1-decyne with catalytic activities up to 5.64 × 106 gPPA mol−1 h−1 and 7.98 × 106 gPA mol−1 h−1, respectively. 2-tttBu also polymerizes the disubstituted acetylene, 1-phenyl-1-propyne. NMR spectroscopic and single crystal X-ray structural studies provide compelling evidence for polymer chain growth via an insertion ring-expansion mechanism.
Co-reporter:M. E. Pascualini, S. A. Stoian, A. Ozarowski, N. V. Di Russo, A. E. Thuijs, K. A. Abboud, G. Christou and A. S. Veige
Dalton Transactions 2015 - vol. 44(Issue 46) pp:NaN20215-20215
Publication Date(Web):2015/10/29
DOI:10.1039/C5DT03960F
High-spin square-planar molecular compounds are rare. In an effort to access this unique combination of geometry and spin state, we report the synthesis of a series of M(II) compounds stabilized by a trianionic pincer-type ligand, highlighting the formation of a high-spin square-planar Co(II) complex. Low-temperature, variable-frequency EPR measurements reveal that the ground electronic state of the Co(II) analogue is a highly anisotropic Kramers doublet (effective g values 7.35, 2.51, 1.48). This doublet can be identified with the lowest doublet of a quartet, S = 3/2 spin state that exhibits a very large ZFS, D ≥ 50 cm−1. The observation of an effective g value considerably greater than the largest spin-only value 6, demonstrates that the orbital angular moment is essentially unquenched along one spatial direction. Density Functional Theory (DFT) and time-dependent DFT calculations reveal the electronic configurations of the ground and excited orbital states. A qualitative crystal field description of the geff tensor shows that it originates from the spin–orbit coupling acting on states obtained through the transfer of a β electron from the doubly occupied xy to the singly-occupied {xz/yz} orbitals.
Co-reporter:Matthew E. O'Reilly, Ion Ghiviriga, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2013 - vol. 42(Issue 10) pp:NaN3336-3336
Publication Date(Web):2012/12/18
DOI:10.1039/C2DT32653A
This report presents the synthesis of the first neutral trianionic ONO pincer-type tungsten alkylidyne complex, [CF3–ONO]WC(tBu)(OEt2) (5) {where CF3–ONO = (MeC6H3[C(CF3)2O])2N3−}. Treating 5 with 1-phenylpropyne, 4,4-dimethyl-2-pentyne, and cyclooctyne yields the corresponding tungstenacyclobutadiene complexes [CF3–ONO]W[κ2-C(tBu)C(Me)C(Ph)] (6), [CF3–ONO]W[κ2-C(tBu)C(Me)C(tBu)] (7), and [CF3–ONO]W[κ2-C(tBu)C(CH2)6C] (8). Complexes 6, 7, and 8 do not undergo retro-[2 + 2]-cycloaddition even at 200 °C or in the presence of PMe3. DFT methods to elucidate the electronic structure of complexes 5 and 6 reveal important electronic factors that contribute to the lack of reactivity for the tungstenacyclobutadienes. An important bonding combination between the pincer N-atom lone pair and the WC bond within 5, termed an inorganic enamine, provides an explanation for the lack of retro-[2 + 2]-cycloaddition from 6, 7, and 8. 15N NMR spectroscopy was used to confirm the computational finding of an inorganic enamine bonding combination. Single crystal X-ray analysis of 5, 6, 7, and 8 provides insight into possible steric inadequacies within the CF3–ONO3− ligand to promote catalytic metathesis.
Co-reporter:Matthew S. Jeletic, Muhammad T. Jan, Ion Ghiviriga, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2009(Issue 15) pp:NaN2776-2776
Publication Date(Web):2009/02/23
DOI:10.1039/B819524B
New iridium and rhodium complexes prepared from C2-symmetric trans-9,10-dihydro-9,10-ethanoanthracene-11,12-bis(1-R)-benzimidazolidine-2-ylidene ligands (R = Me, iPr, and diPh) have been synthesized and characterized. Their catalytic activities have been tested in enantioselective hydrogenation and hydroformylation reactions. The ee’s for the reactions are low. Evidence indicates that even chelating di-N-heterocyclic carbene ligands are susceptible to reductive elimination.
Co-reporter:Andrew R. Powers, Ion Ghiviriga, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2015 - vol. 44(Issue 33) pp:NaN14752-14752
Publication Date(Web):2015/07/20
DOI:10.1039/C5DT02405F
This report outlines the investigation of the iClick mechanism between gold(I)–azides and gold(I)–acetylides to yield digold triazolates. Isolation of digold triazolate complexes offer compelling support for the role of two copper(I) ions in CuAAC. In addition, a kinetic investigation reveals the reaction is first order in both Au(I)–N3 and Au(I)–CC–R, thus second order overall. A Hammett plot with a ρ = 1.02(5) signifies electron-withdrawing groups accelerate the cycloaddition by facilitating the coordination of the second gold ion in a π-complex. Rate inhibition by the addition of free triphenylphosphine to the reaction indicates that ligand dissociation is a prerequisite for the reaction. The mechanistic conclusions mirror those proposed for the CuAAC reaction.
