
No example of a simple uncatalyzed dimerization of a diaminocarbene has been clearly established, so it is timely to ask what factors control the thermodynamics of this reaction, and what mechanisms are responsible for the observed dimerizations? In agreement with qualitative experimental observations, the dimerizations of simple five- and six-membered-ring diaminocarbenes are calculated to be 100 kJ mol−1 less favorable than those of acyclic counterparts. This large difference is semiquantitatively accounted for by bond and torsional angle changes around the carbene centers. Carbenes such as (Et2N)2C are kinetically stable in THF at 25 °C in agreement with calculated energy barriers, but they rapidly dimerize in the presence of the corresponding formamidinium ion. This proton-catalyzed process is probably the most common mechanism for dimer formation, and involves formation of C-protonated dimers, which can be observed in suitable cases. The possibility of alkali-metal-promoted dimerization is raised, and circumstantial evidence for this is presented.
Bisher wurde kein eindeutiger Nachweis für eine einfache, nicht katalysierte Dimerisierung von Diaminocarbenen gefunden. Es ist somit an der Zeit zu hinterfragen, welche Faktoren die Thermodynamik dieser Reaktion steuern und welche Mechanismen für die beobachteten Dimerisierungen verantwortlich sind. In Übereinstimmung mit qualitativen experimentellen Beobachtungen ergab die Berechnung, dass die Dimerisierung von einfachen Fünf- und Sechsringdiaminocarbenen um 100 kJ mol−1 weniger begünstigt ist als die der acyclischen Pendants. Dieser große Unterschied kann halbquantitativ durch die Änderung von Bindungs- und Torsionswinkeln um die Carbenzentren herum erklärt werden. Carbene wie (Et2N)2C sind, in Einklang mit den berechneten Energiebarrieren, in THF bei 25 °C kinetisch stabil, aber sie dimerisieren schnell in Gegenwart des entsprechenden Formamidiniumions. Dieser protonenkatalysierte Prozess ist wahrscheinlich der häufigste Mechanismus der Dimerbildung. Er führt zur Bildung von C-protonierten Dimeren, was in geeigneten Fällen beobachtet werden kann. Es wird auch die Möglichkeit der durch Alkalimetalle geförderten Dimerisierung diskutiert; hierfür werden einige Belege vorgestellt.
![L-arabino-Hex-1-enitol, 1,5-anhydro-2,6-dideoxy-3-O-[(1,1-dimethylethyl)dimethylsilyl]-](http://img.cochemist.com/ccimg/101900/101856-96-6.png)
![L-arabino-Hex-1-enitol, 1,5-anhydro-2,6-dideoxy-3-O-[(1,1-dimethylethyl)dimethylsilyl]-](http://img.cochemist.com/ccimg/101900/101856-96-6_b.png)


bis(2-methyl-2-propanolato)-,(T-4)-](http://img.cochemist.com/ccimg/127000/126949-65-3.png)
bis(2-methyl-2-propanolato)-,(T-4)-](http://img.cochemist.com/ccimg/127000/126949-65-3_b.png)
![5,7-dichloro-3-{2-[(3-methoxy-4-oxocyclohexa-2,5-dien-1-ylidene)methyl]hydrazino}-2H-indol-2-one](http://img.cochemist.com/ccimg/6600/6573-14-4.png)
![5,7-dichloro-3-{2-[(3-methoxy-4-oxocyclohexa-2,5-dien-1-ylidene)methyl]hydrazino}-2H-indol-2-one](http://img.cochemist.com/ccimg/6600/6573-14-4_b.png)
![undecacyclo[9.9.0.0~2,9~.0~3,7~.0~4,20~.0~5,18~.0~6,16~.0~8,15~.0~10,14~.0~12,19~.0~13,17~]icosane (non-preferred name)](/data/chemimg/51900/4493-23-6.png)
![undecacyclo[9.9.0.0~2,9~.0~3,7~.0~4,20~.0~5,18~.0~6,16~.0~8,15~.0~10,14~.0~12,19~.0~13,17~]icosane (non-preferred name)](/data/chemimg/51900/4493-23-6_b.png)
![D-arabino-Hex-1-enitol,1,5-anhydro-2-deoxy-3,6-bis-O-[(1,1-dimethylethyl)dimethylsilyl]-](http://img.cochemist.com/ccimg/111900/111830-53-6.png)
![D-arabino-Hex-1-enitol,1,5-anhydro-2-deoxy-3,6-bis-O-[(1,1-dimethylethyl)dimethylsilyl]-](http://img.cochemist.com/ccimg/111900/111830-53-6_b.png)
![2H-Pyran, 2-[(4-bromo-3-butynyl)oxy]tetrahydro-](http://img.cochemist.com/ccimg/84600/84509-61-5.png)
![2H-Pyran, 2-[(4-bromo-3-butynyl)oxy]tetrahydro-](http://img.cochemist.com/ccimg/84600/84509-61-5_b.png)
















![2-Oxatricyclo[3.3.1.13,7]decane, 5-bromo-](http://img.cochemist.com/ccimg/94100/94053-22-2.png)
![2-Oxatricyclo[3.3.1.13,7]decane, 5-bromo-](http://img.cochemist.com/ccimg/94100/94053-22-2_b.png)
![1,6-Diphosphabicyclo[4.4.4]tetradecane](http://img.cochemist.com/ccimg/50900/50805-71-5.png)
![1,6-Diphosphabicyclo[4.4.4]tetradecane](http://img.cochemist.com/ccimg/50900/50805-71-5_b.png)
![Cyclopenta[b]pyran, octahydro-](http://img.cochemist.com/ccimg/10400/10347-53-2.png)
![Cyclopenta[b]pyran, octahydro-](http://img.cochemist.com/ccimg/10400/10347-53-2_b.png)
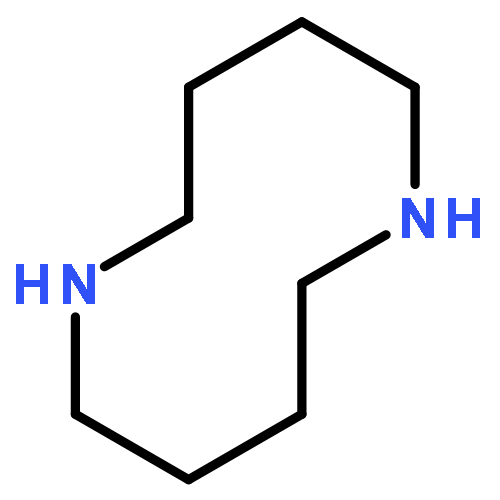
![1,6-Diazabicyclo[4.4.3]tridecane-11-carbonitrile](http://img.cochemist.com/ccimg/90900/90827-77-3.png)
![1,6-Diazabicyclo[4.4.3]tridecane-11-carbonitrile](http://img.cochemist.com/ccimg/90900/90827-77-3_b.png)


