Co-reporter:Ming Yan, Yu Kawamata, and Phil S. Baran
Chemical Reviews November 8, 2017 Volume 117(Issue 21) pp:13230-13230
Publication Date(Web):October 9, 2017
DOI:10.1021/acs.chemrev.7b00397
Electrochemistry represents one of the most intimate ways of interacting with molecules. This review discusses advances in synthetic organic electrochemistry since 2000. Enabling methods and synthetic applications are analyzed alongside innate advantages as well as future challenges of electroorganic chemistry.
Co-reporter:Justine N. deGruyter, Lara R. Malins, Laurin Wimmer, Khalyd J. Clay, Javier Lopez-Ogalla, Tian Qin, Josep Cornella, Zhiqing Liu, Guanda Che, Denghui Bao, Jason M. Stevens, Jennifer X. Qiao, Martin P. Allen, Michael A. Poss, and Phil S. Baran
Organic Letters November 17, 2017 Volume 19(Issue 22) pp:6196-6196
Publication Date(Web):November 8, 2017
DOI:10.1021/acs.orglett.7b03121
Tetrachloro-N-hydroxyphthalimide tetramethyluronium hexafluorophosphate (CITU) is disclosed as a convenient and economical reagent for both acylation and decarboxylative cross-coupling chemistries. Within the former set of reactions, CITU displays reactivity similar to that of common coupling reagents, but with increased safety and reduced cost. Within the latter, increased yields, more rapid conversion, and a simplified procedure are possible across a range of reported decarboxylative transformations.
Co-reporter:Yu Kawamata, Ming Yan, Zhiqing Liu, Deng-Hui Bao, Jinshan Chen, Jeremy T. Starr, and Phil S. Baran
Journal of the American Chemical Society June 7, 2017 Volume 139(Issue 22) pp:7448-7448
Publication Date(Web):May 16, 2017
DOI:10.1021/jacs.7b03539
A practical electrochemical oxidation of unactivated C–H bonds is presented. This reaction utilizes a simple redox mediator, quinuclidine, with inexpensive carbon and nickel electrodes to selectively functionalize “deep-seated” methylene and methine moieties. The process exhibits a broad scope and good functional group compatibility. The scalability, as illustrated by a 50 g scale oxidation of sclareolide, bodes well for immediate and widespread adoption.
Co-reporter:Hang Chu, Joel M. Smith, Jakob Felding, and Phil S. Baran
ACS Central Science January 25, 2017 Volume 3(Issue 1) pp:
Publication Date(Web):December 19, 2016
DOI:10.1021/acscentsci.6b00313
Total syntheses of the complex, highly oxygenated sesquiterpenes thapsigargin (1) and nortrilobolide (2) are presented. Access to analogues of these promising bioactive natural products has been limited to tedious isolation and semisynthetic efforts. Elegant prior total syntheses demonstrated the feasibility of creating these entitites in 36–42 step processes. The currently reported route proceeds in a scalable and more concise fashion by utilizing two-phase terpene synthesis logic. Salient features of the work include application of the classic photosantonin rearrangement and precisely choreographed installation of the multiple oxygenations present on the guaianolide skeleton.
Co-reporter:Julian C. Lo, Dongyoung Kim, Chung-Mao Pan, Jacob T. Edwards, Yuki Yabe, Jinghan Gui, Tian Qin, Sara Gutiérrez, Jessica Giacoboni, Myles W. Smith, Patrick L. Holland, and Phil S. Baran
Journal of the American Chemical Society February 15, 2017 Volume 139(Issue 6) pp:2484-2484
Publication Date(Web):January 17, 2017
DOI:10.1021/jacs.6b13155
This Article details the development of the iron-catalyzed conversion of olefins to radicals and their subsequent use in the construction of C–C bonds. Optimization of a reductive diene cyclization led to the development of an intermolecular cross-coupling of electronically-differentiated donor and acceptor olefins. Although the substitution on the donor olefins was initially limited to alkyl and aryl groups, additional efforts culminated in the expansion of the scope of the substitution to various heteroatom-based functionalities, providing a unified olefin reactivity. A vinyl sulfone acceptor olefin was developed, which allowed for the efficient synthesis of sulfone adducts that could be used as branch points for further diversification. Moreover, this reactivity was extended into an olefin-based Minisci reaction to functionalize heterocyclic scaffolds. Finally, mechanistic studies resulted in a more thorough understanding of the reaction, giving rise to the development of a more efficient second-generation set of olefin cross-coupling conditions.
Co-reporter:Christopher G. Parker, Christian A. Kuttruff, Andrea Galmozzi, Lars Jørgensen, Chien-Hung Yeh, Daniel J. Hermanson, Yujia Wang, Marta Artola, Steven J. McKerrall, Christopher M. Josyln, Bjarne Nørremark, Georg Dünstl, Jakob Felding, Enrique Saez, Phil S. Baran, and Benjamin F. Cravatt
ACS Central Science December 27, 2017 Volume 3(Issue 12) pp:1276-1276
Publication Date(Web):December 6, 2017
DOI:10.1021/acscentsci.7b00420
The diterpenoid ester ingenol mebutate (IngMeb) is the active ingredient in the topical drug Picato, a first-in-class treatment for the precancerous skin condition actinic keratosis. IngMeb is proposed to exert its therapeutic effects through a dual mode of action involving (i) induction of cell death that is associated with mitochondrial dysfunction followed by (ii) stimulation of a local inflammatory response, at least partially driven by protein kinase C (PKC) activation. Although this therapeutic model has been well characterized, the complete set of molecular targets responsible for mediating IngMeb activity remains ill-defined. Here, we have synthesized a photoreactive, clickable analogue of IngMeb and used this probe in quantitative proteomic experiments to map several protein targets of IngMeb in human cancer cell lines and primary human keratinocytes. Prominent among these targets was the mitochondrial carnitine-acylcarnitine translocase SLC25A20, which we show is inhibited in cells by IngMeb and the more stable analogue ingenol disoxate (IngDsx), but not by the canonical PKC agonist 12-O-tetradecanoylphorbol-13-acetate (TPA). SLC25A20 blockade by IngMeb and IngDsx leads to a buildup of cellular acylcarnitines and blockade of fatty acid oxidation (FAO), pointing to a possible mechanism for IngMeb-mediated perturbations in mitochondrial function.
Co-reporter:Ming Yan and Phil S. Baran
Organic Process Research & Development August 18, 2017 Volume 21(Issue 8) pp:1091-1091
Publication Date(Web):August 9, 2017
DOI:10.1021/acs.oprd.7b00208
Co-reporter:Lara R. Malins, Justine N. deGruyter, Kevin J. Robbins, Paul M. Scola, Martin D. Eastgate, M. Reza Ghadiri, and Phil S. Baran
Journal of the American Chemical Society April 12, 2017 Volume 139(Issue 14) pp:5233-5233
Publication Date(Web):March 22, 2017
DOI:10.1021/jacs.7b01624
A thermodynamic approach to peptide macrocyclization inspired by the cyclization of non-ribosomal peptide aldehydes is presented. The method provides access to structurally diverse macrocycles by exploiting the reactivity of transient macrocyclic peptide imines toward inter- and intramolecular nucleophiles. Reactions are performed in aqueous media, in the absence of side chain protecting groups, and are tolerant of all proteinogenic functional groups. Macrocyclic products bearing non-native and rigidifying structural motifs, isotopic labels, and a variety of bioorthogonal handles are prepared, along with analogues of four distinct natural products. Structural interrogation of the linear and macrocyclic peptides using variable-temperature NMR and circular dichroism suggests that preorganization of linear substrates is not a prerequisite for macrocyclization.
Co-reporter:Dr. Chao Li;Dr. Yu Kawamata;Dr. Hugh Nakamura;Julien C. Vantourout;Dr. Zhiqing Liu;Qinglong Hou;Dr. Denghui Bao;Dr. Jeremy T. Starr;Dr. Jinshan Chen;Ming Yan; Dr. Phil S. Baran
Angewandte Chemie International Edition 2017 Volume 56(Issue 42) pp:13088-13093
Publication Date(Web):2017/10/09
DOI:10.1002/anie.201707906
AbstractAlong with amide bond formation, Suzuki cross-coupling, and reductive amination, the Buchwald–Hartwig–Ullmann-type amination of aryl halides stands as one of the most employed reactions in modern medicinal chemistry. The work herein demonstrates the potential of utilizing electrochemistry to provide a complementary avenue to access such critical bonds using an inexpensive nickel catalyst under mild reaction conditions. Of note is the scalability, functional-group tolerance, rapid rate, and the ability to employ a variety of aryl donors (Ar−Cl, Ar−Br, Ar−I, Ar−OTf), amine types (primary and secondary), and even alternative X−H donors (alcohols and amides).
Co-reporter:Dr. Joel M. Smith;Dr. Tian Qin;Rohan R. Merchant;Jacob T. Edwards;Dr. Lara R. Malins;Dr. Zhiqing Liu;Gua Che;Zichao Shen;Dr. Scott A. Shaw;Dr. Martin D. Eastgate; Phil S. Baran
Angewandte Chemie International Edition 2017 Volume 56(Issue 39) pp:11906-11910
Publication Date(Web):2017/09/18
DOI:10.1002/anie.201705107
AbstractThe development of a new decarboxylative cross-coupling method that affords terminal and substituted alkynes from various carboxylic acids is described using both nickel- and iron-based catalysts. The use of N-hydroxytetrachlorophthalimide (TCNHPI) esters is crucial to the success of the transformation, and the reaction is amenable to in situ carboxylic acid activation. Additionally, an inexpensive, commercially available alkyne source is employed in this formal homologation process that serves as a surrogate for other well-established alkyne syntheses. The reaction is operationally simple and broad in scope while providing succinct and scalable avenues to previously reported synthetic intermediates.
Co-reporter:Dr. Chao Li;Dr. Yu Kawamata;Dr. Hugh Nakamura;Julien C. Vantourout;Dr. Zhiqing Liu;Qinglong Hou;Dr. Denghui Bao;Dr. Jeremy T. Starr;Dr. Jinshan Chen;Ming Yan; Dr. Phil S. Baran
Angewandte Chemie 2017 Volume 129(Issue 42) pp:13268-13273
Publication Date(Web):2017/10/09
DOI:10.1002/ange.201707906
AbstractAlong with amide bond formation, Suzuki cross-coupling, and reductive amination, the Buchwald–Hartwig–Ullmann-type amination of aryl halides stands as one of the most employed reactions in modern medicinal chemistry. The work herein demonstrates the potential of utilizing electrochemistry to provide a complementary avenue to access such critical bonds using an inexpensive nickel catalyst under mild reaction conditions. Of note is the scalability, functional-group tolerance, rapid rate, and the ability to employ a variety of aryl donors (Ar−Cl, Ar−Br, Ar−I, Ar−OTf), amine types (primary and secondary), and even alternative X−H donors (alcohols and amides).
Co-reporter:Dr. Joel M. Smith;Dr. Tian Qin;Rohan R. Merchant;Jacob T. Edwards;Dr. Lara R. Malins;Dr. Zhiqing Liu;Gua Che;Zichao Shen;Dr. Scott A. Shaw;Dr. Martin D. Eastgate; Phil S. Baran
Angewandte Chemie 2017 Volume 129(Issue 39) pp:12068-12072
Publication Date(Web):2017/09/18
DOI:10.1002/ange.201705107
AbstractThe development of a new decarboxylative cross-coupling method that affords terminal and substituted alkynes from various carboxylic acids is described using both nickel- and iron-based catalysts. The use of N-hydroxytetrachlorophthalimide (TCNHPI) esters is crucial to the success of the transformation, and the reaction is amenable to in situ carboxylic acid activation. Additionally, an inexpensive, commercially available alkyne source is employed in this formal homologation process that serves as a surrogate for other well-established alkyne syntheses. The reaction is operationally simple and broad in scope while providing succinct and scalable avenues to previously reported synthetic intermediates.
Co-reporter:Shan Yu;Jie Wang;Maoqun Tian;Chao Li;Lisa M. Barton;David S. Peters;Manoj Kumar;Antony W. Yu;Arnab K. Chatterjee;Kristen A. Johnson;Ming Yan
Science 2017 Volume 356(Issue 6342) pp:
Publication Date(Web):09 Jun 2017
DOI:10.1126/science.aam7355
Swapping boron acids for carbon acids
Carbon-bound boronic acids and their esters are widely used as coupling partners to make carbon-carbon bonds. More recently, these chemicals have garnered pharmaceutical interest in their own right. Li et al. report a versatile nickel-catalyzed process to replace carboxylic acids with boronate esters by using a phthalimide activator. The reaction is well suited to late-stage modification of complex molecules. The authors used the approach to produce a potent in vitro inhibitor of human neutrophil elastase, a target of interest in treating inflammatory lung diseases.
Science, this issue p. eaam7355
Co-reporter:Luisruben P. Martinez; Shigenobu Umemiya; Sarah E. Wengryniuk
Journal of the American Chemical Society 2016 Volume 138(Issue 24) pp:7536-7539
Publication Date(Web):June 10, 2016
DOI:10.1021/jacs.6b04816
The structurally intriguing terpenes pallambins C and D have been assembled in only 11 steps from a cheap commodity chemical: furfuryl alcohol. This synthesis, which features a redox-economic approach free of protecting-group manipulations, assembles all four-ring systems via a sequential cyclization strategy. Of these four-ring constructing operations, two are classical (Robinson annulation and Mukaiyama aldol) and two are newly devised. During the course of this work a method for the difunctionalization of enol ethers was developed, and the scope of this transformation was explored.
Co-reporter:Maoqun Tian, Ming Yan, and Phil S. Baran
Journal of the American Chemical Society 2016 Volume 138(Issue 43) pp:14234-14237
Publication Date(Web):October 17, 2016
DOI:10.1021/jacs.6b09701
A concise route to a small family of exotic marine alkaloids known as the araiosamines has been developed, and their absolute configuration has been assigned. The dense array of functionality, high polarity, and rich stereochemistry coupled with equilibrating topologies present an unusual challenge for chemical synthesis and an opportunity for innovation. Key steps involve the use of a new reagent for guanidine installation, a remarkably selective C–H functionalization, and a surprisingly simple final step that intersects a presumed biosynthetic intermediate. Synthetic araiosamines were shown to exhibit potency against Gram-positive and -negative bacteria despite a contrary report of no activity.
Co-reporter:Ming Yan, Julian C. Lo, Jacob T. Edwards, and Phil S. Baran
Journal of the American Chemical Society 2016 Volume 138(Issue 39) pp:12692-12714
Publication Date(Web):September 15, 2016
DOI:10.1021/jacs.6b08856
This Perspective illustrates the defining characteristics of free radical chemistry, beginning with its rich and storied history. Studies from our laboratory are discussed along with recent developments emanating from others in this burgeoning area. The practicality and chemoselectivity of radical reactions enable rapid access to molecules of relevance to drug discovery, agrochemistry, material science, and other disciplines. Thus, these reactive intermediates possess inherent translational potential, as they can be widely used to expedite scientific endeavors for the betterment of humankind.
Co-reporter:Fumihiko Toriyama, Josep Cornella, Laurin Wimmer, Tie-Gen Chen, Darryl D. Dixon, Gardner Creech, and Phil S. Baran
Journal of the American Chemical Society 2016 Volume 138(Issue 35) pp:11132-11135
Publication Date(Web):August 22, 2016
DOI:10.1021/jacs.6b07172
Cross-couplings of alkyl halides and organometallic species based on single electron transfer using Ni and Fe catalyst systems have been studied extensively, and separately, for decades. Here we demonstrate the first couplings of redox-active esters (both isolated and derived in situ from carboxylic acids) with organozinc and organomagnesium species using an Fe-based catalyst system originally developed for alkyl halides. This work is placed in context by showing a direct comparison with a Ni catalyst for >40 examples spanning a range of primary, secondary, and tertiary substrates. This new C–C coupling is scalable and sustainable, and it exhibits a number of clear advantages in several cases over its Ni-based counterpart.
Co-reporter:Artiom Cernijenko; Rune Risgaard
Journal of the American Chemical Society 2016 Volume 138(Issue 30) pp:9425-9428
Publication Date(Web):July 26, 2016
DOI:10.1021/jacs.6b06623
An expedient, practical, and enantioselective route to the highly congested ent-kaurane diterpene maoecrystal V is presented. This route, which has been several years in the making, is loosely modeled after a key pinacol shift in the proposed biosynthesis. Only 11 steps, many of which are strategic in that they build key skeletal bonds and incorporate critical functionalities, are required to access (−)-maoecrystal V. Several unique and unexpected maneuvers are featured in this potentially scalable pathway. Reevaluation of the biological activity calls into question the initial exuberance surrounding this natural product.
Co-reporter:Josep Cornella; Jacob T. Edwards; Tian Qin; Shuhei Kawamura; Jie Wang; Chung-Mao Pan; Ryan Gianatassio; Michael Schmidt; Martin D. Eastgate
Journal of the American Chemical Society 2016 Volume 138(Issue 7) pp:2174-2177
Publication Date(Web):February 2, 2016
DOI:10.1021/jacs.6b00250
A new transformation is presented that enables chemists to couple simple alkyl carboxylic acids with aryl zinc reagents under Ni-catalysis. The success of this reaction hinges on the unique use of redox-active esters that allow one to employ such derivatives as alkyl halides surrogates. The chemistry exhibits broad substrate scope and features a high degree of practicality. The simple procedure and extremely inexpensive nature of both the substrates and pre-catalyst (NiCl2·6H2O, ca. $9.5/mol) bode well for the immediate widespread adoption of this method.
Co-reporter:Matthew T. Villaume, Eran Sella, Garrett Saul, Robert M. Borzilleri, Joseph Fargnoli, Kathy A. Johnston, Haiying Zhang, Mark P. Fereshteh, T. G. Murali Dhar, and Phil S. Baran
ACS Central Science 2016 Volume 2(Issue 1) pp:27
Publication Date(Web):December 23, 2015
DOI:10.1021/acscentsci.5b00345
The fungal-derived Taiwanese natural product antroquinonol A has attracted both academic and commercial interest due to its reported exciting biological properties. This reduced quinone is currently in phase II trials (USA and Taiwan) for the treatment of non-small-cell lung carcinoma (NSCLC) and was recently granted orphan drug status by the FDA for the treatment of pancreatic cancer and acute myeloid leukemia. Pending successful completion of human clinical trials, antroquinonol is expected to be commercialized under the trade name Hocena. A synthesis-enabled biological re-examination of this promising natural product, however, reveals minimal in vitro and in vivo antitumor activity in preclinical models.
Co-reporter:Evan J. Horn, Brandon R. Rosen, and Phil S. Baran
ACS Central Science 2016 Volume 2(Issue 5) pp:302
Publication Date(Web):May 5, 2016
DOI:10.1021/acscentsci.6b00091
While preparative electrolysis of organic molecules has been an active area of research over the past century, modern synthetic chemists have generally been reluctant to adopt this technology. In fact, electrochemical methods possess many benefits over traditional reagent-based transformations, such as high functional group tolerance, mild conditions, and innate scalability and sustainability. In this Outlook we highlight illustrative examples of electrochemical reactions in the context of the synthesis of complex molecules, showcasing the intrinsic benefits of electrochemical reactions versus traditional reagent-based approaches. Our hope is that this field will soon see widespread adoption in the synthetic community.
Co-reporter:Dr. Jie Wang;Dr. Tian Qin;Dr. Tie-Gen Chen;Dr. Laurin Wimmer;Jacob T. Edwards;Dr. Josep Cornella;Benjamin Vokits;Dr. Scott A. Shaw; Phil S. Baran
Angewandte Chemie International Edition 2016 Volume 55( Issue 33) pp:9676-9679
Publication Date(Web):
DOI:10.1002/anie.201605463
Abstract
A transformation analogous in simplicity and functional group tolerance to the venerable Suzuki cross-coupling between alkyl-carboxylic acids and boronic acids is described. This Ni-catalyzed reaction relies upon the activation of alkyl carboxylic acids as their redox-active ester derivatives, specifically N-hydroxy-tetrachlorophthalimide (TCNHPI), and proceeds in a practical and scalable fashion. The inexpensive nature of the reaction components (NiCl2⋅6 H2O—$9.5 mol−1, Et3N) coupled to the virtually unlimited commercial catalog of available starting materials bodes well for its rapid adoption.
Co-reporter:Dr. Jie Wang;Dr. Tian Qin;Dr. Tie-Gen Chen;Dr. Laurin Wimmer;Jacob T. Edwards;Dr. Josep Cornella;Benjamin Vokits;Dr. Scott A. Shaw; Phil S. Baran
Angewandte Chemie 2016 Volume 128( Issue 33) pp:9828-9831
Publication Date(Web):
DOI:10.1002/ange.201605463
Abstract
A transformation analogous in simplicity and functional group tolerance to the venerable Suzuki cross-coupling between alkyl-carboxylic acids and boronic acids is described. This Ni-catalyzed reaction relies upon the activation of alkyl carboxylic acids as their redox-active ester derivatives, specifically N-hydroxy-tetrachlorophthalimide (TCNHPI), and proceeds in a practical and scalable fashion. The inexpensive nature of the reaction components (NiCl2⋅6 H2O—$9.5 mol−1, Et3N) coupled to the virtually unlimited commercial catalog of available starting materials bodes well for its rapid adoption.
Co-reporter:Tian Qin;Josep Cornella;Chao Li;Lara R. Malins;Jacob T. Edwards;Shuhei Kawamura;Brad D. Maxwell;Martin D. Eastgate
Science 2016 Vol 352(6287) pp:801-805
Publication Date(Web):13 May 2016
DOI:10.1126/science.aaf6123
Carbon links without helpful neighbors
It's an irony of modern organic chemistry that the simplest-looking carbon-carbon bonds are often the hardest to make. Most reactions owe their efficiency to neighboring double bonds or oxygen and nitrogen atoms that linger in the products. Qin et al. now present a broadly applicable protocol for making C-C bonds in the absence of such surrounding help. The nickel-catalyzed process couples a zinc-activated carbon center to an ester that's poised to lose CO2. The ready availability of numerous carboxylic acids (which are easily converted to esters) contributes to the reaction's versatility.
Science, this issue p. 801
Co-reporter:Dr. Changxia Yuan;Dr. Yehua Jin;Dr. Nathan C. Wilde;Dr. Phil S. Baran
Angewandte Chemie International Edition 2016 Volume 55( Issue 29) pp:8280-8284
Publication Date(Web):
DOI:10.1002/anie.201602235
Abstract
In the realm of natural product chemistry, few isolates have risen to the level of fame justifiably accorded to Taxol (1) and its chemical siblings. This report describes the most concise route to date for accessing the highly oxidized members of this family. As representative members of taxanes containing five oxygen atoms, decinnamoyltaxinine E (2) and taxabaccatin III (3), have succumbed to enantioselective total synthesis for the first time in only 18 steps from a simple olefin starting material. The strategy holistically mimics nature's approach (two-phase synthesis) and features a carefully choreographed sequence of stereoselective oxidations and a remarkable redox-isomerization to set the key trans-diol present in 2 and 3. This work lays the critical groundwork necessary to access even higher oxidized taxanes such as 1 in a more practical fashion, thus empowering a medicinal chemistry campaign that is not wedded to semi-synthesis.
Co-reporter:Dr. Changxia Yuan;Dr. Yehua Jin;Dr. Nathan C. Wilde;Dr. Phil S. Baran
Angewandte Chemie 2016 Volume 128( Issue 29) pp:8420-8424
Publication Date(Web):
DOI:10.1002/ange.201602235
Abstract
In the realm of natural product chemistry, few isolates have risen to the level of fame justifiably accorded to Taxol (1) and its chemical siblings. This report describes the most concise route to date for accessing the highly oxidized members of this family. As representative members of taxanes containing five oxygen atoms, decinnamoyltaxinine E (2) and taxabaccatin III (3), have succumbed to enantioselective total synthesis for the first time in only 18 steps from a simple olefin starting material. The strategy holistically mimics nature's approach (two-phase synthesis) and features a carefully choreographed sequence of stereoselective oxidations and a remarkable redox-isomerization to set the key trans-diol present in 2 and 3. This work lays the critical groundwork necessary to access even higher oxidized taxanes such as 1 in a more practical fashion, thus empowering a medicinal chemistry campaign that is not wedded to semi-synthesis.
Co-reporter:Lara R. Malins;Chung-Mao Pan;Liher Prieto;Jie Wang;Thomas A. Brandt;Ryan Gianatassio;Justin M. Lopchuk;Michael R. Collins;Gary M. Gallego;Neal W. Sach;Jillian E. Spangler;Huichin Zhu;Jinjiang Zhu
Science 2016 Volume 351(Issue 6270) pp:241-246
Publication Date(Web):15 Jan 2016
DOI:10.1126/science.aad6252
Opening one ring to tack on another
Curious chemists have long sought to learn just how tightly carbon atoms can be bound together. For instance, it's possible to form a bond between two opposite corners of an already strained four-membered ring to make an edge-sharing pair of triangles. Gianatassio et al. have now devised a general use for these and related molecular curiosities. They show that appropriately modified nitrogen centers can pop open the most highly strained bond, leaving the more modestly strained ring motif intact. In this way, small rings can emerge as a convenient diversifying element in compounds, including new pharmaceutical candidates.
Science, this issue p. 241
Co-reporter:Quentin Michaudel, Yoshihiro Ishihara, and Phil S. Baran
Accounts of Chemical Research 2015 Volume 48(Issue 3) pp:712
Publication Date(Web):February 23, 2015
DOI:10.1021/ar500424a
Collaboration between academia and industry is a growing phenomenon within the chemistry community. These sectors have long held strong ties since academia traditionally trains the future scientists of the corporate world, but the recent drastic decrease of public funding is motivating the academic world to seek more private grants. This concept of industrial “sponsoring” is not new, and in the past, some companies granted substantial amounts of money per annum to various academic institutions in exchange for prime access to all their scientific discoveries and inventions. However, academic and industrial interests were not always aligned, and therefore the investment has become increasingly difficult to justify from industry’s point of view. With fluctuating macroeconomic factors, this type of unrestricted grant has become more rare and has been largely replaced by smaller and more focused partnerships. In our view, forging a partnership with industry can be a golden opportunity for both parties and can represent a true symbiosis. This type of project-specific collaboration is engendered by industry’s desire to access very specific academic expertise that is required for the development of new technologies at the forefront of science. Since financial pressures do not allow companies to spend the time to acquire this expertise and even less to explore fundamental research, partnering with an academic laboratory whose research is related to the problem gives them a viable alternative. From an academic standpoint, it represents the perfect occasion to apply “pure science” research concepts to solve problems that benefit humanity. Moreover, it offers a unique opportunity for students to face challenges from the “real world” at an early stage of their career. Although not every problem in industry can be solved by research developments in academia, we argue that there is significant scientific overlap between these two seemingly disparate groups, thereby presenting an opportunity for a symbiosis. This type of partnership is challenging but can be a win–win situation if both parties agree on some general guidelines, including clearly defined goals and deliverables, biweekly meetings to track research progress, and quarterly or annual meetings to recognize overarching, common objectives. This Account summarizes our personal experience concerning collaborations with various industrial groups and the way it impacted the research programs for both sides in a symbiotic fashion.
