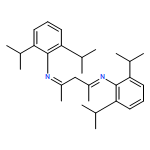
Co-reporter:Philip J. Bailey, Mahmudur Rahman, Simon Parsons, Muhammad R. Azhar and Fraser J. White
Dalton Transactions 2013 vol. 42(Issue 8) pp:2879-2886
Publication Date(Web):19 Dec 2012
DOI:10.1039/C2DT32804F
A simple and convenient route to η5-coordinated Ru and Rh aminofulvene-aldiminate (AFA) complexes is described. The metalloligands [Cp*Ru{η5-(Ph2AFAH)}][BF4] (3), [Cp*Ru{η5-(benzyl2AFAH)}][OTf] (7), [Cp*Rh{η5-(Cy2AFA)H}][BF4]2 (8) and [Cp*Rh{η5-(Cy2AFA)}][BF4] (9) have been synthesised and characterised. The basicity of 9 has been found to be significantly less than its neutral analogue and thus eliminates the need for a deprotonation step to ligate to a second metal in the κ2-N,N′-coordination mode. The reaction of 9 with a palladium precursor provides a mixed-metal complex [Cp*Rh(η5/κ2-Cy2AFA)PdCl2][BF4] (12). Cyclic voltammetry studies of the Ph2AFAH ligand shows an irreversible one electron oxidation peak at +1.0 V (vs. Fc/Fc+). Complex 3 shows an irreversible oxidation at +1.5 V and a reduction peak at −1.0 V. The oxidation of 3 occurs on the AFA ligand backbone whereas the structurally analogous neutral 1,2-bis(imidoyl)pentamethyl-ruthenocene shows reversible oxidation at the Ru center.
Co-reporter:Philip J. Bailey, Nicola L. Bell and Gary S. Nichol
Dalton Transactions 2013 vol. 42(Issue 31) pp:11281-11294
Publication Date(Web):20 Jun 2013
DOI:10.1039/C3DT51150B
The pursuit of heteroleptic ‘half-sandwich’ complexes of boron substituted anionic and charge-neutral tris(methimazolyl)borate ligands is described. The formation of the homoleptic ‘sandwich’ complexes was found to be favoured when the metal precursor contains labile ligands. When unable to form these homoleptic complexes Tm is shown to preferentially coordinate in a κ2-[S,S] fashion. This study reinforces previous assertions that the Tm ligand preferentially adopts the κ2-[S,S] mode when coordinated to electron rich metal centres and demonstrates the strong electron donor properties of substituted Tm ligands.
Co-reporter:Philip J. Bailey, Nicola L. Bell, Gary S. Nichol, Simon Parsons, and Fraser White.
Inorganic Chemistry 2012 Volume 51(Issue 6) pp:3677-3689
Publication Date(Web):February 29, 2012
DOI:10.1021/ic202618p
A number of new charge-neutral zwitterionic tris(methimazolyl)borate ligands have been synthesized, either by substitution of the dimethylamine group in the adduct (dimethylamine)tris(methimazolyl)borane (1) or by insertion into its B–N(dimethylamine) bond by an unsaturated Lewis base. Two new anionic ligands, (thiocyanato)tris(methimazolyl)borate and (cyano)tris(methimazolyl)borate, have also been accessed by this method.
Co-reporter:Philip J. Bailey, Nicola L. Bell, Lim Li Gim, Tai Yucheng, Nicholas Funnell, Fraser White and Simon Parsons
Chemical Communications 2011 vol. 47(Issue 42) pp:11659-11661
Publication Date(Web):30 Sep 2011
DOI:10.1039/C1CC14473A
The synthesis of a new charge-neutral zwitterionic tripodal borate ligand based on 2-hydroxypyridine is reported. (Dimethylaminopyridinium)tris(2-pyridonyl)borate, (DMAP)Thp, has been complexed to copper(I) chloride to give a pseudo-C3 symmetric complex. The propensity for this ligand and other flexible scorpionates to exhibit such helical chirality upon complexation is discussed.