![1H,5H-[1,2]Diphospholo[1,2-a][1,2]diphosphole, tetrahydro-](http://img.cochemist.com/ccimg/66900/66872-86-4.png)
![1H,5H-[1,2]Diphospholo[1,2-a][1,2]diphosphole, tetrahydro-](http://img.cochemist.com/ccimg/66900/66872-86-4_b.png)
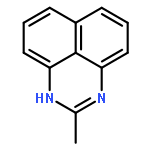
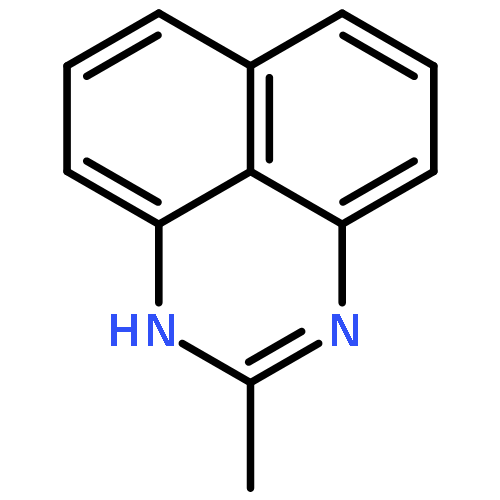
![1H-Naphtho[1,8-bc]-1,5-diazocine, 2,3,4,5-tetrahydro-1,5-dimethyl-](http://img.cochemist.com/ccimg/80800/80703-35-1.png)
![1H-Naphtho[1,8-bc]-1,5-diazocine, 2,3,4,5-tetrahydro-1,5-dimethyl-](http://img.cochemist.com/ccimg/80800/80703-35-1_b.png)



![Cyclohexanol, 1-[1-(phenylthio)cyclopropyl]-](http://img.cochemist.com/ccimg/70100/70042-50-1.png)
![Cyclohexanol, 1-[1-(phenylthio)cyclopropyl]-](http://img.cochemist.com/ccimg/70100/70042-50-1_b.png)
![1,5,9,13-Tetraazatricyclo[11.3.1.15,9]octadecane](http://img.cochemist.com/ccimg/69000/68966-09-6.png)
![1,5,9,13-Tetraazatricyclo[11.3.1.15,9]octadecane](http://img.cochemist.com/ccimg/69000/68966-09-6_b.png)



![1H-Naphtho[1,8-bc][1,5]diazecine, 2,3,4,5,6,7-hexahydro-1,7-dimethyl-](http://img.cochemist.com/ccimg/80800/80703-37-3.png)
![1H-Naphtho[1,8-bc][1,5]diazecine, 2,3,4,5,6,7-hexahydro-1,7-dimethyl-](http://img.cochemist.com/ccimg/80800/80703-37-3_b.png)



![6-Aza-1-azoniabicyclo[4.4.1]undecane, 1-methyl-](http://img.cochemist.com/ccimg/90900/90827-82-0.png)
![6-Aza-1-azoniabicyclo[4.4.1]undecane, 1-methyl-](http://img.cochemist.com/ccimg/90900/90827-82-0_b.png)
![1,5-Diazabicyclo[3.3.3]undecane-2-carbonitrile](http://img.cochemist.com/ccimg/90900/90827-75-1.png)
![1,5-Diazabicyclo[3.3.3]undecane-2-carbonitrile](http://img.cochemist.com/ccimg/90900/90827-75-1_b.png)
![ETHANAMINIUM, N-[(DIETHYLAMINO)METHYLENE]-N-ETHYL-](http://img.cochemist.com/ccimg/45000/44979-14-8.png)
![ETHANAMINIUM, N-[(DIETHYLAMINO)METHYLENE]-N-ETHYL-](http://img.cochemist.com/ccimg/45000/44979-14-8_b.png)

















![Dispiro[cyclohexane-1,2'-[1,3]dioxocane-6',9''-[9H]fluorene]](http://img.cochemist.com/ccimg/203100/203070-79-5.png)
![Dispiro[cyclohexane-1,2'-[1,3]dioxocane-6',9''-[9H]fluorene]](http://img.cochemist.com/ccimg/203100/203070-79-5_b.png)
![9H-Fluorene, 9,9-bis[2-(9H-fluoren-9-yl)ethyl]-](http://img.cochemist.com/ccimg/203100/203070-72-8.png)
![9H-Fluorene, 9,9-bis[2-(9H-fluoren-9-yl)ethyl]-](http://img.cochemist.com/ccimg/203100/203070-72-8_b.png)
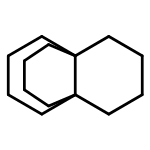






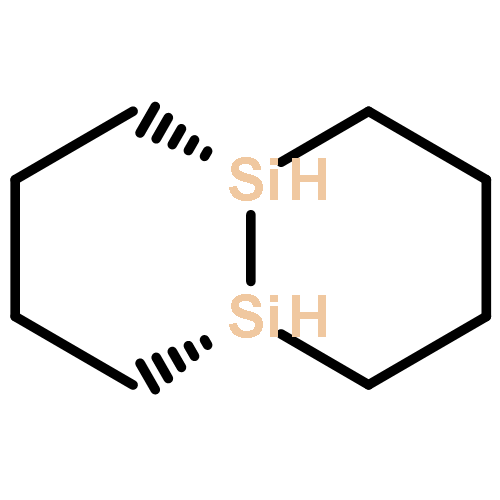


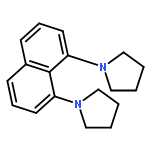



![2H-Cyclopent[b]oxepin, octahydro-](http://img.cochemist.com/ccimg/123100/123061-37-0.png)
![2H-Cyclopent[b]oxepin, octahydro-](http://img.cochemist.com/ccimg/123100/123061-37-0_b.png)
![2(3H)-Thiazolylidene, 3-[2,6-bis(1-methylethyl)phenyl]-4,5-dimethyl-](http://img.cochemist.com/ccimg/188500/188428-94-6.png)
![2(3H)-Thiazolylidene, 3-[2,6-bis(1-methylethyl)phenyl]-4,5-dimethyl-](http://img.cochemist.com/ccimg/188500/188428-94-6_b.png)




