Co-reporter:Jing Zhu, Yi Yuan, Shaozhong Wang, and Zhu-Jun Yao
ACS Omega August 2017? Volume 2(Issue 8) pp:4665-4665
Publication Date(Web):August 18, 2017
DOI:10.1021/acsomega.7b00749
A mild transition-metal-free protocol to prepare 2,3-dialkylated tartaric acid esters has been developed by taking advantage of a visible light photoredox-catalyzed reductive dimerization of α-ketoesters with a combination of an organic dye photocatalyst and a Hantzsch-type 1,4-dihydropyridine hydrogen donor. A broad range of functional groups including cyclopropane, alkene, alkyne, 4-methoxybenzyl ether, acetal, silyl ether, carbamate, cyclic ether, cyclic thioether, bromoalkane, and N-alkoxyphthalimide are well-compatible. By employing the visible light photoredox-catalyzed reductive coupling and the subsequent optical resolution, both enantioenriched diastereomers of 2,3-dialkylated tartaric acid could be acquired conveniently.Topics: Carbonyl compounds (organic); Catalysts; Dimerization; Electrochemical analysis; Photochemical redox reaction; Redox potential;
Co-reporter:Lina Chang, Tao Guo, Ziyi Wang, Shaozhong WangZhu-Jun Yao
The Journal of Organic Chemistry 2017 Volume 82(Issue 3) pp:
Publication Date(Web):January 13, 2017
DOI:10.1021/acs.joc.6b02760
A protocol based on a newly developed N-bromosuccinimide (NBS)-induced cycloisomerization was described to prepare tricyclic azepino[4,5-b]indoles from simple β-enaminoesters or β-enaminones containing an indole unit. A mechanism involving a Pictet–Spengler cyclization, an aziridine ring formation, and a regioselective C–N bond cleavage was proposed to account for the medium-sized ring formation and the migration of electron-withdrawing group (ester, ketone).
Co-reporter:Lei Zhang; Yi Wang; Zhu-Jun Yao; Shaozhong Wang;Zhi-Xiang Yu
Journal of the American Chemical Society 2015 Volume 137(Issue 41) pp:13290-13300
Publication Date(Web):September 25, 2015
DOI:10.1021/jacs.5b05971
In classical transition state theory, a transition state is connected to its reactant(s) and product(s). Recently, chemists found that reaction pathways may bifurcate after a transition state, leading to two or more sets of products. The product distribution for such a reaction containing a bifurcating potential energy surface (bPES) is usually determined by the shape of the bPES and dynamic factors. However, if the bPES leads to two intermediates (other than two products), which then undergo further transformations to give different final products, what factors control the selectivity is still not fully examined. This missing link in transition state theory is founded in the present study. Aiming to develop new methods for the synthesis of azocinoindole derivatives, we found that 2-propargyl-β-tetrahydrocarbolines can undergo ring expansion and spirocyclization under gold catalysis. DFT study revealed that the reaction starts with the intramolecular cyclization of the gold-activated 2-propargyl-β-tetrahydrocarboline with a bPES. The cyclization intermediates can not only interconvert into each other via a [1,5]-alkenyl shift, but also undergo ring expansion (through fragmentation/protodeauration mechanism) or spirocyclization (through deprotonation/protodeauration mechanism). Detailed analysis of the complex PESs for substrates with different substituents indicated that the reaction selectivity is under dynamic control if the interconversion of the intermediates is slower than the ring expansion and spirocyclization processes. Otherwise, the chemical outcome is under typical kinetic control and determined by the relative preference of ring expansion versus spirocyclization pathways. The present study may enrich chemist’s understanding of the determinants for selectivities on bPESs.
Co-reporter:Hao Yin, Naiyu Shan, Shaozhong Wang, and Zhu-Jun Yao
The Journal of Organic Chemistry 2014 Volume 79(Issue 20) pp:9748-9753
Publication Date(Web):September 19, 2014
DOI:10.1021/jo501927e
A common approach to ascididemin-type alkaloids, including ascididemin, bromoleptoclinidinone, neocalliactine acetate, and 11-hydroxyascididemin, based on a Brønsted acid-promoted tandem annulation has been developed. Alkyne building blocks were first designed and then employed in alkaloid synthesis; these building blocks can be accessed by a Sonogashira coupling reaction on a multigram scale.
Co-reporter:Lei Zhang;Lina Chang; Hongwen Hu;Huaqin Wang;Dr. Zhu-Jun Yao;Dr. Shaozhong Wang
Chemistry - A European Journal 2014 Volume 20( Issue 10) pp:2925-2932
Publication Date(Web):
DOI:10.1002/chem.201304524
Abstract
A new methodology taking advantage of gold(I)-catalyzed ring expansion has been developed to assemble tricyclic 1H-azocino[5,4-b]indoles from 2-propargyl-β-tetrahydrocarbolines. The azocinoindoles were obtained in moderate to excellent yields; the structure of which was established by X-ray crystallographic analysis. A mechanism involving regioselective intramolecular hydroarylation, [1,2]-alkenyl migration and carbon–carbon bond-fragmentation was proposed.
Co-reporter:Qiwen Hou, Zhenhua Zhang, Fanji Kong, Shaozhong Wang, Huaqin Wang and Zhu-Jun Yao
Chemical Communications 2013 vol. 49(Issue 7) pp:695-697
Publication Date(Web):23 Nov 2012
DOI:10.1039/C2CC36245G
A tandem reaction initiated by Au(I)-catalyzed C1–C5 cyclization of enediynes bearing an internal carboxy nucleophile has been developed, providing a distinctive methodology for the assembly of fused indenes including indeno[1,2-c]isochromen-5(11H)-ones and indeno[1,2-b]pyran-2(5H)-ones. A mechanism involving the concerted formation of the five- and six-membered rings was proposed.
Co-reporter:Zhenhua Zhang, Lina Chang, Shaozhong Wang, Huaqin Wang and Zhu-Jun Yao
RSC Advances 2013 vol. 3(Issue 40) pp:18446-18452
Publication Date(Web):07 Aug 2013
DOI:10.1039/C3RA43075H
A regio/stereoselective electrophilic addition of 1H-benzotriazole to aromatic alkynes induced by N-iodosuccinimide (NIS) has been achieved, providing unique access to 2-vinyl-2H-benzotriazole derivatives. It was demonstrated that the stereoselectivity is controlled by the nature of the substituents attached to the alkyne terminus. A mechanism involving cyclic iodonium and vinyl cation intermediates was proposed.
Co-reporter:Min Xu, Ke Xu, Shaozhong Wang, Zhu-Jun Yao
Tetrahedron Letters 2013 Volume 54(Issue 35) pp:4675-4678
Publication Date(Web):28 August 2013
DOI:10.1016/j.tetlet.2013.06.079
An efficient protocol to generate indolo[1,2-c]quinazolines from acyclic alkyne substrates by ZnBr2-promoted domino hydroamination–cyclization has been established. The dichotomous properties of zinc salts involving alkynophilicity and oxophilicity assure a one-pot formation of five- and six-membered nitrogen-containing rings in the skeleton.
Co-reporter:Hao Zhang, Wei-Chen Cui, Zhi-Long Hu, Shu-Yan Yu, Shaozhong Wang and Zhu-Jun Yao
RSC Advances 2012 vol. 2(Issue 12) pp:5101-5104
Publication Date(Web):13 Apr 2012
DOI:10.1039/C2RA20095C
The Brønsted acid-promoted dimerization of o-alkynylbenzaldehydes has been discovered and studied in the presence of 45% aq. HBF4 in acetic acid. The developed cascade methodology provides a convenient one-step synthesis of symmetrical 2,3,6,7-dibenzo-9-oxabicyclo[3.3.1]nona-2,6-diene (Kagan's ether) analogues bearing various functionalities.
Co-reporter:Hao Yin, Fanji Kong, Shaozhong Wang, Zhu-Jun Yao
Tetrahedron Letters 2012 Volume 53(Issue 52) pp:7078-7082
Publication Date(Web):26 December 2012
DOI:10.1016/j.tetlet.2012.10.065
A novel methodology taking advantage of a domino reaction initiated by an Au(I)-catalyzed 6-endo-dig cycloisomerization under silver-free condition was developed to prepare pentacyclic pyrido[4,3,2-mn]acridin-8-ones from N-propargylaminoquinones. Triphenylphosphinegold(I) chloride in combination with TFA was firstly employed and displayed excellent catalytic efficiency in the domino reaction.
Co-reporter:Shu-Yan Yu, Zhi-Long Hu, Hao Zhang, Shaozhong Wang, Zhu-Jun Yao
Tetrahedron Letters 2012 Volume 53(Issue 22) pp:2765-2768
Publication Date(Web):30 May 2012
DOI:10.1016/j.tetlet.2012.03.106
Electronically unfavorable reactions between isochromenylium tetrafluoroborates and electron-deficient olefins have been studied and achieved by assistance of a phenolic hydroxyl group in the olefin substrates, providing the corresponding 2-oxabicyclo[3.3.1]nonane derivatives diastereoselectively in moderate to satisfactory yields. The new methodology is initiated by an intermolecular C-1 O-glycosylation, and completed with an intramolecular Michael addition and an aldol condensation in cascade fashion.
Co-reporter:Na Fei, Hao Yin, Shaozhong Wang, Huaqin Wang, and Zhu-Jun Yao
Organic Letters 2011 Volume 13(Issue 16) pp:4208-4211
Publication Date(Web):July 18, 2011
DOI:10.1021/ol201542h
An efficient synthetic methodology was developed to assemble 1-azaanthraquinones from N-propargylaminoquinones by copper(II)-promoted sequential 6-endo-dig chlorocyclization and oxidative aromatization. The approach can be extended to preprare chlorinated alkaloids such as cleistophine and sampangine. A possible mechanism involving carbon–carbon bond formation triggered by regioselective electrophilic activation and carbon–chlorine bond formation via reductive elimination was proposed.
Co-reporter:Na Fei, Qiwen Hou, Shaozhong Wang, Huaqin Wang and Zhu-Jun Yao
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 18) pp:4096-4103
Publication Date(Web):23 Jul 2010
DOI:10.1039/C004896H
An efficient methodology taking advantage of the excellent nucleophilicity of aminoquinone to assemble the azaanthraquinone framework was developed via an iodine-induced 6-endo-dig electrophilic cyclization. Therefore, starting from N-propargylaminoquinones, various 3-iodo-1-azaanthraquinones were obtained in yields ranging from 45% to 90%. The metal-free protocol features facile installation of an iodine atom on the azaanthraquinone ring and benign functional group compatibility.
Co-reporter:Shuheng Li
Journal of Heterocyclic Chemistry 2008 Volume 45( Issue 6) pp:1875-1878
Publication Date(Web):
DOI:10.1002/jhet.5570450651
Abstract
A facile approach was developed on assembly of the 2-pyridone nucleus by ferric chloride promoted [3+3] cycloaddition in propionic acid. The tandem process involves cyclization of Michael adduct followed by aromatization. Thus, different substituted 1,2-dihydro-2-oxo-3-pyridinecarboxylate and 1,2-dihydro-2-oxo-3-pyridinecarboxamide derivatives were prepared in good yields from various enones with malonamic ester and malonamide, respectively
Co-reporter:Qiwen Hou, Zhenhua Zhang, Fanji Kong, Shaozhong Wang, Huaqin Wang and Zhu-Jun Yao
Chemical Communications 2013 - vol. 49(Issue 7) pp:NaN697-697
Publication Date(Web):2012/11/23
DOI:10.1039/C2CC36245G
A tandem reaction initiated by Au(I)-catalyzed C1–C5 cyclization of enediynes bearing an internal carboxy nucleophile has been developed, providing a distinctive methodology for the assembly of fused indenes including indeno[1,2-c]isochromen-5(11H)-ones and indeno[1,2-b]pyran-2(5H)-ones. A mechanism involving the concerted formation of the five- and six-membered rings was proposed.
Co-reporter:Na Fei, Qiwen Hou, Shaozhong Wang, Huaqin Wang and Zhu-Jun Yao
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 18) pp:NaN4103-4103
Publication Date(Web):2010/07/23
DOI:10.1039/C004896H
An efficient methodology taking advantage of the excellent nucleophilicity of aminoquinone to assemble the azaanthraquinone framework was developed via an iodine-induced 6-endo-dig electrophilic cyclization. Therefore, starting from N-propargylaminoquinones, various 3-iodo-1-azaanthraquinones were obtained in yields ranging from 45% to 90%. The metal-free protocol features facile installation of an iodine atom on the azaanthraquinone ring and benign functional group compatibility.