Co-reporter:Philip J. Bailey, Anna Collins, Peter Haack, Simon Parsons, Mahmudur Rahman, Damian Smith and Fraser J. White
Dalton Transactions 2010 vol. 39(Issue 6) pp:1591-1597
Publication Date(Web):04 Dec 2009
DOI:10.1039/B914707A
Bis(N,N′-2,6-diisopropylphenyl)-6-aminofulvene-2-aldimine (4) has been synthesised and characterised. The synthesis and characterisation of two zwitterionic Pd(II) complexes [(Ph2AFA)Pd(Me)DMAP] (1) and [(Ph2AFA)Pd(N,N-dimethylbenzylamine-2-C,N)] (2) are reported. Activation of 1 and 2 for ethene polymerisation with Lewis acids such as BF3 and B(C6F5)3 were not successful. Attempted synthesis of halide-bridged dimers of the form [(Ph2AFA)Pd(μ-X)]2 resulted in formation of bis-chelated complexes [(Cy2AFA)2Pd] (3) and [(tBu2AFA)2Pd] (5).
Co-reporter:PhilipJ. Bailey Dr.;Laura Budd Dr.;FilipaA. Cavaco;Simon Parsons ;Felix Rudolphi;Alejro Sanchez-Perucha Dr. ;FraserJ. White Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 9) pp:2819-2829
Publication Date(Web):
DOI:10.1002/chem.200902763
Abstract
The dimethylamine in the adducts [(HNMe2)B(azolyl)3] (azolyl=methimazolyl, pyrazolyl), obtained by reaction of the azole with B(NMe2)3, can readily be substituted with a range of nitrogen donors to provide new charge-neutral, tripodal ligands in high yield. This observation has led to a revision of an earlier interpretation of the mechanism of the formation of these species. The donor properties of the ligands [(nmi)B(azolyl)3] (nmi=N-methylimidazole) have been compared with their anionic analogues [HB(azolyl)3]− by synthesis of their manganese(I)–tricarbonyl complexes and comparison of their infrared νCO energies. This comparison indicates that the new neutral ligands are only marginally weaker donors than the corresponding anionic hydrotris(azolyl)borate ligands. This may be explained by the ability of the attached nmi ring to stabilize a positive charge remotely from the coordinated metal, which may also account for the fact that the [(nmi)B(pyrazolyl)3] ligand is a substantially stronger donor than the similarly neutral tris(pyrazolyl)methane ligand.
Co-reporter:Philip J. Bailey, Chiara McCormack, Simon Parsons, Felix Rudolphi, Alejandro Sanchez Perucha and Peter Wood
Dalton Transactions 2007 (Issue 4) pp:476-480
Publication Date(Web):16 Oct 2006
DOI:10.1039/B611331A
The activation of tris(dimethylamino)borane towards reaction with a chiral methimazole by N-methylimidazole has been used to prepare the first example of a chiral tris(methimazolyl)borate ligand. Coordination of this neutral ligand to Ru(II) has been achieved by reaction with [(p-cymene)RuCl2]2 to provide a single diastereomer complex in which the chirality of the methimazolyl substituents dictate the chirality of the bicyclo[3.3.3]cage formed by the ligand on coordination to the metal. The alternative approach to chiral tris(methimazolyl)borate ligands involving the introduction of a chiral group onto the boron atom has been explored by replacing N-methylimidazole in the above reaction by chiral oxazolines as activating bases in reaction with simple methimazole. However, although the B(NMe2)3 is activated to reaction with methimazole by these oxazolines, an intramolecular oxazoline ring-opening by a coordinated methimazolyl sulfur occurs and prevents the successful synthesis of these ligands.
Co-reporter:Philip J. Bailey, Caroline M. Dick, Sylvie Fabre, Simon Parsons and Lesley J. Yellowlees
Dalton Transactions 2006 (Issue 13) pp:1602-1610
Publication Date(Web):22 Feb 2006
DOI:10.1039/B514591K
Treatment of dimethylmagnesium with the α-diimine ligands Ar′NC(R)C(R)NAr′ [R = naphth-1,8-diyl (1), H (2), CH3 (3); Ar′ = 2,6-diisopropylphenyl] in diethyl ether provides the neutral methyl-bridged dimeric complexes [(α-diimine−˙)Mg+(µ-CH3)]2via single electron transfer (SET) to the coordinated diimine and elimination of a methyl radical. These biradical species have been characterised by EPR spectroscopy and, for the ligand 1, X-ray crystallography. In the presence of THF the reaction of ligand 1 proceeds to the diamagnetic [(ene-1,2-diamide)Mg(THF)3] complex in which the diimine ligand has been doubly reduced to an ene-diamide by two successive SET processes. Comparison of the structural data for the free ligand 1 with that obtained for the α-diimine radical anion and ene-diamide complexes shows the expected increases in C–N, and decreases in C–C, bond lengths within the N–C–C–N unit consistent with the progressive reduction of the ligand. In the case of ligand 3, reaction at low temperature provides the complex [Mg(µ2-Me){Ar′NC(Me)2C(Me)NAr′}]2 in which methyl transfer to a ligand imine carbon atom has occurred. This species has also been structurally characterised. This contrasts with the formation of the radical species at room temperature, and indicates the involvement of an intermediate in which the radical products of the SET process are held in close proximity by the solvent cage. Two competing processes of methyl radical escape and methyl transfer to the ligand account for the formation of the observed products at different temperatures.