![2H-Furo[1,2-a][1,3]dioxin-5-ium, hexahydro-](http://img.cochemist.com/ccimg/123100/123061-35-8.png)
![2H-Furo[1,2-a][1,3]dioxin-5-ium, hexahydro-](http://img.cochemist.com/ccimg/123100/123061-35-8_b.png)


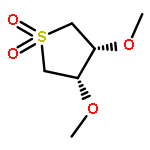





![SPIRO[3.5]NONAN-1-ONE, 7-(1,1-DIMETHYLETHYL)-](http://img.cochemist.com/ccimg/41500/41487-65-4.png)
![SPIRO[3.5]NONAN-1-ONE, 7-(1,1-DIMETHYLETHYL)-](http://img.cochemist.com/ccimg/41500/41487-65-4_b.png)


![1H,5H-Pyrazolo[1,2-a]pyrazolium, tetrahydro-4-methyl-, iodide](http://img.cochemist.com/ccimg/18500/18477-85-5.png)
![1H,5H-Pyrazolo[1,2-a]pyrazolium, tetrahydro-4-methyl-, iodide](http://img.cochemist.com/ccimg/18500/18477-85-5_b.png)

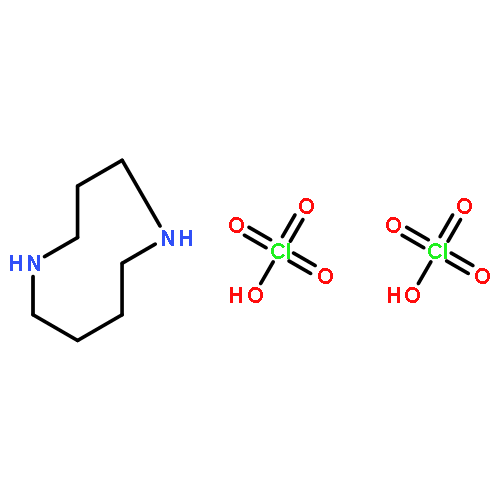




![1,6-DIAZABICYCLO[4.4.4]TETRADECANE-2-CARBONITRILE](http://img.cochemist.com/ccimg/90900/90827-78-4.png)
![1,6-DIAZABICYCLO[4.4.4]TETRADECANE-2-CARBONITRILE](http://img.cochemist.com/ccimg/90900/90827-78-4_b.png)
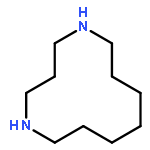

![1,9-Diazabicyclo[7.3.1]tridecane](http://img.cochemist.com/ccimg/112700/112623-88-8.png)
![1,9-Diazabicyclo[7.3.1]tridecane](http://img.cochemist.com/ccimg/112700/112623-88-8_b.png)




![6H-BENZO[C]QUINOLIZIN-6-ONE](http://img.cochemist.com/ccimg/118200/118199-87-4.png)
![6H-BENZO[C]QUINOLIZIN-6-ONE](http://img.cochemist.com/ccimg/118200/118199-87-4_b.png)
![NAPHTHO[1,8-BC][1,5]DIAZONINE, 1,2,3,4,5,6-HEXAHYDRO-1,6-DIMETHYL-](http://img.cochemist.com/ccimg/80800/80703-36-2.png)
![NAPHTHO[1,8-BC][1,5]DIAZONINE, 1,2,3,4,5,6-HEXAHYDRO-1,6-DIMETHYL-](http://img.cochemist.com/ccimg/80800/80703-36-2_b.png)
![NAPHTHO[1,8-EF]-1,4-DIAZEPINE, 1,2,3,4-TETRAHYDRO-1,4-DIMETHYL-](http://img.cochemist.com/ccimg/80800/80703-34-0.png)
![NAPHTHO[1,8-EF]-1,4-DIAZEPINE, 1,2,3,4-TETRAHYDRO-1,4-DIMETHYL-](http://img.cochemist.com/ccimg/80800/80703-34-0_b.png)




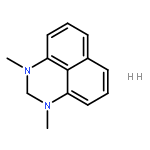
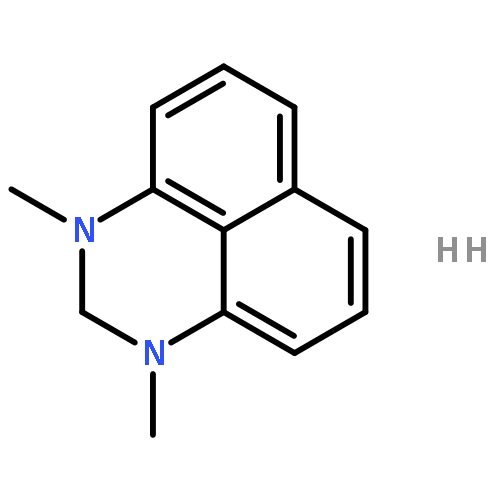
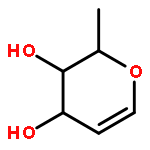
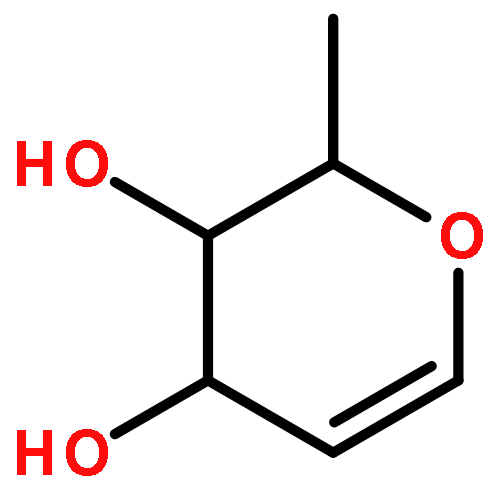
![2H-PYRIMIDO[1,2-A]AZOCINE, 3,4,6,7,8,9,10,11-OCTAHYDRO-](http://img.cochemist.com/ccimg/58400/58379-23-0.png)
![2H-PYRIMIDO[1,2-A]AZOCINE, 3,4,6,7,8,9,10,11-OCTAHYDRO-](http://img.cochemist.com/ccimg/58400/58379-23-0_b.png)






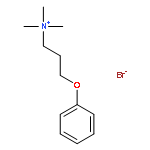





![1,8-Diazabicyclo[6.3.3]tetradecane](http://img.cochemist.com/ccimg/82500/82407-82-7.png)
![1,8-Diazabicyclo[6.3.3]tetradecane](http://img.cochemist.com/ccimg/82500/82407-82-7_b.png)
![1,7-Diazabicyclo[5.5.3]pentadecane](http://img.cochemist.com/ccimg/85000/84905-02-2.png)
![1,7-Diazabicyclo[5.5.3]pentadecane](http://img.cochemist.com/ccimg/85000/84905-02-2_b.png)
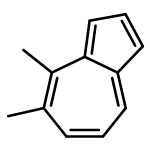