![(E)-3-ethoxy-1-(9-ethyl-3-hydroxy-7-methoxy-1H-pyrrolo[3,4-b]quinolin-2(3H)-yl)prop-2-en-1-one](http://img.cochemist.com/ccimg/1335200/1335104-70-5.png)
![(E)-3-ethoxy-1-(9-ethyl-3-hydroxy-7-methoxy-1H-pyrrolo[3,4-b]quinolin-2(3H)-yl)prop-2-en-1-one](http://img.cochemist.com/ccimg/1335200/1335104-70-5_b.png)


![1H-PYRANO[4'',3'':4',5']PYRIMIDO[1',2':1,2]PYRROLO[3,4-B]QUINOLINE-3,14(4H,12H)-DIONE, 4-ETHYL-4-HYDROXY-, (4S)-](http://img.cochemist.com/ccimg/842200/842131-27-5.png)
![1H-PYRANO[4'',3'':4',5']PYRIMIDO[1',2':1,2]PYRROLO[3,4-B]QUINOLINE-3,14(4H,12H)-DIONE, 4-ETHYL-4-HYDROXY-, (4S)-](http://img.cochemist.com/ccimg/842200/842131-27-5_b.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2_b.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bS)-](/data/chemimg/103700/874948-63-7.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bS)-](/data/chemimg/103700/874948-63-7_b.png)
![4,11-diethyl-9-methoxy-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinolin-14(12H)-one](http://img.cochemist.com/ccimg/1335200/1335104-74-9.png)
![4,11-diethyl-9-methoxy-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinolin-14(12H)-one](http://img.cochemist.com/ccimg/1335200/1335104-74-9_b.png)




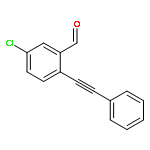
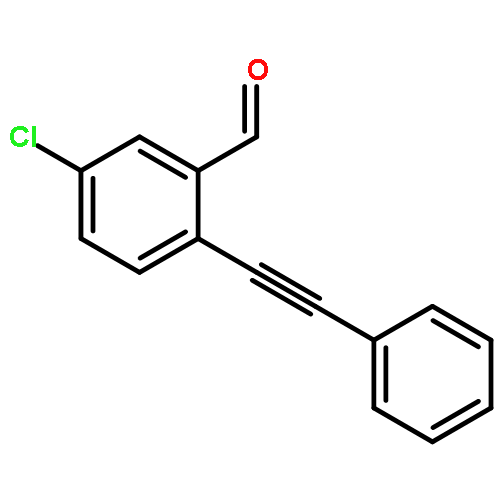
![Benzaldehyde, 2-[2-(4-fluorophenyl)ethynyl]-](/data/chemimg/143500/1042369-43-6.png)
![Benzaldehyde, 2-[2-(4-fluorophenyl)ethynyl]-](/data/chemimg/143500/1042369-43-6_b.png)
![Formamide, N-(4'-methoxy[1,1'-biphenyl]-2-yl)-](http://img.cochemist.com/ccimg/926700/926644-15-7.png)
![Formamide, N-(4'-methoxy[1,1'-biphenyl]-2-yl)-](http://img.cochemist.com/ccimg/926700/926644-15-7_b.png)
![ACETAMIDE, N-[4'-(1,1-DIMETHYLETHYL)[1,1'-BIPHENYL]-2-YL]-](http://img.cochemist.com/ccimg/869700/869631-30-1.png)
![ACETAMIDE, N-[4'-(1,1-DIMETHYLETHYL)[1,1'-BIPHENYL]-2-YL]-](http://img.cochemist.com/ccimg/869700/869631-30-1_b.png)
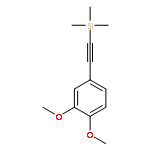

