Co-reporter:Katie L. Strong, Matthew P. Epplin, John Bacsa, Christopher J. Butch, Pieter B. Burger, David S. Menaldino, Stephen F. Traynelis, and Dennis C. Liotta
Journal of Medicinal Chemistry July 13, 2017 Volume 60(Issue 13) pp:5556-5556
Publication Date(Web):June 6, 2017
DOI:10.1021/acs.jmedchem.7b00239
We have identified a series of positive allosteric NMDA receptor (NMDAR) modulators derived from a known class of GluN2C/D-selective tetrahydroisoquinoline analogues that includes CIQ. The prototypical compound of this series contains a single isopropoxy moiety in place of the two methoxy substituents present in CIQ. Modifications of this isopropoxy-containing scaffold led to the identification of analogues with enhanced activity at the GluN2B subunit. We identified molecules that potentiate the response of GluN2B/GluN2C/GluN2D, GluN2B/GluN2C, and GluN2C/GluN2D-containing NMDARs to maximally effective concentrations of agonist. Multiple compounds potentiate the response of NMDARs with submicromolar EC50 values. Analysis of enantiomeric pairs revealed that the S-(−) enantiomer is active at the GluN2B, GluN2C, and/or GluN2D subunits, whereas the R-(+) enantiomer is only active at GluN2C/D subunits. These results provide a starting point for the development of selective positive allosteric modulators for GluN2B-containing receptors.
Co-reporter:Dennis C. Liotta and George R. Painter
Accounts of Chemical Research 2016 Volume 49(Issue 10) pp:2091
Publication Date(Web):October 5, 2016
DOI:10.1021/acs.accounts.6b00274
The HIV/AIDS epidemic, which was first reported on in 1981, progressed in just 10 years to a disease afflicting 10 million people worldwide including 1 million in the US. In 1987, AZT was approved for treating HIV/AIDS. Unfortunately, its clinical usefullness was severly limited by associated toxicities and the emergence of resistance. Three other drugs that were approved in the early 1990s suffered from similar liabilities.In 1990, the Liotta group at Emory University developed a highly diastereoselective synthesis of racemic 3′-thia-2′,3′-dideoxycytidine and 3′-thia-2′,3′-5-fluorodideoxycytidine and demonstrated that these compounds exhibited excellent anti-HIV activity with no apparent cytotoxicity. Subsequently, the enantiomers of these compounds were separated using enzyme-mediated kinetic resolutions and their (−)-enantiomers (3TC and FTC, respectively) were found to have exceptionally attractive preclinical profiles. In addition to their anti-HIV activity, 3TC and FTC potently inhibit the replication of hepatitis B virus.The development of FTC, which was being carried out by Burroughs Wellcome, had many remarkable starts and stops. For example, passage studies indicated that the compound rapidly selected for a single resistant mutant, M184V, and that this strain was 500–1000-fold less sensitive to FTC than was wild-type virus. Fortunately, it was found that combinations of AZT with either 3TC or FTC were synergistic. The effectiveness of AZT–3TC combination therapy was subsequently demonstrated in four independent clinical trials, and in 1997, the FDA approved Combivir, a fixed dose combination of AZT and 3TC.In phase 1 clinical trials, FTC was well tolerated by all subjects with no adverse events observed. However, the development of FTC was halted by the aquistition of Wellcome PLC by Glaxo PLC in January 1995. In 1996, Triangle Pharmaceuticals licensed FTC from Emory and initiated a series of phase I/II clinical studies that demonstrated the safety and efficacy of the drug. In August 1998, FTC was granted “Fast Track” status, based primarily on its potential for once daily dosing. While the outcomes of two subsequent phase III trials were positive, a third phase III clinical trial involving combinations of 3TC or FTC with stavudine and neviripine had to be terminated due to serious liver-related adverse events. Although analysis of the data suggested that the liver toxicity was due to neviripine, the FDA decided that the study could not be used for drug registration. Ultimately, in January 2003, Gilead Sciences acquired Triangle Pharmaceuticals and completed the development of FTC (emtricitabine), which was approved for once a day, oral administration in July 2003. A year later, Truvada, a once a day, oral, fixed dose combination of emtricitabine and tenofovir disoproxyl fumarate received FDA approval and quickly became the accepted first line therapy when used with a third antiretroviral agent. In July 2006, the FDA approved Atripla, a once a day, oral, fixed dose combination of emtricitabine, tenofovir disoproxyl fumarate, and efavirenz, which represented the culmination of two decades of research that had transformed AIDS from a death sentence to a manageable chronic disease.
Co-reporter:Kyle E. Giesler, Jose Marengo, and Dennis C. Liotta
Journal of Medicinal Chemistry 2016 Volume 59(Issue 15) pp:7097-7110
Publication Date(Web):July 13, 2016
DOI:10.1021/acs.jmedchem.6b00428
The therapeutic value of numerous small molecules hinges on their ability to permeate the plasma membrane. This is particularly true for tenofovir (TFV), adefovir, and other antiviral nucleosides that demonstrate potent antiviral activity but poor bioavailability. Using TFV as a model substrate, we hybridized two disparate prodrug strategies to afford novel reduction-sensitive lipid conjugates of TFV that exhibit subnanomolar activity toward HIV-1 and are stable in human plasma for more than 24 h with a therapeutic index approaching 30000. These compounds significantly rival the clinically approved formulation of TFV and revitalize the potential of disulfide-bearing prodrugs which have seen limited in vitro and in vivo success since their debut over 20 years ago. We further demonstrate the utility of these conjugates as a tool to indirectly probe the enzymatic hydrolysis of phosphonomonoesters that may further advance the development of other prodrug strategies for nucleosides, peptides, and beyond.
Co-reporter:Kyle E. Giesler and Dennis C. Liotta
Journal of Medicinal Chemistry 2016 Volume 59(Issue 22) pp:10244-10252
Publication Date(Web):October 19, 2016
DOI:10.1021/acs.jmedchem.6b01292
The pharmacokinetic properties of tenofovir (TFV) and other charged nucleoside analogues are dramatically improved upon conjugation to a lipid prodrug. We previously prepared reduction-sensitive lipid conjugates of TFV that demonstrate superior antiviral activity compared to other lipid conjugates including the clinically approved formulation, tenofovir disoproxil fumarate (TDF). In continuation of that work, we have synthesized next-generation conjugates with reduced cytotoxicity that retain potent antiviral activity against HIV-1 and HBV with a therapeutic index >100000 for our most potent conjugate. We also show that disulfide reduction is not responsible for prodrug cleavage unless 3-exo-tet intramolecular cyclization can occur, suggesting that enzymatic hydrolysis is predominantly responsible for activity of our prodrugs in vitro.
Co-reporter:Eric J. Miller, Suzanne G. Mays, Mark T. Baillie, Randy B. Howard, Deborah G. Culver, Manohar Saindane, Sarah T. Pruett, Jason J. Holt, David S. Menaldino, Taylor J. Evers, G. Prabhakar Reddy, Richard F. Arrendale, Michael G. Natchus, John A. Petros, and Dennis C. Liotta
ACS Medicinal Chemistry Letters 2016 Volume 7(Issue 5) pp:537
Publication Date(Web):March 21, 2016
DOI:10.1021/acsmedchemlett.6b00113
The orally bioavailable 1-deoxy-sphingosine analog, Enigmol, has demonstrated anticancer activity in numerous in vivo settings. However, as no Enigmol analog with enhanced potency in vitro has been identified, a new strategy to improve efficacy in vivo by increasing tumor uptake was adopted. Herein, synthesis and biological evaluation of two novel fluorinated Enigmol analogs, CF3-Enigmol and CF2-Enigmol, are reported. Each analog was equipotent to Enigmol in vitro, but achieved higher plasma and tissue levels than Enigmol in vivo. Although plasma and tissue exposures were anticipated to trend with fluorine content, CF2-Enigmol absorbed into tissue at strikingly higher concentrations than CF3-Enigmol. Using mouse xenograft models of prostate cancer, we also show that CF3-Enigmol underperformed Enigmol-mediated inhibition of tumor growth and elicited systemic toxicity. By contrast, CF2-Enigmol was not systemically toxic and demonstrated significantly enhanced antitumor activity as compared to Enigmol.Keywords: Enigmol; fluorination; prostate cancer; Sphingolipids
Co-reporter:Bryan D. Cox, Anthony R. Prosser, Yongnian Sun, Zhufang Li, Sangil Lee, Ming B. Huang, Vincent C. Bond, James P. Snyder, Mark Krystal, Lawrence J. Wilson, and Dennis C. Liotta
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 7) pp:753
Publication Date(Web):May 6, 2015
DOI:10.1021/acsmedchemlett.5b00036
We report novel anti-HIV-1 agents with combined dual host–pathogen pharmacology. Lead compound 3, composed of a pyrazole-piperidine core, exhibits three concurrent mechanisms of action: (1) non-nucleoside reverse transcriptase inhibition, (2) CCR5-mediated M-tropic viral entry inhibition, and (3) CXCR4-based T-tropic viral entry inhibition that maintains native chemokine ligand binding. This discovery identifies important tool compounds for studying viral infectivity and prototype agents that block HIV-1 entry through dual chemokine receptor ligation.Keywords: chemokine receptors; HIV-1; multitarget drug discovery; non-nucleoside reverse transcriptase inhibitors; viral entry inhibitors
Co-reporter:Anthony R. Prosser, Dennis C. Liotta
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:3005-3007
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2014.10.024
A convenient, single-pot protocol for the transformation of esters into analytically pure ketones is described herein. This method circumvents the need for purification and affords near quantitative yields for all substrates investigated. As a test of its utility, the method is used to improve the yields of a process for preparing a pharmacologically relevant anti-HIV intermediate by nearly 10 fold.
Co-reporter:Brooke M. Katzman, Riley E. Perszyk, Hongjie Yuan, Yesim Altas Tahirovic, Ayodeji E. Sotimehin, Stephen F. Traynelis, Dennis C. Liotta
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 23) pp:5583-5588
Publication Date(Web):1 December 2015
DOI:10.1016/j.bmcl.2015.10.046
NMDA receptors mediate a slow Ca2+-permeable component of excitatory synaptic transmission, and are involved in numerous normal brain functions including learning and memory. NMDA receptor over-activation can lead to cell death and abnormal excitation in ischemia associated with stroke, traumatic brain injury, and epilepsy. We have explored a series of novel noncompetitive allosteric modulators of NMDA receptor function characterized by an iminothiazolidinone ring. Saturating concentrations of these compounds inhibit NMDA receptors to varying maximal extents, raising the possibility that they may attenuate over-activation in pathological situations while preserving some minimal receptor function, which may limit side-effects. The best in class compounds have sub-micromolar IC50 values and show modest preference for GluN2C- and GluN2D-containing receptors.
Co-reporter:Sommer S. Zimmerman ; Alpa Khatri ; Ethel C. Garnier-Amblard ; Praseeda Mullasseril ; Natalie L. Kurtkaya ; Stefka Gyoneva ; Kasper B. Hansen ; Stephen F. Traynelis
Journal of Medicinal Chemistry 2014 Volume 57(Issue 6) pp:2334-2356
Publication Date(Web):February 10, 2014
DOI:10.1021/jm401695d
NMDA receptors are tetrameric complexes composed of GluN1 and GluN2A–D subunits that mediate a slow Ca2+-permeable component of excitatory synaptic transmission. NMDA receptors have been implicated in a wide range of neurological diseases and thus represent an important therapeutic target. We herein describe a novel series of pyrrolidinones that selectively potentiate only NMDA receptors that contain the GluN2C subunit. The most active analogues tested were over 100-fold selective for recombinant GluN2C-containing receptors over GluN2A/B/D-containing NMDA receptors as well as AMPA and kainate receptors. This series represents the first class of allosteric potentiators that are selective for diheteromeric GluN2C-containing NMDA receptors.
Co-reporter:Timothy M. Acker ; Alpa Khatri ; Katie M. Vance ; Cathryn Slabber ; John Bacsa ; James P. Snyder ; Stephen F. Traynelis
Journal of Medicinal Chemistry 2013 Volume 56(Issue 16) pp:6434-6456
Publication Date(Web):August 2, 2013
DOI:10.1021/jm400652r
Here we describe the synthesis and structure–activity relationship for a class of pyrazoline-containing dihydroquinolone negative allosteric modulators of the NMDA receptor that show strong subunit selectivity for GluN2C- and GluN2D-containing receptors over GluN2A- and GluN2B-containing receptors. Several members of this class inhibit NMDA receptor responses in the nanomolar range and are more than 50-fold selective over GluN1/GluN2A and GluN1/GluN2B NMDA receptors, as well as AMPA, kainate, GABA, glycine, nicotinic, serotonin, and purinergic receptors. Analysis of the purified enantiomers of one of the more potent and selective compounds shows that the S-enantiomer is both more potent and more selective than the R-enantiomer. The S-enantiomer had an IC50 of 0.17–0.22 μM at GluN2D- and GluN2C-containing receptors, respectively, and showed over 70-fold selectivity over other NMDA receptor subunits. The subunit selectivity of this class of compounds should be useful in defining the role of GluN2C- and GluN2D-containing receptors in specific brain circuits in both physiological and pathophysiological conditions.
