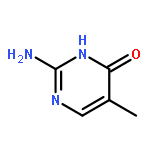
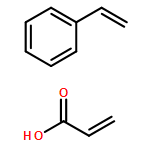
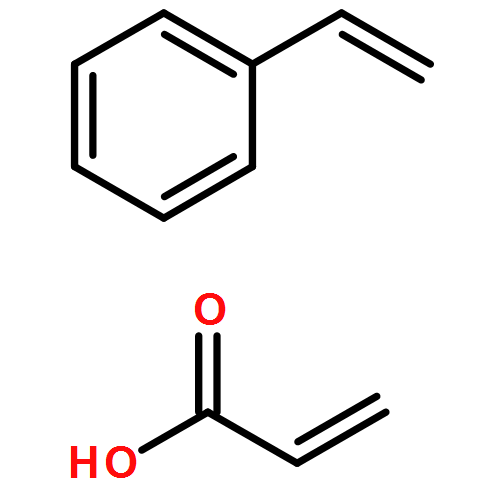
Co-reporter:Mithun Chowdhury, Yunlong Guo, Yucheng Wang, Weston L. Merling, Jayachandra H. Mangalara, David S. Simmons, and Rodney D. Priestley
The Journal of Physical Chemistry Letters March 16, 2017 Volume 8(Issue 6) pp:1229-1229
Publication Date(Web):March 3, 2017
DOI:10.1021/acs.jpclett.7b00214
When geometrically confined to the nanometer length scale, a condition in which a large portion of the material is in the nanoscale vicinity of interfaces, polymers can show astonishing changes in physical properties. In this investigation, we employ a unique noncontact capillary nanoshearing method to directly probe nanoresolved gradients in the rheological response of ultrathin polymer films as a function of temperature and stress. Results show that ultrathin polymer films, in response to an applied shear stress, exhibit a gradient in molecular mobility and viscosity that originates at the interfaces. We demonstrate, via molecular dynamics simulations, that these gradients in molecular mobility reflect gradients in the average segmental relaxation time and the glass-transition temperature.
Co-reporter:Chris Sosa, Victoria E. Lee, Lorena S. Grundy, Mary J. Burroughs, Rui Liu, Robert K. Prud’homme, and Rodney D. Priestley
Langmuir June 13, 2017 Volume 33(Issue 23) pp:5835-5835
Publication Date(Web):June 1, 2017
DOI:10.1021/acs.langmuir.7b01021
In an effort to incorporate increasingly higher levels of functionality into soft nanoparticles, heterogeneously structured particles stand out as a simple means to enhance functionality by tailoring only particle architecture. Various means exist for the fabrication of particles with specific structural configurations; however, the tunability of particle morphology is still a challenging and often laborious task, especially in self-assembled systems where a single equilibrium configuration dominates. Improved strategies for multipatch particle assembly are therefore needed to allow for the tailoring of particle structure via a single, continuous assembly route. One means of accomplishing this is through kinetic trapping of particle morphologies along the path to the final equilibrium configuration in precipitation-induced, phase-separating polymer blends. Here, we demonstrate this capability by using rapid nanoprecipitation to control the overall size, composition, and patch distribution of soft colloids. In particular, we illustrate that polymer feed concentration, blend ratio, and polymer molecular weight can all serve as functional handles with which to consistently alter particle patch distributions in a self-assembling homopolymer system without redesigning the starting materials. We furthermore delineate the role of polymer vitrification in the determination of particle structure.
Co-reporter:Guochang Li;Jian Li;Ziwei Zhou;Congling Li;Chao Cai;Bingkun Guo;Lei Han;Rui Liu
Dalton Transactions 2017 vol. 46(Issue 47) pp:16419-16425
Publication Date(Web):2017/12/06
DOI:10.1039/C7DT03021E
We present the use of silica-polydopamine (SiO2@PDA) core–shell nanoparticles (NPs) as self-confined templates for the fabrication of ultra-stable hollow Pt anchored N-doped carbon nanospheres (Pt/HN-C). SiO2@PDA nanospheres were fabricated by a facile one-pot process, followed by the deposition of Pt NPs onto the outer shell layer. The confinement and adhesion of the PDA framework ensured the distribution and stability of Pt NPs after carbonization at the carbon shell layer. The converted Pt/HN-C exhibited excellent catalytic performance and durability for the oxygen reduction reaction (ORR).
