Co-reporter:Travis R. Helgren and Timothy J. Hagen
Journal of Chemical Education March 14, 2017 Volume 94(Issue 3) pp:345-345
Publication Date(Web):February 13, 2017
DOI:10.1021/acs.jchemed.6b00555
Drug design and discovery remains a popular topic of study to many students interested in visible, real-world applications of the chemical sciences. It is important that laboratory experiments detailing the early stages of drug discovery incorporate both compound design and an exploration of ligand/receptor interactions. Molecular modeling is widely employed in research endeavors seeking to predict the activity of potential compounds prior to synthesis and can therefore be used to illustrate these concepts. The following activity therefore details the use of AutoDock to predict the binding affinity and docked pose of a series of CDK2 inhibitors. Students can then compare their docking output to experimentally determined inhibitory activities and crystal structures. Finally, the AutoDock workflow detailed in this activity can be used in research settings, provided the receptor crystal structure is known.Keywords: Biochemistry; Chemoinformatics; Computational Chemistry; Computer-Based Learning; Drugs/Pharmaceuticals; Graduate Education/Research; Inquiry-Based/Discovery Learning; Medicinal Chemistry; Upper-Division Undergraduate;
Co-reporter:Gashaw M. Goshu, Debarati Ghose, Joy M. Bain, Phillip G. Pierce, Darren W. Begley, Stephen N. Hewitt, Hannah S. Udell, Peter J. Myler, R. Meganathan, Timothy J. Hagen
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 24) pp:5699-5704
Publication Date(Web):15 December 2015
DOI:10.1016/j.bmcl.2015.10.096
The fragment FOL7185 (compound 17) was found to be a hit against IspD and IspE enzymes isolated from bacteria, and a series of analogs containing the pyrazolopyrimidine core were synthesized. The majority of these compounds inhibited the growth of Burkholderia thailandensis (Bt) and Pseudomonas aeruginosa (Pa) in the Kirby–Bauer disk diffusion susceptibility test. Compound 29 shows inhibitory activity at 0.1 mM (32.2 μg/mL), which is comparable to the control compound kanamycin (48.5 μg/mL). Compound 29 also shows inhibitory activity at 0.5 mM against kanamycin resistant P. aeruginosa. Saturation transfer difference NMR (STD-NMR) screening of these compounds against BtIspD and BtIspE indicated that most of these compounds significantly interact with BtIspE, suggesting that the compounds may inhibit the growth of Bt by disrupting isoprenoid biosynthesis. Ligand epitope mapping of compound 29 with BtIspE indicated that hydrogens on 2,4-dichlorophenyl group have higher proximity to the surface of the enzyme than hydrogens on the pyrazolopyrimidine ring.
Co-reporter:Timothy J. Hagen, Xuesheng Mo, Alex B. Burgin, David Fox 3rd, Zheng Zhang, Mark E. Gurney
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 16) pp:4031-4034
Publication Date(Web):15 August 2014
DOI:10.1016/j.bmcl.2014.06.002
In this study we report a series of triazine derivatives that are potent inhibitors of PDE4B. We also provide a series of structure activity relationships that demonstrate the triazine core can be used to generate subtype selective inhibitors of PDE4B versus PDE4D. A high resolution co-crystal structure shows that the inhibitors interact with a C-terminal regulatory helix (CR3) locking the enzyme in an inactive ‘closed’ conformation. The results show that the compounds interact with both catalytic domain and CR3 residues. This provides the first structure-based approach to engineer PDE4B-selective inhibitors.
Co-reporter:Phumvadee Wangtrakuldee, Matthew S. Byrd, Cristine G. Campos, Michael W. Henderson, Zheng Zhang, Michael Clare, Ali Masoudi, Peter J. Myler, James R. Horn, Peggy A. Cotter, and Timothy J. Hagen
ACS Medicinal Chemistry Letters 2013 Volume 4(Issue 8) pp:699-703
Publication Date(Web):July 1, 2013
DOI:10.1021/ml400034m
Evaluation of a series of MetAP inhibitors in an in vitro enzyme activity assay led to the first identification of potent molecules that show significant growth inhibition against Burkholderia pseudomallei. Nitroxoline analogues show excellent inhibition potency in the BpMetAP1 enzyme activity assay with the lowest IC50 of 30 nM and inhibit the growth of B. pseudomallei and B. thailandensis at concentrations ≥31 μM.Keywords: BpMetAP1; Burkholderia pseudomallei; melioidosis; methionine aminopeptidase inhibitor;
Co-reporter:Amy Dalby, Xuesheng Mo, Robert Stoa, Nathaniel Wroblewski, Zheng Zhang, Timothy J. Hagen
Tetrahedron Letters 2013 Volume 54(Issue 21) pp:2737-2739
Publication Date(Web):22 May 2013
DOI:10.1016/j.tetlet.2013.03.063
The optimization and synthesis of biaryl PDE4D allosteric modulator D159687 was achieved on gram scale via a concise two-step process. The synthesis features sequential chemoselective Suzuki coupling reactions taking advantage of different reactivity profiles of benzyl versus aryl halides. The method was then applied to the synthesis of two additional PDE4D allosteric modulators, D159404 and D159153. The efficient synthesis of these PDE4 allosteric modulators will allow for further biological evaluation of these compounds and the method developed will empower rapid analog formation through combinatorial chemical means.