![Thiourea,N-[(1R,2R)-2-aminocyclohexyl]-N'-[3,5-bis(trifluoromethyl)phenyl]-](/data/chemimg/103900/860994-58-7.png)
![Thiourea,N-[(1R,2R)-2-aminocyclohexyl]-N'-[3,5-bis(trifluoromethyl)phenyl]-](/data/chemimg/103900/860994-58-7_b.png)
![1H-INDENO[1,2-B]PYRIDINE-2,5-DIONE, 1-METHYL-4-(2-THIENYL)-](http://img.cochemist.com/ccimg/853300/853261-97-9.png)
![1H-INDENO[1,2-B]PYRIDINE-2,5-DIONE, 1-METHYL-4-(2-THIENYL)-](http://img.cochemist.com/ccimg/853300/853261-97-9_b.png)
![1H-Indeno[1,2-b]pyridine-2,5-dione, 4-(2-furanyl)-1-methyl-](http://img.cochemist.com/ccimg/853300/853261-96-8.png)
![1H-Indeno[1,2-b]pyridine-2,5-dione, 4-(2-furanyl)-1-methyl-](http://img.cochemist.com/ccimg/853300/853261-96-8_b.png)
![1H-INDENO[1,2-B]PYRIDINE-2,5-DIONE, 4-(3-METHOXYPHENYL)-1-METHYL-](http://img.cochemist.com/ccimg/853300/853261-95-7.png)
![1H-INDENO[1,2-B]PYRIDINE-2,5-DIONE, 4-(3-METHOXYPHENYL)-1-METHYL-](http://img.cochemist.com/ccimg/853300/853261-95-7_b.png)
![1H-Indeno[1,2-b]pyridine-2,5-dione, 4-(2-chlorophenyl)-1-methyl-](http://img.cochemist.com/ccimg/853300/853261-93-5.png)
![1H-Indeno[1,2-b]pyridine-2,5-dione, 4-(2-chlorophenyl)-1-methyl-](http://img.cochemist.com/ccimg/853300/853261-93-5_b.png)
![BENZOIC ACID, 2-[1,6-DIHYDRO-1-METHYL-6-OXO-4-(2-THIENYL)-2-PYRIDINYL]-](http://img.cochemist.com/ccimg/853300/853261-92-4.png)
![BENZOIC ACID, 2-[1,6-DIHYDRO-1-METHYL-6-OXO-4-(2-THIENYL)-2-PYRIDINYL]-](http://img.cochemist.com/ccimg/853300/853261-92-4_b.png)
![Benzoic acid, 2-[4-(2-furanyl)-1,6-dihydro-1-methyl-6-oxo-2-pyridinyl]-](http://img.cochemist.com/ccimg/853300/853261-91-3.png)
![Benzoic acid, 2-[4-(2-furanyl)-1,6-dihydro-1-methyl-6-oxo-2-pyridinyl]-](http://img.cochemist.com/ccimg/853300/853261-91-3_b.png)
![Pyrido[2,1-a]isoindole-4,6-dione, 2-(2-thienyl)-](http://img.cochemist.com/ccimg/853300/853261-87-7.png)
![Pyrido[2,1-a]isoindole-4,6-dione, 2-(2-thienyl)-](http://img.cochemist.com/ccimg/853300/853261-87-7_b.png)
![Pyrido[2,1-a]isoindole-4,6-dione, 2-(2-furanyl)-](http://img.cochemist.com/ccimg/853300/853261-86-6.png)
![Pyrido[2,1-a]isoindole-4,6-dione, 2-(2-furanyl)-](http://img.cochemist.com/ccimg/853300/853261-86-6_b.png)
![PYRIDO[2,1-A]ISOINDOLE-4,6-DIONE, 2-(3-METHOXYPHENYL)-](http://img.cochemist.com/ccimg/853300/853261-85-5.png)
![PYRIDO[2,1-A]ISOINDOLE-4,6-DIONE, 2-(3-METHOXYPHENYL)-](http://img.cochemist.com/ccimg/853300/853261-85-5_b.png)
![Pyrido[2,1-a]isoindole-4,6-dione, 2-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/853300/853261-84-4.png)
![Pyrido[2,1-a]isoindole-4,6-dione, 2-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/853300/853261-84-4_b.png)
![PYRIDO[2,1-A]ISOINDOLE-4,6-DIONE, 2-(2-CHLOROPHENYL)-](http://img.cochemist.com/ccimg/853300/853261-83-3.png)
![PYRIDO[2,1-A]ISOINDOLE-4,6-DIONE, 2-(2-CHLOROPHENYL)-](http://img.cochemist.com/ccimg/853300/853261-83-3_b.png)
![Benzoic acid, 2-[1,6-dihydro-6-oxo-4-(2-thienyl)-2-pyridinyl]-](http://img.cochemist.com/ccimg/853300/853261-82-2.png)
![Benzoic acid, 2-[1,6-dihydro-6-oxo-4-(2-thienyl)-2-pyridinyl]-](http://img.cochemist.com/ccimg/853300/853261-82-2_b.png)
![BENZOIC ACID, 2-[4-(2-FURANYL)-1,6-DIHYDRO-6-OXO-2-PYRIDINYL]-](http://img.cochemist.com/ccimg/853300/853261-81-1.png)
![BENZOIC ACID, 2-[4-(2-FURANYL)-1,6-DIHYDRO-6-OXO-2-PYRIDINYL]-](http://img.cochemist.com/ccimg/853300/853261-81-1_b.png)
![Benzoic acid, 2-[1,6-dihydro-4-(3-methoxyphenyl)-6-oxo-2-pyridinyl]-](http://img.cochemist.com/ccimg/853300/853261-80-0.png)
![Benzoic acid, 2-[1,6-dihydro-4-(3-methoxyphenyl)-6-oxo-2-pyridinyl]-](http://img.cochemist.com/ccimg/853300/853261-80-0_b.png)
![BENZOIC ACID, 2-[1,6-DIHYDRO-4-(4-METHOXYPHENYL)-6-OXO-2-PYRIDINYL]-](http://img.cochemist.com/ccimg/853300/853261-79-7.png)
![BENZOIC ACID, 2-[1,6-DIHYDRO-4-(4-METHOXYPHENYL)-6-OXO-2-PYRIDINYL]-](http://img.cochemist.com/ccimg/853300/853261-79-7_b.png)
![Benzoic acid, 2-[4-(2-chlorophenyl)-1,6-dihydro-6-oxo-2-pyridinyl]-](http://img.cochemist.com/ccimg/853300/853261-78-6.png)
![Benzoic acid, 2-[4-(2-chlorophenyl)-1,6-dihydro-6-oxo-2-pyridinyl]-](http://img.cochemist.com/ccimg/853300/853261-78-6_b.png)
![BENZOIC ACID, 2-[1-OXO-3-(2-THIENYL)-2-PROPENYL]-](http://img.cochemist.com/ccimg/853300/853261-77-5.png)
![BENZOIC ACID, 2-[1-OXO-3-(2-THIENYL)-2-PROPENYL]-](http://img.cochemist.com/ccimg/853300/853261-77-5_b.png)
![Benzoic acid, 2-[3-(3-methoxyphenyl)-1-oxo-2-propenyl]-](http://img.cochemist.com/ccimg/853300/853261-76-4.png)
![Benzoic acid, 2-[3-(3-methoxyphenyl)-1-oxo-2-propenyl]-](http://img.cochemist.com/ccimg/853300/853261-76-4_b.png)
![BENZOIC ACID, 2-[3-(4-METHYLPHENYL)-1-OXO-2-PROPENYL]-](http://img.cochemist.com/ccimg/853300/853261-75-3.png)
![BENZOIC ACID, 2-[3-(4-METHYLPHENYL)-1-OXO-2-PROPENYL]-](http://img.cochemist.com/ccimg/853300/853261-75-3_b.png)
![Benzoic acid, 2-[4-(4-chlorophenyl)-1,6-dihydro-6-oxo-2-pyridinyl]-](http://img.cochemist.com/ccimg/853300/853261-74-2.png)
![Benzoic acid, 2-[4-(4-chlorophenyl)-1,6-dihydro-6-oxo-2-pyridinyl]-](http://img.cochemist.com/ccimg/853300/853261-74-2_b.png)
![BENZOIC ACID, 2-[4-(4-FLUOROPHENYL)-1,6-DIHYDRO-6-OXO-2-PYRIDINYL]-](http://img.cochemist.com/ccimg/853300/853261-73-1.png)
![BENZOIC ACID, 2-[4-(4-FLUOROPHENYL)-1,6-DIHYDRO-6-OXO-2-PYRIDINYL]-](http://img.cochemist.com/ccimg/853300/853261-73-1_b.png)
![BENZOIC ACID, 2-[1,6-DIHYDRO-4-(4-METHYLPHENYL)-6-OXO-2-PYRIDINYL]-](http://img.cochemist.com/ccimg/853300/853261-71-9.png)
![BENZOIC ACID, 2-[1,6-DIHYDRO-4-(4-METHYLPHENYL)-6-OXO-2-PYRIDINYL]-](http://img.cochemist.com/ccimg/853300/853261-71-9_b.png)
![Pyrido[2,1-a]isoindole-4,6-dione, 2-(4-chlorophenyl)-](http://img.cochemist.com/ccimg/853300/853261-70-8.png)
![Pyrido[2,1-a]isoindole-4,6-dione, 2-(4-chlorophenyl)-](http://img.cochemist.com/ccimg/853300/853261-70-8_b.png)
![PYRIDO[2,1-A]ISOINDOLE-4,6-DIONE, 2-(4-FLUOROPHENYL)-](http://img.cochemist.com/ccimg/853300/853261-69-5.png)
![PYRIDO[2,1-A]ISOINDOLE-4,6-DIONE, 2-(4-FLUOROPHENYL)-](http://img.cochemist.com/ccimg/853300/853261-69-5_b.png)
![Pyrido[2,1-a]isoindole-4,6-dione, 2-[4-(1-methylethyl)phenyl]-](http://img.cochemist.com/ccimg/853300/853261-68-4.png)
![Pyrido[2,1-a]isoindole-4,6-dione, 2-[4-(1-methylethyl)phenyl]-](http://img.cochemist.com/ccimg/853300/853261-68-4_b.png)
![PYRIDO[2,1-A]ISOINDOLE-4,6-DIONE, 2-(4-METHYLPHENYL)-](http://img.cochemist.com/ccimg/853300/853261-67-3.png)
![PYRIDO[2,1-A]ISOINDOLE-4,6-DIONE, 2-(4-METHYLPHENYL)-](http://img.cochemist.com/ccimg/853300/853261-67-3_b.png)
![1H-INDENO[1,2-B]PYRIDINE-2,5-DIONE, 4-(4-CHLOROPHENYL)-1-METHYL-](http://img.cochemist.com/ccimg/853300/853261-62-8.png)
![1H-INDENO[1,2-B]PYRIDINE-2,5-DIONE, 4-(4-CHLOROPHENYL)-1-METHYL-](http://img.cochemist.com/ccimg/853300/853261-62-8_b.png)
![1H-Indeno[1,2-b]pyridine-2,5-dione, 4-(4-fluorophenyl)-1-methyl-](http://img.cochemist.com/ccimg/853300/853261-61-7.png)
![1H-Indeno[1,2-b]pyridine-2,5-dione, 4-(4-fluorophenyl)-1-methyl-](http://img.cochemist.com/ccimg/853300/853261-61-7_b.png)
![1H-Indeno[1,2-b]pyridine-2,5-dione, 1-methyl-4-(4-methylphenyl)-](http://img.cochemist.com/ccimg/853300/853261-59-3.png)
![1H-Indeno[1,2-b]pyridine-2,5-dione, 1-methyl-4-(4-methylphenyl)-](http://img.cochemist.com/ccimg/853300/853261-59-3_b.png)
![1H-INDENO[1,2-B]PYRIDINE-2,5-DIONE, 1-METHYL-4-PHENYL-](http://img.cochemist.com/ccimg/853300/853261-58-2.png)
![1H-INDENO[1,2-B]PYRIDINE-2,5-DIONE, 1-METHYL-4-PHENYL-](http://img.cochemist.com/ccimg/853300/853261-58-2_b.png)