Co-reporter:Rose M. Santangelo Freel ; Kevin K. Ogden ; Katie L. Strong ; Alpa Khatri ; Kathryn M. Chepiga ; Henrik S. Jensen ; Stephen F. Traynelis
Journal of Medicinal Chemistry 2013 Volume 56(Issue 13) pp:5351-5381
Publication Date(Web):April 29, 2013
DOI:10.1021/jm400177t
We describe here the synthesis and evaluation of a series of tetrahydroisoquinolines that show subunit-selective potentiation of NMDA receptors containing the GluN2C or GluN2D subunits. Bischler–Napieralski conditions were employed in the key step for the conversion of acyclic amides to the corresponding tetrahydroisoquinoline-containing analogs. Compounds were evaluated using both two-electrode voltage clamp recordings from Xenopus laevis oocytes and imaging of mammalian BHK cells loaded with Ca2+-sensitive dyes. The most potent analogues had EC50 values of 300 nM and showed over 2-fold potentiation of the response to maximally effective concentrations of glutamate and glycine but had no effect on responses from NMDA receptors containing the GluN2A or GluN2B subunits AMPA, kainate, and GABA or glycine receptors or a variety of other potential targets. These compounds represent a potent class of small molecule subunit-selective potentiators of NMDA receptors.
Co-reporter:Ethel C. Garnier-Amblard, Suzanne G. Mays, Richard F. Arrendale, Mark T. Baillie, Anatoliy S. Bushnev, Deborah G. Culver, Taylor J. Evers, Jason J. Holt, Randy B. Howard, Lanny S. Liebeskind, David S. Menaldino, Michael G. Natchus, John A. Petros, Harsha Ramaraju, G. Prabhakar Reddy, and Dennis C. Liotta
ACS Medicinal Chemistry Letters 2011 Volume 2(Issue 6) pp:438
Publication Date(Web):March 25, 2011
DOI:10.1021/ml2000164
Enigmol is a synthetic, orally active 1-deoxysphingoid base analogue that has demonstrated promising activity against prostate cancer. In these studies, the pharmacologic roles of stereochemistry and N-methylation in the structure of enigmols were examined. A novel enantioselective synthesis of all four possible 2S-diastereoisomers of enigmol (2-aminooctadecane-3,5-diols) from l-alanine is reported, which features a Liebeskind−Srogl cross-coupling reaction between l-alanine thiol ester and (E)-pentadec-1-enylboronic acid as the key step. In vitro biological evaluation of the four enigmol diastereoisomers and 2S,3S,5S-N-methylenigmol against two prostate cancer cell lines (PC-3 and LNCaP) indicates that all but one diastereomer demonstrate potent oncolytic activity. In nude mouse xenograft models of human prostate cancer, enigmol was equally effective as standard prostate cancer therapies (androgen deprivation or docetaxel), and two of the enigmol diastereomers, 2S,3S,5R-enigmol and 2S,3R,5S-enigmol, also caused statistically significant inhibition of tumor growth. A pharmacokinetic profile of enigmol and N-methylenigmol is also presented.Keywords: 1-deoxysphingoid bases; Enigmol; Liebeskind−Srogl reaction; LNCaP; palladium cross-coupling; PC-3; prostate cancer therapy
Co-reporter:Aizhi Zhu ; Weiqiang Zhan ; Zhongxing Liang ; Younghyoun Yoon ; Hua Yang ; Hans E. Grossniklaus ; Jianguo Xu ; Mauricio Rojas ; Mark Lockwood ; James P. Snyder ; Dennis C. Liotta ;Hyunsuk Shim
Journal of Medicinal Chemistry 2010 Volume 53(Issue 24) pp:8556-8568
Publication Date(Web):November 24, 2010
DOI:10.1021/jm100786g
The C-X-C chemokine receptor type 4 (CXCR4)/stromal cell derived factor-1 (SDF-1 or CXCL12) interaction and the resulting cell signaling cascade play a key role in metastasis and inflammation. On the basis of the previously published CXCR4 antagonist 5 (WZ811), a series of novel nonpeptidic anti-CXCR4 small molecules have been designed and synthesized to improve potency. Following a structure−activity profile around 5, more advanced compounds in the N,N′-(1, 4-phenylenebis(methylene)) dipyrimidin-2-amines series were discovered and shown to possess higher CXCR4 binding potential and specificity than 5. Compound 26 (508MCl) is the lead compound and exhibits subnanomolar potency in three in vitro assays including competitive binding, Matrigel invasion and Gαi cyclic adenosine monophosphate (cAMP) modulation signaling. Furthermore, compound 26 displays promising effects by interfering with CXCR4 function in three mouse models: paw inflammation, Matrigel plug angiogenesis, and uveal melanoma micrometastasis. These data demonstrate that dipyrimidine amines are unique CXCR4 antagonists with high potency and specificity.
Co-reporter:Cara A. Mosley ; Timothy M. Acker ; Kasper B. Hansen ; Praseeda Mullasseril ; Karen T. Andersen ; Phuong Le ; Kimberly M. Vellano ; Hans Bräuner-Osborne ; Dennis C. Liotta ;Stephen F. Traynelis
Journal of Medicinal Chemistry 2010 Volume 53(Issue 15) pp:5476-5490
Publication Date(Web):July 13, 2010
DOI:10.1021/jm100027p
We describe a new class of subunit-selective antagonists of N-methyl d-aspartate (NMDA)-selective ionotropic glutamate receptors that contain the (E)-3-phenyl-2-styrylquinazolin-4(3H)-one backbone. The inhibition of recombinant NMDA receptor function induced by these quinazolin-4-one derivatives is noncompetitive and voltage-independent, suggesting that this family of compounds does not exert action on the agonist binding site of the receptor or block the channel pore. The compounds described here resemble CP-465,022 ((S)-3-(2-chlorophenyl)-2-[2-(6-diethylaminomethyl-pyridin-2-yl)-vinyl]-6-fluoro-3H-quinazolin-4-one), a noncompetitive antagonist of AMPA-selective glutamate receptors. However, modification of ring substituents resulted in analogues with greater than 100-fold selectivity for recombinant NMDA receptors over AMPA and kainate receptors. Furthermore, within this series of compounds, analogues were identified with 50-fold selectivity for recombinant NR2C/D-containing receptors over NR2A/B containing receptors. These compounds represent a new class of noncompetitive subunit-selective NMDA receptor antagonists.
Co-reporter:Christopher J. MacNevin ; Fahim Atif ; Iqbal Sayeed ; Donald G. Stein
Journal of Medicinal Chemistry 2009 Volume 52(Issue 19) pp:6012-6023
Publication Date(Web):September 8, 2009
DOI:10.1021/jm900712n
Preclinical and clinical research findings have revealed that the hormone progesterone, when acutely administered, can dramatically reduce cerebral edema, inflammation, tissue necrosis, and programmed cell death following traumatic brain injury (TBI). The poor aqueous solubility of progesterone, however, limits its potential use as a therapeutic. Several chemically novel analogues of progesterone and its natural metabolite allopregnanolone have been synthesized and screened using both in vitro and whole animal models of TBI. The new derivatives demonstrated greatly improved solubility and select compounds have shown equivalent effectiveness to progesterone in reducing cerebral edema after TBI.
Co-reporter:Cara A. Mosley, Scott J. Myers, Ernest E. Murray, Rose Santangelo, Yesim A. Tahirovic, Natalie Kurtkaya, Praseeda Mullasseril, Hongjie Yuan, Polina Lyuboslavsky, Phuong Le, Lawrence J. Wilson, Manuel Yepes, Ray Dingledine, Stephen F. Traynelis, Dennis C. Liotta
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 17) pp:6463-6480
Publication Date(Web):1 September 2009
DOI:10.1016/j.bmc.2009.05.085
The synthesis and structure–activity relationship analysis of a novel class of amide-based biaryl NR2B-selective NMDA receptor antagonists are presented. Some of the studied compounds are potent, selective, non-competitive, and voltage-independent antagonists of NR2B-containing NMDA receptors. Like the founding member of this class of antagonists (ifenprodil), several interesting compounds of the series bind to the amino terminal domain of the NR2B subunit to inhibit function. Analogue potency is modulated by linker length, flexibility, and hydrogen bonding opportunities. However, unlike previously described classes of NR2B-selective NMDA antagonists that exhibit off-target activity at a variety of monoamine receptors, the compounds described herein show much diminished effects against the hERG channel and α1-adrenergic receptors. Selections of the compounds discussed have acceptable half-lives in vivo and are predicted to permeate the blood–brain barrier. These data together suggest that masking charged atoms on the linker region of NR2B-selective antagonists can decrease undesirable side effects while still maintaining on-target potency.The synthesis and biological activity of a novel series of amide-based NR2B-selective NMDA receptor antagonists is detailed. The compounds herein represent a family of compounds with excellent on-target potency and decreased off-target effects when compared with previously described antagonists.
Co-reporter:Yongfeng Li, Shuli Mao, Michael W. Hager, Kimberlynne D. Becnel, Raymond F. Schinazi, Dennis C. Liotta
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 12) pp:3398-3401
Publication Date(Web):15 June 2007
DOI:10.1016/j.bmcl.2007.03.094
A series of 2′-substituted cyclobutyl nucleoside analogs were efficiently prepared by constructing the core cyclobutyl ring using different [2+2] cycloaddition approaches. The triphosphate derivative of a cyclobutyl nucleoside was also synthesized and evaluated against wild-type and mutant HIV reverse transcriptases (RT). Whereas the nucleoside analogs were inactive against HIV-1 in culture, the nucleotide showed good activity not only against wild-type and recombinant HIV RT (IC50 = 4.7, 6.9 μM), but also against the M184I and M184V mutants (IC50 = 6.1, 6.9 μM) in cell-free assays.Several 2′-substituted cyclobutyl nucleosides were synthesized and evaluated as anti-HIV agents. Whereas the cyclobutyl nucleosides were not active against HIV in culture, the triphosphate forms were quite active against wild-type and mutant forms of HIV reverse transcriptase (RT).
Co-reporter:Shuli Mao, Martin Bouygues, Christopher Welch, Mirlinda Biba, Jen Chilenski, Raymond F. Schinazi, Dennis C. Liotta
Bioorganic & Medicinal Chemistry Letters 2004 Volume 14(Issue 19) pp:4991-4994
Publication Date(Web):4 October 2004
DOI:10.1016/j.bmcl.2004.07.016
The β-d-enantiomer of FDOC (2′,3′-dideoxy-5-fluoro-oxacytidine) exhibits potent anti-HIV-1 activity. It was obtained in optically pure form by employing a tandem kinetic resolution/chiral salt crystallization protocol. In addition, conditions were developed that allowed the unwanted butyrate ester of the l-enantiomer of FDOC to be racemized. This material could then be recycled in future resolutions.The d-enantiomer of FDOC was obtained in optically pure form via a tandem kinetic resolution/chiral salt crystallization protocol. In addition, conditions were developed that allowed the unwanted l-enantiomer to be racemized and recycled.
Co-reporter:Anlys Olivera, Terry W. Moore, Fang Hu, Andrew P. Brown, Aiming Sun, Dennis C. Liotta, James P. Snyder, Younghyoun Yoon, Hyunsuk Shim, Adam I. Marcus, Andrew H. Miller, Thaddeus W.W. Pace
International Immunopharmacology (February 2012) Volume 12(Issue 2) pp:368-377
Publication Date(Web):1 February 2012
DOI:10.1016/j.intimp.2011.12.009
Nuclear factor kappa B (NF-κB) is a key signaling molecule in the elaboration of the inflammatory response. Data indicate that curcumin, a natural ingredient of the curry spice turmeric, acts as a NF-κB inhibitor and exhibits both anti-inflammatory and anti-cancer properties. Curcumin analogs with enhanced activity on NF-κB and other inflammatory signaling pathways have been developed including the synthetic monoketone compound 3,5-Bis(2-fluorobenzylidene)-4-piperidone (EF24). 3,5-Bis(2-pyridinylmethylidene)-4-piperidone (EF31) is a structurally-related curcumin analog whose potency for NF-κB inhibition has yet to be determined. To examine the activity of EF31 compared to EF24 and curcumin, mouse RAW264.7 macrophages were treated with EF31, EF24, curcumin (1–100 μM) or vehicle (DMSO 1%) for 1 h. NF-κB pathway activity was assessed following treatment with lipopolysaccharide (LPS) (1 μg/mL). EF31 (IC50 ~ 5 μM) exhibited significantly more potent inhibition of LPS-induced NF-κB DNA binding compared to both EF24 (IC50 ~ 35 μM) and curcumin (IC50 > 50 μM). In addition, EF31 exhibited greater inhibition of NF-κB nuclear translocation as well as the induction of downstream inflammatory mediators including pro-inflammatory cytokine mRNA and protein (tumor necrosis factor-α, interleukin-1β, and interleukin-6). Regarding the mechanism of these effects on NF-κB, EF31 (IC50 ~ 1.92 μM) exhibited significantly greater inhibition of IκB kinase β compared to EF24 (IC50 ~ 131 μM). Finally, EF31 demonstrated potent toxicity in NF-κB-dependent cancer cell lines while having minimal and reversible toxicity in RAW264.7 macrophages. These data indicate that EF31 is a more potent inhibitor of NF-κB activity than either EF24 or curcumin while exhibiting both anti-inflammatory and anticancer activities. Thus, EF31 represents a promising curcumin analog for further therapeutic development.Highlights► EF31 is a potent inhibitor of Iκβ kinase, NF-κB translocation, and NF-κB DNA-binding activity in RAW264.7 macrophages. ► EF31 blocks the expression of pro-inflammatory cytokine mRNA and protein. ► EF31 inhibits MAPK transcription factors. ► EF31 exhibits potent anti-cancer activity in various cancer cell lines.
.jpg)