Co-reporter:Hyuncheol Jeong, Simone Napolitano, Craig B. Arnold, and Rodney D. Priestley
The Journal of Physical Chemistry Letters 2017 Volume 8(Issue 1) pp:229-234
Publication Date(Web):December 15, 2016
DOI:10.1021/acs.jpclett.6b02573
Matrix-assisted pulsed laser evaporation (MAPLE) provides a gentle means for the quasi-vapor deposition of macromolecules. It offers a unique opportunity for the bottom-up control of polymer crystallization as film growth and crystallization occur simultaneously. Surprisingly, with increasing deposition time, it has been shown that crystallization becomes prohibited despite the availability of polymer via continuous deposition. In this Letter, we investigate the molecular origins of suppressed crystallization in poly(ethylene oxide) films deposited by MAPLE atop silicon substrates. We find that suppressed crystallization results from the formation of an irreversibly adsorbed polymer nanolayer at the substrate that forms during deposition. Substrate temperature is shown to influence the stability of the irreversibly adsorbed nanolayer and, hence, polymer thin film crystallization. Our investigation offers new insight into how temperature and interfacial interactions can serve as a new toolbox to tune polymer film morphology in bottom-up deposition.
Co-reporter:Rui Liu and Rodney D. Priestley
Journal of Materials Chemistry A 2016 vol. 4(Issue 18) pp:6680-6692
Publication Date(Web):05 Feb 2016
DOI:10.1039/C5TA09607C
Core–shell nanoparticles (NPs) have emerged as a type of important nanomaterial for various applications. The challenge in the preparation of core–shell NPs is to find a simple, cost-effective and less time-consuming strategy with minimum environmental impact. The consolidation of multiple preparation steps into one step represents a new green synthesis pathway in chemistry and materials science. In this review, we provide an overview of the recent developments in the fabrication of core–shell NPs through one-step/pot methodologies. A variety of one-step/pot preparation methods are presented, discussed and compared, followed by the summary and outlook of this emerging area.
Co-reporter:Dane Christie;Chuan Zhang;Jie Fu;Bruce Koel
Journal of Polymer Science Part B: Polymer Physics 2016 Volume 54( Issue 17) pp:1776-1783
Publication Date(Web):
DOI:10.1002/polb.24082
ABSTRACT
Recently, there has been significant interest in measuring the glass transition temperature (Tg) of thin polymer films floated atop liquid substrates. However, such films still have intrinsically asymmetric interfaces, that is, a free surface and a liquid–polymer interface. In an effort to analyze the influence of different liquids on the Tg of confined polymers in which there is no interfacial asymmetry, a colloidal suspension of polystyrene (PS) nanoparticles (NPs) was employed. The Tgs of PS NPs suspended in either glycerol or an ionic liquid were characterized using differential scanning calorimetry. Nanoparticles suspended in an ionic liquid showed an invariance of Tg with confinement, that is, decreasing diameter. In contrast, nanoparticles suspended in glycerol showed a slight decrease in Tg with confinement. The dependence of NP Tg on the nature of the surrounding liquid exhibited a positive correlation with the interfacial energy of the liquid–PS interface and no correlation with interfacial softness, as measured by viscosity. A comparison of the results with thin films supported by liquid or solid substrates revealed a nontrivial interplay between interfacial softness and interfacial interactions on the Tg of confined PS. © 2016 Wiley Periodicals, Inc. J. Polym. Sci., Part B: Polym. Phys. 2016, 54, 1776–1783
Co-reporter:Hyuncheol Jeong, Kimberly B. Shepard, Geoffrey E. Purdum, Yunlong Guo, Yueh-Lin Loo, Craig B. Arnold, and Rodney D. Priestley
Macromolecules 2016 Volume 49(Issue 7) pp:2860-2867
Publication Date(Web):March 22, 2016
DOI:10.1021/acs.macromol.5b02675
We demonstrated a polymeric thin film fabrication process in which molecular-scale crystallization proceeds with additive film growth, by employing an innovative vapor-assisted deposition process termed matrix-assisted pulsed laser evaporation (MAPLE). In comparison to solution-casting commonly adopted for the deposition of polymer thin films, this physical vapor deposition (PVD) methodology can prolong the time scale of film formation and allow for the manipulation of temperature during deposition. For the deposition of molecular and atomic systems, such a PVD manner has been demonstrated to facilitate molecular ordering and delicately manipulate crystalline morphology during film growth. Here, using MAPLE, we deposited thin films of a model polymer, poly(ethylene oxide) (PEO), atop a temperature-controlled substrate with an average growth rate of less than 10 nm/h. The mechanism of deposition is sequential addition of nanoscale liquid droplets. We discovered that the deposition process leads to the formation of two-dimensional (2D) PEO crystals, composed of monolamellar crystals laterally grown from larger nucleus droplets. The 2D crystalline coverage and crystal thickness of the films can be manipulated with two processing parameters, deposition time, and temperature.