![PYRIDO[2,1-A]ISOINDOLE-4,6-DIONE, 2-PHENYL-](http://img.cochemist.com/ccimg/853300/853261-56-0.png)
![PYRIDO[2,1-A]ISOINDOLE-4,6-DIONE, 2-PHENYL-](http://img.cochemist.com/ccimg/853300/853261-56-0_b.png)






![5H-PYRANO[4,3-D]PYRIMIDINE, 2-CHLORO-8-ETHYL-4-METHOXY-](http://img.cochemist.com/ccimg/848900/848891-83-8.png)
![5H-PYRANO[4,3-D]PYRIMIDINE, 2-CHLORO-8-ETHYL-4-METHOXY-](http://img.cochemist.com/ccimg/848900/848891-83-8_b.png)


![9H-PYRIDO[3,4-B]INDOLE, 1-(3-BROMOPHENYL)-](http://img.cochemist.com/ccimg/767400/767354-20-1.png)
![9H-PYRIDO[3,4-B]INDOLE, 1-(3-BROMOPHENYL)-](http://img.cochemist.com/ccimg/767400/767354-20-1_b.png)
![[1,1'-BIPHENYL]-2-AMINE, 4'-(1,1-DIMETHYLETHYL)-](http://img.cochemist.com/ccimg/744300/744262-30-4.png)
![[1,1'-BIPHENYL]-2-AMINE, 4'-(1,1-DIMETHYLETHYL)-](http://img.cochemist.com/ccimg/744300/744262-30-4_b.png)






![1H-Pyrido[3,4-b]indol-1-one, 2,9-dihydro-6,7-dimethoxy-3-phenyl-](http://img.cochemist.com/ccimg/735300/735291-05-1.png)
![1H-Pyrido[3,4-b]indol-1-one, 2,9-dihydro-6,7-dimethoxy-3-phenyl-](http://img.cochemist.com/ccimg/735300/735291-05-1_b.png)
![6H-1,3-DIOXOLO[4,5-F]PYRIDO[3,4-B]INDOL-6-ONE, 5,7-DIHYDRO-8-PHENYL-](http://img.cochemist.com/ccimg/735300/735290-99-0.png)
![6H-1,3-DIOXOLO[4,5-F]PYRIDO[3,4-B]INDOL-6-ONE, 5,7-DIHYDRO-8-PHENYL-](http://img.cochemist.com/ccimg/735300/735290-99-0_b.png)
![1H-Pyrido[3,4-b]indol-1-one, 2,9-dihydro-3-(4-methylphenyl)-](http://img.cochemist.com/ccimg/735300/735290-97-8.png)
![1H-Pyrido[3,4-b]indol-1-one, 2,9-dihydro-3-(4-methylphenyl)-](http://img.cochemist.com/ccimg/735300/735290-97-8_b.png)