![dichloropalladium; diphenyl-[(4-phenyl-4,5-dihydrooxazol-2-yl)methyl]phosphonium](http://img.cochemist.com/ccimg/7300/7234-56-2.png)
![dichloropalladium; diphenyl-[(4-phenyl-4,5-dihydrooxazol-2-yl)methyl]phosphonium](http://img.cochemist.com/ccimg/7300/7234-56-2_b.png)
![7-Chloro-6-hydroxybenzo[c]quinolizinium chloride](http://img.cochemist.com/ccimg/191100/191091-58-4.png)
![7-Chloro-6-hydroxybenzo[c]quinolizinium chloride](http://img.cochemist.com/ccimg/191100/191091-58-4_b.png)
![1,6-Diazabicyclo[4.3.1]decane](http://img.cochemist.com/ccimg/25400/25337-75-1.png)
![1,6-Diazabicyclo[4.3.1]decane](http://img.cochemist.com/ccimg/25400/25337-75-1_b.png)

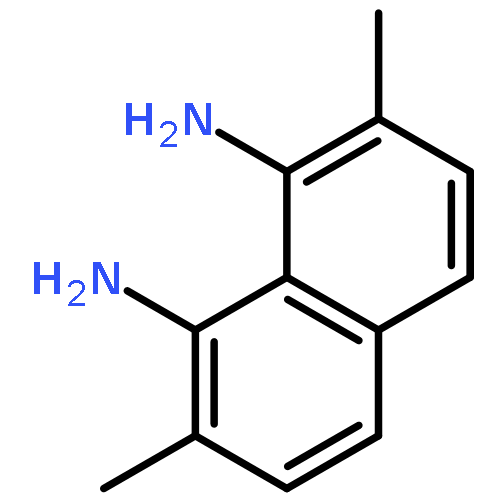

![octahydro-2H-Pyrido[1,2-a]pyrimidine](http://img.cochemist.com/ccimg/19100/19068-79-2.png)
![octahydro-2H-Pyrido[1,2-a]pyrimidine](http://img.cochemist.com/ccimg/19100/19068-79-2_b.png)








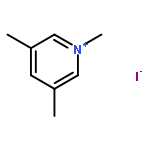
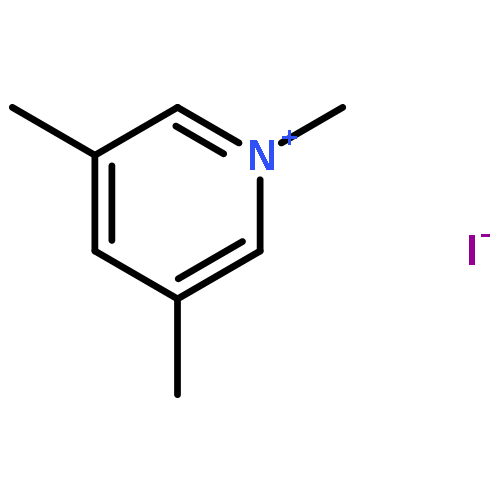
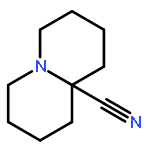
![1-Azabicyclo[4.4.4]tetradecane](http://img.cochemist.com/ccimg/75200/75197-24-9.png)
![1-Azabicyclo[4.4.4]tetradecane](http://img.cochemist.com/ccimg/75200/75197-24-9_b.png)
![1,7-Diazabicyclo[5.5.2]tetradecane](http://img.cochemist.com/ccimg/85000/84905-00-0.png)
![1,7-Diazabicyclo[5.5.2]tetradecane](http://img.cochemist.com/ccimg/85000/84905-00-0_b.png)
![1-Azabicyclo[4.4.4]tetradec-5-ene](http://img.cochemist.com/ccimg/84600/84509-55-7.png)
![1-Azabicyclo[4.4.4]tetradec-5-ene](http://img.cochemist.com/ccimg/84600/84509-55-7_b.png)
![tricyclo[3.3.1.1~3,7~]dec-1-yl](http://img.cochemist.com/ccimg/19800/19740-18-2.png)
![tricyclo[3.3.1.1~3,7~]dec-1-yl](http://img.cochemist.com/ccimg/19800/19740-18-2_b.png)














![1,6-Diazabicyclo[4.4.2]dodecane](http://img.cochemist.com/ccimg/88900/88885-89-6.png)
![1,6-Diazabicyclo[4.4.2]dodecane](http://img.cochemist.com/ccimg/88900/88885-89-6_b.png)




![Diindeno[1,2,3,4-defg:1',2',3',4'-mnop]chrysene](http://img.cochemist.com/ccimg/169400/169331-80-0.png)
![Diindeno[1,2,3,4-defg:1',2',3',4'-mnop]chrysene](http://img.cochemist.com/ccimg/169400/169331-80-0_b.png)
![octahydro-1H-Pyrrolo[1,2-a][1,3]diazepine](http://img.cochemist.com/ccimg/122000/121994-94-3.png)
![octahydro-1H-Pyrrolo[1,2-a][1,3]diazepine](http://img.cochemist.com/ccimg/122000/121994-94-3_b.png)


![3,4-dihydro-2H-1,5-propanonaphtho[1,8-bc][1,5]diazocine](http://img.cochemist.com/ccimg/60000/59950-40-2.png)
![3,4-dihydro-2H-1,5-propanonaphtho[1,8-bc][1,5]diazocine](http://img.cochemist.com/ccimg/60000/59950-40-2_b.png)








![1,6-diazabicyclo[4.3.2]undecane](http://img.cochemist.com/ccimg/82500/82407-81-6.png)
![1,6-diazabicyclo[4.3.2]undecane](http://img.cochemist.com/ccimg/82500/82407-81-6_b.png)
![1,7-diazabicyclo[5.3.3]tridecane](http://img.cochemist.com/ccimg/85000/84904-98-3.png)
![1,7-diazabicyclo[5.3.3]tridecane](http://img.cochemist.com/ccimg/85000/84904-98-3_b.png)