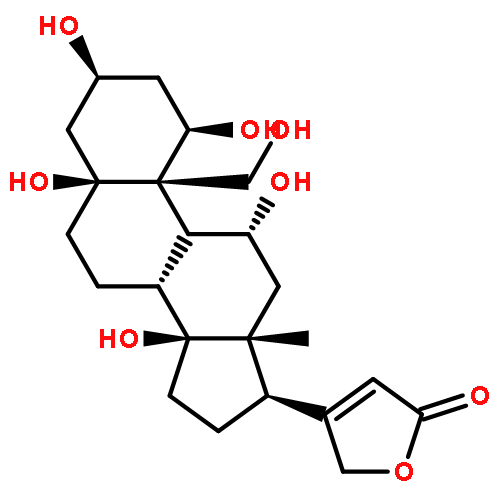
Co-reporter:Hans Renata; Qianghui Zhou; Georg Dünstl; Jakob Felding; Rohan R. Merchant; Chien-Hung Yeh
Journal of the American Chemical Society 2015 Volume 137(Issue 3) pp:1330-1340
Publication Date(Web):January 16, 2015
DOI:10.1021/ja512022r
The natural product ouabagenin is a complex cardiotonic steroid with a highly oxygenated skeleton. This full account describes the development of a concise synthesis of ouabagenin, including the evolution of synthetic strategy to access hydroxylation at the C19 position of a steroid skeleton. In addition, approaches to install the requisite butenolide moiety at the C17 position are discussed. Lastly, methodology developed in this synthesis has been applied in the generation of novel analogues of corticosteroid drugs bearing a hydroxyl group at the C19 position.
Co-reporter:Hai T. Dao; Chao Li; Quentin Michaudel; Brad D. Maxwell
Journal of the American Chemical Society 2015 Volume 137(Issue 25) pp:8046-8049
Publication Date(Web):June 19, 2015
DOI:10.1021/jacs.5b05144
A solution to the classic unsolved problem of olefin hydromethylation is presented. This highly chemoselective method can tolerate labile and reactive chemical functionalities and uses a simple set of reagents. An array of olefins, including mono-, di-, and trisubstituted olefins, are all smoothly hydromethylated. This mild protocol can be used to simplify the synthesis of a specific target or to directly “edit” complex natural products and other advanced materials. The method is also amenable to the simple installation of radioactive and stable labeled methyl groups.
Co-reporter:Yi Yang See; Aaron T. Herrmann; Yoshinori Aihara
Journal of the American Chemical Society 2015 Volume 137(Issue 43) pp:13776-13779
Publication Date(Web):October 14, 2015
DOI:10.1021/jacs.5b09463
Steroids bearing C12 oxidations are widespread in nature, yet only one preparative chemical method addresses this challenge in a low-yielding and not fully understood fashion: Schönecker’s Cu-mediated oxidation. This work shines new light onto this powerful C–H oxidation method through mechanistic investigation, optimization, and wider application. Culminating in a scalable, rapid, high-yielding, and operationally simple protocol, this procedure is applied to the first synthesis of several parent polyoxypregnane natural products, representing a gateway to over 100 family members.
Co-reporter:Yu Feng; Dane Holte; Jochen Zoller; Shigenobu Umemiya; Leah R. Simke
Journal of the American Chemical Society 2015 Volume 137(Issue 32) pp:10160-10163
Publication Date(Web):August 9, 2015
DOI:10.1021/jacs.5b07154
Verruculogen and fumitremorgin A are bioactive alkaloids that contain a unique eight-membered endoperoxide. Although related natural products such as fumitremorgins B and C have been previously synthesized, we report the first synthesis of the more complex, endoperoxide-containing members of this family. A concise route to verruculogen and fumitremorgin A relied not only on a hydroperoxide/indole hemiaminal cyclization, but also on the ability to access the seemingly simple starting material, 6-methoxytryptophan. An iridium-catalyzed C–H borylation/Chan–Lam procedure guided by an N-TIPS group enabled the conversion of a tryptophan derivative into a 6-methoxytryptophan derivative, proving to be a general way to functionalize the C6 position of an N,C3-disubstituted indole for the synthesis of indole-containing natural products and pharmaceuticals.
Co-reporter:Ryan D. Baxter, Yong Liang, Xin Hong, Timothy A. Brown, Richard N. Zare, K. N. Houk, Phil S. Baran, and Donna G. Blackmond
ACS Central Science 2015 Volume 1(Issue 8) pp:456
Publication Date(Web):November 2, 2015
DOI:10.1021/acscentsci.5b00332
Kinetic, spectroscopic, and computational studies of radical C–H arylations highlight the interplay between chemical and physical rate processes in these multiphase reactions. Anomalous concentration dependences observed here may be reconciled by considering the role of phase transfer processes that mediate concentrations in each phase. In addition, understanding interactions through phase boundaries enables their use in optimization of reaction performance.
Co-reporter:Thomas J. Maimone, Yoshihiro Ishihara, Phil S. Baran
Tetrahedron 2015 Volume 71(Issue 22) pp:3652-3665
Publication Date(Web):3 June 2015
DOI:10.1016/j.tet.2014.11.010
The Stigonemataceae family of cyanobacteria produces a class of biogenetically related indole natural products that include hapalindoles and ambiguines. In this full account, a practical route to the tetracyclic hapalindole family is presented by way of an eight-step, enantiospecific, protecting-group-free total synthesis of (−)-hapalindole U that features an oxidative indole–enolate coupling. With gram-scale access to hapalindole U, the first total synthesis of an ambiguine alkaloid, (+)-ambiguine H, was completed via an isonitrile-assisted prenylation of an indole followed by a photofragmentation cascade.
Co-reporter:Dr. Yehua Jin;Dr. Chien-Hung Yeh;Dr. Christian A. Kuttruff;Dr. Lars Jørgensen;Dr. Georg Dünstl;Dr. Jakob Felding;Dr. Swaminathan R. Natarajan;Dr. Phil S. Baran
Angewandte Chemie 2015 Volume 127( Issue 47) pp:14250-14254
Publication Date(Web):
DOI:10.1002/ange.201507977
Abstract
Ingenol derivatives with varying degrees of oxidation were prepared by two-phase terpene synthesis. This strategy has allowed access to analogues that cannot be prepared by semisynthesis from natural ingenol. Complex ingenanes resulting from divergent CH oxidation of a common intermediate were found to interact with protein kinase C in a manner that correlates well with the oxidation state of the ingenane core. Even though previous work on ingenanes has suggested a strong correlation between potential to activate PKCδ and induction of neutrophil oxidative burst, the current study shows that the potential to activate PKCβII is of key importance while interaction with PKCδ is dispensable. Thus, key modifications of the ingenane core allowed PKC isoform selectivity wherein PKCδ-driven activation of keratinocytes is strongly reduced or even absent while PKCβII-driven activation of neutrophils is retained.
Co-reporter:Jinghan Gui;Chung-Mao Pan;Ying Jin;Tian Qin;Julian C. Lo;Bryan J. Lee;Steven H. Spergel;Michael E. Mertzman;William J. Pitts;Thomas E. La Cruz;Michael A. Schmidt;Nitin Darvatkar;Swaminathan R. Natarajan
Science 2015 Volume 348(Issue 6237) pp:886-891
Publication Date(Web):22 May 2015
DOI:10.1126/science.aab0245
Stitching C-N bonds from nitro groups
Numerous compounds in pharmaceutical research have carbon-nitrogen bonds, and chemists are always looking for ways to make them more efficiently. Gui et al. present a method that links the carbon in an olefin to the nitrogen in a nitroaromatic compound (see the Perspective by Kürti). Nitroaromatics are readily available, and the method tolerates a wide range of other chemical groups present on either reacting partner.
Science, this issue p. 886; see also p. 863
Co-reporter:Dr. Yehua Jin;Dr. Chien-Hung Yeh;Dr. Christian A. Kuttruff;Dr. Lars Jørgensen;Dr. Georg Dünstl;Dr. Jakob Felding;Dr. Swaminathan R. Natarajan;Dr. Phil S. Baran
Angewandte Chemie International Edition 2015 Volume 54( Issue 47) pp:14044-14048
Publication Date(Web):
DOI:10.1002/anie.201507977
Abstract
Ingenol derivatives with varying degrees of oxidation were prepared by two-phase terpene synthesis. This strategy has allowed access to analogues that cannot be prepared by semisynthesis from natural ingenol. Complex ingenanes resulting from divergent CH oxidation of a common intermediate were found to interact with protein kinase C in a manner that correlates well with the oxidation state of the ingenane core. Even though previous work on ingenanes has suggested a strong correlation between potential to activate PKCδ and induction of neutrophil oxidative burst, the current study shows that the potential to activate PKCβII is of key importance while interaction with PKCδ is dispensable. Thus, key modifications of the ingenane core allowed PKC isoform selectivity wherein PKCδ-driven activation of keratinocytes is strongly reduced or even absent while PKCβII-driven activation of neutrophils is retained.
Co-reporter:Steven J. McKerrall ; Lars Jørgensen ; Christian A. Kuttruff ; Felix Ungeheuer
Journal of the American Chemical Society 2014 Volume 136(Issue 15) pp:5799-5810
Publication Date(Web):April 8, 2014
DOI:10.1021/ja501881p
The complex diterpenoid (+)-ingenol possesses a uniquely challenging scaffold and constitutes the core of a recently approved anti-cancer drug. This full account details the development of a short synthesis of 1 that takes place in two separate phases (cyclase and oxidase) as loosely modeled after terpene biosynthesis. Initial model studies establishing the viability of a Pauson–Khand approach to building up the carbon framework are recounted. Extensive studies that led to the development of a 7-step cyclase phase to transform (+)-3-carene into a suitable tigliane-type core are also presented. A variety of competitive pinacol rearrangements and cyclization reactions were overcome to develop a 7-step oxidase phase producing (+)-ingenol. The pivotal pinacol rearrangement is further examined through DFT calculations, and implications for the biosynthesis of (+)-ingenol are discussed.
Co-reporter:Julian C. Lo ; Yuki Yabe
Journal of the American Chemical Society 2014 Volume 136(Issue 4) pp:1304-1307
Publication Date(Web):January 15, 2014
DOI:10.1021/ja4117632
A redox-economic method for the direct coupling of olefins that uses an inexpensive iron catalyst and a silane reducing agent is reported. Thus, unactivated olefins can be joined directly to electron-deficient olefins in both intra- and intermolecular settings to generate hindered bicyclic systems, vicinal quaternary centers, and even cyclopropanes in good yield. The reaction is not sensitive to oxygen or moisture and has been performed on gram-scale. Most importantly, it allows access to many compounds that would be difficult or perhaps impossible to access using other methods.
Co-reporter:Nathan C. Wilde ; Minetaka Isomura ; Abraham Mendoza
Journal of the American Chemical Society 2014 Volume 136(Issue 13) pp:4909-4912
Publication Date(Web):March 13, 2014
DOI:10.1021/ja501782r
The first successful effort to replicate the beginning of the Taxol oxidase phase in the laboratory is reported, culminating in the total synthesis of taxuyunnanine D, itself a natural product. Through a combination of computational modeling, reagent screening, and oxidation sequence analysis, the first three of eight C–H oxidations (at the allylic sites corresponding to C-5, C-10, and C-13) required to reach Taxol from taxadiene were accomplished. This work lays a foundation for an eventual total synthesis of Taxol capable of delivering not only the natural product but also analogs inaccessible via bioengineering.
Co-reporter:Jinghan Gui ; Qianghui Zhou ; Chung-Mao Pan ; Yuki Yabe ; Aaron C. Burns ; Michael R. Collins ; Martha A. Ornelas ; Yoshihiro Ishihara
Journal of the American Chemical Society 2014 Volume 136(Issue 13) pp:4853-4856
Publication Date(Web):March 10, 2014
DOI:10.1021/ja5007838
A practical C–H functionalization method for the methylation of heteroarenes is presented. Inspiration from Nature’s methylating agent, S-adenosylmethionine (SAM), allowed for the design and development of zinc bis(phenylsulfonylmethanesulfinate), or PSMS. The action of PSMS on a heteroarene generates a (phenylsulfonyl)methylated intermediate that can be easily separated from unreacted starting material. This intermediate can then be desulfonylated to the methylated product or elaborated to a deuteriomethylated product, and can divergently access medicinally important motifs. This mild, operationally simple protocol that can be conducted in open air at room temperature is compatible with sensitive functional groups for the late-stage functionalization of pharmacologically relevant substrates.
Co-reporter:Klement Foo ; Eran Sella ; Isabelle Thomé ; Martin D. Eastgate
Journal of the American Chemical Society 2014 Volume 136(Issue 14) pp:5279-5282
Publication Date(Web):March 23, 2014
DOI:10.1021/ja501879c
A simple method for direct C–H imidation is reported using a new perester-based self-immolating reagent and a base-metal catalyst. The succinimide products obtained can be easily deprotected in situ (if desired) to reveal the corresponding anilines directly. The scope of the reaction is broad, the conditions are extremely mild, and the reaction is tolerant of oxidizable and acid-labile functionality, multiple heteroatoms, and aryl iodides. Mechanistic studies indicate that ferrocene (Cp2Fe) plays the role of an electron shuttle in the decomposition of the perester reagent, delivering a succinimidyl radical ready to add to an aromatic system.
Co-reporter:Brandon R. Rosen ; Erik W. Werner ; Alexander G. O’Brien
Journal of the American Chemical Society 2014 Volume 136(Issue 15) pp:5571-5574
Publication Date(Web):April 3, 2014
DOI:10.1021/ja5013323
N–N-linked dimeric indole alkaloids represent an unexplored class of natural products for which chemical synthesis has no practical solution. To meet this challenge, an electrochemical oxidative dimerization method was developed, which was applied as the pivotal step of the first total synthesis of dixiamycin B. This method is also general for N–N dimerization of substituted carbazoles and β-carbolines, providing entry into seldom explored chemical space.
Co-reporter:Rodrigo A. Rodriguez ; Chung-Mao Pan ; Yuki Yabe ; Yu Kawamata ; Martin D. Eastgate
Journal of the American Chemical Society 2014 Volume 136(Issue 19) pp:6908-6911
Publication Date(Web):April 23, 2014
DOI:10.1021/ja5031744
Unlike its other halogen atom siblings, the utility of chlorinated arenes and (hetero)arenes are twofold: they are useful in tuning electronic structure as well as acting as points for diversification via cross-coupling. Herein we report the invention of a new guanidine-based chlorinating reagent, CBMG or “Palau’chlor”, inspired by a key chlorospirocyclization en route to pyrrole imidazole alkaloids. This direct, mild, operationally simple, and safe chlorinating method is compatible with a range of nitrogen-containing heterocycles as well as select classes of arenes, conjugated π-systems, sulfonamides, and silyl enol ethers. Comparisons with other known chlorinating reagents revealed CBMG to be the premier reagent.
Co-reporter:Emily C. Cherney ; Justin M. Lopchuk ; Jason C. Green
Journal of the American Chemical Society 2014 Volume 136(Issue 36) pp:12592-12595
Publication Date(Web):August 26, 2014
DOI:10.1021/ja507321j
A unified approach to ent-atisane diterpenes and related atisine and hetidine alkaloids has been developed from ent-kaurane (−)-steviol (1). The conversion of the ent-kaurane skeleton to the ent-atisane skeleton features a Mukaiyama peroxygenation with concomitant cleavage of the C13–C16 bond. Conversion to the atisine skeleton (9) features a C20-selective C–H activation using a Suárez modification of the Hofmann–Löffler–Freytag (HLF) reaction. A cascade sequence involving azomethine ylide isomerization followed by Mannich cyclization forms the C14–C20 bond in the hetidine skeleton (8). Finally, attempts to form the N–C6 bond of the hetisine skeleton (7) with a late-stage HLF reaction are discussed. The synthesis of these skeletons has enabled the completion of (−)-methyl atisenoate (3) and (−)-isoatisine (4).
Co-reporter:Rodrigo A. Rodriguez ; Danielle Barrios Steed ; Yu Kawamata ; Shun Su ; Peter A. Smith ; Tyler C. Steed ; Floyd E. Romesberg
Journal of the American Chemical Society 2014 Volume 136(Issue 43) pp:15403-15413
Publication Date(Web):October 20, 2014
DOI:10.1021/ja508632y
Antibiotic-resistant bacteria present an ongoing challenge to both chemists and biologists as they seek novel compounds and modes of action to out-maneuver continually evolving resistance pathways, especially against Gram-negative strains. The dimeric pyrrole–imidazole alkaloids represent a unique marine natural product class with diverse primary biological activity and chemical architecture. This full account traces the strategy used to develop a second-generation route to key spirocycle 9, culminating in a practical synthesis of the axinellamines and enabling their discovery as broad-spectrum antibacterial agents, with promising activity against both Gram-positive and Gram-negative bacteria. While their detailed mode of antibacterial action remains unclear, the axinellamines appear to cause secondary membrane destabilization and impart an aberrant cellular morphology consistent with the inhibition of normal septum formation. This study serves as a rare example of a natural product initially reported to be devoid of biological activity surfacing as an active antibacterial agent with an intriguing mode of action.
Co-reporter:Christian A. Kuttruff, Martin D. Eastgate and Phil S. Baran
Natural Product Reports 2014 vol. 31(Issue 4) pp:419-432
Publication Date(Web):16 Dec 2013
DOI:10.1039/C3NP70090A
The ability to procure useful quantities of a molecule by simple, scalable routes is emerging as an important goal in natural product synthesis. Approaches to molecules that yield substantial material enable collaborative investigations (such as SAR studies or eventual commercial production) and inherently spur innovation in chemistry. As such, when evaluating a natural product synthesis, scalability is becoming an increasingly important factor. In this Highlight, we discuss recent examples of natural product synthesis from our laboratory and others, where the preparation of gram-scale quantities of a target compound or a key intermediate allowed for a deeper understanding of biological activities or enabled further investigational collaborations.
Co-reporter:Fionn O’Hara ; Aaron C. Burns ; Michael R. Collins ; Deepak Dalvie ; Martha A. Ornelas ; Alfin D. N. Vaz ; Yuta Fujiwara
Journal of Medicinal Chemistry 2014 Volume 57(Issue 4) pp:1616-1620
Publication Date(Web):January 28, 2014
DOI:10.1021/jm4017976
The bioavailability of aromatic azaheterocyclic drugs can be affected by the activity of aldehyde oxidase (AO). Susceptibility to AO metabolism is difficult to predict computationally and can be complicated in vivo by differences between species. Here we report the use of bis(((difluoromethyl)sulfinyl)oxy)zinc (DFMS) as a source of CF2H radical for a rapid and inexpensive chemical “litmus test” for the early identification of heteroaromatic drug candidates that have a high probability of metabolism by AO.
Co-reporter:Will R. Gutekunst and Phil S. Baran
The Journal of Organic Chemistry 2014 Volume 79(Issue 6) pp:2430-2452
Publication Date(Web):February 18, 2014
DOI:10.1021/jo4027148
The application of C–H functionalization logic to target-oriented synthesis provides an exciting new venue for the development of new and useful strategies in organic chemistry. In this article, C–H functionalization reactions are explored as an alternative approach to access pseudodimeric cyclobutane natural products, such as the dictazole and the piperarborenine families. The use of these strategies in a variety of complex settings highlights the subtle geometric, steric, and electronic effects at play in the auxiliary guided C–H functionalization of cyclobutanes.
Co-reporter:Ryan Gianatassio;Dr. Shuhei Kawamura;Cecil L Eprile;Dr. Klement Foo;Jason Ge;Dr. Aaron C. Burns;Dr. Michael R. Collins;Dr. Phil S. Baran
Angewandte Chemie International Edition 2014 Volume 53( Issue 37) pp:9851-9855
Publication Date(Web):
DOI:10.1002/anie.201406622
Abstract
A simple method to convert readily available carboxylic acids into sulfinate salts by employing an interrupted Barton decarboxylation reaction is reported. A medicinally oriented panel of ten new sulfinate reagents was created using this method, including a key trifluoromethylcyclopropanation reagent, TFCS-Na. The reactivity of six of these salts towards CH functionalization was field-tested using several different classes of heterocycles.
Co-reporter:Dr. Phil S. Baran
Angewandte Chemie International Edition 2014 Volume 53( Issue 37) pp:9704-9705
Publication Date(Web):
DOI:10.1002/anie.201407049
Co-reporter:Dr. Alexer G. O'Brien;Dr. Akinobu Maruyama;Dr. Yasuhide Inokuma; Makoto Fujita; Phil S. Baran; Donna G. Blackmond
Angewandte Chemie International Edition 2014 Volume 53( Issue 44) pp:11868-11871
Publication Date(Web):
DOI:10.1002/anie.201407948
Abstract
Electrochemical reactions are shown to be effective for the CH functionalization of a number of heterocyclic substrates that are recalcitrant to conventional peroxide radical initiation conditions. Monitoring reaction progress under electrochemical conditions provides mechanistic insight into the CH functionalization of a series of heterocycles of interest in medicinal chemistry.
Co-reporter:Hai T. Dao ;Dr. Phil S. Baran
Angewandte Chemie International Edition 2014 Volume 53( Issue 52) pp:14382-14386
Publication Date(Web):
DOI:10.1002/anie.201408022
Abstract
The utility of quinone diazides in materials science is vast and well-documented, yet this potentially useful motif has languished in the annals of organic synthesis. Herein we show that modern tools of catalysis can be employed with free or suitably masked quinone diazides to unleash the power of these classic diazo compounds in the context of both inter- and intramolecular olefin cyclopropanation.
Co-reporter:Quentin Michaudel;Dr. Guillaume Journot;Dr. Alicia Regueiro-Ren;Dr. Animesh Goswami;Dr. Zhiwei Guo;Dr. Thomas P. Tully;Dr. Lufeng Zou;Dr. Raghunath O. Ramabhadran;Dr. Kendall N. Houk;Dr. Phil S. Baran
Angewandte Chemie International Edition 2014 Volume 53( Issue 45) pp:12091-12096
Publication Date(Web):
DOI:10.1002/anie.201407016
Abstract
Physicochemical properties constitute a key factor for the success of a drug candidate. Whereas many strategies to improve the physicochemical properties of small heterocycle-type leads exist, complex hydrocarbon skeletons are more challenging to derivatize because of the absence of functional groups. A variety of CH oxidation methods have been explored on the betulin skeleton to improve the solubility of this very bioactive, yet poorly water-soluble, natural product. Capitalizing on the innate reactivity of the molecule, as well as the few molecular handles present on the core, allowed oxidations at different positions across the pentacyclic structure. Enzymatic oxidations afforded several orthogonal oxidations to chemical methods. Solubility measurements showed an enhancement for many of the synthesized compounds.
Co-reporter:Xin Hong, Dane Holte, Daniel C. G. Götz, Phil S. Baran, and K. N. Houk
The Journal of Organic Chemistry 2014 Volume 79(Issue 24) pp:12177-12184
Publication Date(Web):October 17, 2014
DOI:10.1021/jo502219d
Density functional theory (DFT) calculations with B3LYP and M06 functionals elucidated the reactivities of alkynes and Z/E selectivity of cyclodecatriene products in the Ni-catalyzed [4 + 4 + 2] cycloadditions of dienes and alkynes. The Ni-mediated oxidative cyclization of butadienes determines the Z/E selectivity. Only the oxidative cyclization of one s-cis to one s-trans butadiene is facile and exergonic, leading to the observed 1Z,4Z,8E-cyclodecatriene product. The same step with two s-cis or s-trans butadienes is either kinetically or thermodynamically unfavorable, and the 1Z,4E,8E- and 1Z,4Z,8Z-cyclodecatriene isomers are not observed in experiments. In addition, the competition between the desired cooligomerization and [2 + 2 + 2] cycloadditions of alkynes depends on the coordination of alkynes. With either electron-deficient alkynes or alkynes with free hydroxyl groups, the coordination of alkynes is stronger than that of dienes, and alkyne trimerization prevails. With alkyl-substituted alkynes, the generation of alkyne-coordinated nickel complex is much less favorable, and the [4 + 4 + 2] cycloaddition occurs.
Co-reporter:Hai T. Dao ;Dr. Phil S. Baran
Angewandte Chemie 2014 Volume 126( Issue 52) pp:14610-14614
Publication Date(Web):
DOI:10.1002/ange.201408022
Abstract
The utility of quinone diazides in materials science is vast and well-documented, yet this potentially useful motif has languished in the annals of organic synthesis. Herein we show that modern tools of catalysis can be employed with free or suitably masked quinone diazides to unleash the power of these classic diazo compounds in the context of both inter- and intramolecular olefin cyclopropanation.
Co-reporter:Dr. Alexer G. O'Brien;Dr. Akinobu Maruyama;Dr. Yasuhide Inokuma; Makoto Fujita; Phil S. Baran; Donna G. Blackmond
Angewandte Chemie 2014 Volume 126( Issue 44) pp:12062-12065
Publication Date(Web):
DOI:10.1002/ange.201407948
Abstract
Electrochemical reactions are shown to be effective for the CH functionalization of a number of heterocyclic substrates that are recalcitrant to conventional peroxide radical initiation conditions. Monitoring reaction progress under electrochemical conditions provides mechanistic insight into the CH functionalization of a series of heterocycles of interest in medicinal chemistry.
Co-reporter:Dr. Phil S. Baran
Angewandte Chemie 2014 Volume 126( Issue 37) pp:9858-9859
Publication Date(Web):
DOI:10.1002/ange.201407049
Co-reporter:Quentin Michaudel;Dr. Guillaume Journot;Dr. Alicia Regueiro-Ren;Dr. Animesh Goswami;Dr. Zhiwei Guo;Dr. Thomas P. Tully;Dr. Lufeng Zou;Dr. Raghunath O. Ramabhadran;Dr. Kendall N. Houk;Dr. Phil S. Baran
Angewandte Chemie 2014 Volume 126( Issue 45) pp:12287-12292
Publication Date(Web):
DOI:10.1002/ange.201407016
Abstract
Physicochemical properties constitute a key factor for the success of a drug candidate. Whereas many strategies to improve the physicochemical properties of small heterocycle-type leads exist, complex hydrocarbon skeletons are more challenging to derivatize because of the absence of functional groups. A variety of CH oxidation methods have been explored on the betulin skeleton to improve the solubility of this very bioactive, yet poorly water-soluble, natural product. Capitalizing on the innate reactivity of the molecule, as well as the few molecular handles present on the core, allowed oxidations at different positions across the pentacyclic structure. Enzymatic oxidations afforded several orthogonal oxidations to chemical methods. Solubility measurements showed an enhancement for many of the synthesized compounds.
Co-reporter:Ryan Gianatassio;Dr. Shuhei Kawamura;Cecil L Eprile;Dr. Klement Foo;Jason Ge;Dr. Aaron C. Burns;Dr. Michael R. Collins;Dr. Phil S. Baran
Angewandte Chemie 2014 Volume 126( Issue 37) pp:10009-10013
Publication Date(Web):
DOI:10.1002/ange.201406622
Abstract
A simple method to convert readily available carboxylic acids into sulfinate salts by employing an interrupted Barton decarboxylation reaction is reported. A medicinally oriented panel of ten new sulfinate reagents was created using this method, including a key trifluoromethylcyclopropanation reagent, TFCS-Na. The reactivity of six of these salts towards CH functionalization was field-tested using several different classes of heterocycles.