Co-reporter:Zheng Zhang, Sriram Jakkaraju, Joy Blain, Kenneth Gogol, Lei Zhao, Robert C. Hartley, Courtney A. Karlsson, Bart L. Staker, Thomas E. Edwards, Lance J. Stewart, Peter J. Myler, Michael Clare, Darren W. Begley, James R. Horn, Timothy J. Hagen
Bioorganic & Medicinal Chemistry Letters 2013 23(24) pp: 6860-6863
Publication Date(Web):
DOI:10.1016/j.bmcl.2013.09.101
Co-reporter:Travis R. Helgren, Congling Chen, Phumvadee Wangtrakuldee, Thomas E. Edwards, Bart L. Staker, Jan Abendroth, Banumathi Sankaran, Nicole A. Housley, Peter J. Myler, Jonathon P. Audia, James R. Horn, Timothy J. Hagen
Bioorganic & Medicinal Chemistry (1 February 2017) Volume 25(Issue 3) pp:
Publication Date(Web):1 February 2017
DOI:10.1016/j.bmc.2016.11.013
Methionine aminopeptidase (MetAP) is a class of ubiquitous enzymes essential for the survival of numerous bacterial species. These enzymes are responsible for the cleavage of N-terminal formyl-methionine initiators from nascent proteins to initiate post-translational modifications that are often essential to proper protein function. Thus, inhibition of MetAP activity has been implicated as a novel antibacterial target. We tested this idea in the present study by targeting the MetAP enzyme in the obligate intracellular pathogen Rickettsia prowazekii. We first identified potent RpMetAP inhibitory species by employing an in vitro enzymatic activity assay. The molecular docking program AutoDock was then utilized to compare published crystal structures of inhibited MetAP species to docked poses of RpMetAP. Based on these in silico and in vitro screens, a subset of 17 compounds was tested for inhibition of R. prowazekii growth in a pulmonary vascular endothelial cell (EC) culture infection model system. All compounds were tested over concentration ranges that were determined to be non-toxic to the ECs and 8 of the 17 compounds displayed substantial inhibition of R. prowazekii growth. These data highlight the therapeutic potential for inhibiting RpMetAP as a novel antimicrobial strategy and set the stage for future studies in pre-clinical animal models of infection.