Co-reporter:Chris Sosa, Rui Liu, Christina Tang, Fengli Qu, Sunny Niu, Martin Z. Bazant, Robert K. Prud’homme, and Rodney D. Priestley
Macromolecules 2016 Volume 49(Issue 9) pp:3580-3585
Publication Date(Web):April 20, 2016
DOI:10.1021/acs.macromol.6b00708
Soft colloidal particles with multiple surface patches of differing composition are critical to the development of complex macroscopic structures that can serve as interfacial catalysts, macroscale surfactants, electronically responsive materials, and drug delivery vehicles. Here, we present a continuous process for the scalable formation of soft colloidal particles with multiple surface domains that employs well-established principles of polymer precipitation and phase separation to controllably shape particle architectures. Our results illustrate the broad range of particle morphologies, including Janus and Cerberus structures, and surface compositions accessible to our versatile solution-based assembly system. We also identify polymer diffusion, precipitation, and vitrification as the primary determinants of particle structure for the first time.
Co-reporter:Mary J. Burroughs, Simone Napolitano, Daniele Cangialosi, and Rodney D. Priestley
Macromolecules 2016 Volume 49(Issue 12) pp:4647-4655
Publication Date(Web):June 7, 2016
DOI:10.1021/acs.macromol.6b00400
We employ a fluorescence bilayer method to directly measure the glass transition temperature (Tg) of the irreversibly adsorbed layer of polystyrene (PS) buried in bulk films as a function of adsorption time, tads. This bilayer geometry allows for the examination of interfacial effects on Tg of the adsorbed nanolayer. In the presence of a free surface, we observe a substantial reduction in Tg from bulk that lessens with tads as a result of increased chain adsorption at the substrate. Submerging the adsorbed layer and effectively removing the free surface results in a suppression of the Tg deviation at early tads, suggesting chain adsorption dictates Tg at long tads. Annealing in the bilayer geometry promotes recovery of bulk Tg on a time scale reflecting the degree of adsorption. Our data are quantitatively rationalized via the free volume holes diffusion model, which explains adsorbed nanolayer Tg in terms of the diffusion of free volume pockets toward interfacial sinks.
Co-reporter:Rodney D. Priestley, Daniele Cangialosi, Simone Napolitano
Journal of Non-Crystalline Solids 2015 Volume 407() pp:288-295
Publication Date(Web):1 January 2015
DOI:10.1016/j.jnoncrysol.2014.09.048
•The glass transition in confined polymers is reviewed.•Dynamic and pseudo-thermodynamic measurements of Tg are decoupled in confinement.•Free volume diffusion and chain adsorption offer insight into decoupling.Understanding why the glass transition temperature (Tg) of polymers deviates substantially from the bulk with nanoscale confinement has been a 20-year mystery. Ever since the observation in the mid-1990s that the Tg values of amorphous polymer thin films are different from their bulk values, efforts to understand this behavior have intensified, and the topic remains the subject of intense research and debate. This is due to the combined scientific and technological implications of size-dependent glassy properties. Here, we discuss an intriguing aspect of the glassy behavior of confined amorphous polymers. As experimentally assessed, the glass transition is a dynamic event mediated by segmental dynamics. Thus, it seems intuitive to expect that a change in Tg due to confinement necessitates a corresponding change in molecular dynamics, and that such change in dynamics may be predicted based on our understanding of the glass transition. The aim of this perspectives article is to examine whether or not segmental dynamics change in accordance with the value of Tg for confined polymers based on bulk rules. We highlight past and recent findings that have examined the relationship between Tg and segmental dynamics of confined polymers. Within this context, the decoupling between these two aspects of the glass transition in confinement is emphasized. We discuss these results within the framework of our current understanding of the glass transition as well as efforts to resolve this decoupling. Finally, the anomalous decoupling between translational (diffusion) and rotational (segmental) motion taking place in the proximity of attractive interfaces in polymer thin films is discussed.