Co-reporter:Philip J. Bailey Dr.;Daniel Lorono-Gonzales;Chiara McCormack;Frances Millican;Simon Parsons Dr.;Robert Pfeifer Dr.;Pedro P. Pinho Dr.;Felix Rudolphi;Alejro Sanchez Perucha
Chemistry - A European Journal 2006 Volume 12(Issue 20) pp:
Publication Date(Web):10 MAR 2006
DOI:10.1002/chem.200501323
Reaction of 2-mercapto-1-methylimidazole (methimazole) with tris(dimethylamino)borane, B(NMe2)3, provides the tetrahedral dimethylamine adduct of tris(methimazolyl)borane, [(Me2HN)B(methimazolyl)3]. By contrast, imidazole, 2-methylimidazole, 2-chloroimidazole and benzimidazole provide the homoleptic tetra-azolyl systems H[B(azolyl)4], and the same product is obtained even when a substoichiometric quantity of the heterocyle is employed. The change in reaction outcome is correlated with the variation of basic pKa for the heterocycles. A simple acid-base reaction with elimination of HNMe2 is proposed for the reaction with the weakly basic, but more strongly acidic, methimazole. However, for the more strongly basic imidazoles, initial coordination of the heterocycle imine nitrogen to the weakly Lewis acidic boron centre in B(NMe2)3 to form the tetrahedral adduct [(azole)B(NMe2)3] is proposed. The greater availability of the NMe2 lone pairs in this species results in increased basicity and a rapid reaction with further heterocycle to provide the observed H[B(azolyl)4] products. For 2-nitroimidazole, the low basicity (and increased NH acidity) results in the formation of [(HNMe2)B(2-nitroimidazolyl)3] on reaction with B(NMe2)3, analogous to the product formed with methimazole. Both [(HNMe2)B(methimazolyl)3] and H[B(benzimidazolyl)4] have been structurally characterised by single crystal X-ray crystallography. This chemistry has been exploited to provide a new synthesis of borate-centred tripod ligands, whereby N-methylimidazole is used to activate B(NMe2)3 to reaction with methimazole to form the new ligand [(N-methylimidazole)B(methimazolyl)3] in good yield and a complex of this ligand with RuII has been structurally characterised.
Co-reporter:Philip J. Bailey, Robert A. Coxall, Caroline M. Dick, Sylvie Fabre, Simon Parsons and Lesley J. Yellowlees
Chemical Communications 2005 (Issue 36) pp:4563-4565
Publication Date(Web):17 Aug 2005
DOI:10.1039/B505697G
Treatment of dimethylmagnesium with bulky α-diimine ligands provides either the biradical methyl-bridged complexes [(α-diimine−˙)Mg+(μ-CH3)]2via single electron transfer (SET), or the product of methyl transfer to an imine carbon atom depending upon conditions.
Co-reporter:Philip J. Bailey, Daniel Loroño-González and Simon Parsons
Chemical Communications 2003 (Issue 12) pp:1426-1427
Publication Date(Web):16 May 2003
DOI:10.1039/B303540A
Two magnesium complexes of the 6-aminofulvene-2-aldimine (AFA) system bearing cyclohexyl groups on the donor nitrogen atoms have been synthesised; in the first the ligand is coordinated via the two nitrogen donors while in the second it is found to ligate magnesium via the cyclopentadienyl and the imine donors.
