Co-reporter:Yi Yu, Huiyong Sun, Keith Gilmore, Tingjun Hou, Suidong Wang, and Youyong Li
ACS Applied Materials & Interfaces September 27, 2017 Volume 9(Issue 38) pp:32452-32452
Publication Date(Web):September 1, 2017
DOI:10.1021/acsami.7b05478
Single-walled carbon nanotubes (SWCNTs) have attracted considerable attention owing to their applications in various fields such as biotechnology and biomedicine. Recently, aggregated SWCNTs have shown more significant effects on the treatment of methamphetamine addiction (Nat. Nanotech. 2016, 11, 613). However, the mechanisms underlying these actions are unclear. By using all-atom molecular dynamics simulations, we investigate the effects of single and aggregated SWCNTs (single-(10,10)CNT, aggregated-7-(10,10)CNTs, and single-(35,35)CNT with the same diameter as that of the aggregated one) on the activity of dopamine-related proteins [tyrosine hydroxylase (TyrOH) and dopamine transporter (DAT), which are related to the synthesis and transport of dopamine, respectively]. We find that both TyrOH and DAT can adsorb onto these SWCNTs. For TyrOH, the aggregated-7-(10,10)CNTs mainly affect the conformation of the active site of the protein, and hence, they are more effective in inhibiting the expression of TyrOH. For DAT, our results suggest that the aggregated-7-(10,10)CNTs allow DAT to maintain an outward-facing conformation and hence are favorable to the reuptake of dopamine. The binding of a dopamine reuptake inhibitor, [3H]-WIN35,428, to DAT is significantly disrupted by aggregated-7-(10,10)CNTs and hence improve the ability to transport dopamine. Our results provide the dynamic interactions of proteins with single/aggregated SWCNTs, which illustrate the mechanism of aggregated SWCNTs for the treatment of drug addiction.Keywords: conformational changes; dopamine-related proteins; interactions; MD simulations; single-walled carbon nanotubes;
Co-reporter:Yujin Ji, Lifeng Ding, Yuanyuan Cheng, Hao Zhou, Siyuan Yang, Fan Li, and Youyong Li
The Journal of Physical Chemistry C November 2, 2017 Volume 121(Issue 43) pp:24104-24104
Publication Date(Web):October 11, 2017
DOI:10.1021/acs.jpcc.7b08370
Safe and efficient storage and separation of acetylene pose a significant challenge in industry. In this study, we investigated 11 open Cu(II) site (OCS)-based metal–organic frameworks (MOFs) formed by various organic ligands for their C2H2 adsorption capacities and their C2H2/CO2, C2H2/CH4 separation performance using both grand canonical Monte Carlo (GCMC) simulations and density functional theory (DFT) calculations. Our simulations revealed that both OCSs and organic ligands of the MOFs play key roles in promoting C2H2 storage capacity and the separation of C2H2 over CH4 and CO2 under 2 bar. Judicious selection of organic ligands with suitable dimensions and functional sites, such as methyl group, Lewis basic nitrogen site, and fluorine group, can facilitate C2H2 adsorption in addition to OCS and help distinguish C2H2 from CH4 and CO2. Short ligands presented in the MOFs, such as MOF-505 which gives the highest volumetric C2H2 storage capacity under 2 bar, not only increase the density of OCSs but also create overlapped interaction regions for guest molecules. GCMC simulation results suggested that NOTT-106 had the second highest volumetric C2H2 storage capacity because of its methyl functionalized ligands. DFT calculations however suggested that the Lewis basic nitrogen functionalized ligand of ZJU-40 might have a stronger affinity with C2H2 than that of NOTT-106, which indicated that C2H2 storage capacity in ZJU-40 should be better than that in NOTT-106. In contrast, MOF-505, ZJU-40, and NOTT-108 showed excellent C2H2/CH4 separation performance as well as modest C2H2/CO2 separation capability.
Co-reporter:Yunxia Liu;Xiao Yuan;Krisztian Palotas;Haiping Lin;Tingjun Hou;Shuit-Tong Lee
ACS Nano February 28, 2017 Volume 11(Issue 2) pp:2060-2065
Publication Date(Web):January 26, 2017
DOI:10.1021/acsnano.6b08260
The inherent instability of CH3NH3PbX3 remains a major technical barrier for the industrial applications of perovskite materials. Recently, the most stable surface structures of CH3NH3PbX3 have been successfully characterized by using density functional theory (DFT) calculations together with the high-resolution scanning tunneling microscopy (STM) results. The two coexisting phases of the perovskite surfaces have been ascribed to the alternate orientation of the methylammonium (MA) cations. Notably, similar surface defect images (a dark depression at the sites of X atoms) have been observed on surfaces produced with various experimental methods. As such, these defects are expected to be intrinsic to the perovskite crystals and may play an important role in the structural decomposition of perovskite materials. Understanding the nature of such defects should provide some useful information toward understanding the instability of perovskite materials. Thus, we investigate the chemical identity of the surface defects systematically with first-principles density functional theory calculations and STM simulations. The calculated STM images of the Br and Br-MA vacancies are both in good agreement with the experimental measurements. In vacuum conditions, the formation energy of Br-MA is 0.43 eV less than the Br vacancy. In the presence of solvation effects, however, the formation energy of a Br vacancy becomes 0.42 eV lower than the Br-MA vacancy. In addition, at the vacancy sites, the adsorption energies of water, oxygen, and acetonitrile molecules are significantly higher than those on the pristine surfaces. This clearly demonstrated that the structural decomposition of perovskites are much easier to start from these vacancy sites than the pristine surfaces. Combining DFT calculations and STM simulations, this work reveals the chemical identities of the intrinsic defects in the CH3NH3PbX3 perovskite crystals and their effects on the stability of perovskite materials.Keywords: CH3NH3PbBr3; density functional theory; STM simulations; surface defects;
Co-reporter:Yujin Ji;Mingye Yang;Huilong Dong;Tingjun Hou;Lu Wang
Nanoscale (2009-Present) 2017 vol. 9(Issue 25) pp:8608-8615
Publication Date(Web):2017/06/29
DOI:10.1039/C7NR00688H
Full utilization of solar energy to split water into hydrogen and oxygen is an efficient way to solve the current energy problem. Graphene-like germanium monochalcogenides (GeS or GeSe) are proposed here as efficient photocatalysts for water splitting under a broad range from ultraviolet, visible to near-infrared light dependent on their thickness. Compared to traditional photocatalysts, GeS and GeSe possess a large intrinsic dipole and introduce an internal electric field directing from Ge surface to S/Se surface, which causes notable band bending. The band bending means they possess favorable band positions located outside the reduction potential and oxidation potential of water, overcoming the restriction of their band gaps. Moreover, multilayer GeS and GeSe further provide a good separation of electrons and holes, which effectively reduces the probability of their recombination and ensures photocatalytic activity with high efficiency.
Co-reporter:Xiaotian Sun;Yunxia Liu;Zhigang Song;Yongdan Li;Weizhou Wang;Haiping Lin;Lu Wang
Journal of Materials Chemistry C 2017 vol. 5(Issue 17) pp:4159-4166
Publication Date(Web):2017/05/04
DOI:10.1039/C7TC00306D
Defects are unavoidable during the synthesis of materials, especially for two-dimensional (2D) nanomaterials. They are usually seen as detrimental to device properties, but sometimes bring about new beneficial effects. In order to clarify the influence of defects on the structural and electronic properties, we have performed first-principles calculations to systematically investigate the structural stability, mobility and electronic properties of typical point defects in 2D arsenene (h-As), antimonene (h-Sb) and antimony arsenide (h-AsSb), including the Stone–Wales defects, single vacancies (SVs), double vacancies (DVs) and adatoms. To provide visual guidance for experimental observations, scanning tunnelling microscopy (STM) images are simulated. Compared to defects in graphene and silicene, these defects form more easily with lower formation energies, and SVs can diffuse very quickly to the edges with a lower diffusion barrier of less than 1 eV. Monolayer arsenene, antimonene and antimony arsenide are indirect band gap semiconductors, and the defective structures significantly reduce the band gaps. Most of the SV and adatom defects carry magnetic moments due to the dangling bonds resulting from the absent or extra atom. Our present results have demonstrated that the point defects induce significant effects on the electronic properties of pristine arsenene, antimonene and the antimony arsenide alloy, which should be considered in their future applications.