![6,7-Dimethoxy-1-[(4-nitrophenoxy)methyl]-1,2,3,4-tetrahydroisoquinoline](http://img.cochemist.com/ccimg/887300/887201-39-0.png)
![6,7-Dimethoxy-1-[(4-nitrophenoxy)methyl]-1,2,3,4-tetrahydroisoquinoline](http://img.cochemist.com/ccimg/887300/887201-39-0_b.png)
![Benzeneacetamide, N-[2-(aminocarbonyl)-5-nitrophenyl]-](http://img.cochemist.com/ccimg/865900/865837-21-4.png)
![Benzeneacetamide, N-[2-(aminocarbonyl)-5-nitrophenyl]-](http://img.cochemist.com/ccimg/865900/865837-21-4_b.png)
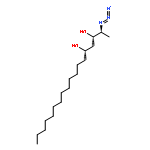
![1,3-Dioxan-2-one, 4-[(1S)-1-hydroxyethyl]-6-tridecyl-, (4R,6S)-](http://img.cochemist.com/ccimg/862500/862472-88-6.png)
![1,3-Dioxan-2-one, 4-[(1S)-1-hydroxyethyl]-6-tridecyl-, (4R,6S)-](http://img.cochemist.com/ccimg/862500/862472-88-6_b.png)