Co-reporter:Philip J Bailey, Daniel J Lorono-Gonzales, Chiara McCormack, Simon Parsons, Matthew Price
Inorganica Chimica Acta 2003 Volume 354() pp:61-67
Publication Date(Web):30 October 2003
DOI:10.1016/S0020-1693(03)00180-4
A number of new complexes of the tris(methimazolyl)hydroborate (Tm) ligand have been synthesised: [(Tm)RuCl(DMSO)2] (1), [(Tm)Ru(p-cymene)]Cl (2), [(Tm)RuCp] (3) and [(Tm)Mn(CO)3] (4). Complexes 2 and 4 have been characterised structurally by X-ray crystallography and a partial crystallographic analysis was obtained for 1. The Tm ligand is found to coordinate as a tripod ligand through its sulphur donors in all complexes except 3, for which there is evidence of bidentate coordination and formation of a 16-electron species.Four new ruthenium and manganese complexes of the tris(methimazolyl)hydroborate (Tm) ligand have been prepared and characterised.
Co-reporter:Philip J. Bailey Dr.;Robert A. Coxall Dr.;Caroline M. Dick Dr.;Sylvie Fabre Dr.;Louise C. Henderson;Christian Herber;Stephen T. Liddle Dr.;Daniel Loroño-González;Andrew Parkin Dr.;Simon Parsons Dr.
Chemistry - A European Journal 2003 Volume 9(Issue 19) pp:
Publication Date(Web):26 SEP 2003
DOI:10.1002/chem.200305053
A new high-yield synthesis of [(PhCH2)2Mg(thf)2] and [{(PhCH2)CH3Mg(thf)}2] via benzylpotassium has allowed a simple entry into benzylmagnesium coordination chemistry. The syntheses and X-ray crystal structures of both [(η2-Me2NCH2CH2NMe2)Mg(CH2Ph)2] and [η2-HC{C(CH3)NAr′}2Mg(CH2Ph)(thf)] (Ar′=2,6-diisopropylphenyl) are reported. The latter β-diketiminate complex reacts with dioxygen to provide a 1:2 mixture of dimeric benzylperoxo and benzyloxo complexes. The benzylperoxo complex [{η2-HC{C(CH3)NAr′}2Mg(μ-η2:η1-OOCH2Ph)}2] is the first example of a structurally characterised Group 2 metal–alkylperoxo complex and contains the benzylperoxo ligands in an unusual μ-η2:η1-coordination mode, linking the two five-coordinate magnesium centres. The OO separation in the benzylperoxo ligands is 1.44(2) Å. Reaction of the benzylperoxo/benzyloxo complex mixture with further [η2-HC{C(CH3)NAr′}2Mg(CH2Ph)(thf)] results in complete conversion of the benzylperoxo species into the benzyloxo complex. This reaction, therefore, establishes the cleavage of dioxygen by this system as a two-step process that involves initial oxygen insertion into the MgCH2Ph bond followed by OO/MgC σ-bond metathesis of the resulting benzylperoxo ligand with a second MgCH2Ph bond. The formation of a 1:2 mixture of the benzylperoxo and benzyloxo species indicates that the rate of the insertion is faster than that of the metathesis, and this is shown to be consistent with a radical mechanism for the insertion process.
Co-reporter:Philip J. Bailey, Tony Barrett, Simon Parsons
Journal of Organometallic Chemistry 2001 Volume 625(Issue 2) pp:236-244
Publication Date(Web):22 April 2001
DOI:10.1016/S0022-328X(01)00674-X
Treatment of the phosphonium salt [PhP(CH2Ph)3]Cl with three molar equivalents of nBuLi gives the dilithiated species [Li2PhP(CHPh)3]. The crystal structure of the monolithiated species as its TMEDA adduct has been determined. Treatment of the dilithiate species with [((η-C5Me5)MX2]2 (M=Cr, X=Br; M=Rh, X=Cl) provides [(η-C5Me5)M{η2-PhP(CH2Ph)(CHPh)2}X] containing chelating phosphoniodiylide ligands which are thought to be the hydrolysis products of the target complexes [{PhP(CHPh)3}M(C5Me5)].
Co-reporter:Philip J. Bailey Dr.;Stephen T. Liddle Dr.;Carole A. Morrison Dr.;Simon Parsons Dr.