Co-reporter:Mingye Yang;Lu Wang;Tingjun Hou
Journal of Materials Chemistry C 2017 vol. 5(Issue 1) pp:201-207
Publication Date(Web):2016/12/22
DOI:10.1039/C6TC04487E
Two-dimensional nanomaterials have attracted extensive interest and their heterostructures possess excellent electronic and optical properties suitable for various applications. Based on first-principles calculations, we investigated the structural stability and electronic properties of WS2 and graphene oxide (GO) heterostructures. We considered three types of GO, including epoxy only, hydroxyl only and both epoxy and hydroxyl on the GO surface. Our results show that the interlayer binding energy per WS2 unit increases from 0.117 eV to 0.214 eV as the surface oxygen concentration of GO increases. The band gap of the WS2/GO heterostructures can be efficiently tuned in a wide range from 0.13 eV to 1.91 eV by changing the oxygen functionalities and the concentration. The spatial separation of the conduction band minimum and valence band maximum is observed, which are distributed in different layers. In addition, the work function of WS2 can also be modulated by GO in the range of 4.09 eV to 6.34 eV, which potentially increases the carrier concentration and broadens the applications of WS2 and other transition metal dichalcogenide materials in optoelectronic devices.
Co-reporter:Junqing Yan;Ping Li;Yujin Ji;Hui Bian;Shengzhong (Frank) Liu
Journal of Materials Chemistry A 2017 vol. 5(Issue 40) pp:21478-21485
Publication Date(Web):2017/10/17
DOI:10.1039/C7TA07208B
The water oxidation reaction (OER) is the key process used for the generation of H2, and new catalyst systems are needed to achieve considerable efficiency in the OER. In the present work, β-FeOOH samples, both bare and doped with earth-abundant elements (Ni and Co), were synthesized and confirmed to be promising semiconductors for solar-driven water oxidation. This is the first time that the direct use of β-FeOOH samples has been reported in a water splitting system, in which β-FeOOH has always been used as a co-catalyst. From a combination of DFT calculations and experimental results, (a) β-FeOOH can play two roles, namely, carrier generation and improving the kinetics of water oxidation, owing to the abundance of surface OH groups; (b) doping with Ni or Co can improve the photocatalytic activity of β-FeOOH, and this enhancement benefits from the separation of photogenerated carriers; (c) samples doped with Ni or Co give better performance in electrocatalytic water oxidation than bare β-FeOOH. After being doped with these earth-abundant elements, β-FeOOH delivered saturated photocurrent densities of 4.71 (for Co) and 2.55 (for Ni) mA cm−2, which are higher than the value of 0.69 mA cm−2 recorded for bare β-FeOOH at a low external bias of 1.0 V versus RHE.
Co-reporter:Yujin Ji;Mingye Yang;Huilong Dong;Lu Wang;Tingjun Hou
Journal of Materials Chemistry A 2017 vol. 5(Issue 4) pp:1734-1741
Publication Date(Web):2017/01/24
DOI:10.1039/C6TA08321H
Exploration of oxygen reduction reaction (ORR) catalysts based on non-precious metals is of great importance to the development of fuel cells. In this work, we theoretically investigate the practical feasibility and catalytic activity of two-dimensional (2D) group IVA monochalcogenides MXs (M = Ge/Sn and X = S/Se) as potential and efficient ORR catalysts. Through first-principles calculations, it is found that these 2D MXs are semiconductors with high adsorption affinity and dissociation activity for oxygen molecules. Furthermore, the free energy diagrams along the four-electron pathway are simulated to elucidate the whole ORR mechanism catalyzed by the monolayer MXs (GeS, GeSe, and SnSe here), with all the energetically favorable intermediates being exothermic in free energy. In the acidic environment, the GeS monolayer exhibits the best performance due to its considerably low overpotential (0.52 V). While in the alkaline environment, there is no overpotential for the ORR catalyzed by the 2D MXs (GeS, GeSe, and SnSe). Our work provides a novel insight into the electrochemical applications of IVA monochalcogenides, which is expected to realize experimentally.
Co-reporter:Xiaohong Tan;Yujin Ji;Huilong Dong;Meiyi Liu;Tingjun Hou
RSC Advances (2011-Present) 2017 vol. 7(Issue 79) pp:50239-50245
Publication Date(Web):2017/10/26
DOI:10.1039/C7RA10305K
Visible-light-driven water-splitting technology has attracted wide attention because of its sustainable and renewable production of hydrogen. The key to photocatalysis field is to search materials with suitable oxidizing and reducing potential. Herein we investigated the novel 2D γ-phosphorus nitride (γ-PN) monolayer as a candidate photocatalyst for reducing water into hydrogen and oxygen from the first-principles and molecular dynamics calculations. Our results show that the 2D γ-PN monolayer is an indirect semiconductor whose conduction and valence band edges matches well with the chemical potential of H+/H2 and O2/H2O. Strain effect is considered to tune the electronic properties of γ-PN. The calculation results reveal that tensile strain decreases the band gap and upshifts the work function of the γ-PN monolayer. The γ-PN monolayer at the 10% tensile strain shows the optimal performance in catalysing the water-splitting due to the low band gap and the better absorption spectrum at the UV-visible light range. Furthermore, the molecular dynamics shows that the γ-PN in the aqueous solution always keep stable after 100 ps simulation. Both the density functional theory (DFT) and molecular dynamics (MD) simulation reflect that the γ-PN monolayer is reliable and promising as a water-splitting photocatalyst.
Co-reporter:Xiao Yuan;Mingye Yang;Lu Wang
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 21) pp:13846-13854
Publication Date(Web):2017/05/31
DOI:10.1039/C7CP01727H
Based on the first-principles calculations, we systematically studied the structures and electronic properties of two-dimensional (2D) transition metal dichalcogenide (TMD) alloys with half-to-half mixing of S and Se. Using the chemical potentials of S and Se, the energetic phase diagrams for both single phases and mixed phases of TMD were constructed. A new heterolayer structure (for Sc and Ti) and alternating structure (for Cr, Mn, Fe, Zr, Mo, and W) were proposed for the first time, which were thermodynamically stable for MSSe alloys under the S-poor (relatively low chemical potential of S) and Se-rich (relatively high chemical potential of Se) conditions, and further compared with the disordered structures. Moreover, band gaps, carrier effective mass, and work functions were calculated for these stable mixed phases. Compared to the single phases of MS2 and MSe2, MSSe alloys showed superior electronic properties including tunable band gaps and work functions. Importantly, the significantly reduced effective mass of the carriers in the MSSe alloys may induce higher carrier mobility, providing better performance of TMD materials in electronic devices.
Co-reporter:Yujin Ji;Huilong Dong;Mingye Yang;Tingjun Hou
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 31) pp:20457-20462
Publication Date(Web):2017/08/09
DOI:10.1039/C7CP04044J
The development of novel cathode catalysts is of great importance to the practical applications of nonaqueous lithium oxygen (Li–O2) batteries. Here by using first-principles calculations, we revealed the catalytic mechanism and evaluated the catalytic activity of monolayer germanium monochalcogenides (2D-GeXs, X = S/Se) as cathode catalytic materials. For 2D-GeXs, Li4O4 with a ring-like structure is the final discharge product. The free energy diagram demonstrates that 2D-GeSe is more energetically favorable than 2D-GeS due to its considerably lower oxygen reduction reaction (ORR) overpotential (0.94 V) and oxygen evolution reaction (OER) overpotential (1.30 V), which originate from its weaker binding with LiO2 and stronger binding with the inserted Li atom. The analyses on electronic properties elucidate that the final product of Li4O4 on 2D-GeSe induces the semiconductor to semi-metal transition. Our results reflect that 2D-GeSe is an excellent candidate as a cathode material in nonaqueous Li–O2 batteries, while 2D-GeS is not appropriate.