![4-(3-hydroxypropyl)tetrahydro-1H,5H-pyrazolo[1,2-a]pyrazol-4-ium](http://img.cochemist.com/ccimg/66400/66314-45-2.png)
![4-(3-hydroxypropyl)tetrahydro-1H,5H-pyrazolo[1,2-a]pyrazol-4-ium](http://img.cochemist.com/ccimg/66400/66314-45-2_b.png)
![5,9-Diaza-1-azoniabicyclo[7.3.1]tridec-1(13)-ene, 5-methyl-, iodide](http://img.cochemist.com/ccimg/111000/110969-92-1.png)
![5,9-Diaza-1-azoniabicyclo[7.3.1]tridec-1(13)-ene, 5-methyl-, iodide](http://img.cochemist.com/ccimg/111000/110969-92-1_b.png)
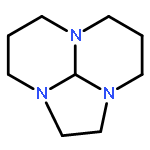
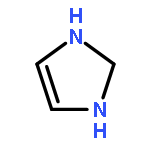
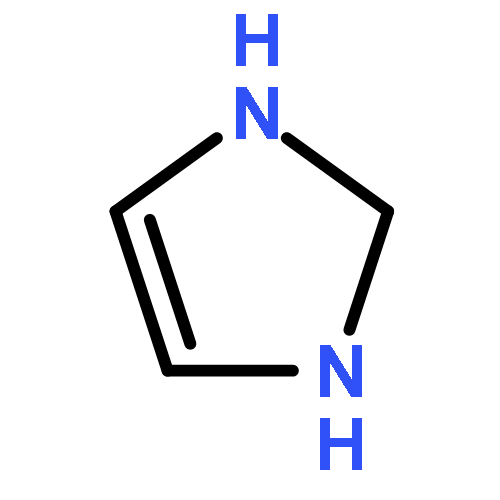
![1H-Imidazo[1,2-a]imidazole, 2,3,5,6-tetrahydro-](http://img.cochemist.com/ccimg/6600/6573-15-5.png)
![1H-Imidazo[1,2-a]imidazole, 2,3,5,6-tetrahydro-](http://img.cochemist.com/ccimg/6600/6573-15-5_b.png)
![Spiro[cyclopropane-1,9'-[9H]fluorene]](http://img.cochemist.com/ccimg/200/167-02-2.png)
![Spiro[cyclopropane-1,9'-[9H]fluorene]](http://img.cochemist.com/ccimg/200/167-02-2_b.png)
![2,3-dihydro-1,4-ethanonaphtho[1,8-ef][1,4]diazepine](http://img.cochemist.com/ccimg/60000/59950-41-3.png)
![2,3-dihydro-1,4-ethanonaphtho[1,8-ef][1,4]diazepine](http://img.cochemist.com/ccimg/60000/59950-41-3_b.png)




![1,5,9-Triazabicyclo[7.3.1]tridecane, 5-methyl-](http://img.cochemist.com/ccimg/139300/139258-70-1.png)
![1,5,9-Triazabicyclo[7.3.1]tridecane, 5-methyl-](http://img.cochemist.com/ccimg/139300/139258-70-1_b.png)




![1,8-diazabicyclo[6.5.3]hexadecane](http://img.cochemist.com/ccimg/85000/84905-04-4.png)
![1,8-diazabicyclo[6.5.3]hexadecane](http://img.cochemist.com/ccimg/85000/84905-04-4_b.png)
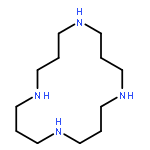

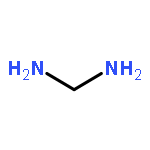



![octahydro-midazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/85000/84905-07-7.png)
![octahydro-midazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/85000/84905-07-7_b.png)


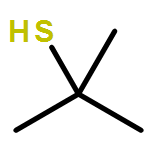





![1,5,10-Triazabicyclo[8.3.1]tetradecane](http://img.cochemist.com/ccimg/65000/64924-82-9.png)
![1,5,10-Triazabicyclo[8.3.1]tetradecane](http://img.cochemist.com/ccimg/65000/64924-82-9_b.png)
![1,6-Diazabicyclo[4.3.3]dodecane](http://img.cochemist.com/ccimg/66400/66314-44-1.png)
![1,6-Diazabicyclo[4.3.3]dodecane](http://img.cochemist.com/ccimg/66400/66314-44-1_b.png)




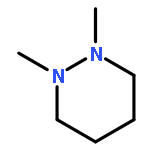









![(4e)-3-methyl-4-[(e)-3-phenylprop-2-enylidene]-1,2-oxazol-5-one](http://img.cochemist.com/ccimg/18000/17975-54-1.png)
![(4e)-3-methyl-4-[(e)-3-phenylprop-2-enylidene]-1,2-oxazol-5-one](http://img.cochemist.com/ccimg/18000/17975-54-1_b.png)
![1,5,9-Triazabicyclo[7.3.1]tridecane](http://img.cochemist.com/ccimg/111600/111560-13-5.png)
![1,5,9-Triazabicyclo[7.3.1]tridecane](http://img.cochemist.com/ccimg/111600/111560-13-5_b.png)



![1,7-Diazabicyclo[5.3.2]dodecane](http://img.cochemist.com/ccimg/85000/84904-97-2.png)
![1,7-Diazabicyclo[5.3.2]dodecane](http://img.cochemist.com/ccimg/85000/84904-97-2_b.png)
![1,7-Diazabicyclo[5.4.2]tridecane](http://img.cochemist.com/ccimg/85000/84904-99-4.png)
![1,7-Diazabicyclo[5.4.2]tridecane](http://img.cochemist.com/ccimg/85000/84904-99-4_b.png)
![1,8-Diazabicyclo[6.3.2]tridecane](http://img.cochemist.com/ccimg/85000/84905-06-6.png)
![1,8-Diazabicyclo[6.3.2]tridecane](http://img.cochemist.com/ccimg/85000/84905-06-6_b.png)
![1,7-Diazabicyclo[5.4.3]tetradecane](http://img.cochemist.com/ccimg/85000/84905-01-1.png)
![1,7-Diazabicyclo[5.4.3]tetradecane](http://img.cochemist.com/ccimg/85000/84905-01-1_b.png)
![1,7-Diazabicyclo[5.4.4]pentadecane](http://img.cochemist.com/ccimg/88900/88885-91-0.png)
![1,7-Diazabicyclo[5.4.4]pentadecane](http://img.cochemist.com/ccimg/88900/88885-91-0_b.png)



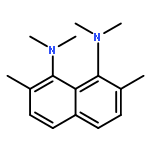



![1,8-Diazabicyclo[6.4.2]tetradecane](http://img.cochemist.com/ccimg/88900/88885-90-9.png)
![1,8-Diazabicyclo[6.4.2]tetradecane](http://img.cochemist.com/ccimg/88900/88885-90-9_b.png)