Co-reporter:Colin C. Neikirk, Jae Woo Chung, Rodney D. Priestley
Polymer 2015 Volume 79() pp:212-220
Publication Date(Web):19 November 2015
DOI:10.1016/j.polymer.2015.10.019
•Synthesized polyacrylate containing H-bonding group ureidopyrimidinone (UPy).•These polymers were blended with UPy functionalized silica nanoparticles (NP).•Higher polymer UPy content led to higher importance of NP surface functionality.•Mechanical properties were correlated with NP aggregate morphology by TEM.•Demonstrated complex interplay between matrix and filler H-bonding.The dispersion, aggregate morphology, and interfacial strength of fillers in polymer nanocomposites are crucial in determining their physical properties. Here, we utilize the quadruple hydrogen bonding group ureidopyrimidinone (UPy) to investigate enthalpic effects on the dispersion of silica nanoparticles in a poly(butyl acrylate) copolymer matrix and to correlate dispersion with mechanical properties. Butyl acrylate was copolymerized with an UPy side chain functional methacrylate monomer and blended with silica nanoparticles containing various degrees of UPy, hexyl, or silanol surface functionality. The tensile and dynamic mechanical responses of these nanocomposites were measured, and the effects of varying surface functionality on mechanical properties are discussed and correlated to dispersion by TEM. The competing effects of filler–filler, matrix–filler, and matrix–matrix hydrogen bonding lead to a system where both the type and amount of filler surface functionality are of critical importance to the material's final strength.Figure optionsDownload full-size imageDownload high-quality image (168 K)Download as PowerPoint slide
Co-reporter:Rui Liu, Chris Sosa, Yao-Wen Yeh, Fengli Qu, Nan Yao, Robert K. Prud'homme and Rodney D. Priestley
Journal of Materials Chemistry A 2014 vol. 2(Issue 41) pp:17286-17290
Publication Date(Web):22 Sep 2014
DOI:10.1039/C4TA04036H
We report a scalable and continuous preparation route for metal nanoparticles (Au or Pt)@nanosphere polymer composites through flash nanoprecipitation (FNP). Uniform metal@nanosphere polymer composites with 2–3 nm metal colloids decorating the surface on the nanosphere are obtained with tunable overall particle size and metal nanoparticle arrangement. The obtained composites show high catalytic ability and stability in the reduction of p-nitrophenol.
Co-reporter:Rui Liu, Yao-Wen Yeh, Vivienne H. Tam, Fengli Qu, Nan Yao and Rodney D. Priestley
Chemical Communications 2014 vol. 50(Issue 65) pp:9056-9059
Publication Date(Web):29 May 2014
DOI:10.1039/C4CC02507E
A facile one-pot Stöber route is used to synthesize high-quality Ag, AgBr–silica–resorcinol formaldehyde polymer core–shell–shell nanospheres. The obtained core–shell–shell templates can be converted to Ag@carbon yolk–shell nanostructures with tunable dimensions.
Co-reporter:Rui Liu, Fengli Qu, Yunlong Guo, Nan Yao and Rodney D. Priestley
Chemical Communications 2014 vol. 50(Issue 4) pp:478-480
Publication Date(Web):21 Nov 2013
DOI:10.1039/C3CC47050D
A facile one-step Stöber route to synthesize high-quality core–shell–shell templates is reported for the fabrication of Au@carbon yolk–shell nanostructures. The converted Au@carbon yolk–shell nanostructures exhibited high catalytic performance as illustrated by the reduction reaction of o-nitrophenol.
Co-reporter:Chuan Zhang, Yunlong Guo, and Rodney D. Priestley
ACS Macro Letters 2014 Volume 3(Issue 6) pp:501
Publication Date(Web):May 13, 2014
DOI:10.1021/mz500204q
We report the effect of isochoric confinement on the characteristic length of the glass transition (ξα) for polystyrene (PS) and poly(4-methylstyrene) (P4MS). Utilizing silica-capped PS and P4MS nanoparticles as model systems, ξα values are determined from the thermal fluctuation model and calorimetric data. With decreasing nanoparticle diameter, ξα decreases, suggesting a reduction in the number of segmental units required for cooperative motion at the glass transition under confinement. Furthermore, a direct correlation is observed between ξα and the isochoric fragility (mv) in confined polymers. Due to a nearly constant ratio of the isochoric to isobaric fragility in confined polymer nanoparticles, a correlation between ξα and mv also implies a correlation between ξα and the volume contribution to the temperature dependence of structural relaxation. Lastly, we observe that when the fragility and characteristic length are varied in the same system the relationship between the two properties appears to be more correlated than that of across different bulk glass-formers.
Co-reporter:Kimberly B. Shepard, Craig B. Arnold, and Rodney D. Priestley
ACS Macro Letters 2014 Volume 3(Issue 10) pp:1046
Publication Date(Web):October 2, 2014
DOI:10.1021/mz500546u
We characterized the transport, i.e., time-of-flight, and nanoscale thermal properties of amorphous polymer nanoglobules fabricated via a laser-deposition technique, Matrix-Assisted Pulsed Laser Deposition (MAPLE). Here, we report the first experimental measurement of the velocity of polymer during MAPLE processing and its connection to nanostructured film formation. A nanoscale dilatometry technique using atomic force microscopy was employed to directly measure the thermal properties of MAPLE-deposited polymer nanoglobules. Similarly to bulk stable polymer glasses deposited by MAPLE, polymer nanoglobules were found to exhibit enhanced thermal stability and low density despite containing only thousands of molecules. By directly connecting the exceptional properties of the nanostructured building blocks to those of bulk stable glasses, we gain insight into the physics of glassy polymeric materials formed via vapor-assisted techniques.