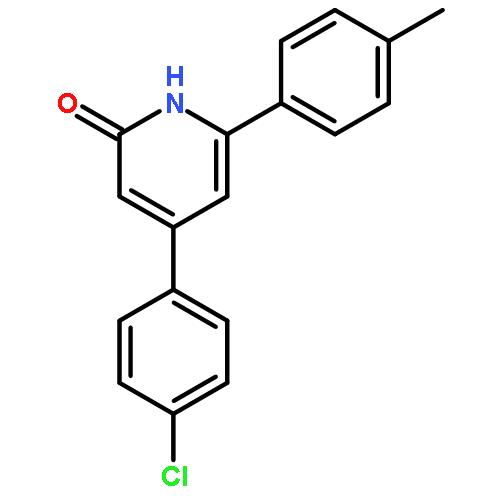









![2-Naphthalenol, 1-[(R)-(hexahydro-1H-azepin-1-yl)phenylmethyl]-](http://img.cochemist.com/ccimg/522000/521960-32-7.png)
![2-Naphthalenol, 1-[(R)-(hexahydro-1H-azepin-1-yl)phenylmethyl]-](http://img.cochemist.com/ccimg/522000/521960-32-7_b.png)
![1-[(r)-phenyl(piperidin-1-yl)methyl]naphthalen-2-ol](http://img.cochemist.com/ccimg/522000/521960-31-6.png)
![1-[(r)-phenyl(piperidin-1-yl)methyl]naphthalen-2-ol](http://img.cochemist.com/ccimg/522000/521960-31-6_b.png)
![2-NAPHTHALENOL, 1-[(R)-PHENYL-1-PYRROLIDINYLMETHYL]-](http://img.cochemist.com/ccimg/522000/521960-29-2.png)
![2-NAPHTHALENOL, 1-[(R)-PHENYL-1-PYRROLIDINYLMETHYL]-](http://img.cochemist.com/ccimg/522000/521960-29-2_b.png)


![2-Naphthalenol, 1-[(S)-(hexahydro-1H-azepin-1-yl)phenylmethyl]-](http://img.cochemist.com/ccimg/500400/500352-93-2.png)
![2-Naphthalenol, 1-[(S)-(hexahydro-1H-azepin-1-yl)phenylmethyl]-](http://img.cochemist.com/ccimg/500400/500352-93-2_b.png)
![2-Naphthalenol, 1-[(S)-phenyl-1-piperidinylmethyl]-](http://img.cochemist.com/ccimg/500400/500352-92-1.png)
![2-Naphthalenol, 1-[(S)-phenyl-1-piperidinylmethyl]-](http://img.cochemist.com/ccimg/500400/500352-92-1_b.png)
![2-Naphthalenol, 1-[(S)-phenyl-1-pyrrolidinylmethyl]-](http://img.cochemist.com/ccimg/500400/500352-91-0.png)
![2-Naphthalenol, 1-[(S)-phenyl-1-pyrrolidinylmethyl]-](http://img.cochemist.com/ccimg/500400/500352-91-0_b.png)
![ETHANOL, 2-[[(2S)-2-(METHOXYMETHOXY)DODECYL]OXY]-](http://img.cochemist.com/ccimg/438100/438037-21-9.png)
![ETHANOL, 2-[[(2S)-2-(METHOXYMETHOXY)DODECYL]OXY]-](http://img.cochemist.com/ccimg/438100/438037-21-9_b.png)
![OXIRANE, [(7S)-7-DECYL-2,5,8,10-TETRAOXAUNDEC-1-YL]-, (2S)-](http://img.cochemist.com/ccimg/438100/438037-19-5.png)
![OXIRANE, [(7S)-7-DECYL-2,5,8,10-TETRAOXAUNDEC-1-YL]-, (2S)-](http://img.cochemist.com/ccimg/438100/438037-19-5_b.png)











![Pyrido[3,2-g]quinoline-5,10-dione, 4-methyl-](http://img.cochemist.com/ccimg/327100/327039-46-3.png)
![Pyrido[3,2-g]quinoline-5,10-dione, 4-methyl-](http://img.cochemist.com/ccimg/327100/327039-46-3_b.png)






![8H-Benzo[c]pyrido[4,3,2-mn]acridin-8-one](http://img.cochemist.com/ccimg/283200/283152-33-0.png)
![8H-Benzo[c]pyrido[4,3,2-mn]acridin-8-one](http://img.cochemist.com/ccimg/283200/283152-33-0_b.png)


![N-[2-[2-(2-AMINOPHENYL)ETHYNYL]PHENYL]-2,2,2-TRIFLUOROACETAMIDE](/data/chemimg/425800/228575-98-2.png)
![N-[2-[2-(2-AMINOPHENYL)ETHYNYL]PHENYL]-2,2,2-TRIFLUOROACETAMIDE](/data/chemimg/425800/228575-98-2_b.png)
![Formamide, N-[1,2-diphenyl-2-(phenylamino)ethyl]-N-phenyl-](http://img.cochemist.com/ccimg/222800/222737-26-0.png)
![Formamide, N-[1,2-diphenyl-2-(phenylamino)ethyl]-N-phenyl-](http://img.cochemist.com/ccimg/222800/222737-26-0_b.png)








![2-Naphthalenol,1-[(S)-aminophenylmethyl]-](/data/chemimg/20700/219897-38-8.png)
![2-Naphthalenol,1-[(S)-aminophenylmethyl]-](/data/chemimg/20700/219897-38-8_b.png)




![Benzaldehyde, 2-[(4-methylphenyl)ethynyl]-](http://img.cochemist.com/ccimg/189100/189008-33-1.png)
![Benzaldehyde, 2-[(4-methylphenyl)ethynyl]-](http://img.cochemist.com/ccimg/189100/189008-33-1_b.png)
![1-(2-methyl-1-propen-1-yl)-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/185200/185140-85-6.png)
![1-(2-methyl-1-propen-1-yl)-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/185200/185140-85-6_b.png)




















![Acetamide, N-(2'-methoxy[1,1'-biphenyl]-2-yl)-](http://img.cochemist.com/ccimg/141600/141540-23-0.png)
![Acetamide, N-(2'-methoxy[1,1'-biphenyl]-2-yl)-](http://img.cochemist.com/ccimg/141600/141540-23-0_b.png)


















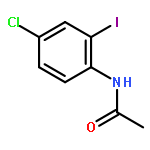
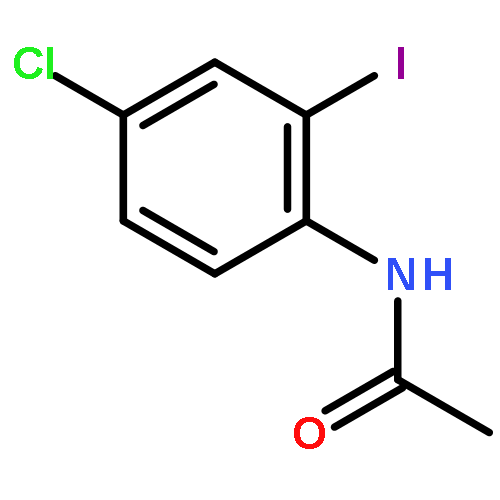






![Benzoic acid, 2-[3-(2-furanyl)-1-oxo-2-propenyl]-](http://img.cochemist.com/ccimg/111700/111660-30-1.png)
![Benzoic acid, 2-[3-(2-furanyl)-1-oxo-2-propenyl]-](http://img.cochemist.com/ccimg/111700/111660-30-1_b.png)
![Benzoic acid, 2-[3-[4-(1-methylethyl)phenyl]-1-oxo-2-propenyl]-](http://img.cochemist.com/ccimg/111700/111660-26-5.png)
![Benzoic acid, 2-[3-[4-(1-methylethyl)phenyl]-1-oxo-2-propenyl]-](http://img.cochemist.com/ccimg/111700/111660-26-5_b.png)
![[3,2':4',3''-Terpyridin]-6'(1'H)-one](http://img.cochemist.com/ccimg/110400/110357-83-0.png)
![[3,2':4',3''-Terpyridin]-6'(1'H)-one](http://img.cochemist.com/ccimg/110400/110357-83-0_b.png)