Co-reporter:S. A. Gonsales, M. E. Pascualini, I. Ghiviriga and A. S. Veige
Chemical Communications 2015 - vol. 51(Issue 69) pp:NaN13407-13407
Publication Date(Web):2015/07/21
DOI:10.1039/C5CC04851F
The trianionic pincer supported tungsten–vinyl complex [CF3-ONO]W(O){(CH3)3CCC(CH3)2} (3syn) undergoes facile double bond rotation at ambient temperature. The degenerate methyl exchange rates were measured via selective inversion-recovery experiments. DFT computations in conjunction with experimentally determined rate constants support a double bond rotation that proceeds via a Zwiterionic transition state.
Co-reporter:Mary E. Garner, Weijia Niu, Xigao Chen, Ion Ghiviriga, Khalil A. Abboud, Weihong Tan and Adam S. Veige
Dalton Transactions 2015 - vol. 44(Issue 4) pp:NaN1923-1923
Publication Date(Web):2014/11/14
DOI:10.1039/C4DT02850C
This work describes several synthetic approaches to append organic functional groups to gold and silver N-heterocyclic carbene (NHC) complexes suitable for applications in biomolecule conjugation. Carboxylate appended NHC ligands (3) lead to unstable AuI complexes that convert into bis-NHC species (4). A benzyl protected carboxylate NHC–AuI complex 2 was synthesized but deprotection to produce the carboxylic acid functionality could not be achieved. A small library of new alkyne functionalized NHC proligands were synthesized and used for subsequent silver and gold metalation reactions. The alkyne appended NHC gold complex 13 readily reacts with benzyl azide in a copper catalyzed azide–alkyne cycloaddition reaction to form the triazole appended NHC gold complex 14. Cell cytotoxicity studies were performed on DLD-1 (colorectal adenocarcinoma), Hep-G2 (hepatocellular carcinoma), MCF-7 (breast adenocarcinoma), CCRF-CEM (human T-Cell leukemia), and HEK (human embryonic kidney). Complete spectroscopic characterization of the ligands and complexes was achieved using 1H and 13C NMR, gHMBC, ESI-MS, and combustion analysis.
Co-reporter:Andrew R. Powers, Xi Yang, Trevor J. Del Castillo, Ion Ghiviriga, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2013 - vol. 42(Issue 42) pp:NaN14966-14966
Publication Date(Web):2013/09/04
DOI:10.1039/C3DT52105B
Metal-azide–metal-acetylide cycloaddition (iClick) reactions to synthesize heterotrimetallics and an unexpected novel tetranuclear gold(I) complex, are described. In addition, a discussion regarding the connection between traditional azide–alkyne cycloaddition reactions and iClick is presented focusing on applications towards linking multiple metal ions.
Co-reporter:Matthew E. O'Reilly and Adam S. Veige
Chemical Society Reviews 2014 - vol. 43(Issue 17) pp:NaN6369-6369
Publication Date(Web):2014/06/13
DOI:10.1039/C4CS00111G
Trianionic pincer and pincer-type ligands are the focus of this review. Metal ions from across the periodic table, from main group elements, transition metals, and the rare earths, are combined with trianionic pincer ligands to produce some of the most interesting complexes to appear in the literature over the past decade. This review provides a comprehensive examination of the synthesis, characterization, properties, and catalytic applications of trianionic pincer metal complexes. Some of the interesting applications employing trianionic pincer and pincer-type complexes include: (1) catalyzed aerobic oxidation, (2) alkene isomerization, (3) alkene and alkyne polymerization, (4) nitrene and carbene group transfer, (5) fundamental transformations such as oxygen-atom transfer, (6) nitrogen-atom transfer, (7) O2 activation, (8) C–H bond activation, (9) disulfide reduction, and (10) ligand centered storage of redox equivalents (i.e. redox active ligands). Expansion of the architecture, type of donor atoms, chelate ring size, and steric and electronic properties of trianionic pincer ligands has occurred rapidly over the past ten years. This review is structured according to the type of pincer donor atoms that bind to the metal ion. The type of donor atoms within trianionic pincer and pincer-type ligands to be discussed include: NCN3−, OCO3−, CCC3−, redox active NNN3−, NNN3−, redox active ONO3−, ONO3−, and SNS3−. Since this is the first review of trianionic pincer and pincer-type ligands, an emphasis is placed on providing the reader with in-depth discussion of synthetic methods, characterization data, and highlights of these complexes as catalysts.
Co-reporter:Matthew S. Jeletic, Ion Ghiviriga, Khalil A. Abboud and Adam S. Veige
Dalton Transactions 2010 - vol. 39(Issue 28) pp:NaN6394-6394
Publication Date(Web):2010/06/21
DOI:10.1039/C0DT00268B
This report details the synthesis and characterization of the first chiral diNHC cyclophane ligand and its palladium allyl complex. The complex was tested for 1,4-conjugate addition of phenylboronic acid to 2-cyclohexen-1-one and provides R-3-phenylcyclohexanone in 50% enantiomeric excess.