Co-reporter:Zhaoran Wang, Huilong Dong, Xiaohui Yu, Yujin Ji, ... Youyong Li
Current Applied Physics 2017 Volume 17, Issue 12(Volume 17, Issue 12) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.cap.2017.08.019
•2D PPc can efficiently separate NH3 from H2 and N2 after ammonia synthesis.•Only the newly formed pore in 2D porous PPc can be used for gas separation.•2D PPc offers good selectivity (107) of (H2, N2)/NH3 at room temperatures.•MD simulations on selectivity also give good accordance with DFT results.Porous monolayer materials have been proven potential for gas separation and purification, because of their natural pathways of controllable sizes and well-ordered distribution. In this work, a novel material, two-dimensional (2D) porous polyphthalocyanine (PPc) is investigated by density functional theory (DFT) simulations for separating NH3 from H2 and N2 during ammonia synthesis process. Based on the calculated diffusion barriers through transition state search, we demonstrate that 2D PPc is able to offer high selectivity (107) of (H2, N2)/NH3 at room temperature. Further molecular dynamics (MD) simulation also indicates that the 2D PPc can effectively separate NH3 from H2 and N2. Thus the 2D PPc is promising for the practical applications of synthetic ammonia process.
Co-reporter:Huilong Dong, Liujiang Zhou, Thomas Frauenheim, Tingjun Hou, Shuit-Tong Lee and Youyong Li
Nanoscale 2016 vol. 8(Issue 13) pp:6994-6999
Publication Date(Web):08 Mar 2016
DOI:10.1039/C6NR00046K
The SiC7 siligraphene (g-SiC7) is a novel 2D nanomaterial with a graphene-like structure. Based on theoretical calculations, we have systematically investigated the structure, stability, electronic and optical properties of g-SiC7 siligraphene. The calculated results reveal that g-SiC7 siligraphene is a semiconductor with a direct band gap of 1.13 eV, which can be easily tuned by applying biaxial strain or a perpendicular electric field. Such a g-SiC7 siligraphene shows superior sunlight optical absorbance and is better than g-SiC2 siligraphene and single-layer black phosphorus (phosphorene) in near infrared and visible photon ranges, thus holding great potential for photovoltaics applications as a light donor material.
Co-reporter:Yunxia Liu, Yao-Jun Dong, Zeyuan Tang, Xue-Feng Wang, Lu Wang, Tingjun Hou, Haiping Lin and Youyong Li
Journal of Materials Chemistry A 2016 vol. 4(Issue 26) pp:6380-6385
Publication Date(Web):07 Jun 2016
DOI:10.1039/C6TC01328G
Recently, borophene was reported to be produced on silver surfaces. We employed density functional theory and electronic transport calculations to investigate the stabilities, electronic structures and transport properties of borophene nanoribbons. The stability of a borophene nanoribbon increases with its width and only the lined-edged borophene nanoribbons are stable in the free-standing form. Such anisotropic stability dependence is ascribed to the large scale delocalization of π electrons along the boron rows. In particular, all line-edge borophene nanoribbons undergo edge reconstructions, in which the out-of-plane bulking edge atoms are reconstructed to form quasi planar edge structures. Such edge reconstructions have not yet been reported, which is critical for the formation of boron nanostructrues. Subsequent electronic transport calculations based on a non-equilibrium Green’s function indicated that line-edge borophene nanoribbons exhibit low-resistivity Ohmic conductance. Our results indicated that the line-edge borophene nanoribbons present remarkable properties (high thermal stabilities, Ohmic conductance with low electrical resistivity and good rigidities) and are promising for applications as one-dimensional electrical connections in compact nanoscale circuits.
Co-reporter:Xiaotian Kong, Huiyong Sun, Peichen Pan, Sheng Tian, Dan Li, Youyong Li and Tingjun Hou
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 3) pp:2034-2046
Publication Date(Web):02 Dec 2015
DOI:10.1039/C5CP05622E
Due to the high sequence identity of the binding pockets of cyclin-dependent kinases (CDKs), designing highly selective inhibitors towards a specific CDK member remains a big challenge. 4-(thiazol-5-yl)-2-(phenylamino) pyrimidine derivatives are effective inhibitors of CDKs, among which the most promising inhibitor 12u demonstrates high binding affinity to CDK9 and attenuated binding affinity to other homologous kinases, such as CDK2. In this study, in order to rationalize the principle of the binding preference towards CDK9 over CDK2 and to explore crucial information that may aid the design of selective CDK9 inhibitors, MM/GBSA calculations based on conventional molecular dynamics (MD) simulations and enhanced sampling simulations (umbrella sampling and steered MD simulations) were carried out on two representative derivatives (12u and 4). The calculation results show that the binding specificity of 12u to CDK9 is primarily controlled by conformational change of the G-loop and variation of the van der Waals interactions. Furthermore, the enhanced sampling simulations revealed the different reaction coordinates and transient interactions of inhibitors 12u and 4 as they dissociate from the binding pockets of CDK9 and CDK2. The physical principles obtained from this study may facilitate the discovery and rational design of novel and specific inhibitors of CDK9.
Co-reporter:Lu Wang, Huilong Dong, Zhenyu Guo, Liling Zhang, Tingjun Hou, and Youyong Li
The Journal of Physical Chemistry C 2016 Volume 120(Issue 31) pp:17427-17434
Publication Date(Web):July 18, 2016
DOI:10.1021/acs.jpcc.6b04639
The development of carbon-based metal-free electrocatalysts for oxygen reduction reaction (ORR) is essential for large-scale commercial applications of fuel cells. Using density functional theory computations, we explore the potentials of a novel boron-doped graphene nanoribbon (BGNR) as an excellent electrocatalyst for ORR in an acidic environment. The plausible reaction pathways are studied, and the optimal reaction mechanism is identified. Our results show that ORR at BGNR prefers to proceed through a four-electron OOH pathway. The overpotential for ORR on BGNR is calculated to be 0.38 V, which is lower than that on the Pt-based catalysts (0.45 V). For comparison, we study the catalytic activity of the single B-doped graphene nanoribbon (S-BGNR) and B-doped graphene (BG) for ORR. Remarkably, the para-B distribution on BGNR leads to high affinity for O2 adsorption and excellent catalytic activity, which is superior to S-BGNR and BG. Our results indicate that BGNR is a promising metal-free ORR catalyst for fuel cells.
Co-reporter:Robin Ohmann; Luis K. Ono; Hui-Seon Kim; Haiping Lin; Michael V. Lee; Youyong Li; Nam-Gyu Park;Yabing Qi
Journal of the American Chemical Society 2015 Volume 137(Issue 51) pp:16049-16054
Publication Date(Web):December 6, 2015
DOI:10.1021/jacs.5b08227
Organic–inorganic perovskite is a promising class of materials for photovoltaic applications and light emitting diodes. However, so far commercialization is still impeded by several drawbacks. Atomic-scale effects have been suggested to be possible causes, but an unequivocal experimental view at the atomic level is missing. Here, we present a low-temperature scanning tunneling microscopy study of single crystal methylammonium lead bromide CH3NH3PbBr3. Topographic images of the in situ cleaved perovskite surface reveal the real-space atomic structure. Compared to the bulk we observe modified arrangements of atoms and molecules on the surface. With the support of density functional theory we explain these by surface reconstruction and a substantial interplay of the orientation of the polar organic cations (CH3NH3)+ with the position of the hosting anions. This leads to structurally and electronically distinct domains with ferroelectric and antiferroelectric character. We further demonstrate local probing of defects, which may also impact device performance.