Co-reporter:Rui Liu, Yunlong Guo, Gloria Odusote, Fengli Qu, and Rodney D. Priestley
ACS Applied Materials & Interfaces 2013 Volume 5(Issue 18) pp:9167
Publication Date(Web):September 6, 2013
DOI:10.1021/am402585y
We present the synthesis and multifunctional utilization of core–shell Fe3O4 polydopamine nanoparticles (Fe3O4@PDA NPs) to serve as the enabling platform for a range of applications including responsive drug delivery, recyclable catalyst support, and adsorbent. Magnetite Fe3O4 NPs formed in a one-pot process by the hydrothermal approach were coated with a polydopamine shell layer of ∼20 nm in thickness. The as prepared Fe3O4@PDA NPs were used for the controlled drug release in a pH-sensitive manner via reversible bonding between catechol and boronic acid groups of PDA and the anticancer drug bortezomib (BTZ), respectively. The facile deposition of Au NPs atop Fe3O4@PDA NPs was achieved by utilizing PDA as both the reducing agent and the coupling agent. The nanocatalysts exhibited high catalytic performance for the reduction of o-nitrophenol. Furthermore, the recovery and reuse of the catalyst was demonstrated 10 times without any detectible loss in activity. Finally, the PDA layers were converted into carbon to obtain Fe3O4@C and used as an adsorbent for the removal of Rhodamine B from an aqueous solution. The synergistic combination of unique features of PDA and magnetic nanoparticles establishes these core–shell NPs as a versatile platform for multiple applications.Keywords: core-shell nanoparticles; dopamine; Fe3O4; multi-functional; polydopamine;
Co-reporter:Chuan Zhang and Rodney D. Priestley
Soft Matter 2013 vol. 9(Issue 29) pp:7076-7085
Publication Date(Web):17 May 2013
DOI:10.1039/C3SM50171J
It is now well established that the glass transition temperature (Tg) of polymers can deviate substantially from the bulk with nanoscale confinement. Understanding the impact of confinement on the Tg of polymers is important but it does not provide information about the temperature dependence of the cooperative segmental dynamics near the Tg, a phenomenon commonly referred to as the dynamic fragility. Here, we measure the dynamic fragility index (m) as well as the Tg of confined poly(4-methylstyrene) (P4MS) under isobaric and isochoric conditions. We accomplish this via variable cooling rate differential scanning calorimetry (DSC) studies on aqueous-suspended and silica-capped P4MS nanoparticles. We observe that both the isobaric (mp) and isochoric (mv) fragilities decrease with confinement. However, Tg decreases and remains constant with isobaric and isochoric confinement, respectively. The importance of interfaces in the observed trends as well as comparisons to prior studies is discussed.
Co-reporter:Jae Woo Chung, Colin Neikirk, Rodney D. Priestley
Journal of Colloid and Interface Science 2013 Volume 396() pp:16-22
Publication Date(Web):15 April 2013
DOI:10.1016/j.jcis.2013.01.038
The effect of coumarin molecules on the formation of polymeric nanoparticles is examined using a model polymer, poly(methyl methacrylate) (PMMA), functionalized with varying amounts of coumarin pendant groups (PCM). PCM nanoparticles are prepared in a continuous manner by Flash NanoPrecipitation (FNP). PCM forms spherical nanoparticles in water, while the PMMA without coumarin functionality fails to form nanoparticles. As the amount of coumarin functionality increases, the nanoparticle size and size polydispersity are decreased and the nanoparticle stability in water is enhanced. In particular, well-isolated spherical nanoparticles are generated from PCM with 20 mol% coumarin side chain functionality. These results can be explained by an observed increase in the negative surface charge with increasing coumarin content in the polymer.Graphical abstractHighlights► We investigate the impact of coumarin on the formation of polymer nanoparticles. ► Polymer nanoparticles are prepared by flash nanoprecipitation. ► Addition of coumarin to polymer side chain significantly improves nanoparticle stability. ► Increasing coumarin content leads to a decrease in the zeta potential of nanoparticles.
Co-reporter:Colin C. Neikirk, Jae Woo Chung and Rodney D. Priestley
RSC Advances 2013 vol. 3(Issue 37) pp:16686-16696
Publication Date(Web):09 Aug 2013
DOI:10.1039/C3RA42031K
Supramolecular polymer nanocomposites represent an attractive alternative to traditional polymers for advanced materials that exhibit stimuli-responsive and self-healing properties. Here, we investigate the effects of specific hydrogen bonding interactions between surface functionalized silica nanoparticles and ureidopyrimidinone (UPy) based hydrogen bonded supramolecular poly(ε-caprolactone) in a supramolecular polymer nanocomposite. The effect of varying levels of nanoparticle UPy surface functionalization is considered. In addition to the anticipated improvements in Young's modulus (∼50%) and storage modulus (∼2×) with silica loading, increases in strain at breaking point (∼25%) with silica loading were observed and attributed to particle–matrix hydrogen bonding. However, increasing the extent of UPy surface functionality at a constant nanoparticle loading level led to a marked decrease in storage modulus relative to nanocomposites prepared with as-received silica nanoparticles. TEM investigation of these nanocomposites show an increase in nanoparticle aggregation. Nanoparticle aggregation provides both an explanation for the observed storage modulus reduction and evidence of particle–particle interactions. These results give interesting insight into the competing effects of specific supramolecular interactions in supramolecular polymer nanocomposite materials.