Co-reporter:Hans Renata;Qianghui Zhou
Science 2013 Vol 339(6115) pp:59-63
Publication Date(Web):04 Jan 2013
DOI:10.1126/science.1230631
Co-reporter:Qianghui Zhou ; Jinghan Gui ; Chung-Mao Pan ; Earl Albone ; Xin Cheng ; Edward M. Suh ; Luigi Grasso ; Yoshihiro Ishihara
Journal of the American Chemical Society 2013 Volume 135(Issue 35) pp:12994-12997
Publication Date(Web):August 19, 2013
DOI:10.1021/ja407739y
A general C–H functionalization method for the tagging of natural products and pharmaceuticals is described. An azide-containing sulfinate reagent allows the appendage of azidoalkyl chains onto heteroaromatics, the product of which can then be attached to a monoclonal antibody by a “click” reaction. This strategy expands the breadth of bioactive small molecules that can be linked to macromolecules in a manner that is beyond the scope of existing methods in bioconjugation to permit tagging of the “seemingly untaggable”.
Co-reporter:Fionn O’Hara, Donna G. Blackmond, and Phil S. Baran
Journal of the American Chemical Society 2013 Volume 135(Issue 32) pp:12122-12134
Publication Date(Web):July 17, 2013
DOI:10.1021/ja406223k
Radical addition processes can be ideally suited for the direct functionalization of heteroaromatic bases, yet these processes are only sparsely used due to the perception of poor or unreliable control of regiochemistry. A systematic investigation of factors affecting the regiochemistry of radical functionalization of heterocycles using alkylsulfinate salts revealed that certain types of substituents exert consistent and additive effects on the regioselectivity of substitution. This allowed us to establish guidelines for predicting regioselectivity on complex π-deficient heteroarenes, including pyridines, pyrimidines, pyridazines, and pyrazines. Since the relative contribution from opposing directing factors was dependent on solvent and pH, it was sometimes possible to tune the regiochemistry to a desired result by modifying reaction conditions. This methodology was applied to the direct, regioselective introduction of isopropyl groups into complex, biologically active molecules, such as diflufenican (44) and nevirapine (45).
Co-reporter:Sarah E. Wengryniuk, Andreas Weickgenannt, Christopher Reiher, Neil A. Strotman, Ke Chen, Martin D. Eastgate, and Phil S. Baran
Organic Letters 2013 Volume 15(Issue 4) pp:792-795
Publication Date(Web):January 25, 2013
DOI:10.1021/ol3034675
A mild method for the regioselective C2-bromination of fused azine N-oxides is presented, employing tosic anhydride as the activator and tetra-n-butylammonium bromide as the nucleophilic bromide source. The C2-brominated compounds are produced in moderate to excellent yields and with excellent regioselectivity in most cases. The potential extension of this method to other halogens, effecting C2-chlorination with Ts2O/TBACl is also presented. Finally, this method could be incorporated into a viable one-pot oxidation/bromination process, using methyltrioxorhenium/urea hydropgen peroxide as the oxidant.
Co-reporter:Ippei Usui, David W. Lin, Takeshi Masuda, and Phil S. Baran
Organic Letters 2013 Volume 15(Issue 9) pp:2080-2083
Publication Date(Web):April 11, 2013
DOI:10.1021/ol400709f
The first synthesis of members of the sarcodonin family, phellodonin and sarcodonin ε, is reported herein. This verifies that the unprecedented and seemingly unstable N,N-dioxide-containing benzodioxazine framework can be constructed in the laboratory and lends further support to the proposed structures. The key step in the synthesis involves a biomimetic hetero-Diels–Alder reaction between a pyrazine N-oxide and an ortho-quinone.
Co-reporter:Dr. Qianghui Zhou;Alessro Ruffoni;Ryan Gianatassio;Dr. Yuta Fujiwara;Dr. Eran Sella;Dr. Doron Shabat;Dr. Phil S. Baran
Angewandte Chemie International Edition 2013 Volume 52( Issue 14) pp:3949-3952
Publication Date(Web):
DOI:10.1002/anie.201300763
Co-reporter:Yoshihiro Ishihara, Abraham Mendoza, Phil S. Baran
Tetrahedron 2013 69(27–28) pp: 5685-5701
Publication Date(Web):
DOI:10.1016/j.tet.2013.04.028
Co-reporter:Bron R. Rosen;Leah R. Simke;Peter S. Thuy-Boun;Dr. Darryl D. Dixon;Dr. Jin-Quan Yu;Dr. Phil S. Baran
Angewandte Chemie International Edition 2013 Volume 52( Issue 28) pp:7317-7320
Publication Date(Web):
DOI:10.1002/anie.201303838
Co-reporter:Emily C. Cherney;Dr. Jason C. Green;Dr. Phil S. Baran
Angewandte Chemie 2013 Volume 125( Issue 34) pp:9189-9192
Publication Date(Web):
DOI:10.1002/ange.201304609
Co-reporter:Bron R. Rosen;Leah R. Simke;Peter S. Thuy-Boun;Dr. Darryl D. Dixon;Dr. Jin-Quan Yu;Dr. Phil S. Baran
Angewandte Chemie 2013 Volume 125( Issue 28) pp:7458-7461
Publication Date(Web):
DOI:10.1002/ange.201303838
Co-reporter:Dr. Qianghui Zhou;Alessro Ruffoni;Ryan Gianatassio;Dr. Yuta Fujiwara;Dr. Eran Sella;Dr. Doron Shabat;Dr. Phil S. Baran
Angewandte Chemie 2013 Volume 125( Issue 14) pp:4041-4044
Publication Date(Web):
DOI:10.1002/ange.201300763
Co-reporter:Emily C. Cherney;Dr. Jason C. Green;Dr. Phil S. Baran
Angewandte Chemie International Edition 2013 Volume 52( Issue 34) pp:9019-9022
Publication Date(Web):
DOI:10.1002/anie.201304609
Co-reporter:Jakob Felding;Christian A. Kuttruff;Steven J. McKerrall;Felix Ungeheuer;Lars Jørgensen
Science 2013 Volume 341(Issue 6148) pp:878-882
Publication Date(Web):23 Aug 2013
DOI:10.1126/science.1241606
Ingenol Ingenuity
The diterpenoid i ngenol is the core structure of a topical drug recently commercialized to treat actinic keratosis, a precancerous skin condition. Sourcing the compound from the Euphorbia plants that produce it is relatively inefficient, so Jørgensen et al. (p. 878, published online 1 August) devised a chemical synthesis starting from the comparatively simple and inexpensive monoterpene chiral (+)-3-carene. The synthetic sequence involves 14 steps—less than half as long as prior chemical routes to the target—and relies on a two-stage approach, inspired by the posited biosynthetic pathway, in which preliminary assembly of the fused ring framework precedes hydroxylation of the periphery.
Co-reporter:Tobias Brückl, Ryan D. Baxter, Yoshihiro Ishihara, and Phil S. Baran
Accounts of Chemical Research 2012 Volume 45(Issue 6) pp:826
Publication Date(Web):October 21, 2011
DOI:10.1021/ar200194b
The combustion of organic matter is perhaps the oldest and most common chemical transformation utilized by mankind. The generation of a C–O bond at the expense of a C–H bond during this process may be considered the most basic form of C–H functionalization. This illustrates the extreme generality of the term “C–H functionalization”, because it can describe the conversion of literally any C–H bond into a C–X bond (X being anything except H). Therefore, it may be of use to distinguish between what, in our view, are two distinct categories of C–H functionalization logic: “guided” and “innate”. Guided C–H functionalizations, as the name implies, are guided by external reagents or directing groups (covalently or fleetingly bound) to install new functional groups at the expense of specifically targeted C–H bonds. Conversely, innate C–H functionalizations may be broadly defined as reactions that exchange C–H bonds for new functional groups based solely on natural reactivity patterns in the absence of other directing forces.Two substrates that illustrate this distinction are dihydrojunenol and isonicotinic acid. The C–H functionalization processes of hydroxylation or arylation, respectively, can take place at multiple locations on each molecule. Innate functionalizations lead to substitution patterns that are dictated by the inherent bias (steric or electronic) of the substrate undergoing C–H cleavage, whereas guided functionalizations lead to substitution patterns that are controlled by external directing forces such as metal complexation or steric bias of the reagent. Although the distinction between guided and innate C–H functionalizations may not always be clear in cases that do not fit neatly into a single category, it is a useful convention to consider when analyzing reactivity patterns and strategies for synthesis. We must emphasize that although a completely rigorous distinction between guided and innate C–H functionalization may not be practical, we have nonetheless found it to be a useful tool at the planning stage of synthesis.In this Account, we trace our own studies in the area of C–H functionalization in synthesis through the lens of “guided” and “innate” descriptors. We show how harnessing innate reactivity can be beneficial for achieving unique bond constructions between heterocycles and carbonyl compounds, enabling rapid and scalable total syntheses. Guided and innate functionalizations were used synergistically to create an entire family of terpenes in a controlled fashion. We continue with a discussion of the synthesis of complex alkaloids with high nitrogen content, which required the invention of a uniquely chemoselective innate C–H functionalization protocol. These findings led us to develop a series of innate C–H functionalization reactions for forging C–C bonds of interest to the largest body of practicing organic chemists: medicinal chemists. Strategic use of C–H functionalization logic can have a dramatically positive effect on the efficiency of synthesis, whether guided or innate.
Co-reporter:Yuta Fujiwara ; Janice A. Dixon ; Rodrigo A. Rodriguez ; Ryan D. Baxter ; Darryl D. Dixon ; Michael R. Collins ; Donna G. Blackmond
Journal of the American Chemical Society 2012 Volume 134(Issue 3) pp:1494-1497
Publication Date(Web):January 9, 2012
DOI:10.1021/ja211422g
Molecular scaffolds containing alkylfluorine substituents are desired in many areas of chemical research from materials to pharmaceuticals. Herein, we report the invention of a new reagent (Zn(SO2CF2H)2, DFMS) for the innate difluoromethylation of organic substrates via a radical process. This mild, operationally simple, chemoselective, and scalable difluoromethylation method is compatible with a range of nitrogen-containing heteroarene substrates of varying complexity as well as select classes of conjugated π-systems and thiols. Regiochemical comparisons suggest that the CF2H radical generated from the new reagent possesses nucleophilic character.
Co-reporter:Darryl D. Dixon ; Jonathan W. Lockner ; Qianghui Zhou
Journal of the American Chemical Society 2012 Volume 134(Issue 20) pp:8432-8435
Publication Date(Web):May 14, 2012
DOI:10.1021/ja303937y
A scalable, divergent synthesis of bioactive meroterpenoids has been developed. A key component of this work is the invention of “borono-sclareolide”, a terpenyl radical precursor that enables gram-scale preparation of (+)-chromazonarol. Subsequent synthetic operations on this key intermediate permit rapid access to a variety of related meroterpenoids, many of which possess important biological activity.
Co-reporter:Quentin Michaudel ; Damien Thevenet
Journal of the American Chemical Society 2012 Volume 134(Issue 5) pp:2547-2550
Publication Date(Web):January 24, 2012
DOI:10.1021/ja212020b
Intermolecular Ritter-type C–H amination of unactivated sp3 carbons has been developed. This new reaction proceeds under mild conditions using readily available reagents and an inexpensive source of nitrogen (acetonitrile). A broad scope of substrates can be aminated with this method since many functional groups are tolerated. This reaction also allows for the direct, innate C–H amination of a variety of hydrocarbons such as cyclohexane without the need of prefunctionalization or installation of a directing group.
Co-reporter:Klement Foo;Ippei Usui;Daniel C. G. Götz;Erik W. Werner;Dane Holte
Angewandte Chemie 2012 Volume 124( Issue 46) pp:11659-11663
Publication Date(Web):
DOI:10.1002/ange.201206904
Co-reporter:Will R. Gutekunst;Ryan Gianatassio ;Dr. Phil S. Baran
Angewandte Chemie 2012 Volume 124( Issue 30) pp:
Publication Date(Web):
DOI:10.1002/ange.201203897
Co-reporter:Dane Holte, Daniel C. G. Götz, and Phil S. Baran
The Journal of Organic Chemistry 2012 Volume 77(Issue 2) pp:825-842
Publication Date(Web):January 3, 2012
DOI:10.1021/jo202314a
Artificially mimicking the cyclase phase of terpene biosynthesis inspires the invention of new methodologies, since working with carbogenic frameworks containing minimal functionality limits the chemist’s toolbox of synthetic strategies. For example, the construction of terpene skeletons from five-carbon building blocks would be an exciting pathway to mimic in the laboratory. Nature oligomerizes, cyclizes, and then oxidizes γ,γ-dimethylallyl pyrophosphate (DMAPP) and isopentenyl pyrophosphate (IPP) to all of the known terpenes. Starting from isoprene, the goal of this work was to mimic Nature’s approach for rapidly building molecular complexity. In principle, the controlled oligomerization of isoprene would drastically simplify the synthesis of terpenes used in the medicine, perfumery, flavor, and materials industries. This article delineates our extensive efforts to cooligomerize isoprene or butadiene with alkynes in a controlled fashion by zerovalent nickel catalysis building off the classic studies by Wilke and co-workers.
Co-reporter:Klement Foo;Ippei Usui;Daniel C. G. Götz;Erik W. Werner;Dane Holte
Angewandte Chemie International Edition 2012 Volume 51( Issue 46) pp:11491-11495
Publication Date(Web):
DOI:10.1002/anie.201206904
Co-reporter:Will R. Gutekunst;Ryan Gianatassio ;Dr. Phil S. Baran
Angewandte Chemie International Edition 2012 Volume 51( Issue 30) pp:
Publication Date(Web):
DOI:10.1002/anie.201203897
Co-reporter:Will R. Gutekunst and Phil S. Baran
Chemical Society Reviews 2011 vol. 40(Issue 4) pp:1976-1991
Publication Date(Web):07 Feb 2011
DOI:10.1039/C0CS00182A
In this critical review, the strategic and economic benefits of C–H functionalization logic will be analyzed through the critical lens of total synthesis. In order to illustrate the dramatically simplifying effects this type of logic can potentially have on synthetic planning, we take the reader through a series of case studies in which it has already been successfully applied. In the first section, a chronological look at key historical syntheses will be examined, leading into modern day examples. In the second section, our own experience with applying and executing synthesis with a C–H functionalization “mindset” will be discussed (114 references).
Co-reporter:Yuta Fujiwara ; Victoriano Domingo ; Ian B. Seiple ; Ryan Gianatassio ; Matthew Del Bel
Journal of the American Chemical Society 2011 Volume 133(Issue 10) pp:3292-3295
Publication Date(Web):February 22, 2011
DOI:10.1021/ja111152z
A direct functionalization of a variety of quinones with several boronic acids has been developed. This scalable reaction proceeds readily at room temperature in an open flask using inexpensive reagents: catalytic silver(I) nitrate in the presence of a persulfate co-oxidant. The scope with respect to quinones is broad, with a variety of alkyl- and arylboronic acids undergoing efficient cross-coupling. The mechanism is presumed to proceed through a nucleophilic radical addition to the quinone with in situ reoxidation of the resulting dihydroquinone. This method has been applied to complex substrates, including a steroid derivative and a farnesyl natural product.
Co-reporter:Jun Shi ; Georg Manolikakes ; Chien-Hung Yeh ; Carlos A. Guerrero ; Ryan A. Shenvi ; Hiroki Shigehisa
Journal of the American Chemical Society 2011 Volume 133(Issue 20) pp:8014-8027
Publication Date(Web):May 3, 2011
DOI:10.1021/ja202103e
Full details are provided for an improved synthesis of cortistatin A and related structures as well as the underlying logic and evolution of strategy. The highly functionalized cortistatin A-ring embedded with a key heteroadamantane was synthesized by a simple and scalable five-step sequence. A chemoselective, tandem geminal dihalogenation of an unactivated methyl group, a reductive fragmentation/trapping/elimination of a bromocyclopropane, and a facile chemoselective etherification reaction afforded the cortistatin A core, dubbed “cortistatinone”. A selective Δ16-alkene reduction with Raney Ni provided cortistatin A. With this scalable and practical route, copious quantities of cortistatinone, Δ16-cortistatin A (the equipotent direct precursor to cortistatin A), and its related analogues were prepared for further biological studies.
Co-reporter:Shun Su ; Rodrigo A. Rodriguez
Journal of the American Chemical Society 2011 Volume 133(Issue 35) pp:13922-13925
Publication Date(Web):August 16, 2011
DOI:10.1021/ja206191g
The development of a simple, efficient, scalable, and stereocontrolled synthesis of a common intermediate en route to the axinellamines, massadines, and palau’amine is reported. This completely new route was utilized to prepare the axinellamines on a gram scale. In a more general sense, three distinct and enabling methodological advances were made during these studies: (1) an ethylene glycol-assisted Pauson–Khand cycloaddition reaction, (2) a Zn/In-mediated Barbier-type reaction, and (3) a TfNH2-assisted chlorination–spirocyclization.
Co-reporter:Ian B. Seiple ; Shun Su ; Ian S. Young ; Akifumi Nakamura ; Junichiro Yamaguchi ; Lars Jørgensen ; Rodrigo A. Rodriguez ; Daniel P. O’Malley ; Tanja Gaich ; Matthias Köck
Journal of the American Chemical Society 2011 Volume 133(Issue 37) pp:14710-14726
Publication Date(Web):August 23, 2011
DOI:10.1021/ja2047232
Dimeric pyrrole–imidazole alkaloids represent a rich and topologically unique class of marine natural products. This full account will follow the progression of efforts that culminated in the enantioselective total syntheses of the most structurally ornate members of this family: the axinellamines, the massadines, and palau’amine. A bio-inspired approach capitalizing on the pseudo-symmetry of the members of this class is recounted, delivering a deschloro derivative of the natural product core. Next, the enantioselective synthesis of the chlorocyclopentane core featuring a scalable, catalytic, enantioselective Diels–Alder reaction of a 1-siloxydiene is outlined in detail. Finally, the successful divergent conversion of this core to each of the aforementioned natural products, and the ensuing methodological developments, are described.
Co-reporter:Will R. Gutekunst
Journal of the American Chemical Society 2011 Volume 133(Issue 47) pp:19076-19079
Publication Date(Web):November 8, 2011
DOI:10.1021/ja209205x
A strategy for the construction of unsymmetrical cyclobutanes using C–H functionalization logic is demonstrated in the total synthesis of piperarborenine B and piperarborenine D (reported structure). These syntheses feature a new preparation of cis-cyclobutane dicarboxylates from commercially available coumalate starting materials and a divergent approach to the controlled cis or trans installation of the two distinct aryl rings found in the natural products using the first example of cyclobutane C–H arylation. The structure of piperarborenine D is reassigned to a head-to-head dimer, which was synthesized using an intramolecular [2+2] photocycloaddition strategy.
Co-reporter:Jonathan W. Lockner, Darryl D. Dixon, Rune Risgaard, and Phil S. Baran
Organic Letters 2011 Volume 13(Issue 20) pp:5628-5631
Publication Date(Web):September 16, 2011
DOI:10.1021/ol2023505
Practical radical cyclizations using organoboronic acids and trifluoroborates take place in water, open to air, and in a scalable fashion employing catalytic silver nitrate and stoichiometric potassium persulfate. Both Pschorr-type cyclizations and tandem radical cyclization/trap cascades are described, illustrating the utility of these mild conditions for the generation of polycyclic scaffolds.
Co-reporter:Emily C. Cherney
Israel Journal of Chemistry 2011 Volume 51( Issue 3-4) pp:391-405
Publication Date(Web):
DOI:10.1002/ijch.201100005
Abstract
Terpenes and alkaloids are ever-growing classes of natural products that provide new molecular structures that inspire chemists and possess a broad range of biological activity. Terpenoid-alkaloids originate from the same prenyl units that construct terpene skeletons. However, during biosynthesis, a nitrogen atom (or atoms) is introduced in the form of β-aminoethanol, ethylamine, or methylamine. Nitrogen incorporation can occur either before, during, or after the cyclase phase. The outcome of this unique biosynthesis is the formation of natural products containing unprecedented structures. These complex structural motifs expose current limitations in organic chemistry, thus providing opportunities for invention. This review focuses on total syntheses of terpenoid-alkaloids and unique issues presented by this class of natural products. More specifically, it examines how these syntheses relate to the way terpenoid-alkaloids are made in Nature. Developments in chemistry that have facilitated these syntheses are emphasized, as well as chemical technology needed to conquer those that evade synthesis.
Co-reporter:Yining Ji;Tobias Brueckl;Yuta Fujiwara;Ryan D. Baxter;Shun Su;Ian B. Seiple;Donna G. Blackmond
PNAS 2011 Volume 108 (Issue 35 ) pp:
Publication Date(Web):2011-08-30
DOI:10.1073/pnas.1109059108
Direct methods for the trifluoromethylation of heteroaromatic systems are in extremely high demand in nearly every sector
of chemical industry. Here we report the discovery of a general procedure using a benchtop stable trifluoromethyl radical
source that functions broadly on a variety of electron deficient and rich heteroaromatic systems and demonstrates high functional
group tolerance. This C-H trifluoromethylation protocol is operationally simple (avoids gaseous CF3I), scalable, proceeds at ambient temperature, can be used directly on unprotected molecules, and is demonstrated to proceed
at the innately reactive positions of the substrate. The unique and orthogonal reactivity of the trifluoromethyl radical relative
to aryl radicals has also been investigated on both a complex natural product and a pharmaceutical agent. Finally, preliminary
data suggest that the regioselectivity of C-H trifluoromethylation can be fine-tuned simply by judicious solvent choice.
Co-reporter:Timothy Newhouse ;Dr. Phil S. Baran
Angewandte Chemie International Edition 2011 Volume 50( Issue 15) pp:3362-3374
Publication Date(Web):
DOI:10.1002/anie.201006368
Abstract
CH oxidation has a long history and an ongoing presence in research at the forefront of chemistry and interrelated fields. As such, numerous highly useful articles and reviews have been written on this subject. Logically, these are generally written from the perspective of the scope and limitations of the reagents employed. This Minireview instead attempts to emphasize chemoselectivity imposed by the nature of the substrate. Consequently, many landmark discoveries in the field of CH oxidation are not discussed, but hopefully the perspective taken herein will allow CH oxidation reactions to be more readily incorporated into synthetic planning.
Co-reporter:Klement Foo;Timothy Newhouse;Ikue Mori; Hiromitsu Takayama; Phil S. Baran
Angewandte Chemie 2011 Volume 123( Issue 12) pp:2768-2771
Publication Date(Web):
DOI:10.1002/ange.201008048
Co-reporter:Timothy Newhouse ;Dr. Phil S. Baran
Angewandte Chemie 2011 Volume 123( Issue 15) pp:3422-3435
Publication Date(Web):
DOI:10.1002/ange.201006368
Abstract
Die C-H-Oxidation hat eine lange Geschichte und ist seit jeher Teil der Spitzenforschung in der Chemie und verwandten Gebieten. Folgerichtig wurde bereits eine große Zahl sehr nützlicher Originalbeiträge und Übersichten zu diesem Thema veröffentlicht. Logischerweise wurden diese Artikel im Allgemeinen mit Blick auf die Anwendungsmöglichkeiten und Grenzen der verwendeten Reagentien geschrieben. Ziel dieses Kurzaufsatzes ist es hingegen, stattdessen die durch die Eigenschaften der Substrate hervorgerufene Chemoselektivität herauszuarbeiten. Dies bringt es mit sich, dass viele bahnbrechende Entdeckungen auf dem Gebiet der C-H-Oxidationen nicht behandelt werden; die hier eingenommene Sichtweise wird aber hoffentlich eine schnellere Aufnahme von C-H-Oxidationsreaktionen in Syntheseplanungen ermöglichen.
Co-reporter:David W. Lin, Takeshi Masuda, Moritz B. Biskup, Jonathan D. Nelson, and Phil S. Baran
The Journal of Organic Chemistry 2011 Volume 76(Issue 4) pp:1013-1030
Publication Date(Web):January 20, 2011
DOI:10.1021/jo102228j
A sweeping structural revision of the sarcodonin natural product family (published structures 1a−13a) is proposed after extensive studies aimed at their chemical synthesis. Key features of revised structure 1b include replacement of the N,N-dioxide moiety with an oxime, ring-opening of the central diketopiperazine, and transposition of the terphenyl wing from the 1β−2β position of 1a to the 2β−3β position of 1b. This structure revision arose from the serendipitous synthesis of a benzodioxane aminal (44) whose structure was unambiguously determined by X-ray crystallography and whose spectral properties bore considerable resemblance to the published data for the sarcodonins. A versatile new method for O-arylation of hydroxamic acids is also reported herein, as well as a manganese(III)-mediated α-oxidation of hydroxamic acids to aminals.
Co-reporter:Klement Foo;Timothy Newhouse;Ikue Mori; Hiromitsu Takayama; Phil S. Baran
Angewandte Chemie International Edition 2011 Volume 50( Issue 12) pp:2716-2719
Publication Date(Web):
DOI:10.1002/anie.201008048
Co-reporter:Ian B. Seiple ; Shun Su ; Rodrigo A. Rodriguez ; Ryan Gianatassio ; Yuta Fujiwara ; Adam L. Sobel
Journal of the American Chemical Society 2010 Volume 132(Issue 38) pp:13194-13196
Publication Date(Web):September 2, 2010
DOI:10.1021/ja1066459
A direct arylation of a variety of electron-deficient heterocycles with arylboronic acids has been developed. This new reaction proceeds readily at room temperature using inexpensive reagents: catalytic silver(I) nitrate in the presence of persulfate co-oxidant. The scope with respect to heterocycle and boronic acid coupling partner is broad, and sensitive functional groups are tolerated. This method allows for rapid access to a variety of arylated heterocycles that would be more difficult to access with traditional methods.
Co-reporter:IanB. Seiple;Shun Su Dr.;IanS. Young Dr.;ChadA. Lewis Dr.;Junichiro Yamaguchi Dr. ;PhilS. Baran Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 6) pp:1095-1098
Publication Date(Web):
DOI:10.1002/anie.200907112
Co-reporter:Tanja Gaich and Phil S. Baran
The Journal of Organic Chemistry 2010 Volume 75(Issue 14) pp:4657-4673
Publication Date(Web):June 11, 2010
DOI:10.1021/jo1006812
The field of total synthesis has a rich history and a vibrant future. Landmark advances and revolutionary strides in the logic of synthesis have put the practicing chemist in the enviable position of being able to create nearly any molecule with enough time and effort. The stage is now set for organic chemists to aim for “ideality” in the way molecules are synthesized. This perspective presents a simple and informative definition of “ideality” and demonstrates its use during the self-evaluation of several syntheses from our laboratory.
Co-reporter:Ke Chen, Yoshihiro Ishihara, María Morón Galán, Phil S. Baran
Tetrahedron 2010 66(26) pp: 4738-4744
Publication Date(Web):
DOI:10.1016/j.tet.2010.02.088
Co-reporter:IanB. Seiple;Shun Su Dr.;IanS. Young Dr.;ChadA. Lewis Dr.;Junichiro Yamaguchi Dr. ;PhilS. Baran Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 6) pp:
Publication Date(Web):
DOI:10.1002/anie.200907224
Co-reporter:IanB. Seiple;Shun Su Dr.;IanS. Young Dr.;ChadA. Lewis Dr.;Junichiro Yamaguchi Dr. ;PhilS. Baran Dr.
Angewandte Chemie 2010 Volume 122( Issue 6) pp:
Publication Date(Web):
DOI:10.1002/ange.200907224
Co-reporter:IanB. Seiple;Shun Su Dr.;IanS. Young Dr.;ChadA. Lewis Dr.;Junichiro Yamaguchi Dr. ;PhilS. Baran Dr.