Co-reporter:Chunmiao Du, Haiping Lin, Bin Lin, Zeyao Ma, Tingjun Hou, Jianxin Tang and Youyong Li
Journal of Materials Chemistry A 2015 vol. 3(Issue 46) pp:23113-23119
Publication Date(Web):29 Oct 2015
DOI:10.1039/C5TA05084G
Late transition metals, such as Rh, Ir, Pd and Pt, have a strong tendency to form a square-planar 16-electron complex. Although this feature has been widely used in organometallics to develop homogeneous catalysts, a single-atom heterogeneous analogue has not yet been reported. In this work, we show that a 16-electron complex may act as an important transition state in the CO oxidation over a single Pt atom supported by a MoS2 monolayer (Pt/MoS2). The catalytic oxidation reaction prefers to start with the Langmuir–Hinshelwood (L–H) reaction, where the CO and O2 molecules are first co-adsorbed on the Pt atom, then cross a small barrier of 0.40 eV to form a square-planar 16-electron intermediate state, and subsequently the first CO2 is released. The activation barrier of the following Eley–Rideal (E–R) reaction is only 0.23 eV. The superior catalytic reactivity of the Pt/MoS2 surface can be explained by the quantum confinement effect of the Pt-5d orbitals and the stability of the square-planar 16-electron transition state. In addition, MoS2 may serve as a defect-free two dimensional anchoring substrate for Pt atomic adsorption. It provides not only a very large surface-to-volume ratio, but also a well-defined structure with a uniform distribution of anchoring points. The square-planar 16-electron intermediate state of the L–H reaction, together with the new MoS2 anchoring substrate, may provide a new opportunity for the design of single-atom catalysts on two-dimensional surfaces.
Co-reporter:Min Li, Lu Wang, Ningning Yu, Xiaotian Sun, Tingjun Hou and Youyong Li
Journal of Materials Chemistry A 2015 vol. 3(Issue 15) pp:3645-3649
Publication Date(Web):25 Feb 2015
DOI:10.1039/C5TC00209E
Based on density functional theory (DFT) calculations, we have constructed and investigated different types of single-side hydrogenated graphene (SSHG) structures from their structural motifs. The structural stability and electronic properties of these SSHG structures are extensively analyzed and compared with the reported structures. The single-side hydrogenation causes a severe bending in graphene at high H coverage, which leads to a greater formation energy with increasing H coverage. Among the SSHG structures that we have considered, the configurations with H attached along the armchair direction show the lowest formation energies due to a relatively small buckling compared to other configurations. Moreover, only the armchair hydrogenated graphene opens a band gap near the Fermi level, and the band gap can be modulated from zero to 1.44 eV by varying the H coverage from zero to 50%. Our results suggest an efficient way to prepare graphene-based materials and devices with suitable band gaps.
Co-reporter:Kai Yao, Pengli Tan, Yinchan Luo, Liangzhu Feng, Ligeng Xu, Zhuang Liu, Youyong Li, and Rui Peng
ACS Applied Materials & Interfaces 2015 Volume 7(Issue 22) pp:12270
Publication Date(Web):May 19, 2015
DOI:10.1021/acsami.5b03118
In the past few years, graphene and its derivative, graphene oxide (GO), have been extensively studied for their applications in biotechnology. In our previous work, we reported certain PEGylated GOs (GO-PEGs) can selectively promote trypsin activity and enhance its thermostability. To further explore this, here we synthesized a series of GO-PEGs with varying PEGylation degrees. Enzymatic activity assay shows that both GO and GO-PEGs can protect trypsin, but not chymotrypsin, from thermal denaturation at high temperature. Surprisingly, the lower the PEGylation degree, the better the protection, and GO as well as the GO-PEG with the lowest PEGylation degree show the highest protection efficiency (∼70% retained activity at 70 °C). Fluorescence spectroscopy analysis shows that GO/GO-PEGs have strong interactions with trypsin. Molecular Dynamics (MD) simulation results reveal that trypsin is adsorbed onto the surface of GO through its cationic residues and hydrophilic residues. Different from chymotrypsin adsorbed on GO, the active site of trypsin is covered by GO. MD simulation at high temperature shows that, through such interaction with GO, trypsin’s active site is therefore stabilized and protected by GO. Our work not only illustrates the promising potential of GO/GO-PEGs as efficient, selective modulators for trypsin, but also provides the interaction mechanism of GO with specific proteins at the nano–bio interface.Keywords: enzyme thermostability; graphene oxide; molecular dynamics simulation; nano−bio interface; trypsin;
Co-reporter:Huilong Dong, Bin Lin, Keith Gilmore, Tingjun Hou, Shuit-Tong Lee, Youyong Li
Journal of Power Sources 2015 Volume 299() pp:371-379
Publication Date(Web):20 December 2015
DOI:10.1016/j.jpowsour.2015.09.014
•SiC2 siligraphene is promising as a metal-free ORR catalyst.•The g-SiC2 shows higher adsorption affinity for ORR intermediates than Pt (111).•The g-SiC2 shows a very low activation barrier (0.16 eV) when in alkaline media.•The g-SiC2 shows no overpotential at the 0.4 V bias in alkaline environment.The design and discovery of high-performance metal-free catalytic materials for oxygen reduction reaction (ORR) electrocatalysis is vital for the development of fuel cells. By performing density functional theory (DFT) calculations, we investigate the potential applications of SiC2 siligraphene (g-SiC2) as metal-free ORR catalyst. The g-SiC2 exhibits higher adsorption affinity for the O2 molecule and other ORR intermediates than the traditional Pt (111), and shows good tolerance against CO poisoning. The detailed LH and ER mechanisms in catalyzing ORR by g-SiC2 are simulated and discussed, both in acidic and alkaline environment. We find that, in alkaline environment, the g-SiC2 presents a very low activation barrier (0.16 eV) for the rate determining step (RDS) and shows no overpotential at the equilibrium potential. Our theoretical simulations validate that the siligraphene with alternatively arranged Si and C atoms holds great potential as ORR catalyst in alkaline environment.
Co-reporter:Ningning Yu, Lu Wang, Min Li, Xiaotian Sun, Tingjun Hou and Youyong Li
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 17) pp:11700-11704
Publication Date(Web):25 Mar 2015
DOI:10.1039/C5CP00161G
Molybdenum disulfide (MoS2), a kind of graphene-like, two-dimensional material, has attracted great interest because of its unique properties and potential applications in electronics and sensors. In this paper, first-principle calculations and grand canonical Monte Carlo (GCMC) simulations are performed and used to show that the MoS2 layer is efficient at absorbing non-polar gases. Compared with the popular gas sorbents (metal organic frameworks and carbon-based materials), MoS2 has additional advantages, including large surface to volume ratio and tunable properties. The non-polar gas [carbon dioxide (CO2) and methane (CH4)] adsorption on the MoS2 layer with and without vacancies has been systematically studied. The perfect MoS2 shows little or no adsorption for CO2 and CH4 molecules, but the MoS2 with a single S vacancy and double S vacancies exhibits an excellent adsorption ability for CO2 and CH4 gases. The adsorption energies were 65 kJ mol−1 for CO2 and 47 kJ mol−1 for CH4 (van der Waals-D2), respectively. An orbital coupling between the p orbital of the CO2 (or CH4) molecule and the d orbital of the Mo atom was observed. GCMC simulation results show that MoS2 with a single S vacancy could absorb 42.1 wt% of CO2 and 37.6 wt% of CH4 under a pressure of 80 bar at room temperature. The results given in this paper indicate that monolayer MoS2 with defects is a highly efficient absorbent for non-polar gases.