Co-reporter:Kimberly B. Shepard
Macromolecular Chemistry and Physics 2013 Volume 214( Issue 8) pp:862-872
Publication Date(Web):
DOI:10.1002/macp.201200621
Co-reporter:Kimberly B. Shepard
Macromolecular Chemistry and Physics 2013 Volume 214( Issue 8) pp:
Publication Date(Web):
DOI:10.1002/macp.201370027
Co-reporter:Chuan Zhang;Yunlong Guo
Journal of Polymer Science Part B: Polymer Physics 2013 Volume 51( Issue 7) pp:574-586
Publication Date(Web):
DOI:10.1002/polb.23268
Abstract
For nearly the past two decades, significant effort has been devoted to pursuing an understanding of the glass transition temperature and associated dynamics of polymers confined to the nanoscale. Without question, we know more about the glassy properties of confined polymers today than we knew two decades ago or even a decade ago. Much of our understanding has been obtained via studies on thin polymer films, as they are facile to process and are of substantial technological importance. Nevertheless, studies on polymers confined to other geometries are becoming increasingly more important as we pursue questions difficult to address using thin films and as technology demands the use of confined polymers beyond thin films. In this feature article, we highlight the impact of nanoscale confinement on the glassy properties of polymer nanoparticles. Although the emphasis is placed on contributions from our work, a discussion of the related literature is also presented. Our aim is to elucidate commonalities or fundamental differences in the deviations of glassy properties from the bulk for polymers confined to different geometries. © 2013 Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys, 2013
Co-reporter:Chuan Zhang, Yunlong Guo, Kimberly B. Shepard, and Rodney D. Priestley
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 3) pp:431-436
Publication Date(Web):January 17, 2013
DOI:10.1021/jz302002v
We report the effect of isochoric confinement on the dynamic fragility of a polymeric glass-former, that is, polystyrene (PS). Utilizing silica-capped PS nanospheres as a model system, the fictive temperature (Tf) and the isochoric heat capacity (Cv) are measured as a function of diameter via differential scanning calorimetry. By examining Tf as a function of cooling rate for each sample, the isochoric fragility (mv) is obtained, which decreases significantly as the diameter of the nanospheres is reduced from 260 to 129 nm. Hence, the temperature dependence of structural relaxation near the glass transition is weakened with isochoric confinement.Keywords: confinement; fragility; glass transition; nanoparticles; polystyrene;
Co-reporter:Chuan Zhang, Virginie M. Boucher, Daniele Cangialosi, Rodney D. Priestley
Polymer 2013 Volume 54(Issue 1) pp:230-235
Publication Date(Web):8 January 2013
DOI:10.1016/j.polymer.2012.11.036
Recent studies have illustrated a decoupling between cooperative segmental mobility and the glass transition temperature (Tg) of thin polymer films and nanocomposites. Here, we use dielectric spectroscopy to probe the cooperative segmental mobility and capacitive dilatometry to determine the Tg of films of polystyrene nanospheres with diameters (d) less than 400 nm. We find that both capacitive dilatometry and calorimetry revealed nearly identical suppressions in Tg as the size of the nanospheres was reduced. While Tg was impacted by confinement, in the range 130 nm ≤ d ≤ 400 nm, in stark contrast, the cooperative segmental mobility, i.e., the peak position of the α-relaxation process was not. Furthermore, when d ≤ 200 nm, an additional molecular relaxation process, not observed in bulk, was present. We interpret these findings as evidence of a decoupling between Tg and cooperative segmental mobility in nanospheres. That is, the latter may be impacted by confinement under conditions in which the former is not.
Co-reporter:Jae Woo Chung, Yunlong Guo, Seung-Yeop Kwak and Rodney D. Priestley
Journal of Materials Chemistry A 2012 vol. 22(Issue 13) pp:6017-6026
Publication Date(Web):16 Feb 2012
DOI:10.1039/C2JM16118D
Recently, we discovered that cyclodextrin (CD)-stabilized gold nanoparticles could be synthesized in an aqueous medium from a self-assembled supramolecular structure of CD and gold salt. We showed that the self-assembled structure of the CD complex induced by the gold salt acted as a solid template for the formation of nanoconfined gold seeds and that gold seeds grew into CD-stabilized gold nanoparticles in water without the necessity of other reducing or stabilizing agents. Here, we extensively investigate the supramolecular self-assembled structure of the CD/gold salt complex under various synthetic conditions, the mechanism of the α-CD-stabilized gold nanoparticles formation, and the processing parameters for controlling the size of gold nanoparticles. We demonstrate that gold salts were confined between two different crystalline phases of the supramolecular CD solid template via a gold salt-induced molecular self-assembly process and that thermal treatment of the CD/gold salt complex led to the formation of nanosized gold seeds geometrically confined within the crystalline interface region. Placement of the thermally treated complex in water without the addition of any supplementary additives proliferated the growth of CD-stabilized gold nanoparticles via stabilization of the growing gold seed intermediates by CD molecules. In addition, various processing parameters such as Au salt concentration are shown to affect the size of AuNPs.