Angewandte Chemie 2010 Volume 122( Issue 6) pp:1113-1116
Publication Date(Web):
DOI:10.1002/ange.200907112
Co-reporter:Timothy Newhouse, Phil S. Baran and Reinhard W. Hoffmann
Chemical Society Reviews 2009 vol. 38(Issue 11) pp:3010-3021
Publication Date(Web):21 Aug 2009
DOI:10.1039/B821200G
In this tutorial review the economies of synthesis are analysed from both detailed and macroscopic perspectives, using case-studies from complex molecule synthesis. Atom, step, and redox economy are more than philosophical constructs, but rather guidelines, which enable the synthetic chemist to design and execute an efficient synthesis. Students entering the field of synthesis might find this tutorial helpful for understanding the subtle differences between these economic principles and also see real-world situations where such principles are put into practice.
Co-reporter:Ryan A. Shenvi, Daniel P. O’Malley and Phil S. Baran
Accounts of Chemical Research 2009 Volume 42(Issue 4) pp:530
Publication Date(Web):January 30, 2009
DOI:10.1021/ar800182r
The true creator is necessity, who is the mother of our invention. — Plato IUPAC defines chemoselectivity as “the preferential reaction of a chemical reagent with one of two or more different functional groups”, a definition that describes in rather understated terms the single greatest obstacle to complex molecule synthesis. Indeed, efforts to synthesize natural products often become case studies in the art and science of chemoselective control, a skill that nature has practiced deftly for billions of years but man has yet to master. Confrontation of one or perhaps a collection of functional groups that are either promiscuously reactive or stubbornly inert has the potential to unravel an entire strategic design. One could argue that the degree to which chemists can control chemoselectivity pales in comparison to the state of the art in stereocontrol. In this Account, we hope to illustrate how the combination of necessity and tenacity leads to the invention of chemoselective chemistry for the construction of complex molecules. In our laboratory, a premium is placed upon selecting targets that would be difficult or impossible to synthesize using traditional techniques. The successful total synthesis of such molecules demands a high degree of innovation, which in turn enables the discovery of new reactivity and principles for controlling chemoselectivity. In devising an approach to a difficult target, we choose bond disconnections that primarily maximize skeletal simplification, especially when the proposed chemistry is poorly precedented or completely unknown. By choosing such a strategy—rather than adapting an approach to fit known reactions—innovation and invention become the primary goal of the total synthesis. Delivery of the target molecule in a concise and convergent manner is the natural consequence of such endeavors, and invention becomes a prerequisite for success.
Co-reporter:Noah Z. Burns ; Irina N. Krylova ; Rami N. Hannoush
Journal of the American Chemical Society 2009 Volume 131(Issue 26) pp:9172-9173
Publication Date(Web):June 15, 2009
DOI:10.1021/ja903745s
A total synthesis of the complex, bent aromatic ring-containing marine alkaloid haouamine A is achieved through a route in which every step (with the exception of the final deprotection) is performed on a gram-scale. This is accomplished through the development of a method for the dehydrogenation of cyclohexenones that allows for point-to-planar chirality transfer. This strategy makes it possible to program the desired atropisomeric outcome from a simple chiral cyclohexenone. By synthesizing atrop-haouamine A, this work has firmly established that natural haouamine exists as a single, nonequilibrating atropisomer. Finally, biological investigations demonstrate that the bent aromatic ring of this natural product is critical for anticancer activity against PC3 cells.
Co-reporter:Thomas J. Maimone ; Jun Shi ; Shinji Ashida
Journal of the American Chemical Society 2009 Volume 131(Issue 47) pp:17066-17067
Publication Date(Web):October 30, 2009
DOI:10.1021/ja908194b
The longstanding challenge posed by the complex diterpene vinigrol has been answered for the first time. The notorious difficulty in synthesizing vinigrol stems from its unprecedented decahydro-1,5-butanonaphthalene ring system, eight contiguous stereocenters, and highly congested functionality. This Communication delineates a stereocontrolled 23-step route to vinigrol that is scalable (>5 g prepared of a late-stage intermediate), minimally reliant on protecting group chemistry, and facilitated by a number of unique and chemoselective transformations.
Co-reporter:Timothy Newhouse ; Chad A. Lewis
Journal of the American Chemical Society 2009 Volume 131(Issue 18) pp:6360-6361
Publication Date(Web):April 17, 2009
DOI:10.1021/ja901573x
Gram-scale, enantiospecific total syntheses of the anti-leukemia marine natural products kapakahines B and F have been completed. Two powerfully simplifying transformations have enabled the gram-scale synthesis of this natural product family: (1) an oxidative N−C bond-forming reaction that sets a key quaternary center with complete stereocontrol and (2) a unique strategy for the construction of twisted macrocycles via dynamic equilibration of complex late-stage intermediates.
Co-reporter:Paul J. Krawczuk, Niklas Schöne and Phil S. Baran
Organic Letters 2009 Volume 11(Issue 21) pp:4774-4776
Publication Date(Web):October 1, 2009
DOI:10.1021/ol901963v
An enantioselective synthesis of the maoecrystal V (1) carbon skeleton is described. The key transformations include arylation of a 1,3-dicarbonyl compound with a triarylbismuth(V) dichloride species, oxidative dearomatization of a phenol, and a subsequent intramolecular Diels−Alder reaction.
Co-reporter:Benjamin D. Hafensteiner, María Escribano, Elena Petricci, Phil S. Baran
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 14) pp:3808-3810
Publication Date(Web):15 July 2009
DOI:10.1016/j.bmcl.2009.04.045
A new method is reported for the synthesis of the α,β-unsaturated nitrone moiety characteristic of the stephacidin/avrainvillamide family of bioactive prenylated indole alkaloids. Application to the synthesis of stephacidin analogs and a potential biological probe are showcased.
Co-reporter:Ke Chen
&
Phil S. Baran
Nature 2009 459(7248) pp:824
Publication Date(Web):2009-05-13
DOI:10.1038/nature08043
Although organic chemists often devise synthetic routes for molecules by mimicking enzyme reactions, such syntheses generally target individual compounds; here, a strategy is reported that targets a whole family of compounds. The approach mimics the biosynthesis of terpenes to efficiently prepare five compounds from the eudesmane family of terpenes, and provides a framework for the synthesis of other such compounds.
Co-reporter:MichaelR. Luzung Dr.;ChadA. Lewis Dr. ;PhilS. Baran
Angewandte Chemie 2009 Volume 121( Issue 38) pp:7159-7163
Publication Date(Web):
DOI:10.1002/ange.200902761
Co-reporter:NoahZ. Burns;PhilS. Baran Dr.;ReinhardW. Hoffmann Dr.
Angewandte Chemie 2009 Volume 121( Issue 16) pp:2896-2910
Publication Date(Web):
DOI:10.1002/ange.200806086
Co-reporter:Noah Z. Burns, Mikkel Jessing, Phil S. Baran
Tetrahedron 2009 65(33) pp: 6600-6610
Publication Date(Web):
DOI:10.1016/j.tet.2009.05.075
Co-reporter:Kyle Eastman, Phil S. Baran
Tetrahedron 2009 65(16) pp: 3149-3154
Publication Date(Web):
DOI:10.1016/j.tet.2008.09.028
Co-reporter:MichaelR. Luzung Dr.;ChadA. Lewis Dr. ;PhilS. Baran
Angewandte Chemie International Edition 2009 Volume 48( Issue 38) pp:7025-7029
Publication Date(Web):
DOI:10.1002/anie.200902761
Co-reporter:Ke Chen Dr.;Albert Eschenmoser ;PhilS. Baran
Angewandte Chemie International Edition 2009 Volume 48( Issue 51) pp:9705-9708
Publication Date(Web):
DOI:10.1002/anie.200904474
Co-reporter:NoahZ. Burns;PhilS. Baran Dr.;ReinhardW. Hoffmann Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 16) pp:2854-2867
Publication Date(Web):
DOI:10.1002/anie.200806086
Co-reporter:Jun Shi;Hiroki Shigehisa Dr.;CarlosA. Guerrero;RyanA. Shenvi;Chuang-Chuang Li Dr. ;PhilS. Baran
Angewandte Chemie International Edition 2009 Volume 48( Issue 24) pp:4328-4331
Publication Date(Web):
DOI:10.1002/anie.200901116
Co-reporter:Jun Shi;Hiroki Shigehisa Dr.;CarlosA. Guerrero;RyanA. Shenvi;Chuang-Chuang Li Dr. ;PhilS. Baran
Angewandte Chemie 2009 Volume 121( Issue 24) pp:4392-4395
Publication Date(Web):
DOI:10.1002/ange.200901116
Co-reporter:ThomasJ. Maimone;Ana-Florina Voica ;PhilS. Baran
Angewandte Chemie 2008 Volume 120( Issue 16) pp:3097-3099
Publication Date(Web):
DOI:10.1002/ange.200800167
Co-reporter:DanielP. O'Malley;Junichiro Yamaguchi Dr.;IanS. Young Dr.;IanB. Seiple;PhilS. Baran Dr.
Angewandte Chemie 2008 Volume 120( Issue 19) pp:3637-3639
Publication Date(Web):
DOI:10.1002/ange.200801138
Co-reporter:Junichiro Yamaguchi Dr.;IanB. Seiple;IanS. Young Dr.;DanielP. O'Malley;Michael Maue Dr. ;PhilS. Baran Dr.
Angewandte Chemie 2008 Volume 120( Issue 19) pp:3634-3636
Publication Date(Web):
DOI:10.1002/ange.200705913
Co-reporter:DanielP. O'Malley;Junichiro Yamaguchi Dr.;IanS. Young Dr.;IanB. Seiple;PhilS. Baran Dr.
Angewandte Chemie 2008 Volume 120( Issue 19) pp:
Publication Date(Web):
DOI:10.1002/ange.200890085
Co-reporter:ThomasJ. Maimone;Ana-Florina Voica ;PhilS. Baran
Angewandte Chemie International Edition 2008 Volume 47( Issue 16) pp:3054-3056
Publication Date(Web):
DOI:10.1002/anie.200800167
Co-reporter:DanielP. O'Malley;Junichiro Yamaguchi Dr.;IanS. Young Dr.;IanB. Seiple;PhilS. Baran Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 19) pp:3581-3583
Publication Date(Web):
DOI:10.1002/anie.200801138
Co-reporter:Junichiro Yamaguchi Dr.;IanB. Seiple;IanS. Young Dr.;DanielP. O'Malley;Michael Maue Dr. ;PhilS. Baran Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 19) pp:3578-3580
Publication Date(Web):
DOI:10.1002/anie.200705913
Co-reporter:DanielP. O'Malley;Junichiro Yamaguchi Dr.;IanS. Young Dr.;IanB. Seiple;PhilS. Baran Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 19) pp:
Publication Date(Web):
DOI:10.1002/anie.200890085
Co-reporter:Matthias Köck Priv.-Doz. Dr.;Achim Grube Dr.;Ian B. Seiple;Phil S. Baran Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 35) pp:
Publication Date(Web):28 AUG 2007
DOI:10.1002/anie.200701798
Since its discovery in 1993, the marine natural product palau'amine has intrigued natural product chemists. Its exotic molecular architecture and purported bioactivity made it an ideal target for synthesis. However, as the years went by and related marine alkaloids were isolated, a skeptical eye was cast on the structure of palau'amine; recently these suspicions were confirmed and the structure of palau'amine revised. This Minireview gives a careful overview of the structural revision and its ramifications to both its biogenesis and total synthesis.
Co-reporter:Achim Grube;Stefan Immel Priv.-Doz. Dr.;Phil S. Baran Dr.;Matthias Köck Priv.-Doz. Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 35) pp:
Publication Date(Web):28 AUG 2007
DOI:10.1002/anie.200790172
The dimeric pyrrole–imidazole alkaloid massadine chloride was isolated from the Caribbean sponge Stylissa caribica. M. Köck, P. S. Baran, and co-workers describe in their Communication on page 6721 ff. how they were able to elucidate the structure of this natural product. The compound was unambiguously demonstrated to be the biosynthetic precursor of massadine. Results of experiments and calculations give evidence that the biosynthesis of other complex pyrrole–imidazole alkaloids run through a common intermediate.
Co-reporter:NoahZ. Burns ;PhilS. Baran
Angewandte Chemie International Edition 2007 Volume 47( Issue 1) pp:205-208
Publication Date(Web):
DOI:10.1002/anie.200704576
Co-reporter:Achim Grube;Stefan Immel Priv.-Doz. Dr.;Phil S. Baran Dr.;Matthias Köck Priv.-Doz. Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 43) pp:
Publication Date(Web):26 OCT 2007
DOI:10.1002/anie.200790221
Co-reporter:Achim Grube;Stefan Immel Priv.-Doz. Dr.;Phil S. Baran Dr.;Matthias Köck Priv.-Doz. Dr.
Angewandte Chemie 2007 Volume 119(Issue 35) pp:
Publication Date(Web):28 AUG 2007
DOI:10.1002/ange.200790172
Das dimere Pyrrol-Imidazol-Alkaloid Massadinchlorid wurde aus dem karibischen Schwamm Stylissa caribica isoliert. M. Köck, P. S. Baran und Mitarbeiter beschreiben in ihrer Zuschrift auf S. 6842 ff., wie es ihnen gelang, die Struktur des Naturstoffs aufzuklären. Die Verbindung wurde darüber hinaus als Vorstufe in der Biosynthese von Massadin identifiziert. Ergebnisse aus Experimenten und Rechnungen deuten darauf hin, dass die Biosynthese verschiedener komplexer Pyrrol-Imidazol-Alkaloide über eine gemeinsame Vorstufe verläuft.
Co-reporter:Matthias Köck Priv.-Doz. Dr.;Achim Grube Dr.;Ian B. Seiple;Phil S. Baran Dr.
Angewandte Chemie 2007 Volume 119(Issue 35) pp:
Publication Date(Web):28 AUG 2007
DOI:10.1002/ange.200701798
Seit der Entdeckung im Jahr 1993 hat Palau'amin die Naturstoffchemiker fasziniert. Seine ungewöhnliche Molekülstruktur und die beschriebene biologische Aktivität machen diesen marinen Naturstoff zu einem lohnenden Ziel für die organische Synthese. Im Laufe der Jahre und im Zusammenhang mit der Isolierung verwandter mariner Alkaloide wurde die postulierte chemische Struktur von Palau'amin angezweifelt, und vor kurzem wurde die relative Konfiguration von Palau'amin schließlich revidiert. Dieser Kurzaufsatz gibt eine sorgfältige Übersicht über die Strukturrevision und deren Bedeutung für die Biosynthese sowie die Totalsynthese.
Co-reporter:NoahZ. Burns ;PhilS. Baran
Angewandte Chemie 2007 Volume 120( Issue 1) pp:211-215
Publication Date(Web):
DOI:10.1002/ange.200704576
Co-reporter:Achim Grube;Stefan Immel Priv.-Doz. Dr.;Phil S. Baran Dr.;Matthias Köck Priv.-Doz. Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 35) pp:
Publication Date(Web):8 AUG 2007
DOI:10.1002/anie.200701935
The dimeric pyrrole–imidazole alkaloid massadine chloride (1, see scheme) was isolated from the Caribbean sponge Stylissa caribica. The structure elucidation of massadine chloride and its transformation into massadine (3) via an intermediate “massadine aziridine” (2) is discussed.
Co-reporter:Achim Grube;Stefan Immel Priv.-Doz. Dr.;Phil S. Baran Dr.;Matthias Köck Priv.-Doz. Dr.
Angewandte Chemie 2007 Volume 119(Issue 35) pp:
Publication Date(Web):8 AUG 2007
DOI:10.1002/ange.200701935
Das dimere Pyrrol-Imidazol-Alkaloid Massadinchlorid (1) wurde aus dem karibischen Schwamm Stylissa caribica isoliert. Die Aufklärung seiner Struktur und seine Umsetzung zu Massadin (3) über die Zwischenstufe „Massadinaziridin“ (2) werden diskutiert.
Co-reporter:Achim Grube;Stefan Immel Priv.-Doz. Dr.;Phil S. Baran Dr.;Matthias Köck Priv.-Doz. Dr.
Angewandte Chemie 2007 Volume 119(Issue 43) pp:
Publication Date(Web):26 OCT 2007
DOI:10.1002/ange.200790221
Co-reporter:Phil S. Baran,
Thomas J. Maimone
and
Jeremy M. Richter
Nature 2007 446(7134) pp:404
Publication Date(Web):2007-03-22
DOI:10.1038/nature05569
The field of organic synthesis has made phenomenal advances in the past fifty years, yet chemists still struggle to design synthetic routes that will enable them to obtain sufficient quantities of complex molecules for biological and medical studies. Total synthesis is therefore increasingly focused on preparing natural products in the most efficient manner possible. Here we describe the preparative-scale, enantioselective, total syntheses of members of the hapalindole, fischerindole, welwitindolinone and ambiguine families, each constructed without the need for protecting groups—the use of such groups adds considerably to the cost and complexity of syntheses. As a consequence, molecules that have previously required twenty or more steps to synthesize racemically in milligram amounts can now be obtained as single enantiomers in significant quantities in ten steps or less. Through the extension of the general principles demonstrated here, it should be possible to access other complex molecular architectures without using protecting groups.
Co-reporter:Phil S. Baran, Ke Li, Daniel P. O'Malley,Christos Mitsos
Angewandte Chemie International Edition 2006 45(2) pp:249-252
Publication Date(Web):
DOI:10.1002/anie.200503374
Co-reporter:Brian H. Northrop;Daniel P. O'Malley;Alexros L. Zografos Dr. ;K. N. Houk
Angewandte Chemie International Edition 2006 Volume 45(Issue 25) pp:
Publication Date(Web):16 MAY 2006
DOI:10.1002/anie.200600514
Naturally radical: The microwave-induced rearrangement of sceptrin to natural products ageliferin and nagelamide E results in approximately the same ratio as they are isolated from natural sources. Computational investigations show that the vinylcyclobutane–cyclohexene rearrangement occurs via diradical intermediates and support the involvement of this rearrangement in the biosynthesis of the natural products.
Co-reporter:Brian H. Northrop;Daniel P. O'Malley;Alexros L. Zografos Dr. ;K. N. Houk
Angewandte Chemie 2006 Volume 118(Issue 25) pp:
Publication Date(Web):16 MAY 2006
DOI:10.1002/ange.200600514
Natürlich radikal: Die Mikrowellen-induzierte Umlagerung von Sceptrin in die Naturstoffe Ageliferin und Nagelamid E liefert die Verbindungen etwa im gleichen Verhältnis, wie sie aus natürlichen Quellen isoliert werden. Nach Berechnungen verläuft die Vinylcyclobutan-Cyclohexen-Umlagerung über Diradikalintermediate, und es spricht alles dafür, dass diese Umlagerung auch an der Biosynthese der Naturstoffe beteiligt ist.
Co-reporter:Phil S. Baran ;Michael P. DeMartino
Angewandte Chemie 2006 Volume 118(Issue 42) pp:
Publication Date(Web):29 SEP 2006
DOI:10.1002/ange.200603024
Ganz einfach: Erstmals gelang eine präparativ nützliche intermolekulare oxidative Heterokupplung von Imiden und Oxindolen an Ester, Ketone und Lactone. Das strategisch geschickte Nutzen der ursprünglichen Oxidationsstufe der Reaktanten ermöglicht den Verzicht auf Präfunktionalisierungsschritte (Halogenierung, Enolsilan-Bildung) und erlaubt so eine rasche Synthese unsymmetrischer Lignane wie (−)-Bursehernin (1).
Co-reporter:Phil S. Baran Dr.;Ke Li Dr.;Daniel P. O'Malley;Christos Mitsos Dr.
Angewandte Chemie 2006 Volume 118(Issue 2) pp:
Publication Date(Web):30 NOV 2005
DOI:10.1002/ange.200503374
Ganz ohne Helfer: In nicht auf Auxiliaren basierenden enantioselektiven Synthesen wurden die beiden Enantiomere der dimeren Pyrrol-Imidazol-Alkaloide Sceptrin und Ageliferin erhalten, was die Bestimmung der absoluten Konfiguration von natürlichem Ageliferin ermöglichte. Die „Programmierung“ der Oxaquadricyclan-Fragmentierung zu enantiomerenreinen tetrasubstituierten Cyclobutanen ist der entscheidende Syntheseschritt.
Co-reporter:Phil S. Baran ;Michael P. DeMartino
Angewandte Chemie International Edition 2006 Volume 45(Issue 42) pp:
Publication Date(Web):29 SEP 2006
DOI:10.1002/anie.200603024
No priming necessary: The intermolecular oxidative heterocoupling of imides and oxindoles to esters, ketones, and lactones is shown for the first time to be synthetically viable. Strategic use of the initial oxidation state of the reactants allows for the excision of prefunctionalization steps (halogenation, enol-silane formation) and allows for the concise synthesis of unsymmetrical lignans, such as (−)-bursehernin (1).
Co-reporter:Phil S. Baran Dr.;Carlos A. Guerrero;Benjamin D. Hafensteiner;Narendra B. Ambhaikar Dr.
Angewandte Chemie International Edition 2005 Volume 44(Issue 25) pp:
Publication Date(Web):18 MAY 2005
DOI:10.1002/anie.200500655
Enantioselective total syntheses of the naturally occurring forms of the highly oxidized indole alkaloids avrainvillamide (CJ-17,665) and stephacidin B (see formula) are reported, facilitated by a remarkable oxidation of stephacidin A. A streamlined route to stephacidin A is also reported along with the absolute configuration determination of this family of natural products.
Co-reporter:Phil S. Baran Dr.;Jeremy M. Richter;David W. Lin
Angewandte Chemie International Edition 2005 Volume 44(Issue 4) pp:
Publication Date(Web):6 DEC 2004
DOI:10.1002/anie.200462048
An odd couple: The union of pyrroles and carbonyl compounds (ketones, amides, esters, lactones, lactams, see scheme) is described, and by the use of an intramolecular variant of this new method, the nonsteroidal, anti-inflammatory drug (S)-ketorolac has been prepared in a short enantioselective synthesis. Mechanistic underpinnings of this reaction, which couples unfunctionalized C(sp2) and C(sp3) atoms, are also discussed.
Co-reporter:Phil S. Baran Dr.;Carlos A. Guerrero;Narendra B. Ambhaikar Dr.;Benjamin D. Hafensteiner
Angewandte Chemie International Edition 2005 Volume 44(Issue 4) pp:
Publication Date(Web):7 DEC 2004
DOI:10.1002/anie.200461864
A concise route to the heptacyclic indole alkaloid stephacidin A (1) includes a simple method for the gram-scale synthesis of substituted tryptophans, a remarkable indole annulation, and the first enolate coupling of an amide to an ester. This synthesis secures the relative configuration of the natural product and paves a potential path to several of its congeners.
Co-reporter:Phil S. Baran Dr.;Ryan A. Shenvi;Christos A. Mitsos Dr.
Angewandte Chemie International Edition 2005 Volume 44(Issue 24) pp:
Publication Date(Web):18 MAY 2005
DOI:10.1002/anie.200500522
One-of-a-kind. A bromine-induced rearrangement has been designed for the formation of the spiro-β-lactam ring present in the structurally unique chartelline alkaloids (see picture of chartelline A). This method is used in combination with others to provide rapid access to the carbocyclic skeletons of the chartelline, securine, and securamine alkaloids.
Co-reporter:Phil S. Baran Dr.;Ryan A. Shenvi;Christos A. Mitsos Dr.
Angewandte Chemie 2005 Volume 117(Issue 24) pp:
Publication Date(Web):18 MAY 2005
DOI:10.1002/ange.200500522
Einzigartig: Eine Brom-induzierte Umlagerung wurde mit dem Ziel entwickelt, den Spiro-β-lactamring in den strukturell einzigartigen Chartellinalkaloiden (siehe Strukturformel von Chartellin A) zu erzeugen. Die Methode bietet in Kombination mit anderen einen schnellen Zugang zu den carbocyclischen Gerüsten der Chartellin-, Securin- und Securaminalkaloide.
Co-reporter:Phil S. Baran Dr.;Carlos A. Guerrero;Narendra B. Ambhaikar Dr.;Benjamin D. Hafensteiner
Angewandte Chemie 2005 Volume 117(Issue 4) pp:
Publication Date(Web):7 DEC 2004
DOI:10.1002/ange.200461864
Ein attraktiver Weg zum heptacyclischen Indolalkaloid Stephacidin A (1) umfasst eine einfache Methode für die Synthese substituierter Tryptophane im Gramm-Maßstab, eine bemerkenswerte Methode für die Indolanellierung und die erste Enolatkupplung zwischen einem Amid und einem Ester. Diese Synthese stellt die relative Konfiguration des Naturstoffs sicher und weist einen möglichen Weg zu einigen seiner Analoga.
Co-reporter:Phil S. Baran Dr.;Jeremy M. Richter;David W. Lin
Angewandte Chemie 2005 Volume 117(Issue 4) pp:
Publication Date(Web):6 DEC 2004
DOI:10.1002/ange.200462048
Nicht aktivierte C(sp2)- und C(sp3)-Atome werden bei der beschriebenen Kupplung von Pyrrolen mit Carbonylverbindungen (Ketonen, Amiden, Estern, Lactonen, Lactamen) verknüpft (siehe Schema). Eine intramolekulare Variante dieser neuen Methode machte den nichtsteroidalen entzündungshemmenden Wirkstoff (S)-Ketorolac in einer kurzen enantioselektiven Synthese zugänglich. Zudem werden Überlegungen zum Mechanismus dieser Reaktion vorgestellt.
Co-reporter:Phil S. Baran Dr.;Carlos A. Guerrero;Benjamin D. Hafensteiner;Narendra B. Ambhaikar Dr.
Angewandte Chemie 2005 Volume 117(Issue 25) pp:
Publication Date(Web):18 MAY 2005
DOI:10.1002/ange.200500655
Die enantioselektiven Totalsynthesen der natürlich vorkommenden hoch oxidierten Indol-Alkaloide Avrainvillamid (CJ-17,665) und Stephacidin B (siehe Formel) werden durch eine bemerkenswerte Oxidation von Stephacidin A erleichtert. Eine verbesserte Route zu Stephacidin A wird ebenso beschrieben wie die Bestimmung der Absolutkonfiguration dieser Naturstoff-Familie.
Co-reporter:Phil S. Baran Dr.;Daniel P. O'Malley;Alexros L. Zografos Dr.
Angewandte Chemie 2004 Volume 116(Issue 20) pp:
Publication Date(Web):5 MAY 2004
DOI:10.1002/ange.200453937
Magische Mikrowellen: Allgemein nimmt man an, dass die Biosynthese des antiviralen marinen Alkaloids Ageliferin (1) über eine Diels-Alder-Reaktion verläuft – eine experimentelle Bestätigung steht allerdings noch aus. Eine Totalsynthese von 1 ausgehend von Sceptrin führte nun zu einer alternativen Hypothese für die Biosynthese von 1 und anderen dimeren Pyrrol-Imidazol-Alkaloiden.
Co-reporter:Phil S. Baran Dr.;Daniel P. O'Malley;Alexros L. Zografos Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 20) pp:
Publication Date(Web):5 MAY 2004
DOI:10.1002/anie.200453937
Microwave-induced magic: There is a widely held conviction that the antiviral marine alkaloid ageliferin 1 arises biosynthetically from a Diels–Alder reaction which, although possible, has yet to materialize in the laboratory. A total synthesis of 1 from sceptrin is now reported that has led to a new hypothesis for how 1 and other dimeric pyrrole-imidazole alkaloids might be formed in nature.
Co-reporter:Timothy Newhouse ; Chad A. Lewis ; Kyle J. Eastman
Journal of the American Chemical Society () pp:
Publication Date(Web):April 28, 2010
DOI:10.1021/ja1009458
This report details the invention of a method to enable syntheses of psychotrimine (1) and the kapakahines F and B (2, 3) on a gram scale and in a minimum number of steps. Mechanistic inquiries are presented for the key enabling quaternization of indole at the C3 position by electrophilic attack of an activated aniline species. Excellent chemo-, regio-, and diastereoselectivities are observed for reactions with o-iodoaniline, an indole cation equivalent. Additionally, the scope of this reaction is broad with respect to the tryptamine and aniline components. The anti-cancer profiles of 1−3 have also been evaluated.
Co-reporter:Timothy Newhouse, Phil S. Baran and Reinhard W. Hoffmann
Chemical Society Reviews 2009 - vol. 38(Issue 11) pp:NaN3021-3021
Publication Date(Web):2009/08/21
DOI:10.1039/B821200G
In this tutorial review the economies of synthesis are analysed from both detailed and macroscopic perspectives, using case-studies from complex molecule synthesis. Atom, step, and redox economy are more than philosophical constructs, but rather guidelines, which enable the synthetic chemist to design and execute an efficient synthesis. Students entering the field of synthesis might find this tutorial helpful for understanding the subtle differences between these economic principles and also see real-world situations where such principles are put into practice.
Co-reporter:Will R. Gutekunst and Phil S. Baran
Chemical Society Reviews 2011 - vol. 40(Issue 4) pp:NaN1991-1991
Publication Date(Web):2011/02/07
DOI:10.1039/C0CS00182A
In this critical review, the strategic and economic benefits of C–H functionalization logic will be analyzed through the critical lens of total synthesis. In order to illustrate the dramatically simplifying effects this type of logic can potentially have on synthetic planning, we take the reader through a series of case studies in which it has already been successfully applied. In the first section, a chronological look at key historical syntheses will be examined, leading into modern day examples. In the second section, our own experience with applying and executing synthesis with a C–H functionalization “mindset” will be discussed (114 references).
.jpg)
![Acetic acid, [(4-methylphenyl)thio][(4-nitrobenzoyl)oxy]-, ethyl ester, (-)-](http://img.cochemist.com/ccimg/188500/188488-85-9.png)
![Acetic acid, [(4-methylphenyl)thio][(4-nitrobenzoyl)oxy]-, ethyl ester, (-)-](http://img.cochemist.com/ccimg/188500/188488-85-9_b.png)