Angewandte Chemie International Edition 2001 Volume 40(Issue 23) pp:
Publication Date(Web):28 NOV 2001
DOI:10.1002/1521-3773(20011203)40:23<4463::AID-ANIE4463>3.0.CO;2-2
The cyclic hexanuclear species [{HC[C(tBu)NAr′]2Mg(C3H5)}6] (see structure) in which the allyl group exhibits μ-η1:η1 bonding is formed on thermal treatment of [HC{C(tBu)NAr′}2Mg(C3H5)(thf)] under vacuum. Ar′=2,6-diisopropylphenyl.
Co-reporter:Philip J. Bailey Dr.;Stephen T. Liddle Dr.;Carole A. Morrison Dr.;Simon Parsons Dr.
Angewandte Chemie 2001 Volume 113(Issue 23) pp:
Publication Date(Web):28 NOV 2001
DOI:10.1002/1521-3757(20011203)113:23<4595::AID-ANGE4595>3.0.CO;2-X
Die cyclische sechskernige Verbindung [{HC[C(tBu)NAr′]2Mg(C3H5)}6] (siehe Struktur) mit einer μ-η1:η1-gebundenen Allylgruppe erhält man durch Erhitzen von [HC{C(tBu)NAr′}2Mg(C3H5)(thf)] unter Vakuum. Ar′=2,6-Diisopropylphenyl.
Co-reporter:Philip J. Bailey, Caroline M. E. Dick, Sylvie Fabre and Simon Parsons
Dalton Transactions 2000 (Issue 10) pp:1655-1661
Publication Date(Web):27 Apr 2000
DOI:10.1039/B001316L
Treatment of the Grignard reagent MeMgCl with the lithiates Li[L–X] (Li[L–X] = lithium β-diketiminate [HC{C(Me)NAr′}2Li] (Ar′ = 2,6-diisopropylphenyl) or lithium N,N′-diisopropylaminotroponiminate, Li[(iPr2)ATI]) in THF provided four-co-ordinate methylmagnesium complexes [Mg(η2-L–X)Me(THF)]. The β-diketiminate complex has been characterised by X-ray crystallography, however the aminotroponiminate complex is an oil. Both complexes readily react with oxygen to provide methoxide-bridged dimeric complexes [Mg(μ-OMe)(η2-L–X)]2 and the complex [Mg(μ-OMe){η2-(iPr2)ATI}]2 has structurally been characterised. The methyl-bridged dimeric complex [Mg(μ-Me){HC[C(Me)NAr′]}2]2 may be obtained by removal of THF from the adduct under vacuum at 150 °C or by treatment of the β-diketimine (L–XH) with dimethylmagnesium in toluene with elimination of methane, and has also been characterised crystallographically. In contrast to this, treatment of MgMe2 with the aminotriponimine H[(iPr2)ATI] provides only the bis-chelate complex [Mg{(iPr2)ATI}2] which has also been characterised structurally. However the methyl bridged dimer [Mg(μ-Me){η2-(iPr2)ATI}]2 may be formed by removal of THF from [MgMe{η2-(iPr2)ATI}(THF)] at 110 °C under vacuum.
Co-reporter:Philip J. Bailey, Chiara McCormack, Simon Parsons, Felix Rudolphi, Alejandro Sanchez Perucha and Peter Wood
Dalton Transactions 2007(Issue 4) pp:NaN480-480
Publication Date(Web):2006/10/16
DOI:10.1039/B611331A
The activation of tris(dimethylamino)borane towards reaction with a chiral methimazole by N-methylimidazole has been used to prepare the first example of a chiral tris(methimazolyl)borate ligand. Coordination of this neutral ligand to Ru(II) has been achieved by reaction with [(p-cymene)RuCl2]2 to provide a single diastereomer complex in which the chirality of the methimazolyl substituents dictate the chirality of the bicyclo[3.3.3]cage formed by the ligand on coordination to the metal. The alternative approach to chiral tris(methimazolyl)borate ligands involving the introduction of a chiral group onto the boron atom has been explored by replacing N-methylimidazole in the above reaction by chiral oxazolines as activating bases in reaction with simple methimazole. However, although the B(NMe2)3 is activated to reaction with methimazole by these oxazolines, an intramolecular oxazoline ring-opening by a coordinated methimazolyl sulfur occurs and prevents the successful synthesis of these ligands.