Co-reporter:Chuan Zhang;Jae Woo Chung
Macromolecular Rapid Communications 2012 Volume 33( Issue 20) pp:1798-1803
Publication Date(Web):
DOI:10.1002/marc.201200335
Abstract
Using a facile dialysis nanoprecipitation method, nanoparticles of several hundred nanometers have been successfully generated from a “traditional,” non-biodegradable polymer, that is, polystyrene. The effect of initial polymer concentration inside the dialysis membrane, as well as the polymer/solvent system and the ionic strength (electrolyte concentration) of the dialysis solution, on nanoparticle size is examined. A nucleation-aggregation mechanism has been provided to explain the observed trends. Furthermore, we determine the zeta potential as a function of ionic strength for the generated nanoparticles and show that anionic charging may be present in the system.
Co-reporter:Chuan Zhang, Vikram J. Pansare, Robert K. Prud'homme and Rodney D. Priestley
Soft Matter 2012 vol. 8(Issue 1) pp:86-93
Publication Date(Web):06 Oct 2011
DOI:10.1039/C1SM06182H
Aside from polymerization techniques, polymer nanoparticles can be generated through the displacement of a solvent with a nonsolvent, i.e., nanoprecipitation. In this study, we utilize a facile process termed Flash NanoPrecipitation (FNP) to generate polystyrene (PS) nanoparticles of several different molecular weights. As compared to PS nanoparticles synthesized by surfactant free emulsion polymerization, nanoparticles prepared by FNP show comparable size distributions when the diameter is less than 150 nm. Furthermore, we illustrate that the sizes of PS nanoparticles prepared by FNP can be fine-tuned by changing the polymer and/or electrolyte concentration. The stabilized nanoparticles contain only the radically polymerized polymer chains, which have sulfate anions at the chain termini and no additional external stabilizers. Calculations of the mechanism of particle formation and stabilization show that the size-dependent electrostatic repulsions between nanoparticles and single collapsed polymer chains control assembly and monodispersity. The ability to independently vary polymer molecular weight and nanoparticle size will enable fundamental studies of the effect of confinement on polymer dynamics in a way not easily achievable by other techniques.
Co-reporter:Chuan Zhang, Yunlong Guo, and Rodney D. Priestley
Macromolecules 2011 Volume 44(Issue 10) pp:4001-4006
Publication Date(Web):April 19, 2011
DOI:10.1021/ma1026862
When confined to the nanoscale, the glass transition temperature (Tg) of polymer films can deviate substantially from the bulk, i.e., the Tg-confinement effect. Due to ease of processing most studies have focused on the thickness-dependent Tg of thin films, while few have focused on extending investigations beyond thin films to other geometries. As polymers confined to higher geometrical dimensionalities become the enabling material in technologies ranging from drug delivery to plastic electronics to ultrafiltration, a greater understanding of size effects on the Tg is warranted. Here, we investigate the effects of three-dimensional confinement on the Tg of polymer nanoparticles under soft and hard confinement and quantitatively compare our results to those of thin films to explore commonalities or differences between the Tg-confinement effect for polymers confined to different geometries. Via modulated differential scanning calorimetry, we show that Tg decreases with size for polystyrene (PS) nanoparticles suspended in an aqueous solution, in agreement with the corresponding freestanding films. Furthermore, capping of PS nanoparticles with a hard silica shell leads to a size invariant Tg. These results suggest that the free surface is a key factor in Tg reductions of confined polymer, irrespective of geometry.