![Bicyclo[2.2.1]heptan-2-one,1-(hydroxymethyl)-7,7-dimethyl-, (1R,4R)-](http://img.cochemist.com/ccimg/107300/107243-99-2.png)
![Bicyclo[2.2.1]heptan-2-one,1-(hydroxymethyl)-7,7-dimethyl-, (1R,4R)-](http://img.cochemist.com/ccimg/107300/107243-99-2_b.png)


![2-CYCLOPENTEN-1-ONE, 4-BUTYL-4-[(TRIMETHYLSILYL)OXY]-](http://img.cochemist.com/ccimg/104300/104274-33-1.png)
![2-CYCLOPENTEN-1-ONE, 4-BUTYL-4-[(TRIMETHYLSILYL)OXY]-](http://img.cochemist.com/ccimg/104300/104274-33-1_b.png)
![Magnesium, bromo[1-[(trimethylsilyl)methyl]ethenyl]-](http://img.cochemist.com/ccimg/103600/103559-91-7.png)
![Magnesium, bromo[1-[(trimethylsilyl)methyl]ethenyl]-](http://img.cochemist.com/ccimg/103600/103559-91-7_b.png)



![Tert-butyl-[2-(1h-indol-3-yl)ethoxy]-dimethylsilane](http://img.cochemist.com/ccimg/101100/101079-48-5.png)
![Tert-butyl-[2-(1h-indol-3-yl)ethoxy]-dimethylsilane](http://img.cochemist.com/ccimg/101100/101079-48-5_b.png)