Co-reporter:Xiaotian Kong, Peichen Pan, Dan Li, Sheng Tian, Youyong Li and Tingjun Hou
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 8) pp:6098-6113
Publication Date(Web):27 Jan 2015
DOI:10.1039/C4CP05440G
Anaplastic lymphoma kinase (ALK) has gained increased attention as an attractive therapeutic target for the treatment of various cancers, especially non-small-cell lung cancer (NSCLC). Recently, piperidine carboxamides were reported as Type I1/2 inhibitors of ALK, which occupy both the ATP binding site and the back ATP hydrophobic cavity in DFG-in conformation. Due to the dynamic behavior of ALK in the binding of Type I1/2 inhibitors, the accurate predictions of the binding structures and relative binding potencies of these inhibitors are quite challenging. In this study, different modeling techniques, including molecular docking, ensemble docking based on multiple receptor conformations, molecular dynamics simulations and free energy calculations, were utilized to explore the binding mechanisms of piperidine carboxamides. Our predictions show that the conventional docking protocols are not sufficient to predict the relative binding potencies of the studied inhibitors with high accuracy, but incorporating protein flexibility before or after docking is quite effective to improve the prediction accuracy. Notably, the binding free energies predicted by MM/GBSA or MM/PBSA based on the MD simulations for the docked poses give the highest correlation with the experimental data, highlighting the importance of the inclusion of receptor flexibility for the accurate predictions of the binding potencies for Type I1/2 inhibitors of ALK. Furthermore, the comprehensive analysis of several pairs of representative inhibitors demonstrates the importance of hydrophobic interactions in improving the binding affinities of the inhibitors with the hot-spot residues surrounding the binding pocket. This work is expected to provide valuable clues for further rational design of novel and potent Type I1/2 ALK inhibitors.
Co-reporter:Chongqian Zhang;Chunmiao Du;Zhiwei Feng;Jingyu Zhu
Chemical Biology & Drug Design 2015 Volume 85( Issue 2) pp:119-136
Publication Date(Web):
DOI:10.1111/cbdd.12377
CXCR4 plays a crucial role as a co-receptor with CCR5 for HIV-1 anchoring to mammalian cell membrane and is implicated in cancer metastasis and inflammation. In the current work, we study the relationship of structure and activity of AMD11070 derivatives and other inhibitors of CXCR4 using HQSAR, docking and molecular dynamics (MD) simulations. We obtain an HQSAR model (q2 = 0.779), and the HQSAR result illustrates that AMD11070 shows a high antiretroviral activity. As HQSAR only provides 2D information, we perform docking and MD to study the interaction of It1t, AMD3100, and AMD3465 with CXCR4. Our results illustrate that the binding are affected by two crucial residues Asp97 and Glu288. The butyl amine moiety of AMD11070 contributes to its high antiretroviral activity. Without a butyl amine moiety, (2,7a-Dihydro-1H-benzoimidazol-2-ylmethyl)-methyl-(5,6,7,8-tetrahydro-quinolin-8-yl)-amine (compound 5a) shows low antiretroviral activity. Our results provide structural details about the interactions between the inhibitors and CXCR4, which are useful for rational drug design of CXCR4.
Co-reporter:Xiaojuan Xu, Yujin Ji, Chunmiao Du, Tingjun Hou and Youyong Li
RSC Advances 2015 vol. 5(Issue 87) pp:70939-70948
Publication Date(Web):11 Aug 2015
DOI:10.1039/C5RA12318F
The efficiency of bulk heterojunction (BHJ) solar cells depends strongly on the morphology of the electron donors and electron acceptors in the active layer. Here we use Dissipative Particle Dynamics (DPD) simulation to predict the donor–acceptor morphology and graph theory to predict the efficiency of small molecular organic solar cells (SM OSCs). We focus on a recently reported small molecular organic solar cell based on a new molecule donor, DTS(PTTh2)2 and three molecules DR3TBDTT, DR3TBDTT-HD, and DR3TBD2T with a benzo[1,2-b:4,5-b′]dithiophene (BDT) unit. With our theoretical approach, we are able to study the critical factors affecting the morphology and efficiency such as, the chemical structure of the conjugated molecular, fullerene functional group, solvents, additives, blend ratio, and processing conditions (e.g., annealing temperature). Our results are consistent with experimental conclusions and provide useful guidelines for improving the efficiency of small organic solar cells.
Co-reporter:Shuo Deng
The Journal of Physical Chemistry C 2015 119(52) pp: 28783-28788
Publication Date(Web):December 2, 2015
DOI:10.1021/acs.jpcc.5b10354
On the basis of first-principles calculations, we have systematically studied the adsorption and diffusion of Li ions on monolayer MnO2 and compared with other transition metal dichalcogenides (TMDs) and transition metal dioxides (TMOs). Monolayer MnO2 shows a relatively high Li adsorption energy of 4.37 eV and low Li diffusion barrier of 0.148 eV. The electronic analysis indicates that the electron transferred from Li to the empty orbital of the O atom and there is some orbital coupling between the s orbital of the Li atom and the pz orbital of the O atom in MnO2. Due to Li adsorption on both sides of the MnO2 layer, the theoretical Li storage capacity reaches as high as 616 mAh/g. Our results demonstrated that, compared to other two-dimensional (2D) nanomaterials, monolayer or few-layer MnO2 exhibits excellent performance on Li storage capacity and diffusion rate and is believed to be a promising electrode material for high-capacity Li ion batteries.
Co-reporter:Huilong Dong, Keith Gilmore, Bin Lin, Tingjun Hou, Shuit-Tong Lee, Zhenyu Guo, Youyong Li
Carbon 2015 89() pp: 249-259
Publication Date(Web):
DOI:10.1016/j.carbon.2015.03.046
Co-reporter:Guangbao Yang;Hua Gong;Xiaoxin Qian;Pengli Tan;Zhiwei Li;Teng Liu
Nano Research 2015 Volume 8( Issue 3) pp:751-764
Publication Date(Web):2015 March
DOI:10.1007/s12274-014-0558-0
Mesoporous silica nanoparticles (MSNs) have attracted tremendous attention in recent years as drug delivery carriers due to their large surface areas, tunable sizes, facile modification and considerable biocompatibility. In this work, we fabricate an interesting type of MSNs which are intrinsically doped with photosensitizing molecules, chlorin e6 (Ce6). By increasing the amount of Ce6 doped inside the silica matrix, it is found that the morphology of MSNs changes from spheres to rod-like shapes. The obtained Ce6-doped mesoporous silica nanorods (CMSNRs) are not only able to produce singlet oxygen for photodynamic therapy, but can also serve as a drug delivery platform with high drug loading capacity by utilizing their mesoporous structure. Compared to spherical nanoparticles, it is found that CMSNRs with a larger aspect ratio show much faster uptake by cancer cells. With doxorubicin (DOX) employed as a model drug, the combined photodynamic and chemotherapy is carried out, achieving synergistic anti-tumor effects both in vitro and in vivo. Our study presents a new design of an MSN-based drug delivery platform, which intrinsically is fluorescent and able to serve as a photodynamic agent, promising for future imaging-guided combination therapy of cancer.