Co-reporter:Yunlong Guo, Chuan Zhang, Christine Lai, Rodney D. Priestley, Maria D’Acunzi, and George Fytas
ACS Nano 2011 Volume 5(Issue 7) pp:5365
Publication Date(Web):June 30, 2011
DOI:10.1021/nn201751m
We have measured the glassy-state structural relaxation of aqueous suspended polystyrene (PS) nanoparticles (the case of soft confinement) and the corresponding silica-capped PS nanoparticles (the case of hard confinement) via differential scanning calorimetry. Suspended and capped PS nanoparticles undergo physical aging under isobaric and isochoric conditions, respectively. With decreasing diameter, suspended and capped PS nanoparticles exhibited reduced and bulk glass transition temperatures (Tg), respectively. To account for Tg changes with confinement, all physical aging measurements were performed at a constant value of Tg – Ta, where Ta is the aging temperature. With decreasing diameter, aqueous suspended PS nanoparticles exhibited enhanced physical aging rates in comparison to bulk PS. Due to differences in thermodynamic conditions during aging and interfacial effects from nanoconfinement, at all values of Tg – Ta investigated, capped PS nanoparticles aged at reduced rates compared to the corresponding aqueous suspended PS nanoparticles. We captured the physical aging behavior of all nanoparticles via the Tool, Narayanaswamy, and Moynihan model of structural relaxation.Keywords: confinement; core−shell nanoparticles; glass transition; nanoparticles; physical aging; structural relaxation; TNM model
Co-reporter:Rui Liu, Fengli Qu, Yunlong Guo, Nan Yao and Rodney D. Priestley
Chemical Communications 2014 - vol. 50(Issue 4) pp:NaN480-480
Publication Date(Web):2013/11/21
DOI:10.1039/C3CC47050D
A facile one-step Stöber route to synthesize high-quality core–shell–shell templates is reported for the fabrication of Au@carbon yolk–shell nanostructures. The converted Au@carbon yolk–shell nanostructures exhibited high catalytic performance as illustrated by the reduction reaction of o-nitrophenol.
Co-reporter:Jae Woo Chung, Yunlong Guo, Seung-Yeop Kwak and Rodney D. Priestley
Journal of Materials Chemistry A 2012 - vol. 22(Issue 13) pp:
Publication Date(Web):
DOI:10.1039/C2JM16118D
Co-reporter:Rui Liu, Chris Sosa, Yao-Wen Yeh, Fengli Qu, Nan Yao, Robert K. Prud'homme and Rodney D. Priestley
Journal of Materials Chemistry A 2014 - vol. 2(Issue 41) pp:NaN17290-17290
Publication Date(Web):2014/09/22
DOI:10.1039/C4TA04036H
We report a scalable and continuous preparation route for metal nanoparticles (Au or Pt)@nanosphere polymer composites through flash nanoprecipitation (FNP). Uniform metal@nanosphere polymer composites with 2–3 nm metal colloids decorating the surface on the nanosphere are obtained with tunable overall particle size and metal nanoparticle arrangement. The obtained composites show high catalytic ability and stability in the reduction of p-nitrophenol.
Co-reporter:Rui Liu and Rodney D. Priestley
Journal of Materials Chemistry A 2016 - vol. 4(Issue 18) pp:NaN6692-6692
Publication Date(Web):2016/02/05
DOI:10.1039/C5TA09607C
Core–shell nanoparticles (NPs) have emerged as a type of important nanomaterial for various applications. The challenge in the preparation of core–shell NPs is to find a simple, cost-effective and less time-consuming strategy with minimum environmental impact. The consolidation of multiple preparation steps into one step represents a new green synthesis pathway in chemistry and materials science. In this review, we provide an overview of the recent developments in the fabrication of core–shell NPs through one-step/pot methodologies. A variety of one-step/pot preparation methods are presented, discussed and compared, followed by the summary and outlook of this emerging area.
Co-reporter:Rui Liu, Yao-Wen Yeh, Vivienne H. Tam, Fengli Qu, Nan Yao and Rodney D. Priestley
Chemical Communications 2014 - vol. 50(Issue 65) pp:NaN9059-9059
Publication Date(Web):2014/05/29
DOI:10.1039/C4CC02507E
A facile one-pot Stöber route is used to synthesize high-quality Ag, AgBr–silica–resorcinol formaldehyde polymer core–shell–shell nanospheres. The obtained core–shell–shell templates can be converted to Ag@carbon yolk–shell nanostructures with tunable dimensions.

![2-Propenoic acid, 2-methyl-, 2-[[[(1,6-dihydro-4-methyl-6-oxo-2-pyrimidinyl)amino]carbonyl]amino]ethyl ester](/data/chemimg/3782000/474434-71-4.png)
![2-Propenoic acid, 2-methyl-, 2-[[[(1,6-dihydro-4-methyl-6-oxo-2-pyrimidinyl)amino]carbonyl]amino]ethyl ester](/data/chemimg/3782000/474434-71-4_b.png)




![Acetyl chloride, [(2-oxo-2H-1-benzopyran-7-yl)oxy]-](http://img.cochemist.com/ccimg/125600/125572-56-7.png)
![Acetyl chloride, [(2-oxo-2H-1-benzopyran-7-yl)oxy]-](http://img.cochemist.com/ccimg/125600/125572-56-7_b.png)




![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4.png)
![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4_b.png)