Co-reporter:Philip J. Bailey, Nicola L. Bell, Lim Li Gim, Tai Yucheng, Nicholas Funnell, Fraser White and Simon Parsons
Chemical Communications 2011 - vol. 47(Issue 42) pp:NaN11661-11661
Publication Date(Web):2011/09/30
DOI:10.1039/C1CC14473A
The synthesis of a new charge-neutral zwitterionic tripodal borate ligand based on 2-hydroxypyridine is reported. (Dimethylaminopyridinium)tris(2-pyridonyl)borate, (DMAP)Thp, has been complexed to copper(I) chloride to give a pseudo-C3 symmetric complex. The propensity for this ligand and other flexible scorpionates to exhibit such helical chirality upon complexation is discussed.
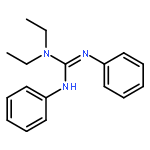
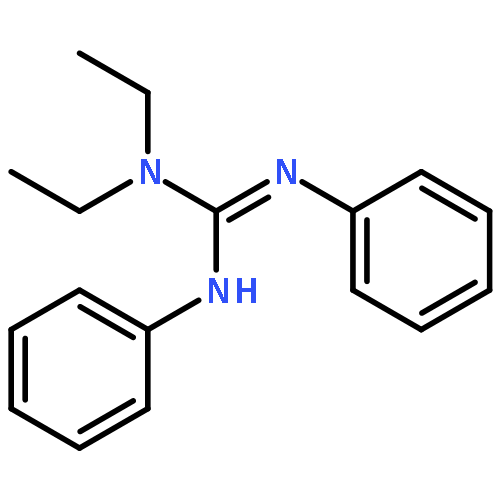
Co-reporter:Philip J. Bailey, Keith J. Grant, Lindsey A. Mitchell, Stuart Pace, Andrew Parkin and Simon Parsons
Dalton Transactions 2000(Issue 12) pp:NaN1891-1891
Publication Date(Web):2000/05/22
DOI:10.1039/B001992P
Treatment of the dimeric ruthenium halide complex [Ru(η-C6H6)Cl2]2 with 1,2,3-triphenylguanidine provided the tris-chelate [Ru{η2-(NPh)2CNHPh}3] with loss of the aromatic ligand and metal oxidation. This product may also be prepared by treatment of the previously reported [Ru(η-p-PriC6H4Me){η2-(NPh)2CNHPh}Cl] with bases (KOH, LiNiPr2, NEt3). Complexes containing chelating guanidinate ligands are also available by means of metathesis of the monolithiated ligands Li[C(NR)2NR2] with metal halide complexes, as exemplified by the synthesis of the square planar bis-chelate [Pt{η2-(NPh)2CNHPh}2] from [Pt(PhCN)2Cl2] and [Ti{η2-(NPh)2CN(Et)2}2Cl2] from [TiCl4(THF)2]. The characterisation of these complexes allows a comparison of the co-ordination properties of chelating guanidinates with early, middle and late transition metals.
Co-reporter:Philip J. Bailey, Anna Collins, Peter Haack, Simon Parsons, Mahmudur Rahman, Damian Smith and Fraser J. White
Dalton Transactions 2010 - vol. 39(Issue 6) pp:NaN1597-1597
Publication Date(Web):2009/12/04
DOI:10.1039/B914707A
Bis(N,N′-2,6-diisopropylphenyl)-6-aminofulvene-2-aldimine (4) has been synthesised and characterised. The synthesis and characterisation of two zwitterionic Pd(II) complexes [(Ph2AFA)Pd(Me)DMAP] (1) and [(Ph2AFA)Pd(N,N-dimethylbenzylamine-2-C,N)] (2) are reported. Activation of 1 and 2 for ethene polymerisation with Lewis acids such as BF3 and B(C6F5)3 were not successful. Attempted synthesis of halide-bridged dimers of the form [(Ph2AFA)Pd(μ-X)]2 resulted in formation of bis-chelated complexes [(Cy2AFA)2Pd] (3) and [(tBu2AFA)2Pd] (5).
Co-reporter:Philip J. Bailey, Nicola L. Bell and Gary S. Nichol
Dalton Transactions 2013 - vol. 42(Issue 31) pp:NaN11294-11294
Publication Date(Web):2013/06/20
DOI:10.1039/C3DT51150B
The pursuit of heteroleptic ‘half-sandwich’ complexes of boron substituted anionic and charge-neutral tris(methimazolyl)borate ligands is described. The formation of the homoleptic ‘sandwich’ complexes was found to be favoured when the metal precursor contains labile ligands. When unable to form these homoleptic complexes Tm is shown to preferentially coordinate in a κ2-[S,S] fashion. This study reinforces previous assertions that the Tm ligand preferentially adopts the κ2-[S,S] mode when coordinated to electron rich metal centres and demonstrates the strong electron donor properties of substituted Tm ligands.