Co-reporter:Xuejie Dang, Huilong Dong, Lu Wang, Yanfei Zhao, Zhenyu Guo, Tingjun Hou, Youyong Li, and Shuit-Tong Lee
ACS Nano 2015 Volume 9(Issue 8) pp:8562
Publication Date(Web):July 27, 2015
DOI:10.1021/acsnano.5b03722
Graphene is a semimetal with zero band gap, which makes it impossible to turn electric conduction off below a certain limit. Transformation of graphene into a semiconductor has attracted wide attention. Owing to compatibility with Si technology, graphene adsorbed on a Si substrate is particularly attractive for future applications. However, to date there is little theoretical work on band gap engineering in graphene and its integration with Si technology. Employing first-principles calculations, we study the electronic properties of monolayer and bilayer graphene adsorbed on clean and hydrogen (H)-passivated Si (111)/Si (100) surfaces. Our calculation shows that the interaction between monolayer graphene and a H-passivated Si surface is weak, with the band gap remaining negligible. For bilayer graphene adsorbed onto a H-passivated Si surface, the band gap opens up to 108 meV owing to asymmetry introduction. In contrast, the interaction between graphene and a clean Si surface is strong, leading to formation of chemical bonds and a large band gap of 272 meV. Our results provide guidance for device designs based on integrating graphene with Si technology.Keywords: band gap; graphene; graphene−Si interaction; silicon;
Co-reporter:Xiaotian Sun, Zhiwei Feng, Tingjun Hou, and Youyong Li
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 10) pp:7153
Publication Date(Web):May 6, 2014
DOI:10.1021/am500167c
Graphene and its water-soluble derivative, graphene oxide (GO), have attracted huge attention because of their interesting physical and chemical properties, and they have shown wide applications in various fields including biotechnology and biomedicine. Recently, GO has been shown to be the most efficient inhibitor for α-chymotrypsin (ChT) compared with all other artificial inhibitors. However, how GO interacts with bioactive proteins and its potential in enzyme engineering have been rarely explored. In this study, we investigate the interactions between ChT and graphene/GO by using molecular dynamics (MD) simulation. We find that ChT is adsorbed onto the surface of GO or graphene during 100 ns MD simulations. The α-helix of ChT plays as an important anchor to interact with GO. The cationic and hydrophobic residues of ChT form strong interactions with GO, which leads to the deformation of the active site of ChT and the inhibition of ChT. In comparison, the active site of ChT is only slightly affected after ChT adsorbed onto the graphene surface. In addition, the secondary structure of ChT is not affected after it is adsorbed onto GO or graphene surface. Our results illustrate the mechanism of the interaction between GO/graphene and enzyme and provide guidelines for designing efficient artificial inhibitors.Keywords: enzyme; graphene oxide; inhibitor; molecular dynamics;
Co-reporter:Zhihong Si, Lihua Qiu, Huilong Dong, Fenglou Gu, Youyong Li, and Feng Yan
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 6) pp:4346
Publication Date(Web):February 26, 2014
DOI:10.1021/am500022c
Imidazolium cations with butyl groups at various substitution positions (N1-, C2-, and N3-), 1-butyl-2,3-dimethylimidazolium ([N1-BDMIm]+), 2-butyl-1,3-dimethylimidazolium ([C2-BDMIm]+), and 3-butyl-1,2-dimethylimidazolium ([N3-BDMIm]+), were synthesized. Quantitative 1H NMR spectra and density functional theory calculation were applied to investigate the chemical stability of the imidazolium cations in alkaline solutions. The results suggested that the alkaline stability of the imidazolium cations was drastically affected by the C2-substitution groups. The alkaline stability of imidazolium cations with various substitution groups at the C2-position, including 2-ethyl-1-butyl-3-methylimidazolium ([C2-EBMIm]+), 1,2-dibutyl-3-methylimidazolium ([C2-BBMIm]+), and 2-hydroxymethyl-1-butyl-3-methylimidazolium ([C2-HMBMIm]+), was further studied. The butyl group substituted imidazolium cation ([C2-BBMIm]+) exhibited the highest alkaline stability at the elevated temperatures. The synthesized anion-exchange membranes based on the [C2-BBMIm]+ cation showed promising alkaline stability. These observations should pave the way to the practical application of imidazolium-based anion exchange membrane fuel cells.Keywords: alkaline stability; anion exchange membranes; imidazolium cations; substitution; theory calculation;
Co-reporter:Sheng Tian, Huiyong Sun, Peichen Pan, Dan Li, Xuechu Zhen, Youyong Li, and Tingjun Hou
Journal of Chemical Information and Modeling 2014 Volume 54(Issue 10) pp:2664-2679
Publication Date(Web):September 18, 2014
DOI:10.1021/ci500414b
In this study, to accommodate receptor flexibility, based on multiple receptor conformations, a novel ensemble docking protocol was developed by using the naïve Bayesian classification technique, and it was evaluated in terms of the prediction accuracy of docking-based virtual screening (VS) of three important targets in the kinase family: ALK, CDK2, and VEGFR2. First, for each target, the representative crystal structures were selected by structural clustering, and the capability of molecular docking based on each representative structure to discriminate inhibitors from non-inhibitors was examined. Then, for each target, 50 ns molecular dynamics (MD) simulations were carried out to generate an ensemble of the conformations, and multiple representative structures/snapshots were extracted from each MD trajectory by structural clustering. On average, the representative crystal structures outperform the representative structures extracted from MD simulations in terms of the capabilities to separate inhibitors from non-inhibitors. Finally, by using the naïve Bayesian classification technique, an integrated VS strategy was developed to combine the prediction results of molecular docking based on different representative conformations chosen from crystal structures and MD trajectories. It was encouraging to observe that the integrated VS strategy yields better performance than the docking-based VS based on any single rigid conformation. This novel protocol may provide an improvement over existing strategies to search for more diverse and promising active compounds for a target of interest.
Co-reporter:Zhenyu Guo, Oleg V. Prezhdo, Tingjun Hou, Xue Chen, Shuit-Tong Lee, and Youyong Li
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 10) pp:1642-1647
Publication Date(Web):April 21, 2014
DOI:10.1021/jz500565v
Highly ordered TiO2 nanotube arrays can be grown by simple electrochemical anodization of a titanium metal sheet, stimulating intense research and applications to solar cells and fuels. TiO2 nanotubes were expected to exhibit better electron transport than nanocrystal films. However, experiments showed that nanotubes provided little advantage over nanoparticles. Using nonadiabatic molecular dynamics, we demonstrate that oxygen vacancies, which are common in TiO2, accelerate nonradiative energy losses by an order of magnitude. Oxygen vacancies produce localized Ti3+ states hundreds of millielectronvolts below the TiO2 conduction band. The states lower the nanotube band gap, trap excited electrons, induce stronger electron–phonon couplings, and facilitate the relaxation. Our results rationalize the unforeseen experimental observations and provide the atomistic basis for improving the structure and charge transport by TiO2 nanotubes.Keywords: charge transport; energy relaxation; nonadiabatic dynamics; time-domain density functional theory; TiO2 nanotube;
Co-reporter:Qian Chen, Chao Wang, Zhixiong Zhan, Weiwei He, Zhenping Cheng, Youyong Li, Zhuang Liu
Biomaterials 2014 35(28) pp: 8206-8214
Publication Date(Web):
DOI:10.1016/j.biomaterials.2014.06.013
Co-reporter:Huilong Dong;Fenglou Gu;Min Li;Bencai Lin;Zhihong Si; Tingjun Hou; Feng Yan;Dr. Shuit-Tong Lee
ChemPhysChem 2014 Volume 15( Issue 14) pp:3006-3014
Publication Date(Web):
DOI:10.1002/cphc.201402262
Abstract
Imidazolium cations are promising candidates for preparing anion-exchange membranes because of their good alkaline stability. Substitution of imidazolium cations is an efficient way to improve their alkaline stability. By combining density functional theory calculations with experimental results, it is found that the LUMO energy correlates with the alkaline stability of imidazolium cations. The results indicate that alkyl groups are the most suitable substituents for the N3 position of imidazolium cations, and the LUMO energies of alkyl-substituted imidazolium cations depend on the electron-donating effect and the hyperconjugation effect. Comparing 1,2-dimethylimidazolium cations (1,2-DMIm+) and 1,3-dimethylimidazolium cations (1,3-DMIm+) with the same substituents reveals that the hyperconjugation effect is more significant in influencing the LUMO energy of 1,3-DMIms. This investigation reveals that LUMO energy is a helpful aid in predicting the alkaline stability of imidazolium cations.
Co-reporter:Huiyong Sun, Youyong Li, Dan Li, and Tingjun Hou
Journal of Chemical Information and Modeling 2013 Volume 53(Issue 9) pp:2376-2389
Publication Date(Web):August 18, 2013
DOI:10.1021/ci400188q
As a safe and efficacious drug, crizotinib was approved by the U.S. Food and Drug Administration (FDA) in 2011 for the treatment of advanced fusion-type nonsmall-cell lung cancer. Although high response ratio was detected from the patients treated with crizotinib, the cancer has eventually conferred resistance to crizotinib. Several drug resistance mutations have been found in the anaplastic lymphoma kinase (ALK) tyrosine kinase domain as the target for crizotinib, but the drug resistance mechanisms remain unclear. Therefore, in this study, the adaptive biasing force (ABF) method and two-end-state free energy calculation approaches were employed to elucidate the resistance mechanisms of crizotinib induced by the mutations L1152R, G1202R, and S1206Y. The ABF simulation results suggest that the reaction coordinates for the unbinding processes of crizotinib from the binding pockets of the mutated ALKs is different from that of the wild type ALK. The potentials of mean force for the crizotinib unbinding and the binding free energies predicted by the two-end-state free energy calculations are consistent with the experimental data. Our results indicate that the three mutations weaken the binding affinity of crizotinib obviously and lead to drug resistance. The free energy decomposition analysis illustrates the importance of the loss of two important H-bonds in the L1152R and S1206Y mutants on drug resistance. The entropy analysis shows that the entropy term plays a critical role in the substantial change of the conformational entropies of G1202R and L1152R. Our results reveal the mechanisms of drug resistance and provide vital clues for the development of new inhibitors to combat drug resistance.