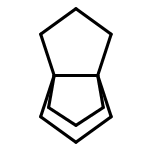

![Hexahydropyrazolo[1,2-a]pyrazole](http://img.cochemist.com/ccimg/5400/5397-67-1.png)
![Hexahydropyrazolo[1,2-a]pyrazole](http://img.cochemist.com/ccimg/5400/5397-67-1_b.png)
![1,6-Diazabicyclo[4.4.3]tridecane](http://img.cochemist.com/ccimg/79500/79401-85-7.png)
![1,6-Diazabicyclo[4.4.3]tridecane](http://img.cochemist.com/ccimg/79500/79401-85-7_b.png)


![5,9-Diaza-1-azoniabicyclo[7.3.1]tridec-1(13)-ene, 5-methyl-](http://img.cochemist.com/ccimg/111000/110969-91-0.png)
![5,9-Diaza-1-azoniabicyclo[7.3.1]tridec-1(13)-ene, 5-methyl-](http://img.cochemist.com/ccimg/111000/110969-91-0_b.png)
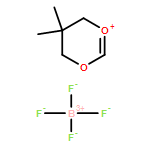
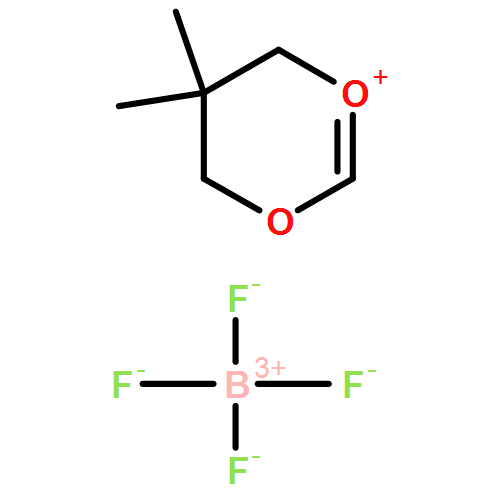
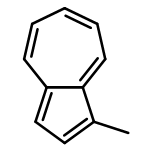



![1,5,9-Triazacyclododecane, 1-methyl-5-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/141100/141081-92-7.png)
![1,5,9-Triazacyclododecane, 1-methyl-5-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/141100/141081-92-7_b.png)





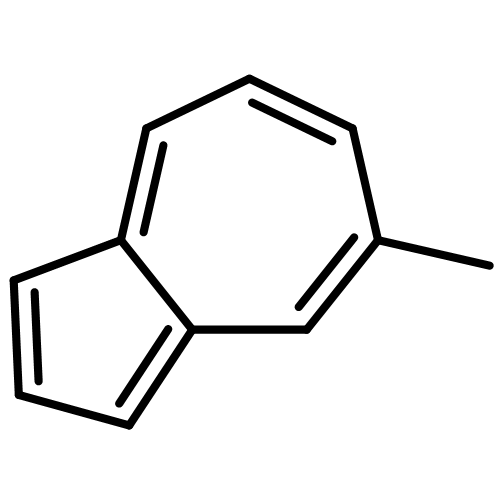


![2H-Pyrimido[1,2-a]pyrimidine, 1,3,4,6,7,8-hexahydro-1-(methylethenyl)-](http://img.cochemist.com/ccimg/141100/141081-91-6.png)
![2H-Pyrimido[1,2-a]pyrimidine, 1,3,4,6,7,8-hexahydro-1-(methylethenyl)-](http://img.cochemist.com/ccimg/141100/141081-91-6_b.png)

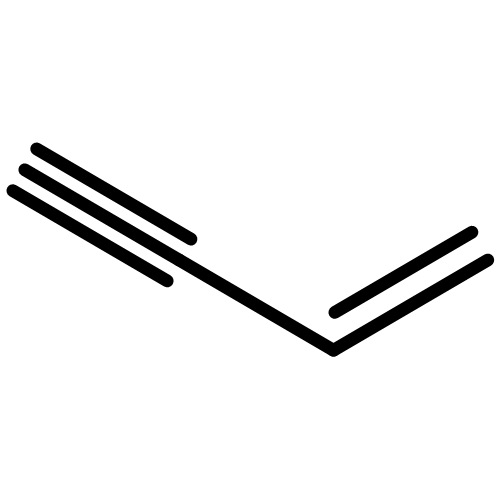


![1,6-DIAZABICYCLO[4.4.4]TETRADECANE](http://img.cochemist.com/ccimg/71100/71058-67-8.png)
![1,6-DIAZABICYCLO[4.4.4]TETRADECANE](http://img.cochemist.com/ccimg/71100/71058-67-8_b.png)
![2,4,8,10-Tetraoxaspiro[5.5]undecane, 3,3,9,9-tetramethyl-](http://img.cochemist.com/ccimg/29300/29280-21-5.png)
![2,4,8,10-Tetraoxaspiro[5.5]undecane, 3,3,9,9-tetramethyl-](http://img.cochemist.com/ccimg/29300/29280-21-5_b.png)
![1,5,10,14-Tetraoxadispiro[5.2.5.2]hexadecane, 3,3,12,12-tetramethyl-](http://img.cochemist.com/ccimg/29300/29280-23-7.png)
![1,5,10,14-Tetraoxadispiro[5.2.5.2]hexadecane, 3,3,12,12-tetramethyl-](http://img.cochemist.com/ccimg/29300/29280-23-7_b.png)
![1,5-Diazabicyclo[3.3.2]decane](http://img.cochemist.com/ccimg/300/283-52-3.png)
![1,5-Diazabicyclo[3.3.2]decane](http://img.cochemist.com/ccimg/300/283-52-3_b.png)

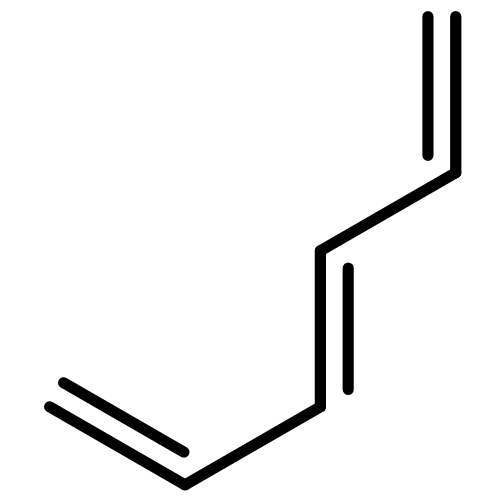
![8-Azabicyclo[3.2.1]octane,3-chloro-8-methyl-, (3-exo)-](http://img.cochemist.com/ccimg/2300/2292-12-8.png)
![8-Azabicyclo[3.2.1]octane,3-chloro-8-methyl-, (3-exo)-](http://img.cochemist.com/ccimg/2300/2292-12-8_b.png)
![Benz[f]azulene](http://img.cochemist.com/ccimg/300/267-00-5.png)
![Benz[f]azulene](http://img.cochemist.com/ccimg/300/267-00-5_b.png)