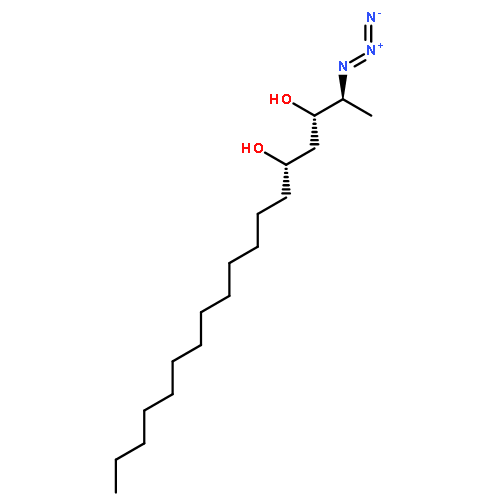
![1,3-DIOXAN-2-ONE, 4-[(1S)-1-AZIDOETHYL]-6-TRIDECYL-, (4S,6R)-](http://img.cochemist.com/ccimg/862500/862472-81-9.png)
![1,3-DIOXAN-2-ONE, 4-[(1S)-1-AZIDOETHYL]-6-TRIDECYL-, (4S,6R)-](http://img.cochemist.com/ccimg/862500/862472-81-9_b.png)
![1,3-Dioxan-2-one, 4-[(1R)-1-hydroxyethyl]-6-tridecyl-, (4S,6R)-](http://img.cochemist.com/ccimg/862500/862472-80-8.png)
![1,3-Dioxan-2-one, 4-[(1R)-1-hydroxyethyl]-6-tridecyl-, (4S,6R)-](http://img.cochemist.com/ccimg/862500/862472-80-8_b.png)
![1,3-DIOXAN-2-ONE, 4-[(1S)-1-AZIDOETHYL]-6-TRIDECYL-, (4S,6S)-](http://img.cochemist.com/ccimg/862500/862472-77-3.png)
![1,3-DIOXAN-2-ONE, 4-[(1S)-1-AZIDOETHYL]-6-TRIDECYL-, (4S,6S)-](http://img.cochemist.com/ccimg/862500/862472-77-3_b.png)
![1,3-DIOXAN-2-ONE, 4-[(1R)-1-HYDROXYETHYL]-6-TRIDECYL-, (4S,6S)-](http://img.cochemist.com/ccimg/862500/862472-75-1.png)
![1,3-DIOXAN-2-ONE, 4-[(1R)-1-HYDROXYETHYL]-6-TRIDECYL-, (4S,6S)-](http://img.cochemist.com/ccimg/862500/862472-75-1_b.png)
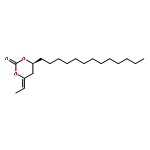
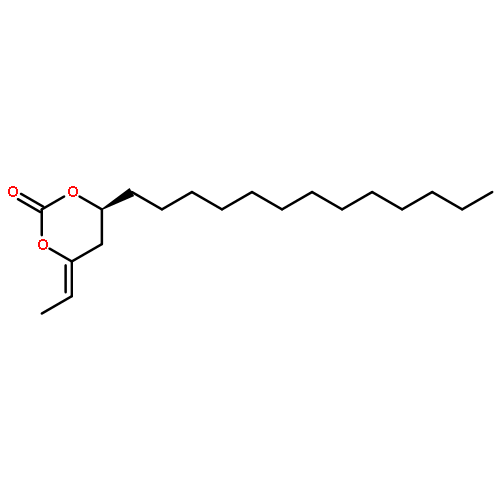
![1,3-Dioxan-2-one, 4-[(1R)-1-iodoethyl]-6-tridecyl-, (4S,6S)-](http://img.cochemist.com/ccimg/862500/862472-72-8.png)
![1,3-Dioxan-2-one, 4-[(1R)-1-iodoethyl]-6-tridecyl-, (4S,6S)-](http://img.cochemist.com/ccimg/862500/862472-72-8_b.png)