Co-reporter:Zhiwei Feng, Tingjun Hou and Youyong Li
Molecular BioSystems 2013 vol. 9(Issue 8) pp:2142-2153
Publication Date(Web):03 May 2013
DOI:10.1039/C3MB70126C
Nucleosides are required for DNA and RNA synthesis, and the nucleoside adenosine has a function in a variety of signaling processes. Nucleosides require a specialized class of integral membrane proteins, known as nucleoside transporters (NTs), for specific transport across cell membranes. NTs are also important determinants for the transport of nucleoside-derived drugs across cell membranes. Recently, the crystal structure of the vcCNT (Vibrio cholerae Concentrative Nucleoside Transporter) was reported. Here we perform molecular dynamics (MD) simulations for the vcCNT structure in the presence of various sodium gradients, since CNTs are sodium-coupled transporters. The results highlight the important role of sodium bound to the vcCNT in the transport of uridine. Our MD simulations show that, without NaCl, uridine remains stable in the binding pocket of the vcCNT. In the presence of 20 mM NaCl, uridine moves from the binding pocket and approaches the entrance of the intracellular side. In the presence of 100 mM NaCl, uridine passes through most part of the entrance and approaches the intracellular side. The polar/charged amino acids in the binding pocket are important in the transport process. They first “fix” the ribose and allow the uracil base of uridine to approach the entrance of the intracellular side, and then “release” the ribose to allow uridine to move freely into the intracellular side coupled with the movement of sodium ions and HP1b. Finally, we propose a detailed mechanism of the nucleoside transport from the binding pocket to the intracellular side of the vcCNT.
Co-reporter:Xiaohui Yu, Tingjun Hou, Xuhui Sun, Youyong Li
Solid State Communications 2013 Volume 162() pp:28-33
Publication Date(Web):May 2013
DOI:10.1016/j.ssc.2013.03.012
•First time study the crystal structures/ phase transition of In2Se3 by DFT.•We identify the stable structures and phase transition mechanism of In2Se3.•Thermal annealing merges the layer structures and improves conductivity of In2Se3.In2Se3 has potential application in photovoltaic cell, solid-state batteries, phase change memories, and in the manufacture of detectors of ionizing radiation. Here we study the crystal structures and phase transition of In2Se3 by using DFT calculations. Crystal structures of In2Se3 include layered structures and vacancy-ordered-in-screw-form structures. Combining with SR-XRD techniques, our studies show that thermal annealing will merge the layer structures of In2Se3 and improves conductivity of In2Se3. Our results provide structural information for different phases and phase transition mechanism of In2Se3.Graphical abstract
Co-reporter:Zhiwei Feng, Tingjun Hou, Youyong Li
Journal of Molecular Graphics and Modelling 2013 Volume 39() pp:1-12
Publication Date(Web):February 2013
DOI:10.1016/j.jmgm.2012.10.003
Histamine H4 receptor (H4R), a member of histamine receptor family, which belongs to class A of G-protein coupled receptors (GPCRs), has been reported to play a critical role in histamine-induced chemotaxis in mast cells and eosinophils. Recently, the crystal structure of human histamine H1 receptor (H1R) was reported, which facilitates structure-based drug discovery of histamine receptor significantly. In the current work, the homology models of H4R and H3R are first constructed based on the crystal structure of H1R. Clobenpropit is then docked into the binding pocket of H4R and two different binding modes can be identified. In order to select a reasonable binding mode, several other ligands including agonists and antagonists are docked into H4R, and the results reveal that all ligands share one preferable binding mode: the protonated NH tightly interacts with Asp3.32 and the imidazole NH interacts with Glu5.46. By comparing H3R and H4R, we find that Glu5.20 and Thr6.55 in H4R involve in the selectivity of H4R. Then, we perform molecular dynamics (MD) simulations for H4R in complex with its compounds. MD results indicate that the preferable docking mode is more stable. Finally, we dock agonist histamine into H1R and H4R, and then perform 20 ns MD simulations for the complexes. H1R or H4R bound with histamine show strong conformational changes from TM5, TM6 and TM7, outward movement of intracellular part of TM6, and conformational change of Tyr7.53, which is consistent with the recent crystal structures of active GPCRs. Our results reveal the mechanism of selectivity and activation for H4R, which is important for developing selective antagonists and agonists for H4R.Graphical abstract20 ns molecular dynamics is performed for H4R with its agonist and antagonist with lipid and water. (a) By comparing H3R and H4R, we find that Glu5.20 and Thr6.55 in H4R involve in the selectivity of H4R. (b) H4R bound with histamine (in magenta color) shows strong conformational changes from TM5, TM6, and TM7, outward movement of intracellular part of TM6, and conformational change of Tyr7.53, which is consistent with the recent crystal structures (in yellow color) of active GPCRs. Our results reveal the mechanism of selectivity and activation for H4R, which is important for developing selective antagonists and agonists for H4R. The arrow indicates movement of TM5, TM6, and TM7 in H4R with agonist bound.Highlights► Based on recent H1R crystal structure, we develop homology models of H4R/H3R. ► We perform MD study of H1R, H3R and H4R bound with ligands in lipid and water. ► We identify the favorable binding mode and important residues involved in binding. ► Our results reveal the important residues for selectivity for H4R. ► Our MD results reveal the conformational changes of H1R and H4R.
Co-reporter:Liang Wu, Tingjun Hou, Youyong Li, K. S. Chan, and Shuit-Tong Lee
The Journal of Physical Chemistry C 2013 Volume 117(Issue 33) pp:17066-17072
Publication Date(Web):July 22, 2013
DOI:10.1021/jp405130c
Graphene is a promising material due to its outstanding properties. Point defects can be created artificially and tailor/improve the relative properties of pristine graphene. Defective graphene is potential material for electronic devices and sensors. Under irradiation or heat treatment, defects may diffuse and aggregate together. Here we perform density functional theory (DFT) to illustrate the migration and coalescence processes of the point defects. We find that the presence of single-vacancy (SV) defect stimulates the migration of another SV defect to bring them together and form the adjacent single vacancy defects. The adjacent single vacancy defects can combine into a divacancy defect, and we study the path. We also study the structural rearrangement of divacancy defect and conclude the relative stability of different types of divacancy defects. In addition, we find that divacancy defect [V2 (5-8-5) defect] is ready to be healed by a neighboring adatom defect. In comparison, divacancy defect [V2 (555-777) defect] cannot be healed by an adatom defect directly. Our results provide the mechanism of migration and coalescence processes of point defects in graphene, which is useful for nanoengineering of graphene with defect.
Co-reporter:Yaguang Zhao, Huilong Dong, Youyong Li and Xuefeng Fu
Chemical Communications 2012 vol. 48(Issue 29) pp:3506-3508
Publication Date(Web):17 Feb 2012
DOI:10.1039/C2CC00114D
Living radical polymerization (LRP) of organic and water soluble acrylates and acrylamides was mediated by a versatile cobalt porphyrin complex (TMP-OH)Co. The capability of this cobalt complex to mediate LRP in both polar and non-polar media permits the direct synthesis of useful and previously difficult to prepare functional block copolymers.