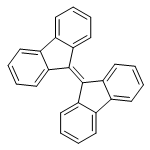
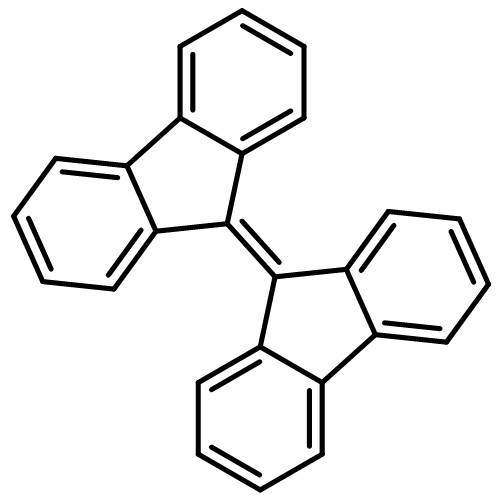


![Methanaminium,N-[(dimethylamino)methylene]-N-methyl-](http://img.cochemist.com/ccimg/38600/38571-87-8.png)
![Methanaminium,N-[(dimethylamino)methylene]-N-methyl-](http://img.cochemist.com/ccimg/38600/38571-87-8_b.png)



![Methylene, [bis[bis(1-methylethyl)amino]phosphino](trimethylsilyl)-](http://img.cochemist.com/ccimg/122100/122048-59-3.png)
![Methylene, [bis[bis(1-methylethyl)amino]phosphino](trimethylsilyl)-](http://img.cochemist.com/ccimg/122100/122048-59-3_b.png)






![[5-(HYDROXYMETHYL)-2,2-DIMETHYL-1,3-DIOXAN-5-YL]METHANOL](http://img.cochemist.com/ccimg/800/770-74-1.png)
![[5-(HYDROXYMETHYL)-2,2-DIMETHYL-1,3-DIOXAN-5-YL]METHANOL](http://img.cochemist.com/ccimg/800/770-74-1_b.png)




























![1,8-Diazabicyclo[6.4.3]pentadecane(9CI)](http://img.cochemist.com/ccimg/85000/84905-03-3.png)
![1,8-Diazabicyclo[6.4.3]pentadecane(9CI)](http://img.cochemist.com/ccimg/85000/84905-03-3_b.png)


![1,7-Diazabicyclo[5.5.4]hexadecane(9CI)](http://img.cochemist.com/ccimg/88900/88885-92-1.png)
![1,7-Diazabicyclo[5.5.4]hexadecane(9CI)](http://img.cochemist.com/ccimg/88900/88885-92-1_b.png)

![1,4,8,11-TETRAAZATRICYCLO[9.3.1.1(4,8)]HEXADECANE](http://img.cochemist.com/ccimg/76000/75920-10-4.png)
![1,4,8,11-TETRAAZATRICYCLO[9.3.1.1(4,8)]HEXADECANE](http://img.cochemist.com/ccimg/76000/75920-10-4_b.png)






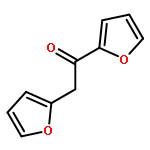
![2-[2-(FURAN-2-YL)ETHYL]FURAN](http://img.cochemist.com/ccimg/36800/36707-31-0.png)
![2-[2-(FURAN-2-YL)ETHYL]FURAN](http://img.cochemist.com/ccimg/36800/36707-31-0_b.png)








![1-[4-(4-FLUOROPHENOXY)BUTYL]-4-(2-METHOXYPHENYL)PIPERAZIN-1-IUM](http://img.cochemist.com/ccimg/5800/5703-88-8.png)
![1-[4-(4-FLUOROPHENOXY)BUTYL]-4-(2-METHOXYPHENYL)PIPERAZIN-1-IUM](http://img.cochemist.com/ccimg/5800/5703-88-8_b.png)








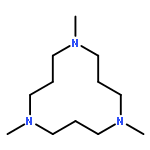
![1,3-Propanediamine, N,N'-bis[2-(dimethylamino)ethyl]-N,N'-dimethyl-](http://img.cochemist.com/ccimg/139300/139258-71-2.png)
![1,3-Propanediamine, N,N'-bis[2-(dimethylamino)ethyl]-N,N'-dimethyl-](http://img.cochemist.com/ccimg/139300/139258-71-2_b.png)
![1,5-Diazabicyclo[3.3.3]undecane(8CI,9CI)](http://img.cochemist.com/ccimg/300/283-58-9.png)
![1,5-Diazabicyclo[3.3.3]undecane(8CI,9CI)](http://img.cochemist.com/ccimg/300/283-58-9_b.png)

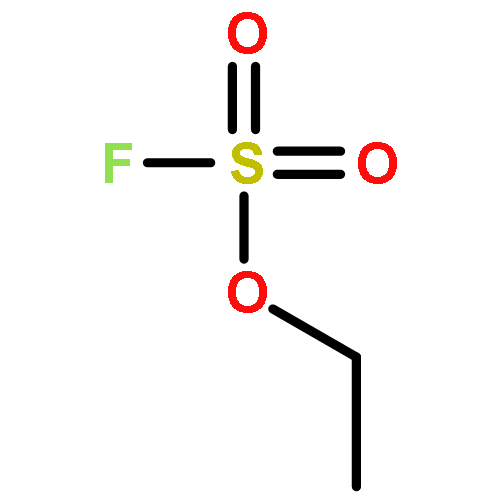
![1,2,3,4,6,7,8,9-OCTAHYDROPYRIDAZINO[1,2-A]PYRIDAZINE](http://img.cochemist.com/ccimg/3700/3661-15-2.png)
![1,2,3,4,6,7,8,9-OCTAHYDROPYRIDAZINO[1,2-A]PYRIDAZINE](http://img.cochemist.com/ccimg/3700/3661-15-2_b.png)
![1,5-Diazabicyclo[3.3.1]nonane](http://img.cochemist.com/ccimg/300/281-17-4.png)
![1,5-Diazabicyclo[3.3.1]nonane](http://img.cochemist.com/ccimg/300/281-17-4_b.png)