![Thieno[3,2-c]pyridine-5(4H)-aceticacid, a-(2-chlorophenyl)-6,7-dihydro-,methyl ester](http://img.cochemist.com/ccimg/90100/90055-48-4.png)
![Thieno[3,2-c]pyridine-5(4H)-aceticacid, a-(2-chlorophenyl)-6,7-dihydro-,methyl ester](http://img.cochemist.com/ccimg/90100/90055-48-4_b.png)


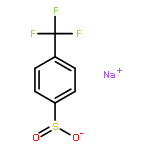
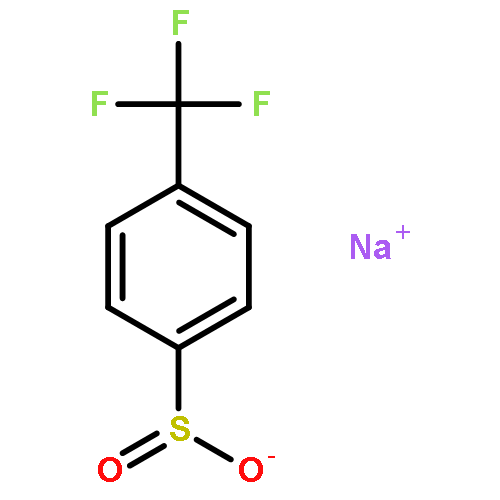




![Cyclohexanone, 3-[1-[(trimethylsilyl)methyl]ethenyl]-](http://img.cochemist.com/ccimg/83700/83650-13-9.png)
![Cyclohexanone, 3-[1-[(trimethylsilyl)methyl]ethenyl]-](http://img.cochemist.com/ccimg/83700/83650-13-9_b.png)


![Bicyclo[1.1.1]pentane-1,3-dicarboxylic acid, monomethyl ester](http://img.cochemist.com/ccimg/83300/83249-10-9.png)
![Bicyclo[1.1.1]pentane-1,3-dicarboxylic acid, monomethyl ester](http://img.cochemist.com/ccimg/83300/83249-10-9_b.png)
![Bicyclo[1.1.1]pentane-1-carboxylic acid, 3-phenyl-, methyl ester](http://img.cochemist.com/ccimg/83300/83249-09-6.png)
![Bicyclo[1.1.1]pentane-1-carboxylic acid, 3-phenyl-, methyl ester](http://img.cochemist.com/ccimg/83300/83249-09-6_b.png)