![Isoquinoline, 3,4-dihydro-6,7-dimethoxy-1-[(3-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/608200/608141-10-2.png)
![Isoquinoline, 3,4-dihydro-6,7-dimethoxy-1-[(3-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/608200/608141-10-2_b.png)
![N-[4-[3-[2-(3,4-DICHLOROPHENYL)ETHYLAMINO]-2-HYDROXYPROPOXY]PHENYL]METHANESULFONAMIDE](http://img.cochemist.com/ccimg/457900/457897-92-6.png)
![N-[4-[3-[2-(3,4-DICHLOROPHENYL)ETHYLAMINO]-2-HYDROXYPROPOXY]PHENYL]METHANESULFONAMIDE](http://img.cochemist.com/ccimg/457900/457897-92-6_b.png)
![6-METHYL-3-(BETA-D-2-DEOXY-RIBOFURANOSYL)FURANO[2,3-D]PYRIMIDIN-2-ONE](http://img.cochemist.com/ccimg/383900/383897-60-7.png)
![6-METHYL-3-(BETA-D-2-DEOXY-RIBOFURANOSYL)FURANO[2,3-D]PYRIMIDIN-2-ONE](http://img.cochemist.com/ccimg/383900/383897-60-7_b.png)
![(3Z,5Z)-3,5-BIS[(2-FLUOROPHENYL)METHYLIDENE]PIPERIDIN-4-ONE;HYDROCHLORIDE](http://img.cochemist.com/ccimg/342900/342808-40-6.png)
![(3Z,5Z)-3,5-BIS[(2-FLUOROPHENYL)METHYLIDENE]PIPERIDIN-4-ONE;HYDROCHLORIDE](http://img.cochemist.com/ccimg/342900/342808-40-6_b.png)