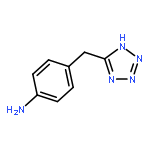
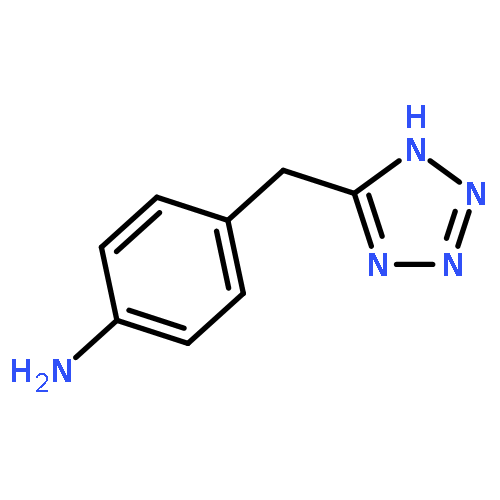
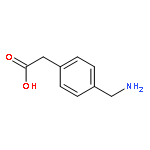
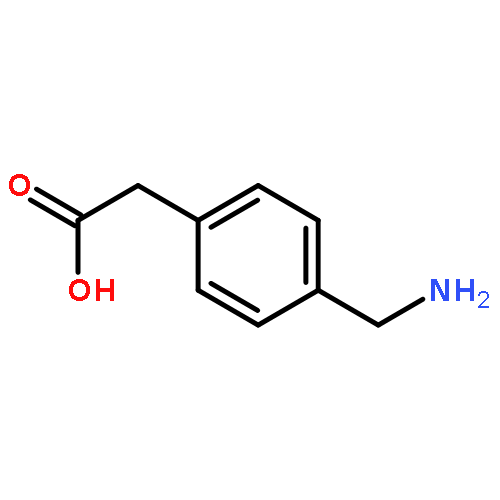
![1-methyl-N-(propan-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine](http://img.cochemist.com/ccimg/116100/116035-77-9.png)
![1-methyl-N-(propan-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine](http://img.cochemist.com/ccimg/116100/116035-77-9_b.png)
![1H-Pyrazolo[3,4-d]pyrimidin-4-amine, 1-methyl-N-(2-phenylethyl)-](http://img.cochemist.com/ccimg/102400/102353-77-5.png)
![1H-Pyrazolo[3,4-d]pyrimidin-4-amine, 1-methyl-N-(2-phenylethyl)-](http://img.cochemist.com/ccimg/102400/102353-77-5_b.png)
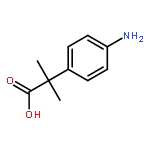
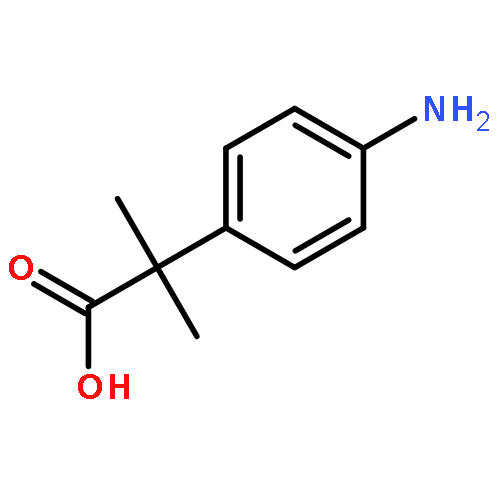
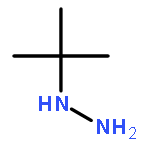
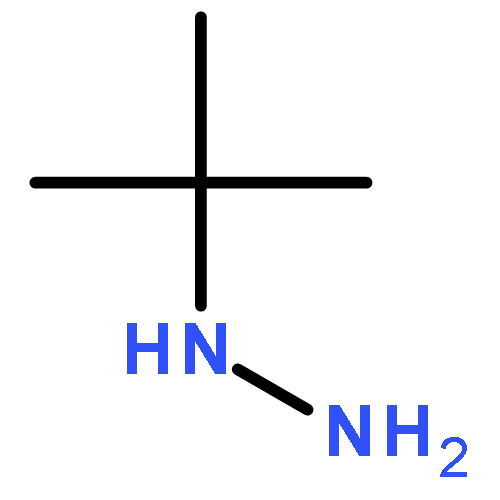


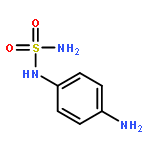

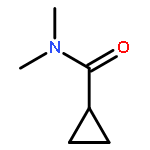
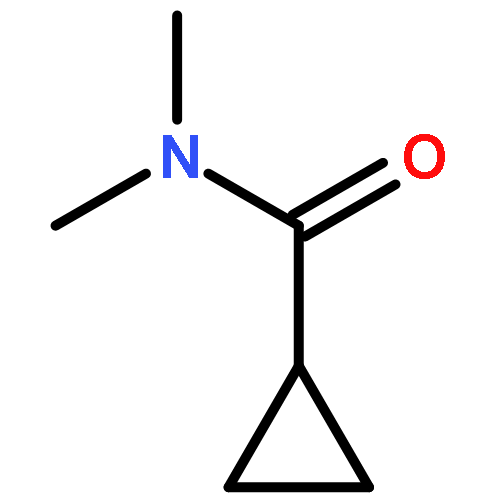
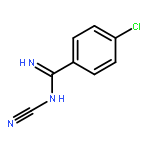
![N-(4-chlorobenzyl)-1-methyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine](http://img.cochemist.com/ccimg/5500/5441-46-3.png)
![N-(4-chlorobenzyl)-1-methyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine](http://img.cochemist.com/ccimg/5500/5441-46-3_b.png)
![2-[(1-methyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino]ethanol](http://img.cochemist.com/ccimg/5400/5334-71-4.png)
![2-[(1-methyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino]ethanol](http://img.cochemist.com/ccimg/5400/5334-71-4_b.png)
![4-(furfurylamino)-1-methyl-1h-pyrazolo[3,4-d]pyrimidine](http://img.cochemist.com/ccimg/5400/5334-58-7.png)
![4-(furfurylamino)-1-methyl-1h-pyrazolo[3,4-d]pyrimidine](http://img.cochemist.com/ccimg/5400/5334-58-7_b.png)
![4-Chloro-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine](http://img.cochemist.com/ccimg/5400/5334-48-5.png)
![4-Chloro-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine](http://img.cochemist.com/ccimg/5400/5334-48-5_b.png)