![1-Oxaspiro[4.5]dec-6-en-8-one, 2,6,10,10-tetramethyl-, (2R,5S)-rel-](/data/chemimg/1322400/77841-36-2.png)
![1-Oxaspiro[4.5]dec-6-en-8-one, 2,6,10,10-tetramethyl-, (2R,5S)-rel-](/data/chemimg/1322400/77841-36-2_b.png)
![Methanesulfonic acid, trifluoro-, 4-[(1,1-dimethylethyl)amino]phenylester](http://img.cochemist.com/ccimg/76500/76472-76-9.png)
![Methanesulfonic acid, trifluoro-, 4-[(1,1-dimethylethyl)amino]phenylester](http://img.cochemist.com/ccimg/76500/76472-76-9_b.png)







![7-OXO-1,2,3,4,5,6-HEXAHYDROTRICYCLO[2.2.1.0<SUP>2,6</SUP>]HEPTANE-3-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/70800/70748-53-7.png)
![7-OXO-1,2,3,4,5,6-HEXAHYDROTRICYCLO[2.2.1.0<SUP>2,6</SUP>]HEPTANE-3-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/70800/70748-53-7_b.png)


![(3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-(acetyloxy)-4-(butanoyloxy)-3,3a-dihydroxy-3,6,9-trimethyl-8-{[(2Z)-2-methylbut-2-enoyl]oxy}-2-oxo-2,3,3a,4,5,6,6a,7,8,9b-decahydroazuleno[4,5-b]furan-7-yl octanoate](http://img.cochemist.com/ccimg/67600/67526-95-8.png)
![(3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-(acetyloxy)-4-(butanoyloxy)-3,3a-dihydroxy-3,6,9-trimethyl-8-{[(2Z)-2-methylbut-2-enoyl]oxy}-2-oxo-2,3,3a,4,5,6,6a,7,8,9b-decahydroazuleno[4,5-b]furan-7-yl octanoate](http://img.cochemist.com/ccimg/67600/67526-95-8_b.png)








![Oxirane, [2-(phenylsulfonyl)ethyl]-](http://img.cochemist.com/ccimg/60800/60788-39-8.png)
![Oxirane, [2-(phenylsulfonyl)ethyl]-](http://img.cochemist.com/ccimg/60800/60788-39-8_b.png)




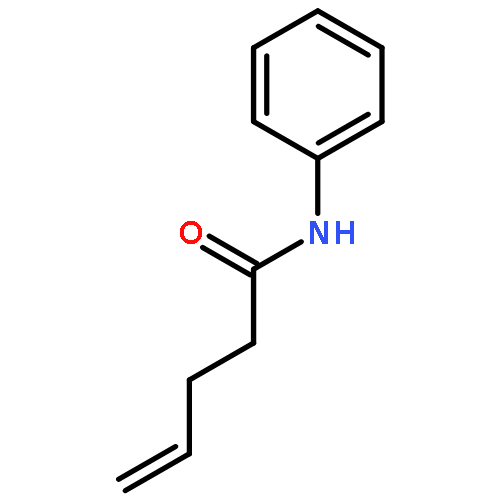









![BICYCLO[2.2.2]OCTANE, 2-PHENYL-](http://img.cochemist.com/ccimg/56200/56139-28-7.png)
![BICYCLO[2.2.2]OCTANE, 2-PHENYL-](http://img.cochemist.com/ccimg/56200/56139-28-7_b.png)
![5-(2-Chlorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine](http://img.cochemist.com/ccimg/55200/55142-85-3.png)
![5-(2-Chlorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine](http://img.cochemist.com/ccimg/55200/55142-85-3_b.png)






![3-[(1E)-2-nitroethenyl]-1H-Indole](http://img.cochemist.com/ccimg/51900/51870-94-1.png)
![3-[(1E)-2-nitroethenyl]-1H-Indole](http://img.cochemist.com/ccimg/51900/51870-94-1_b.png)


![1-Oxaspiro[4.5]dec-6-ene, 2,6,10,10-tetramethyl-, (2R,5S)-rel-](http://img.cochemist.com/ccimg/43200/43126-21-2.png)
![1-Oxaspiro[4.5]dec-6-ene, 2,6,10,10-tetramethyl-, (2R,5S)-rel-](http://img.cochemist.com/ccimg/43200/43126-21-2_b.png)
![(3S,8R,9S,10R,13S,14S)-3-((tert-butyldimethylsilyl)oxy)-10,13-dimethyl-1,2,3,4,7,8,9,10,11,12,13,14,15,16-tetradecahydro-17H-cyclopenta[a]phenanthren-17-one](http://img.cochemist.com/ccimg/42200/42151-23-5.png)
![(3S,8R,9S,10R,13S,14S)-3-((tert-butyldimethylsilyl)oxy)-10,13-dimethyl-1,2,3,4,7,8,9,10,11,12,13,14,15,16-tetradecahydro-17H-cyclopenta[a]phenanthren-17-one](http://img.cochemist.com/ccimg/42200/42151-23-5_b.png)


![Bicyclo[3.1.1]hept-2-ene-2-ethanol,6,6-dimethyl-, 2-acetate, (1R,5S)-](http://img.cochemist.com/ccimg/35900/35836-72-7.png)
![Bicyclo[3.1.1]hept-2-ene-2-ethanol,6,6-dimethyl-, 2-acetate, (1R,5S)-](http://img.cochemist.com/ccimg/35900/35836-72-7_b.png)
![[1.1.1]Propellane](http://img.cochemist.com/ccimg/35700/35634-10-7.png)
![[1.1.1]Propellane](http://img.cochemist.com/ccimg/35700/35634-10-7_b.png)


![2-METHYL-9,10-DIHYDRO-4H-BENZO[5,6]CYCLOHEPTA[1,2-D][1,3]OXAZOL-4<WBR />-OL](http://img.cochemist.com/ccimg/31000/30960-39-5.png)
![2-METHYL-9,10-DIHYDRO-4H-BENZO[5,6]CYCLOHEPTA[1,2-D][1,3]OXAZOL-4<WBR />-OL](http://img.cochemist.com/ccimg/31000/30960-39-5_b.png)






![Bicyclo[2.2.2]octane-2-carboxylicacid](http://img.cochemist.com/ccimg/29300/29221-25-8.png)
![Bicyclo[2.2.2]octane-2-carboxylicacid](http://img.cochemist.com/ccimg/29300/29221-25-8_b.png)
















![Bicyclo[2.2.2]octane-1-carboxylic acid, 4-phenyl-, methyl ester](http://img.cochemist.com/ccimg/23100/23062-52-4.png)
![Bicyclo[2.2.2]octane-1-carboxylic acid, 4-phenyl-, methyl ester](http://img.cochemist.com/ccimg/23100/23062-52-4_b.png)

















![1-azabicyclo[1.1.0]butane](http://img.cochemist.com/ccimg/19600/19540-05-7.png)
![1-azabicyclo[1.1.0]butane](http://img.cochemist.com/ccimg/19600/19540-05-7_b.png)





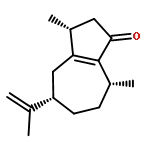
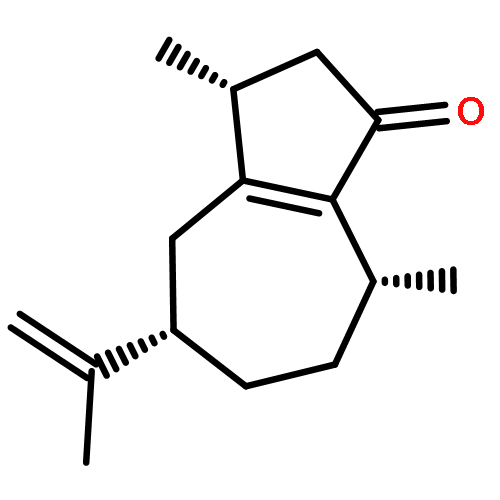
![Bicyclo[3.1.1]hept-3-en-2-one,4,6,6-trimethyl-, (1R,5R)-](http://img.cochemist.com/ccimg/18400/18309-32-5.png)
![Bicyclo[3.1.1]hept-3-en-2-one,4,6,6-trimethyl-, (1R,5R)-](http://img.cochemist.com/ccimg/18400/18309-32-5_b.png)












![5,6-Dihydro[1,1'-biphenyl]-3(4H)-one](http://img.cochemist.com/ccimg/10400/10345-87-6.png)
![5,6-Dihydro[1,1'-biphenyl]-3(4H)-one](http://img.cochemist.com/ccimg/10400/10345-87-6_b.png)









![7-Oxabicyclo[4.1.0]heptane,1-methyl-4-(1-methylethenyl)-, (1S,4R,6R)-](http://img.cochemist.com/ccimg/7000/6909-30-4.png)
![7-Oxabicyclo[4.1.0]heptane,1-methyl-4-(1-methylethenyl)-, (1S,4R,6R)-](http://img.cochemist.com/ccimg/7000/6909-30-4_b.png)
![Benzene,[(pentyloxy)methyl]-](http://img.cochemist.com/ccimg/6400/6382-14-5.png)
![Benzene,[(pentyloxy)methyl]-](http://img.cochemist.com/ccimg/6400/6382-14-5_b.png)








![1-[(4-methylphenyl)sulfonyl]piperidine](http://img.cochemist.com/ccimg/4800/4703-22-4.png)
![1-[(4-methylphenyl)sulfonyl]piperidine](http://img.cochemist.com/ccimg/4800/4703-22-4_b.png)





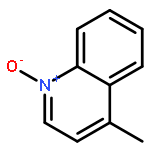
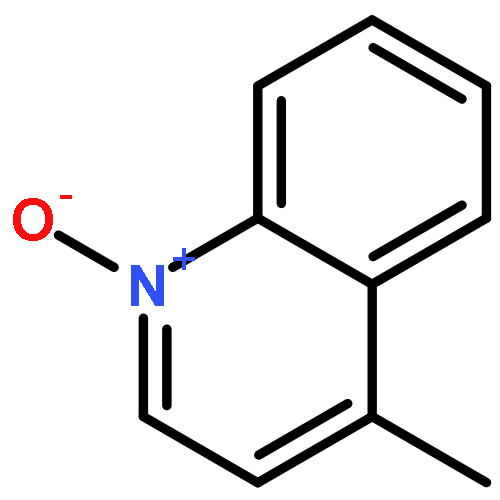










![Bicyclo[2.2.2]octane-1-carboxylic acid, methyl ester](http://img.cochemist.com/ccimg/2100/2064-04-2.png)
![Bicyclo[2.2.2]octane-1-carboxylic acid, methyl ester](http://img.cochemist.com/ccimg/2100/2064-04-2_b.png)

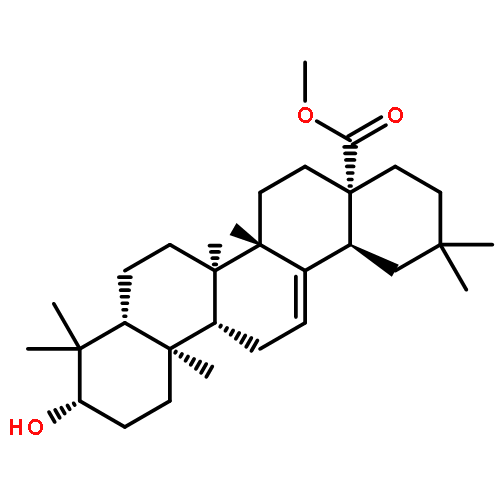


![Azuleno[4,5-b]furan-2,8(3H,4H)-dione,3a,5,6,6a,7,9b-hexahydro-6-hydroxy-3,6,9-trimethyl-, (3S,3aS,6R,6aR,9bS)-](http://img.cochemist.com/ccimg/1700/1618-98-0.png)
![Azuleno[4,5-b]furan-2,8(3H,4H)-dione,3a,5,6,6a,7,9b-hexahydro-6-hydroxy-3,6,9-trimethyl-, (3S,3aS,6R,6aR,9bS)-](http://img.cochemist.com/ccimg/1700/1618-98-0_b.png)



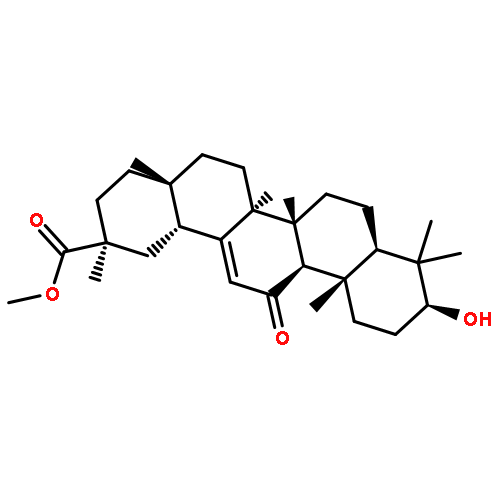








![Bicyclo[3.1.1]hept-3-en-2-ol,4,6,6-trimethyl-](http://img.cochemist.com/ccimg/500/473-67-6.png)
![Bicyclo[3.1.1]hept-3-en-2-ol,4,6,6-trimethyl-](http://img.cochemist.com/ccimg/500/473-67-6_b.png)


![BICYCLO[1.1.1]PENTANE](http://img.cochemist.com/ccimg/400/311-75-1.png)
![BICYCLO[1.1.1]PENTANE](http://img.cochemist.com/ccimg/400/311-75-1_b.png)
![8-Azabicyclo[3.2.1]octane](http://img.cochemist.com/ccimg/300/280-05-7.png)
![8-Azabicyclo[3.2.1]octane](http://img.cochemist.com/ccimg/300/280-05-7_b.png)


![5-Azulenemethanol, 1,2,3,4,5,6,7,8-octahydro-α,α,3,8-tetramethyl-, acetate, [3S-(3α,5α,8α)]-](http://img.cochemist.com/ccimg/200/134-28-1.png)
![5-Azulenemethanol, 1,2,3,4,5,6,7,8-octahydro-α,α,3,8-tetramethyl-, acetate, [3S-(3α,5α,8α)]-](http://img.cochemist.com/ccimg/200/134-28-1_b.png)








![PIPERIDINE, 4-[[(1,1-DIMETHYLETHYL)DIPHENYLSILYL]OXY]-](http://img.cochemist.com/ccimg/396100/396091-35-3.png)
![PIPERIDINE, 4-[[(1,1-DIMETHYLETHYL)DIPHENYLSILYL]OXY]-](http://img.cochemist.com/ccimg/396100/396091-35-3_b.png)








![Carbamic acid, methyl[3-(methylamino)propyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/123200/123183-72-2.png)
![Carbamic acid, methyl[3-(methylamino)propyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/123200/123183-72-2_b.png)


![BICYCLO[1.1.0]BUTANE, 1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/86600/86537-30-6.png)
![BICYCLO[1.1.0]BUTANE, 1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/86600/86537-30-6_b.png)


![Bicyclo[1.1.0]butane, 1-(phenylsulfonyl)-](http://img.cochemist.com/ccimg/81000/80989-84-0.png)
![Bicyclo[1.1.0]butane, 1-(phenylsulfonyl)-](http://img.cochemist.com/ccimg/81000/80989-84-0_b.png)






![Benzenemethanamine, N-[2-(phenylmethoxy)ethyl]-](http://img.cochemist.com/ccimg/38400/38336-06-0.png)
![Benzenemethanamine, N-[2-(phenylmethoxy)ethyl]-](http://img.cochemist.com/ccimg/38400/38336-06-0_b.png)

![Bicyclo[1.1.1]pentan-1-amine hydrochloride](http://img.cochemist.com/ccimg/22300/22287-35-0.png)
![Bicyclo[1.1.1]pentan-1-amine hydrochloride](http://img.cochemist.com/ccimg/22300/22287-35-0_b.png)


![5H-Cyclopropa[3,4]benz[1,2-e]azulen-5-one,1,1a,1b,4,4a,7a,7b,8,9,9a-decahydro-4a,7b,9,9a-tetrahydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-,(1aR,1bS,4aR,7aS,7bS,8R,9R,9aS)-](http://img.cochemist.com/ccimg/17700/17673-25-5.png)
![5H-Cyclopropa[3,4]benz[1,2-e]azulen-5-one,1,1a,1b,4,4a,7a,7b,8,9,9a-decahydro-4a,7b,9,9a-tetrahydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-,(1aR,1bS,4aR,7aS,7bS,8R,9R,9aS)-](http://img.cochemist.com/ccimg/17700/17673-25-5_b.png)






















![Propanoic acid, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-](http://img.cochemist.com/ccimg/116100/116022-80-1.png)
![Propanoic acid, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-](http://img.cochemist.com/ccimg/116100/116022-80-1_b.png)












![Pentacyclo[4.2.0.02,5.03,8.04,7]octane-1,4-dicarboxylicacid, 1-methyl ester](http://img.cochemist.com/ccimg/24600/24539-28-4.png)
![Pentacyclo[4.2.0.02,5.03,8.04,7]octane-1,4-dicarboxylicacid, 1-methyl ester](http://img.cochemist.com/ccimg/24600/24539-28-4_b.png)













![L-Valine, N-[(4-methylphenyl)sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/22000/21957-66-4.png)
![L-Valine, N-[(4-methylphenyl)sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/22000/21957-66-4_b.png)


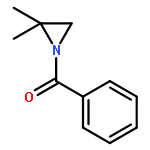






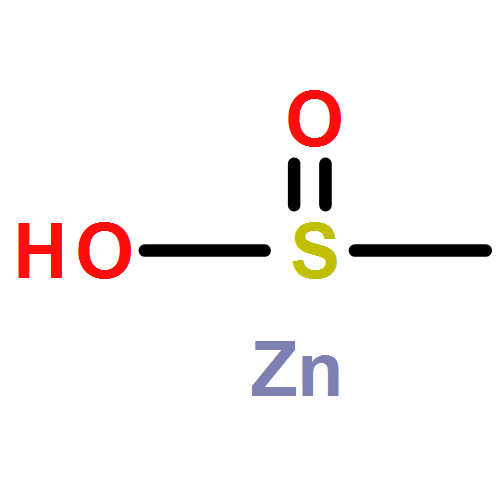








![3,6-Methano-8H-1,5,7-trioxacyclopenta[ij]cycloprop[a]azulene-4,8(3H)-dione,hexahydro-2a-hydroxy-8b-methyl-9-(1-methylethyl)-,(1aR,2aR,3S,6R,6aS,8aS,8bR,9S)-](http://img.cochemist.com/ccimg/17700/17617-46-8.png)
![3,6-Methano-8H-1,5,7-trioxacyclopenta[ij]cycloprop[a]azulene-4,8(3H)-dione,hexahydro-2a-hydroxy-8b-methyl-9-(1-methylethyl)-,(1aR,2aR,3S,6R,6aS,8aS,8bR,9S)-](http://img.cochemist.com/ccimg/17700/17617-46-8_b.png)
![3,6-Methano-8H-1,5,7-trioxacyclopenta[ij]cycloprop[a]azulene-4,8(3H)-dione,hexahydro-2a-hydroxy-8b-methyl-9-(1-methylethenyl)-,(1aR,2aR,3S,6R,6aS,8aS,8bR,9R)-](http://img.cochemist.com/ccimg/17700/17617-45-7.png)
![3,6-Methano-8H-1,5,7-trioxacyclopenta[ij]cycloprop[a]azulene-4,8(3H)-dione,hexahydro-2a-hydroxy-8b-methyl-9-(1-methylethenyl)-,(1aR,2aR,3S,6R,6aS,8aS,8bR,9R)-](http://img.cochemist.com/ccimg/17700/17617-45-7_b.png)
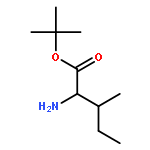