![1,5-Diazabicyclo[3.2.1]octane](http://img.cochemist.com/ccimg/300/280-28-4.png)
![1,5-Diazabicyclo[3.2.1]octane](http://img.cochemist.com/ccimg/300/280-28-4_b.png)
![Spiro[3.5]nonan-1-one](http://img.cochemist.com/ccimg/29900/29800-45-1.png)
![Spiro[3.5]nonan-1-one](http://img.cochemist.com/ccimg/29900/29800-45-1_b.png)



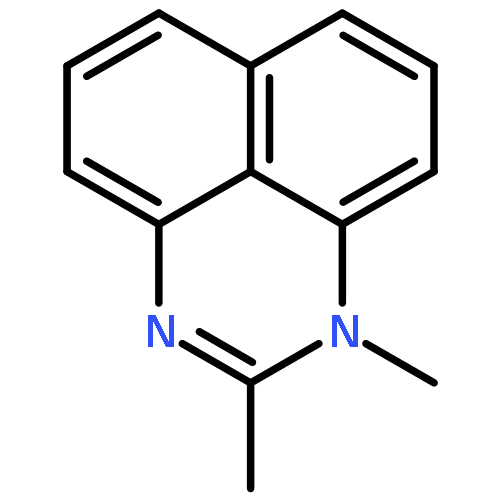
![1-AZABICYCLO[3.3.3]UNDECANE](http://img.cochemist.com/ccimg/31100/31023-92-4.png)
![1-AZABICYCLO[3.3.3]UNDECANE](http://img.cochemist.com/ccimg/31100/31023-92-4_b.png)


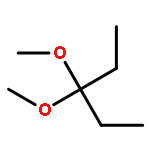
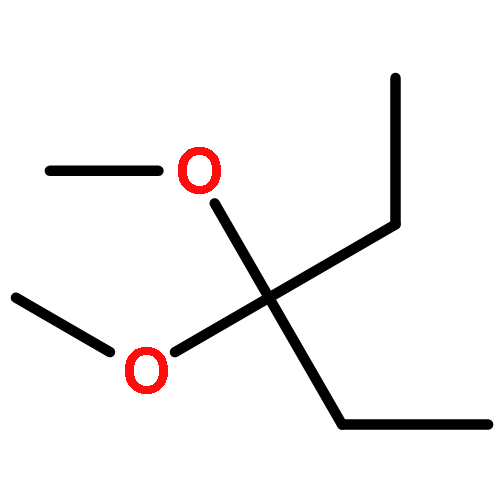




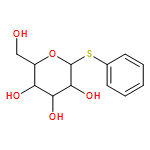
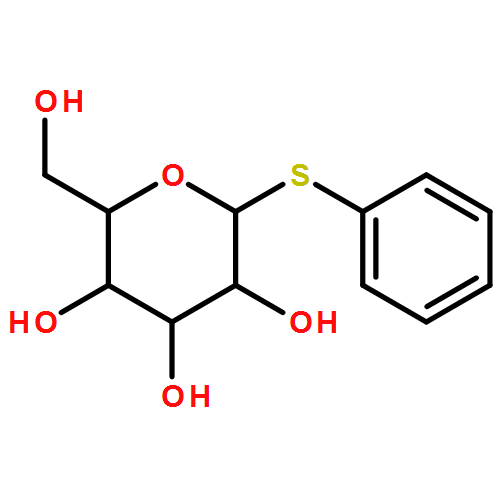








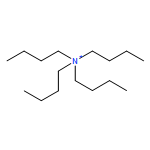
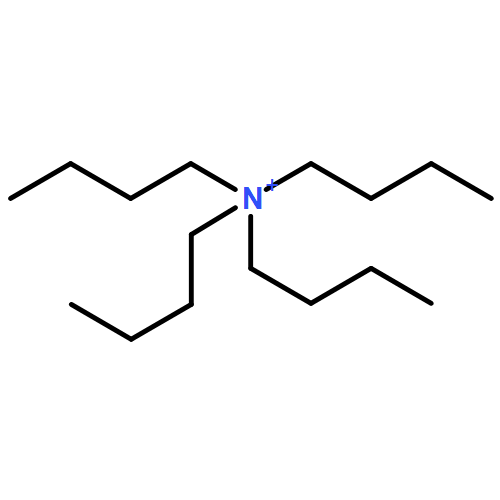




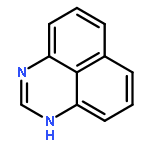
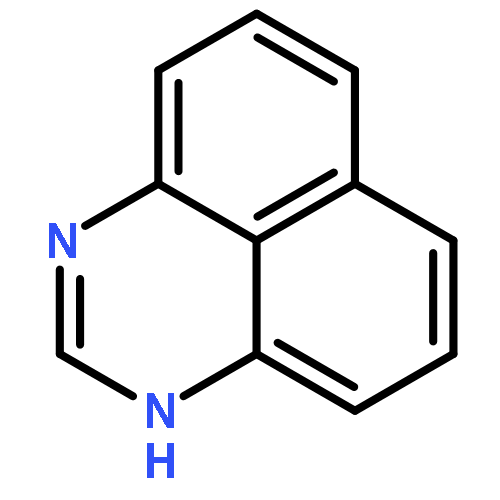




![Methylene, bis[bis(1-methylethyl)amino]-](/data/chemimg/114000/178925-49-0.png)
![Methylene, bis[bis(1-methylethyl)amino]-](/data/chemimg/114000/178925-49-0_b.png)




![1,7-Diazabicyclo[5.3.1]undecane(9CI)](http://img.cochemist.com/ccimg/100100/100098-21-3.png)
![1,7-Diazabicyclo[5.3.1]undecane(9CI)](http://img.cochemist.com/ccimg/100100/100098-21-3_b.png)
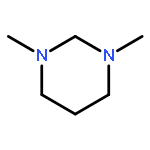

![1,8-Diazabicyclo[6.3.1]dodecane(9CI)](http://img.cochemist.com/ccimg/100100/100098-22-4.png)
![1,8-Diazabicyclo[6.3.1]dodecane(9CI)](http://img.cochemist.com/ccimg/100100/100098-22-4_b.png)




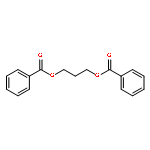
![3-acetylcyclohepta[b]furan-2-one](http://img.cochemist.com/ccimg/22500/22460-76-0.png)
![3-acetylcyclohepta[b]furan-2-one](http://img.cochemist.com/ccimg/22500/22460-76-0_b.png)