![Benzeneethanamine, 3,4-dimethoxy-N-[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/182100/182055-88-5.png)
![Benzeneethanamine, 3,4-dimethoxy-N-[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/182100/182055-88-5_b.png)






![1,1'-[1,4-Phenylenebis(methylene)]bis[1,4,8,11-tetraazacyclotetradecane]](http://img.cochemist.com/ccimg/155200/155148-31-5.png)
![1,1'-[1,4-Phenylenebis(methylene)]bis[1,4,8,11-tetraazacyclotetradecane]](http://img.cochemist.com/ccimg/155200/155148-31-5_b.png)
![4-(4-Fluorophenyl)-2-[4-(methylsulfinyl)phenyl]-5-(4-pyridyl)-1H-imidazole](http://img.cochemist.com/ccimg/152200/152121-47-6.png)
![4-(4-Fluorophenyl)-2-[4-(methylsulfinyl)phenyl]-5-(4-pyridyl)-1H-imidazole](http://img.cochemist.com/ccimg/152200/152121-47-6_b.png)
![2(1H)-Pyrimidinone,4-amino-5-fluoro-1-[(2R,4R)-2-(hydroxymethyl)-1,3-dioxolan-4-yl]-](http://img.cochemist.com/ccimg/145500/145417-33-0.png)
![2(1H)-Pyrimidinone,4-amino-5-fluoro-1-[(2R,4R)-2-(hydroxymethyl)-1,3-dioxolan-4-yl]-](http://img.cochemist.com/ccimg/145500/145417-33-0_b.png)
![1,3-Dioxolan-4-one,2-[[[(1,1-dimethylethyl)diphenylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/145400/145397-22-4.png)
![1,3-Dioxolan-4-one,2-[[[(1,1-dimethylethyl)diphenylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/145400/145397-22-4_b.png)