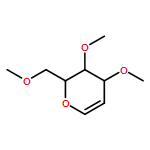



![Benzene, 1-chloro-4-[[(chloromethyl)thio]methyl]-](http://img.cochemist.com/ccimg/15900/15894-25-4.png)
![Benzene, 1-chloro-4-[[(chloromethyl)thio]methyl]-](http://img.cochemist.com/ccimg/15900/15894-25-4_b.png)






















![5H,12H-3,4-Dioxa-5a,11a,15a-triazacyclooct[lm]indeno[5,6-b]fluorene-11,15(2H,13H)-dione, 1,10,10a,14,14a,15b-hexahydro-10a-hydroxy-7-methoxy-](/data/chemimg/446800/12626-18-5.png)
![5H,12H-3,4-Dioxa-5a,11a,15a-triazacyclooct[lm]indeno[5,6-b]fluorene-11,15(2H,13H)-dione, 1,10,10a,14,14a,15b-hexahydro-10a-hydroxy-7-methoxy-](/data/chemimg/446800/12626-18-5_b.png)
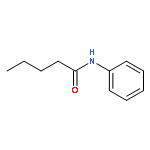





![5H-Cyclohepta[b]pyridine,6,7,8,9-tetrahydro-](http://img.cochemist.com/ccimg/7200/7197-96-8.png)
![5H-Cyclohepta[b]pyridine,6,7,8,9-tetrahydro-](http://img.cochemist.com/ccimg/7200/7197-96-8_b.png)


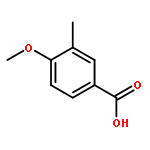

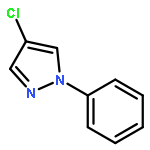
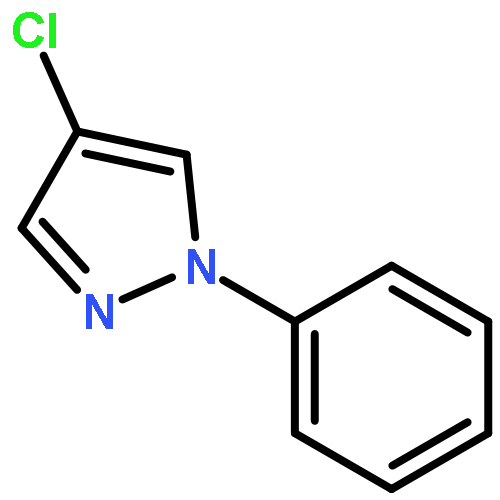


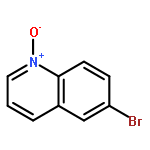
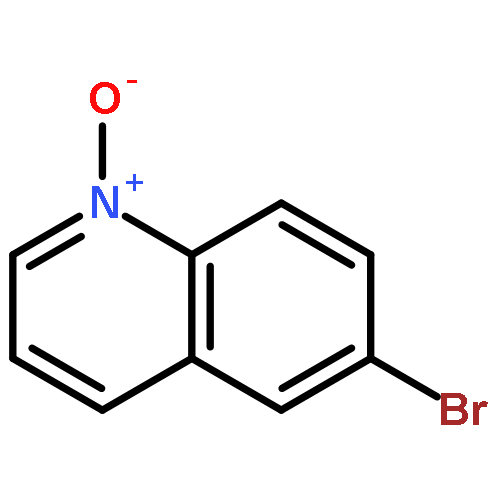





![{[(2-methylprop-2-en-1-yl)oxy]methyl}benzene](http://img.cochemist.com/ccimg/5700/5658-46-8.png)
![{[(2-methylprop-2-en-1-yl)oxy]methyl}benzene](http://img.cochemist.com/ccimg/5700/5658-46-8_b.png)


![9H-Indeno[2,1-c]pyridin-9-one](http://img.cochemist.com/ccimg/5100/5061-91-6.png)
![9H-Indeno[2,1-c]pyridin-9-one](http://img.cochemist.com/ccimg/5100/5061-91-6_b.png)


















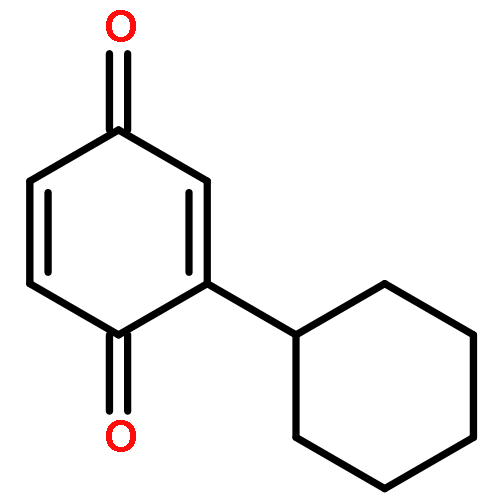







































![Benzothiazole,2-[(difluoromethyl)thio]-](http://img.cochemist.com/ccimg/1000/943-08-8.png)
![Benzothiazole,2-[(difluoromethyl)thio]-](http://img.cochemist.com/ccimg/1000/943-08-8_b.png)












![(1AR,4R,4AR,7S,7AS,7BR)-1,1,4,7-TETRAMETHYLDECAHYDRO-1H-CYCLOPROP<WBR />A[E]AZULENE-4,7-DIOL](http://img.cochemist.com/ccimg/800/763-10-0.png)
![(1AR,4R,4AR,7S,7AS,7BR)-1,1,4,7-TETRAMETHYLDECAHYDRO-1H-CYCLOPROP<WBR />A[E]AZULENE-4,7-DIOL](http://img.cochemist.com/ccimg/800/763-10-0_b.png)






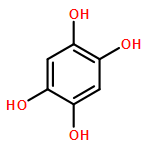
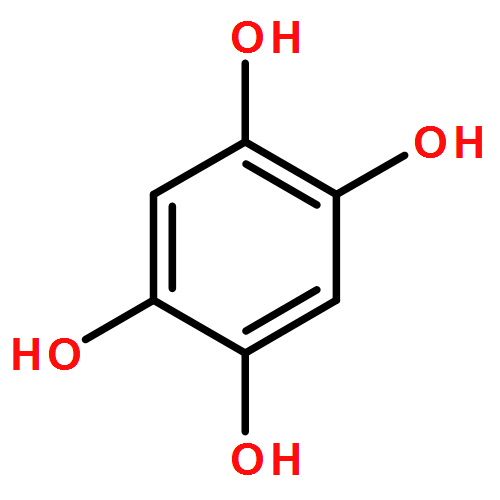


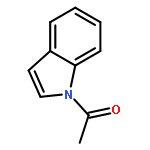
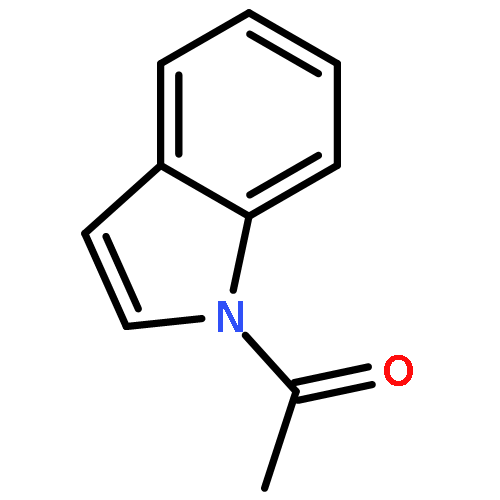
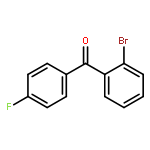
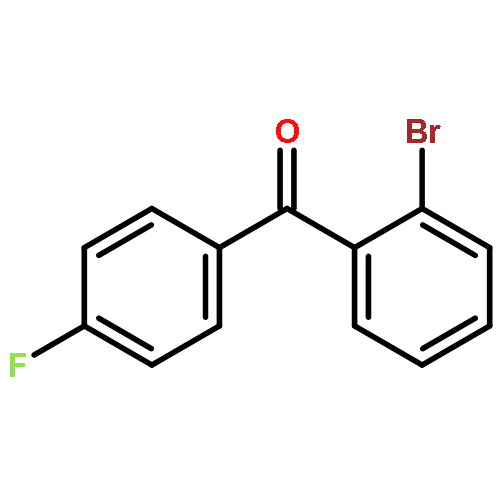






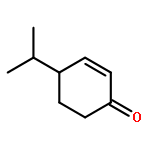
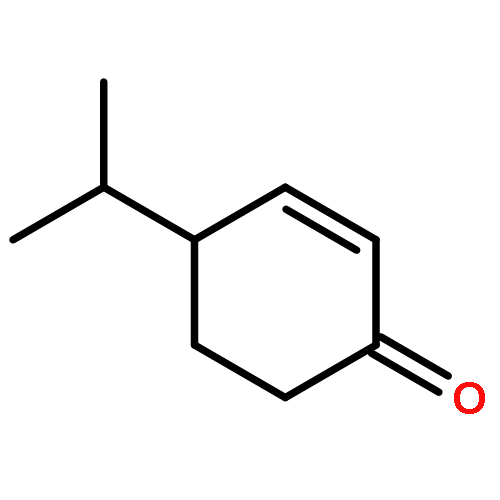
![9H-Pyrido[3,4-b]indole,1-methyl-](http://img.cochemist.com/ccimg/500/486-84-0.png)
![9H-Pyrido[3,4-b]indole,1-methyl-](http://img.cochemist.com/ccimg/500/486-84-0_b.png)


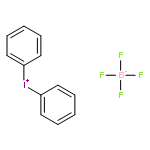
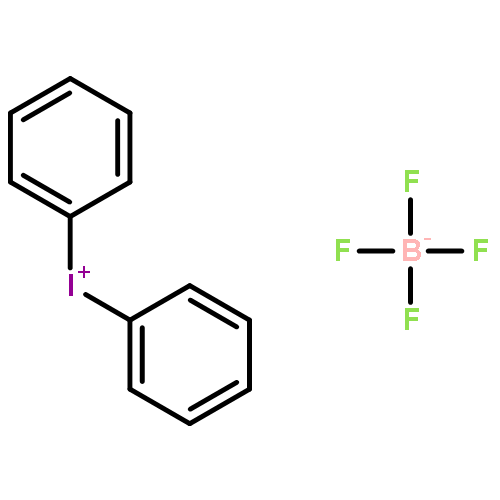
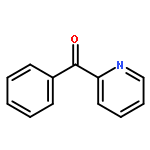
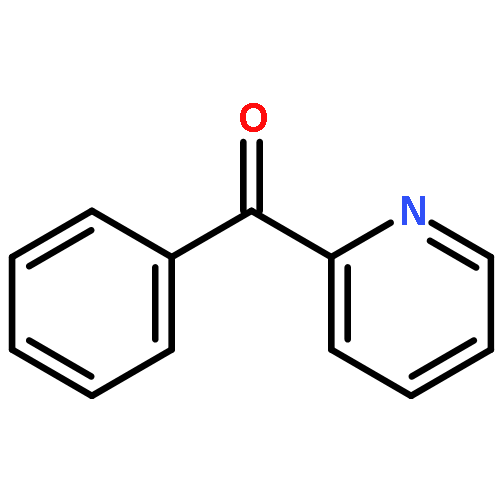
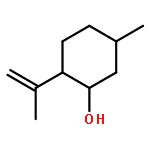
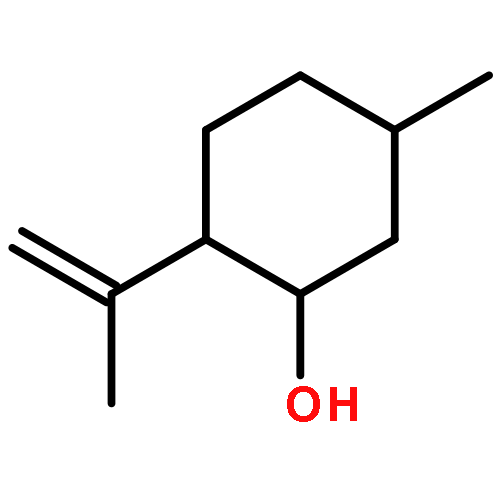



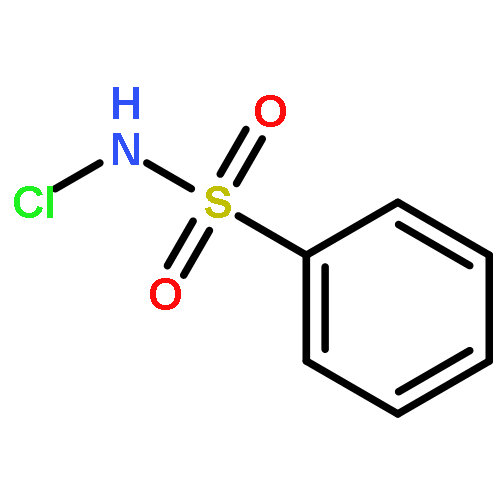
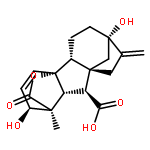











![Carbamic acid, N-[(chloroamino)[(methoxycarbonyl)amino]methylene]-, methyl ester](/data/chemimg/3736000/1596379-00-8.png)
![Carbamic acid, N-[(chloroamino)[(methoxycarbonyl)amino]methylene]-, methyl ester](/data/chemimg/3736000/1596379-00-8_b.png)






![2-[(3-METHYLPHENYL)METHYLAMINO]ETHANOL](http://img.cochemist.com/ccimg/1211600/1211528-71-0.png)
![2-[(3-METHYLPHENYL)METHYLAMINO]ETHANOL](http://img.cochemist.com/ccimg/1211600/1211528-71-0_b.png)






![Carbamic acid, [4-(trimethylsilyl)-3-butynyl]-, methyl ester](http://img.cochemist.com/ccimg/247300/247232-80-0.png)
![Carbamic acid, [4-(trimethylsilyl)-3-butynyl]-, methyl ester](http://img.cochemist.com/ccimg/247300/247232-80-0_b.png)
![1,3-Benzodioxole, 5-[(1E)-2-iodoethenyl]-](http://img.cochemist.com/ccimg/203000/202998-56-9.png)
![1,3-Benzodioxole, 5-[(1E)-2-iodoethenyl]-](http://img.cochemist.com/ccimg/203000/202998-56-9_b.png)






![Carbamic acid, [2-(1H-indol-3-yl)ethyl]-, methyl ester](http://img.cochemist.com/ccimg/58700/58635-45-3.png)
![Carbamic acid, [2-(1H-indol-3-yl)ethyl]-, methyl ester](http://img.cochemist.com/ccimg/58700/58635-45-3_b.png)






![Benzene, [(1E)-2-iodoethenyl]-](http://img.cochemist.com/ccimg/42600/42599-24-6.png)
![Benzene, [(1E)-2-iodoethenyl]-](http://img.cochemist.com/ccimg/42600/42599-24-6_b.png)














![Card-20(22)-enolide,3-[(6-deoxy-a-L-mannopyranosyl)oxy]-1,5,11,14,19-pentahydroxy-,(1b,3b,5b,11a)-](http://img.cochemist.com/ccimg/700/630-60-4.png)
![Card-20(22)-enolide,3-[(6-deoxy-a-L-mannopyranosyl)oxy]-1,5,11,14,19-pentahydroxy-,(1b,3b,5b,11a)-](http://img.cochemist.com/ccimg/700/630-60-4_b.png)


![[2-[(8s,9s,10r,13s,14s,17r)-17-hydroxy-10,13-dimethyl-3,11-dioxo-1,2,6,7,8,9,12,14,15,16-decahydrocyclopenta[a]phenanthren-17-yl]-2-oxoethyl] Acetate](http://img.cochemist.com/ccimg/100/50-04-4.png)
![[2-[(8s,9s,10r,13s,14s,17r)-17-hydroxy-10,13-dimethyl-3,11-dioxo-1,2,6,7,8,9,12,14,15,16-decahydrocyclopenta[a]phenanthren-17-yl]-2-oxoethyl] Acetate](http://img.cochemist.com/ccimg/100/50-04-4_b.png)
![(3S,3aS,6aR,7R,8R,9R,9aR,10aR,12R,12aS)-8-((tert-butyldimethylsilyl)oxy)-2,7,10,10,12-pentamethyl-5,13-dioxo-6a,7,9,9a,10,10a,11,12-octahydro-3H,8H-9,12a-methanocyclopenta[1,10]cyclopropa[6,7]cyclodeca[1,2-d][1,3]dioxol-3-yl acetate](http://img.cochemist.com/ccimg/1453300/1453222-06-4.png)
![(3S,3aS,6aR,7R,8R,9R,9aR,10aR,12R,12aS)-8-((tert-butyldimethylsilyl)oxy)-2,7,10,10,12-pentamethyl-5,13-dioxo-6a,7,9,9a,10,10a,11,12-octahydro-3H,8H-9,12a-methanocyclopenta[1,10]cyclopropa[6,7]cyclodeca[1,2-d][1,3]dioxol-3-yl acetate](http://img.cochemist.com/ccimg/1453300/1453222-06-4_b.png)
![tert-butyl(((1R,2R)-1-((1R,2R,3R,4R,6R)-3-ethynyl-4,7,7-trimethyl-3-((trimethylsilyl)oxy)bicyclo[4.1.0]heptan-2-yl)-2-methylpenta-3,4-dien-1-yl)oxy)dimethylsilane](http://img.cochemist.com/ccimg/1453300/1453222-02-0.png)
![tert-butyl(((1R,2R)-1-((1R,2R,3R,4R,6R)-3-ethynyl-4,7,7-trimethyl-3-((trimethylsilyl)oxy)bicyclo[4.1.0]heptan-2-yl)-2-methylpenta-3,4-dien-1-yl)oxy)dimethylsilane](http://img.cochemist.com/ccimg/1453300/1453222-02-0_b.png)
![(1R,2R,4R,6R)-2-((1R,2R)-1-hydroxy-2-methyl-penta-3,4-dien-1-yl)-4,7,7-trimethylbicyclo[4.1.0]heptan-3-one](http://img.cochemist.com/ccimg/1453300/1453222-00-8.png)
![(1R,2R,4R,6R)-2-((1R,2R)-1-hydroxy-2-methyl-penta-3,4-dien-1-yl)-4,7,7-trimethylbicyclo[4.1.0]heptan-3-one](http://img.cochemist.com/ccimg/1453300/1453222-00-8_b.png)








![Bicyclo[4.1.0]heptan-3-one, 7,7-dimethyl-, (1S,6R)-](http://img.cochemist.com/ccimg/22400/22327-37-3.png)
![Bicyclo[4.1.0]heptan-3-one, 7,7-dimethyl-, (1S,6R)-](http://img.cochemist.com/ccimg/22400/22327-37-3_b.png)






![(1aR,1bR,2R,3R,7bR,8R,9aR)-2-((tert-butyldimethylsilyl)oxy)-1,1,3,8-tetramethyl-7b-((trimethylsilyl)oxy)-1,1a,1b,2,3,5,7b,8,9,9a-decahydro-6H-cyclopropa[3,4]benzo[1,2-e]azulen-6-one](http://img.cochemist.com/ccimg/1453300/1453222-03-1.png)
![(1aR,1bR,2R,3R,7bR,8R,9aR)-2-((tert-butyldimethylsilyl)oxy)-1,1,3,8-tetramethyl-7b-((trimethylsilyl)oxy)-1,1a,1b,2,3,5,7b,8,9,9a-decahydro-6H-cyclopropa[3,4]benzo[1,2-e]azulen-6-one](http://img.cochemist.com/ccimg/1453300/1453222-03-1_b.png)