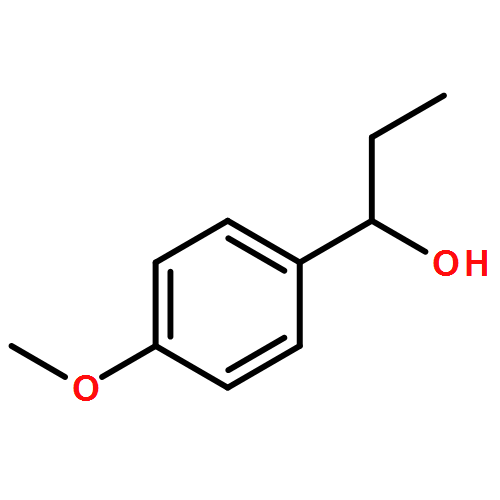






![Benzamide, 3-bromo-N-[2-(3,4-dimethoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/98300/98215-55-5.png)
![Benzamide, 3-bromo-N-[2-(3,4-dimethoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/98300/98215-55-5_b.png)


![4-methylbenzo[g]quinoline-5,10-dione](http://img.cochemist.com/ccimg/96900/96889-94-0.png)
![4-methylbenzo[g]quinoline-5,10-dione](http://img.cochemist.com/ccimg/96900/96889-94-0_b.png)


![Acetamide, N-(3'-methoxy[1,1'-biphenyl]-2-yl)-](http://img.cochemist.com/ccimg/94100/94028-76-9.png)
![Acetamide, N-(3'-methoxy[1,1'-biphenyl]-2-yl)-](http://img.cochemist.com/ccimg/94100/94028-76-9_b.png)
![5,8-QUINOLINEDIONE, 6-[(4-METHOXYPHENYL)AMINO]-](http://img.cochemist.com/ccimg/91700/91613-49-9.png)
![5,8-QUINOLINEDIONE, 6-[(4-METHOXYPHENYL)AMINO]-](http://img.cochemist.com/ccimg/91700/91613-49-9_b.png)


















![[1,1'-Biphenyl]-2-amine, 3',4'-dimethoxy-](/data/chemimg/2293300/78617-18-2.png)
![[1,1'-Biphenyl]-2-amine, 3',4'-dimethoxy-](/data/chemimg/2293300/78617-18-2_b.png)
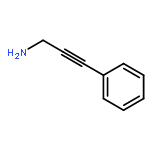
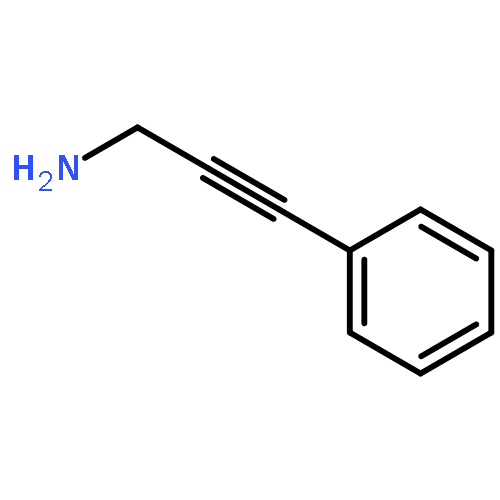
![Acetamide, N-(4'-methyl[1,1'-biphenyl]-2-yl)-](http://img.cochemist.com/ccimg/76500/76472-82-7.png)
![Acetamide, N-(4'-methyl[1,1'-biphenyl]-2-yl)-](http://img.cochemist.com/ccimg/76500/76472-82-7_b.png)


![Benzoic acid, 2-[3-(4-methoxyphenyl)-1-oxo-2-propenyl]-](http://img.cochemist.com/ccimg/68800/68775-78-0.png)
![Benzoic acid, 2-[3-(4-methoxyphenyl)-1-oxo-2-propenyl]-](http://img.cochemist.com/ccimg/68800/68775-78-0_b.png)
![N-[2-(3,4-dimethoxyphenyl)ethyl]benzamide](http://img.cochemist.com/ccimg/67700/67616-16-4.png)
![N-[2-(3,4-dimethoxyphenyl)ethyl]benzamide](http://img.cochemist.com/ccimg/67700/67616-16-4_b.png)


![5,11-Epoxydibenzo[a,e]cyclooctene, 5,6,11,12-tetrahydro-](http://img.cochemist.com/ccimg/66400/66365-45-5.png)
![5,11-Epoxydibenzo[a,e]cyclooctene, 5,6,11,12-tetrahydro-](http://img.cochemist.com/ccimg/66400/66365-45-5_b.png)
![N-[2-(1H-indol-3-yl)ethyl]-Propanamide](http://img.cochemist.com/ccimg/65700/65601-06-1.png)
![N-[2-(1H-indol-3-yl)ethyl]-Propanamide](http://img.cochemist.com/ccimg/65700/65601-06-1_b.png)


![1,4-Naphthalenedione,2-[(4-methoxyphenyl)amino]-](http://img.cochemist.com/ccimg/64600/64505-52-8.png)
![1,4-Naphthalenedione,2-[(4-methoxyphenyl)amino]-](http://img.cochemist.com/ccimg/64600/64505-52-8_b.png)












![Benzamide, N-[2-(1H-indol-2-yl)phenyl]-](http://img.cochemist.com/ccimg/59000/58995-87-2.png)
![Benzamide, N-[2-(1H-indol-2-yl)phenyl]-](http://img.cochemist.com/ccimg/59000/58995-87-2_b.png)


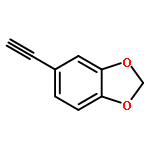
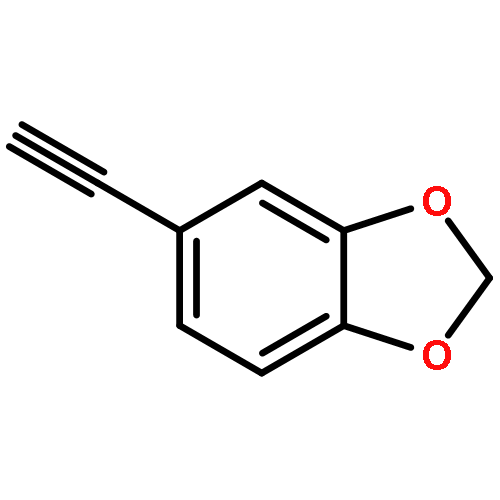







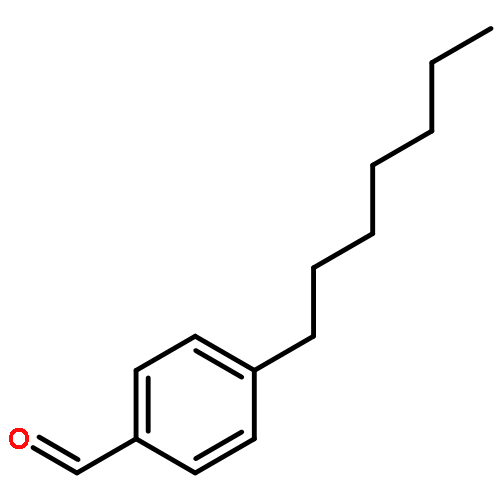






















![Benzamide, N-[2-(1H-indol-3-yl)ethyl]-2-nitro-](http://img.cochemist.com/ccimg/33500/33405-85-5.png)
![Benzamide, N-[2-(1H-indol-3-yl)ethyl]-2-nitro-](http://img.cochemist.com/ccimg/33500/33405-85-5_b.png)


![2-Propenamide,N-[2-(3,4-dimethoxyphenyl)ethyl]-3-phenyl-, (2E)-](http://img.cochemist.com/ccimg/30000/29946-61-0.png)
![2-Propenamide,N-[2-(3,4-dimethoxyphenyl)ethyl]-3-phenyl-, (2E)-](http://img.cochemist.com/ccimg/30000/29946-61-0_b.png)