![Poly[1,3-cyclopentanediyl-(1Z)-1,2-ethenediyl]](http://img.cochemist.com/ccimg/65600/65556-06-1.png)
![Poly[1,3-cyclopentanediyl-(1Z)-1,2-ethenediyl]](http://img.cochemist.com/ccimg/65600/65556-06-1_b.png)









![11,12-Bis[N-(i-propyl)-1H-benzimidazolium-3-methylene]-9,10-dihydro-9,10-ethanoanthracene bis(trifluoromethanesulfonate)](http://img.cochemist.com/ccimg/958100/958004-12-1.png)
![11,12-Bis[N-(i-propyl)-1H-benzimidazolium-3-methylene]-9,10-dihydro-9,10-ethanoanthracene bis(trifluoromethanesulfonate)](http://img.cochemist.com/ccimg/958100/958004-12-1_b.png)
![11,12-Bis[1,3-dihydro-3-(i-propyl)-2H-benzimidazol-2-ylidene-3-methylene]-9,10-dihydro-9,10-ethanoanthracene](http://img.cochemist.com/ccimg/958100/958004-05-2.png)
![11,12-Bis[1,3-dihydro-3-(i-propyl)-2H-benzimidazol-2-ylidene-3-methylene]-9,10-dihydro-9,10-ethanoanthracene](http://img.cochemist.com/ccimg/958100/958004-05-2_b.png)


![1H-Imidazole, 1-[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/844700/844658-92-0.png)
![1H-Imidazole, 1-[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/844700/844658-92-0_b.png)
![1,3-Benzenedimethanamine, N,N'-bis[2,6-bis(1-methylethyl)phenyl]-](http://img.cochemist.com/ccimg/500900/500876-60-8.png)
![1,3-Benzenedimethanamine, N,N'-bis[2,6-bis(1-methylethyl)phenyl]-](http://img.cochemist.com/ccimg/500900/500876-60-8_b.png)
-](http://img.cochemist.com/ccimg/185700/185685-83-0.png)
-](http://img.cochemist.com/ccimg/185700/185685-83-0_b.png)
![1H-Benzimidazole, 1,1'-[1,2-phenylenebis(methylene)]bis-](http://img.cochemist.com/ccimg/171100/171072-77-8.png)
![1H-Benzimidazole, 1,1'-[1,2-phenylenebis(methylene)]bis-](http://img.cochemist.com/ccimg/171100/171072-77-8_b.png)
![1,2-DIMETHOXYETHANE;[2,6-DI(PROPAN-2-YL)PHENYL]IMINO-(2-METHYL-2-PHENYLPROPYLIDENE)MOLYBDENUM(2+);TRIFLUOROMETHANESULFONATE](http://img.cochemist.com/ccimg/127000/126949-63-1.png)
![1,2-DIMETHOXYETHANE;[2,6-DI(PROPAN-2-YL)PHENYL]IMINO-(2-METHYL-2-PHENYLPROPYLIDENE)MOLYBDENUM(2+);TRIFLUOROMETHANESULFONATE](http://img.cochemist.com/ccimg/127000/126949-63-1_b.png)

























![2,8-Dimethyl-2,3,4,4a,5,9b-hexahydro-1H-pyrido[4,3-b]indole](http://img.cochemist.com/ccimg/17500/17411-19-7.png)
![2,8-Dimethyl-2,3,4,4a,5,9b-hexahydro-1H-pyrido[4,3-b]indole](http://img.cochemist.com/ccimg/17500/17411-19-7_b.png)
























-](http://img.cochemist.com/ccimg/185200/185119-00-0.png)
-](http://img.cochemist.com/ccimg/185200/185119-00-0_b.png)