![2-Pentanone, 3-[bis(phenylmethyl)amino]-4-methyl-, (S)-](http://img.cochemist.com/ccimg/131900/131817-82-8.png)
![2-Pentanone, 3-[bis(phenylmethyl)amino]-4-methyl-, (S)-](http://img.cochemist.com/ccimg/131900/131817-82-8_b.png)






![1,2,3-Propanetricarboxylicacid,1,1'-[(1S,2R)-1-[(2S,4R,9R,11S,12S)-12-amino-4,9,11-trihydroxy-2-methyltridecyl]-2-[(1R)-1-methylpentyl]-1,2-ethanediyl]ester, (2R,2'R)-](http://img.cochemist.com/ccimg/116400/116355-83-0.png)
![1,2,3-Propanetricarboxylicacid,1,1'-[(1S,2R)-1-[(2S,4R,9R,11S,12S)-12-amino-4,9,11-trihydroxy-2-methyltridecyl]-2-[(1R)-1-methylpentyl]-1,2-ethanediyl]ester, (2R,2'R)-](http://img.cochemist.com/ccimg/116400/116355-83-0_b.png)
![Acetaldehyde, [[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/115900/115869-43-7.png)
![Acetaldehyde, [[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/115900/115869-43-7_b.png)
amine](http://img.cochemist.com/ccimg/114200/114105-51-0.png)
amine](http://img.cochemist.com/ccimg/114200/114105-51-0_b.png)
![4-Pyrimidinamine, 5-fluoro-N-(trimethylsilyl)-2-[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/111900/111878-21-8.png)
![4-Pyrimidinamine, 5-fluoro-N-(trimethylsilyl)-2-[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/111900/111878-21-8_b.png)


![Isoquinoline, 1,2,3,4-tetrahydro-6,7-dimethoxy-1-[2-(4-methoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/108800/108704-90-1.png)
![Isoquinoline, 1,2,3,4-tetrahydro-6,7-dimethoxy-1-[2-(4-methoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/108800/108704-90-1_b.png)




![1,4-Dioxaspiro[4.5]deca-6,9-dien-8-one, 7,9-dimethyl-](http://img.cochemist.com/ccimg/85300/85268-20-8.png)
![1,4-Dioxaspiro[4.5]deca-6,9-dien-8-one, 7,9-dimethyl-](http://img.cochemist.com/ccimg/85300/85268-20-8_b.png)
![Pyrimidine, 2,4-dimethoxy-5-[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/83400/83355-89-9.png)
![Pyrimidine, 2,4-dimethoxy-5-[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/83400/83355-89-9_b.png)












































![Isoquinoline,3,4-dihydro-6,7-dimethoxy-1-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/72600/72527-29-8.png)
![Isoquinoline,3,4-dihydro-6,7-dimethoxy-1-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/72600/72527-29-8_b.png)







![N-[2-(3,4-dimethoxyphenyl)ethyl]-2-(3-methoxyphenyl)acetamide](http://img.cochemist.com/ccimg/67300/67237-63-2.png)
![N-[2-(3,4-dimethoxyphenyl)ethyl]-2-(3-methoxyphenyl)acetamide](http://img.cochemist.com/ccimg/67300/67237-63-2_b.png)
![Acetamide, 2-chloro-N-[2-(2,3-dimethoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/65800/65713-30-6.png)
![Acetamide, 2-chloro-N-[2-(2,3-dimethoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/65800/65713-30-6_b.png)


