![9H-Pyrido[3,4-b]indole, 1-(2-nitrophenyl)-](http://img.cochemist.com/ccimg/24300/24243-42-3.png)
![9H-Pyrido[3,4-b]indole, 1-(2-nitrophenyl)-](http://img.cochemist.com/ccimg/24300/24243-42-3_b.png)
![2-[(phenylmethyl)amino]-1,4-Naphthalenedione](http://img.cochemist.com/ccimg/24200/24137-17-5.png)
![2-[(phenylmethyl)amino]-1,4-Naphthalenedione](http://img.cochemist.com/ccimg/24200/24137-17-5_b.png)








![1-ethyl-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/20200/20127-61-1.png)
![1-ethyl-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/20200/20127-61-1_b.png)


![1H-PYRIDO[3,4-B]INDOL-1-ONE, 2,9-DIHYDRO-3-PHENYL-](http://img.cochemist.com/ccimg/18500/18480-82-5.png)
![1H-PYRIDO[3,4-B]INDOL-1-ONE, 2,9-DIHYDRO-3-PHENYL-](http://img.cochemist.com/ccimg/18500/18480-82-5_b.png)
![1H-Pyrido[3,4-b]indol-1-one, 3-(4-chlorophenyl)-2,9-dihydro-](http://img.cochemist.com/ccimg/18500/18480-81-4.png)
![1H-Pyrido[3,4-b]indol-1-one, 3-(4-chlorophenyl)-2,9-dihydro-](http://img.cochemist.com/ccimg/18500/18480-81-4_b.png)


![1-phenyl-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/16800/16765-79-0.png)
![1-phenyl-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/16800/16765-79-0_b.png)




![Benzamide, N-[2-(3,4-dimethoxyphenyl)ethyl]-4-methoxy-](http://img.cochemist.com/ccimg/15600/15547-66-7.png)
![Benzamide, N-[2-(3,4-dimethoxyphenyl)ethyl]-4-methoxy-](http://img.cochemist.com/ccimg/15600/15547-66-7_b.png)






![Benzamide, N-[2-(3,4-dimethoxyphenyl)ethyl]-4-nitro-](http://img.cochemist.com/ccimg/10300/10268-50-5.png)
![Benzamide, N-[2-(3,4-dimethoxyphenyl)ethyl]-4-nitro-](http://img.cochemist.com/ccimg/10300/10268-50-5_b.png)




![4-[(4-AMINOBENZOYL)AMINO]-5-HYDROXYNAPHTHALENE-2,7-DISULPHONIC ACID](http://img.cochemist.com/ccimg/7300/7299-46-9.png)
![4-[(4-AMINOBENZOYL)AMINO]-5-HYDROXYNAPHTHALENE-2,7-DISULPHONIC ACID](http://img.cochemist.com/ccimg/7300/7299-46-9_b.png)


![Acetamide, N-[2-(3,4-dimethoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/6300/6275-29-2.png)
![Acetamide, N-[2-(3,4-dimethoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/6300/6275-29-2_b.png)
![Benzoic acid, 2-[3-(2-chlorophenyl)-1-oxo-2-propenyl]-](http://img.cochemist.com/ccimg/6300/6261-69-4.png)
![Benzoic acid, 2-[3-(2-chlorophenyl)-1-oxo-2-propenyl]-](http://img.cochemist.com/ccimg/6300/6261-69-4_b.png)
![Benzoic acid, 2-[3-(4-chlorophenyl)-1-oxo-2-propenyl]-](http://img.cochemist.com/ccimg/6300/6261-68-3.png)
![Benzoic acid, 2-[3-(4-chlorophenyl)-1-oxo-2-propenyl]-](http://img.cochemist.com/ccimg/6300/6261-68-3_b.png)

















![N-[2-(1H-indol-3-yl)ethyl]-Benzamide](http://img.cochemist.com/ccimg/4800/4753-09-7.png)
![N-[2-(1H-indol-3-yl)ethyl]-Benzamide](http://img.cochemist.com/ccimg/4800/4753-09-7_b.png)




![6-methyl-11H-indolo[3,2-c]quinoline](http://img.cochemist.com/ccimg/4300/4295-28-7.png)
![6-methyl-11H-indolo[3,2-c]quinoline](http://img.cochemist.com/ccimg/4300/4295-28-7_b.png)




![benzo[g]quinoline-5,10-dione](http://img.cochemist.com/ccimg/3800/3712-09-2.png)
![benzo[g]quinoline-5,10-dione](http://img.cochemist.com/ccimg/3800/3712-09-2_b.png)








![Benzenamine,N-[(4-chlorophenyl)methylene]-](http://img.cochemist.com/ccimg/2400/2362-79-0.png)
![Benzenamine,N-[(4-chlorophenyl)methylene]-](http://img.cochemist.com/ccimg/2400/2362-79-0_b.png)
![Benzenamine,N-[(4-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/2400/2362-77-8.png)
![Benzenamine,N-[(4-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/2400/2362-77-8_b.png)
![Acetamide,N-[1,1'-biphenyl]-2-yl-](http://img.cochemist.com/ccimg/2200/2113-47-5.png)
![Acetamide,N-[1,1'-biphenyl]-2-yl-](http://img.cochemist.com/ccimg/2200/2113-47-5_b.png)
![Acetamide, N-(4'-methoxy[1,1'-biphenyl]-2-yl)-](http://img.cochemist.com/ccimg/1300/1217-87-4.png)
![Acetamide, N-(4'-methoxy[1,1'-biphenyl]-2-yl)-](http://img.cochemist.com/ccimg/1300/1217-87-4_b.png)
![[1,1'-Biphenyl]-2-amine, 2'-methoxy-](http://img.cochemist.com/ccimg/1300/1206-76-4.png)
![[1,1'-Biphenyl]-2-amine, 2'-methoxy-](http://img.cochemist.com/ccimg/1300/1206-76-4_b.png)
![[1,1'-Biphenyl]-2-amine, 4'-methyl-](/data/chemimg/370200/1204-43-9.png)
![[1,1'-Biphenyl]-2-amine, 4'-methyl-](/data/chemimg/370200/1204-43-9_b.png)





![N-[2-(1H-INDOL-3-YL)ETHYL]ACETAMIDE](http://img.cochemist.com/ccimg/1100/1016-47-3.png)
![N-[2-(1H-INDOL-3-YL)ETHYL]ACETAMIDE](http://img.cochemist.com/ccimg/1100/1016-47-3_b.png)






![9H-Pyrido[3,4-b]indole,1-methyl-](http://img.cochemist.com/ccimg/500/486-84-0.png)
![9H-Pyrido[3,4-b]indole,1-methyl-](http://img.cochemist.com/ccimg/500/486-84-0_b.png)


![11H-Indolo[3,2-c]quinoline](http://img.cochemist.com/ccimg/300/239-09-8.png)
![11H-Indolo[3,2-c]quinoline](http://img.cochemist.com/ccimg/300/239-09-8_b.png)

















![2-Propen-1-one,1-[1,1'-biphenyl]-4-yl-3-phenyl-](http://img.cochemist.com/ccimg/2500/2453-44-3.png)
![2-Propen-1-one,1-[1,1'-biphenyl]-4-yl-3-phenyl-](http://img.cochemist.com/ccimg/2500/2453-44-3_b.png)



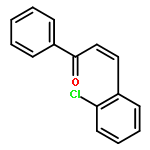
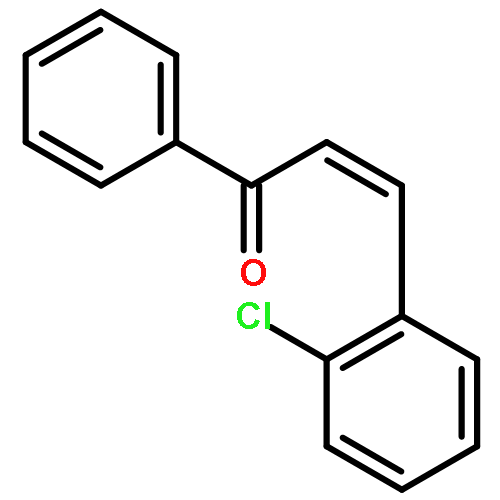