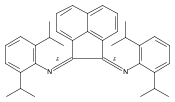
Co-reporter:Philip J. Bailey, Mahmudur Rahman, Simon Parsons, Muhammad R. Azhar and Fraser J. White
Dalton Transactions 2013 - vol. 42(Issue 8) pp:NaN2886-2886
Publication Date(Web):2012/12/19
DOI:10.1039/C2DT32804F
A simple and convenient route to η5-coordinated Ru and Rh aminofulvene-aldiminate (AFA) complexes is described. The metalloligands [Cp*Ru{η5-(Ph2AFAH)}][BF4] (3), [Cp*Ru{η5-(benzyl2AFAH)}][OTf] (7), [Cp*Rh{η5-(Cy2AFA)H}][BF4]2 (8) and [Cp*Rh{η5-(Cy2AFA)}][BF4] (9) have been synthesised and characterised. The basicity of 9 has been found to be significantly less than its neutral analogue and thus eliminates the need for a deprotonation step to ligate to a second metal in the κ2-N,N′-coordination mode. The reaction of 9 with a palladium precursor provides a mixed-metal complex [Cp*Rh(η5/κ2-Cy2AFA)PdCl2][BF4] (12). Cyclic voltammetry studies of the Ph2AFAH ligand shows an irreversible one electron oxidation peak at +1.0 V (vs. Fc/Fc+). Complex 3 shows an irreversible oxidation at +1.5 V and a reduction peak at −1.0 V. The oxidation of 3 occurs on the AFA ligand backbone whereas the structurally analogous neutral 1,2-bis(imidoyl)pentamethyl-ruthenocene shows reversible oxidation at the Ru center.

![Ethanaminium, N-[(benzoylamino)methyl]-N,N-diethyl-, chloride](http://img.cochemist.com/ccimg/78200/78134-74-4.png)
![Ethanaminium, N-[(benzoylamino)methyl]-N,N-diethyl-, chloride](http://img.cochemist.com/ccimg/78200/78134-74-4_b.png)
![Benzamide, N,N',N''-[nitrilotris(methylene)]tris-](http://img.cochemist.com/ccimg/52000/51912-07-3.png)
![Benzamide, N,N',N''-[nitrilotris(methylene)]tris-](http://img.cochemist.com/ccimg/52000/51912-07-3_b.png)
![2H-Imidazole-2-thione, 1,3-dihydro-1-[(1S)-1-phenylethyl]-](http://img.cochemist.com/ccimg/928400/928331-18-4.png)
![2H-Imidazole-2-thione, 1,3-dihydro-1-[(1S)-1-phenylethyl]-](http://img.cochemist.com/ccimg/928400/928331-18-4_b.png)








![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)




![(2,6,6-trimethylbicyclo[3.1.1]heptan-3-yl)borane](http://img.cochemist.com/ccimg/64300/64234-27-1.png)
![(2,6,6-trimethylbicyclo[3.1.1]heptan-3-yl)borane](http://img.cochemist.com/ccimg/64300/64234-27-1_b.png)







![Benzenamine,N-[[5-[(phenylamino)methylene]-1,3-cyclopentadien-1-yl]methylene]-](http://img.cochemist.com/ccimg/29900/29836-96-2.png)
![Benzenamine,N-[[5-[(phenylamino)methylene]-1,3-cyclopentadien-1-yl]methylene]-](http://img.cochemist.com/ccimg/29900/29836-96-2_b.png)


![Borane, dichloro[(1R,2S,3R,5R)-2,6,6-trimethylbicyclo[3.1.1]hept-3-yl]-](http://img.cochemist.com/ccimg/121400/121326-42-9.png)
![Borane, dichloro[(1R,2S,3R,5R)-2,6,6-trimethylbicyclo[3.1.1]hept-3-yl]-](http://img.cochemist.com/ccimg/121400/121326-42-9_b.png)