![2-CHLORO-N-[2-(3-METHOXYPHENYL)ETHYL]ACETAMIDE](http://img.cochemist.com/ccimg/34200/34162-12-4.png)
![2-CHLORO-N-[2-(3-METHOXYPHENYL)ETHYL]ACETAMIDE](http://img.cochemist.com/ccimg/34200/34162-12-4_b.png)
![Acetamide, N-[2-(1,3-benzodioxol-5-yl)ethyl]-2-chloro-](http://img.cochemist.com/ccimg/33200/33100-03-7.png)
![Acetamide, N-[2-(1,3-benzodioxol-5-yl)ethyl]-2-chloro-](http://img.cochemist.com/ccimg/33200/33100-03-7_b.png)


![Benzenepropanamide, N-[2-(3,4-dimethoxyphenyl)ethyl]-4-methoxy-](http://img.cochemist.com/ccimg/30100/30002-00-7.png)
![Benzenepropanamide, N-[2-(3,4-dimethoxyphenyl)ethyl]-4-methoxy-](http://img.cochemist.com/ccimg/30100/30002-00-7_b.png)
























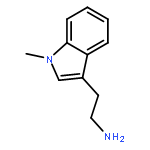
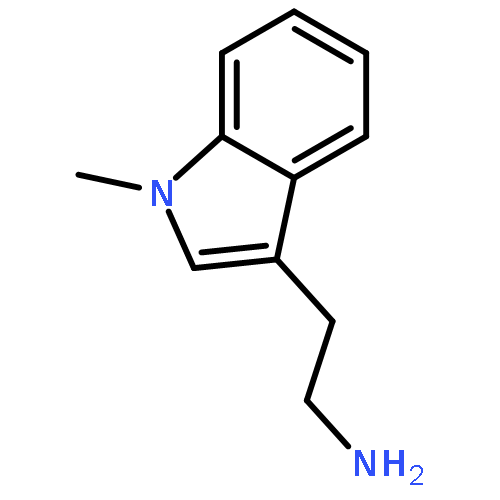



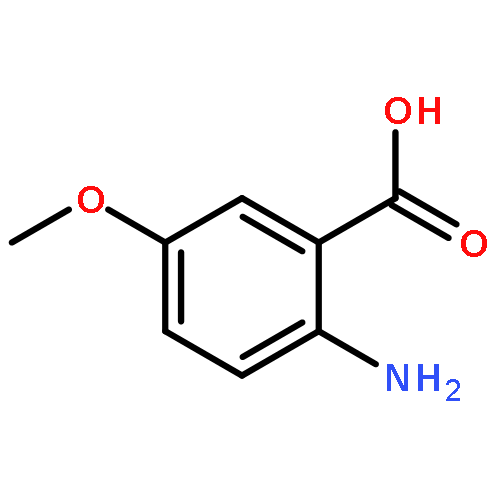
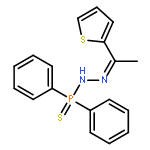







![N-[2-(3,4-DIMETHOXYPHENYL)ETHYL]-2-(4-METHOXYPHENYL)ACETAMIDE](http://img.cochemist.com/ccimg/4100/4078-65-3.png)
![N-[2-(3,4-DIMETHOXYPHENYL)ETHYL]-2-(4-METHOXYPHENYL)ACETAMIDE](http://img.cochemist.com/ccimg/4100/4078-65-3_b.png)








![(NE)-N-[(2-METHYLPHENYL)METHYLIDENE]HYDROXYLAMINE](http://img.cochemist.com/ccimg/3400/3376-32-7.png)
![(NE)-N-[(2-METHYLPHENYL)METHYLIDENE]HYDROXYLAMINE](http://img.cochemist.com/ccimg/3400/3376-32-7_b.png)
![ACETAMIDE, N-[2-(3,4-DIMETHOXYPHENYL)ETHYL]-2-PHENOXY-](http://img.cochemist.com/ccimg/3400/3341-36-4.png)
![ACETAMIDE, N-[2-(3,4-DIMETHOXYPHENYL)ETHYL]-2-PHENOXY-](http://img.cochemist.com/ccimg/3400/3341-36-4_b.png)








![Benzenepropanamide, N-[2-(3,4-dimethoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/2900/2878-60-6.png)
![Benzenepropanamide, N-[2-(3,4-dimethoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/2900/2878-60-6_b.png)









![(3-chlorophenyl){6,7-dimethoxy-1-[(4-methoxyphenoxy)methyl]-3,4-d Ihydro-2(1h)-isoquinolinyl}methanone](http://img.cochemist.com/ccimg/486500/486427-17-2.png)
![(3-chlorophenyl){6,7-dimethoxy-1-[(4-methoxyphenoxy)methyl]-3,4-d Ihydro-2(1h)-isoquinolinyl}methanone](http://img.cochemist.com/ccimg/486500/486427-17-2_b.png)





























![2(3H)-Furanone,5-[[[(1,1-dimethylethyl)diphenylsilyl]oxy]methyl]dihydro-, (S)-](http://img.cochemist.com/ccimg/102800/102717-29-3.png)
![2(3H)-Furanone,5-[[[(1,1-dimethylethyl)diphenylsilyl]oxy]methyl]dihydro-, (S)-](http://img.cochemist.com/ccimg/102800/102717-29-3_b.png)











![2-BUTANONE, 3-[BIS(PHENYLMETHYL)AMINO]-, (S)-](http://img.cochemist.com/ccimg/150400/150357-17-8.png)
![2-BUTANONE, 3-[BIS(PHENYLMETHYL)AMINO]-, (S)-](http://img.cochemist.com/ccimg/150400/150357-17-8_b.png)