![Gold,[[1,1'-biphenyl]-2-ylbis(1,1-dimethylethyl)phosphine]chloro-](http://img.cochemist.com/ccimg/854100/854045-93-5.png)
![Gold,[[1,1'-biphenyl]-2-ylbis(1,1-dimethylethyl)phosphine]chloro-](http://img.cochemist.com/ccimg/854100/854045-93-5_b.png)


![Benzene, 1-[(1E)-3-bromo-1-propen-1-yl]-4-(trifluoromethyl)-](/data/chemimg/117000/762247-01-8.png)
![Benzene, 1-[(1E)-3-bromo-1-propen-1-yl]-4-(trifluoromethyl)-](/data/chemimg/117000/762247-01-8_b.png)
![Dichloro[(±)−BINAP]digold(I)](http://img.cochemist.com/ccimg/685200/685138-48-1.png)
![Dichloro[(±)−BINAP]digold(I)](http://img.cochemist.com/ccimg/685200/685138-48-1_b.png)


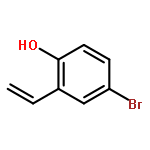
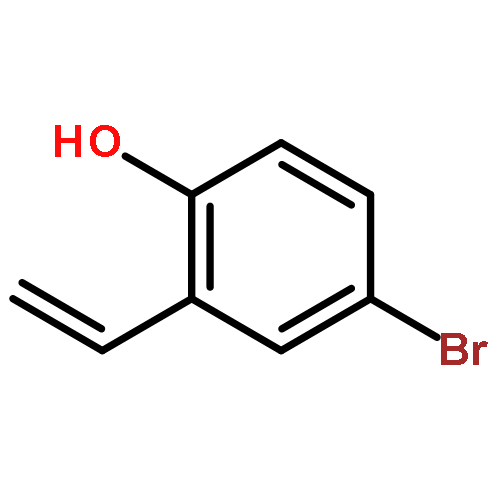






![5-methoxy-9H-quino[4,3,2-de][1,10]phenanthrolin-9-one](http://img.cochemist.com/ccimg/143400/143370-23-4.png)
![5-methoxy-9H-quino[4,3,2-de][1,10]phenanthrolin-9-one](http://img.cochemist.com/ccimg/143400/143370-23-4_b.png)
![9H-Benzo[b]pyrido[4,3,2-mn]acridin-9-one](http://img.cochemist.com/ccimg/143100/143091-80-9.png)
![9H-Benzo[b]pyrido[4,3,2-mn]acridin-9-one](http://img.cochemist.com/ccimg/143100/143091-80-9_b.png)

![9H-QUINO[4,3,2-DE][1,10]PHENANTHROLIN-9-ONE, 5-(ACETYLOXY)-](http://img.cochemist.com/ccimg/131000/130932-74-0.png)
![9H-QUINO[4,3,2-DE][1,10]PHENANTHROLIN-9-ONE, 5-(ACETYLOXY)-](http://img.cochemist.com/ccimg/131000/130932-74-0_b.png)


![Benzene, [(1E)-4-bromo-1-methyl-1-butenyl]-](http://img.cochemist.com/ccimg/129000/128902-50-1.png)
![Benzene, [(1E)-4-bromo-1-methyl-1-butenyl]-](http://img.cochemist.com/ccimg/129000/128902-50-1_b.png)
![Benzene, [(1Z)-3-bromo-1-propenyl]-](http://img.cochemist.com/ccimg/115200/115117-88-9.png)
![Benzene, [(1Z)-3-bromo-1-propenyl]-](http://img.cochemist.com/ccimg/115200/115117-88-9_b.png)
![9H-quino[4,3,2-de][1,10]phenanthrolin-9-one](http://img.cochemist.com/ccimg/114700/114622-04-7.png)
![9H-quino[4,3,2-de][1,10]phenanthrolin-9-one](http://img.cochemist.com/ccimg/114700/114622-04-7_b.png)
![9H-Quino[4,3,2-de][1,10]phenanthrolin-9-one,6-bromo-](http://img.cochemist.com/ccimg/109900/109802-17-7.png)
![9H-Quino[4,3,2-de][1,10]phenanthrolin-9-one,6-bromo-](http://img.cochemist.com/ccimg/109900/109802-17-7_b.png)
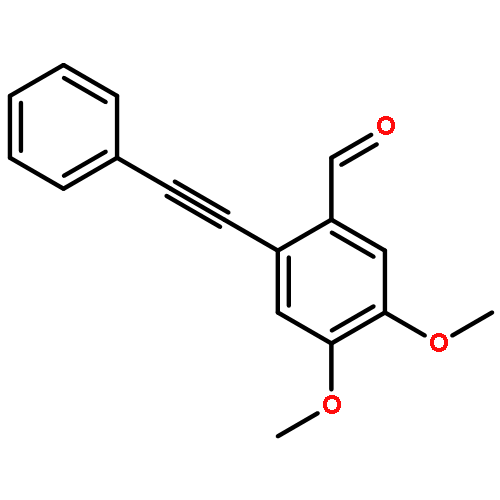






![1H-Pyrido[3,4-b]indole-1-acetic acid, 2,3,4,9-tetrahydro-, methyl ester](http://img.cochemist.com/ccimg/61800/61756-61-4.png)
![1H-Pyrido[3,4-b]indole-1-acetic acid, 2,3,4,9-tetrahydro-, methyl ester](http://img.cochemist.com/ccimg/61800/61756-61-4_b.png)








![Benzene, [(1E)-3-bromo-1-methyl-1-propen-1-yl]-](/data/chemimg/3760200/39171-59-0.png)
![Benzene, [(1E)-3-bromo-1-methyl-1-propen-1-yl]-](/data/chemimg/3760200/39171-59-0_b.png)


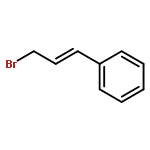
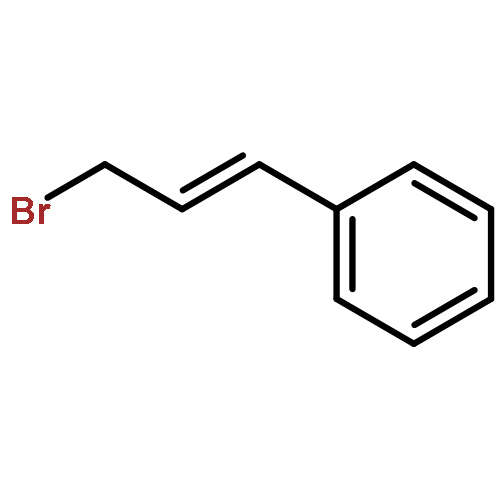
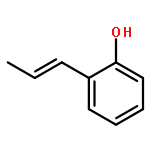
![1H-Pyrido[3,4-b]indole, 2,3,4,9-tetrahydro-9-methyl-1-phenyl-](http://img.cochemist.com/ccimg/17000/16979-84-3.png)
![1H-Pyrido[3,4-b]indole, 2,3,4,9-tetrahydro-9-methyl-1-phenyl-](http://img.cochemist.com/ccimg/17000/16979-84-3_b.png)
![2,3,4,9-tetrahydro-9-methyl-1H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/16600/16502-02-6.png)
![2,3,4,9-tetrahydro-9-methyl-1H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/16600/16502-02-6_b.png)
![Benzene, 1-[(1E)-3-bromo-1-propen-1-yl]-4-methoxy-](/data/chemimg/926100/16034-99-4.png)
![Benzene, 1-[(1E)-3-bromo-1-propen-1-yl]-4-methoxy-](/data/chemimg/926100/16034-99-4_b.png)























{kind=link}