Co-reporter:Zhiwei Feng, Tingjun Hou, and Youyong Li
Journal of Chemical Information and Modeling 2012 Volume 52(Issue 4) pp:1005-1014
Publication Date(Web):March 10, 2012
DOI:10.1021/ci200594d
The β2 adrenergic receptor (β2AR) plays a key role in the control of smooth muscle relaxation in airways, the therapy of asthma, and a series of other basic physiological functions. Recently, the crystal structure of the β2AR–Gs protein complex was reported, which facilitates study of the activation mechanism of the β2AR and G-protein-coupled receptors (GPCRs). In this work, we perform 20 ns molecular dynamics (MD) simulations of the β2AR–Gs protein complex with its agonist in an explicit lipid and water environment to investigate the activation mechanism of β2AR. We find that during 20 ns MD simulation with a nanobody bound the interaction between the β2AR and the Gs protein is stable and the whole system is equilibrated within 6 ns. However, without a nanobody stabilizing the complex, the agonist triggers conformational changes of β2AR sequentially from the extracellular region to the intracellular region, especially the intracellular parts of TM3, TM5, TM6, and TM7, which directly interact with the Gs protein. Our results show that the β2AR–Gs protein complex makes conformational changes in the following sequence: (1) an agonist-bound part of β2AR, (2) the intracellular region of β2AR, and (3) the Gs protein.
Co-reporter:Zhiwei Feng, Tingjun Hou, and Youyong Li
Journal of Chemical Information and Modeling 2012 Volume 52(Issue 8) pp:2119-2131
Publication Date(Web):July 2, 2012
DOI:10.1021/ci300250q
FocA, a member of the formate-nitrite transporter (FNT) family, transports formate and nitrite across biological membranes in cellular organisms. The export and uptake of formate in bacteria are both mediated by FocA, which undergoes a pH-dependent functional switch. Recently, the crystal structures of Escherichia coli FocA (EcFocA), Vibrio cholerae FocA (VcFocA), and Salmonella typhimurium FocA (StFocA) were reported. We performed molecular dynamics (MD) on StFocA and EcFocA with different states of His209 (protonated and unprotonated), representing different pH conditions of FocA. The N-terminal helix in each protomer of StFocA covers and blocks the formate channel. At neutral or high pH (MD simulations with unprotonated His209), the concerted movement of the N-terminal helices of pairs of protomers of StFocA opens its formate channel. At low pH (MD simulations with protonated His209), protonated His209 interacts tightly with its neighboring residue Asn262, and the channel becomes narrower, so that the formate can hardly pass through the channel. We obtained similar results for EcFocA. Our study shows that pairs of protomers of FocA move in a concerted way to achieve its pH-dependent gating function, which provides information on the dynamics of the gating mechanism of FNT proteins and aquaporins.
Co-reporter:Zhiwei Feng, Tingjun Hou and Youyong Li
Molecular BioSystems 2012 vol. 8(Issue 10) pp:2699-2709
Publication Date(Web):06 Jul 2012
DOI:10.1039/C2MB25184A
Tripartite complex AcrB–ToIC, the major efflux system in Escherichia coli, is the principal multidrug transporter in Gram-negative bacteria, which is important in antibiotic drug tolerance. AcrB is a homotrimer that acts as a tripartite complex with the outer membrane channel ToIC and the membrane fusion protein AcrA. Recently, the crystal structures of AcrB bound to the high-molecular-mass drugs rifampicin and erythromycin were reported. Here we performed 20 ns molecular dynamics (MD) simulations of the AcrB–rifampicin–minocycline complex in a lipid bilayer and explicit water. We found that the bound drugs, rifampicin and erythromycin, made a unidirectional peristaltic movement towards the extrusion funnel of ToIC, which was facilitated by the water efflux in the channel of AcrB. With a shift of the Phe-617 loop, rifampicin in the access monomer moved towards the entrance of the distal binding pocket. Minocycline in the binding monomer moved from the distal binding pocket towards the gate of the central funnel. The channel between the entrance and the gate made a concerted opening during the MD simulations, which was helpful for the peristaltic movement. Our results showed that the mutations of Gly616Pro and Gly619Pro prevented the movement of the Phe-617 loop, which indicated the critical role of the flexibility of the Phe-617 loop. In addition, three putative proton translocation channels were proposed based on our results. Our study provided dynamical information and important residues for the peristaltic movement in AcrB, which were critical for substrate uptake and extrusion function.
Co-reporter:Liang Wu, Tingjun Hou, Yi Wang, Yanfei Zhao, Zhenyu Guo, Youyong Li, Shuit-Tong Lee
Journal of Alloys and Compounds 2012 Volume 541() pp:250-255
Publication Date(Web):15 November 2012
DOI:10.1016/j.jallcom.2012.06.091
Zinc oxide (ZnO) is a promising material for its wide application in solid-state devices. With the pressure raised from an ambient condition, ZnO transforms from fourfold wurtzite (B4) to sixfold coordinated rocksalt (B1) structure. Doping is an efficient approach to improve the structures and properties of materials. Here we use density-functional theory (DFT) to study doped ZnO and find that the transition pressure from B4 phase to B1 phase of ZnO always decreases with different types of transition metal (V, Cr, Mn, Fe, Co, or Ni) doped, but the phase transition path is not affected by doping. This is consistent with the available experimental results for Mn-doped ZnO and Co-doped ZnO. Doping in ZnO causes the lattice distortion, which leads to the decrease of the bulk modulus and accelerates the phase transition. Mn-doped ZnO shows the strongest magnetic moment due to its half filled d orbital. For V-doped ZnO and Cr-doped ZnO, the magnetism is enhanced by phase transition from B4 to B1. But for Mn-doped ZnO, Fe-doped ZnO, Co-doped ZnO, and Ni-doped ZnO, B1 phase shows weaker magnetic moment than B4 phase. These results can be explained by the amount of charge transferred from the doped atom to O atom. Our results provide a theoretical basis for the doping approach to change the structures and properties of ZnO.Graphical abstractHighlights► We study the doping effect on B4, B1 structures and phase transition of ZnO. ► We calculate the phase transition barrier and phase transition path of doped ZnO. ► The transition metal doping decreases the bulk modulus and phase transition pressure. ► The magnetic properties are influenced by the phase transition process.
Co-reporter:Huilong Dong, Tingjun Hou, Yaguang Zhao, Xuefeng Fu, Youyong Li
Computational and Theoretical Chemistry 2012 Volume 1001() pp:51-59
Publication Date(Web):1 December 2012
DOI:10.1016/j.comptc.2012.10.010
We design versatile cobalt porphyrin complexes for living radical polymerization (LRP) of olefins and perform density functional theory (DFT) calculations to investigate the efficiency of cobalt porphyrin complexes. Our calculation results demonstrate that (TMPOH)CoR is efficient due to less steric hindrance. By comparing the bond dissociation energies (BDEs) and reaction pathways, our calculation results show that the efficiency of cobalt porphyrin complexes is in the following sequence: (TPFP)CoR > (TMPOH)CoR > (TMP)CoR.We find that electronic effect and steric hindrance are important for bond dissociation energies of cobalt porphyrin complexes. Among the chosen monomers, CoCR BDEs of VAc and AN are the highest, and tBA’s is the lowest, which determines their performance in LRP. The polydispersity index (PDI) of these polymerized olefins we obtained is consistent with our calculated BDE. We conclude that the activation barrier of the radical pair correlates with the catalytic efficiency. Low activation barrier corresponds to efficient catalytic reaction. Our results show that the modified species (TMPOH)CoII and (TPFP)CoII are better LRP catalysts for olefins than (TMP)CoII.Graphical abstractEfficient versatile cobalt prophyrin complex with reduced steric hindrance for polymerization of acrylates and acrylamides.Highlights► We design versatile cobalt porphyrin complexes for LRP of olefins. ► Electronic effect and steric hindrance determine the efficiency of the catalysts. ► Calculation and experimental results are consistent for the catalytic efficiency.
Co-reporter:Zhiwei Feng;Tingjun Hou
Journal of Molecular Modeling 2012 Volume 18( Issue 12) pp:5051-5063
Publication Date(Web):2012 December
DOI:10.1007/s00894-012-1509-x
D3 receptor, a member of dopamine (DA) D2-like receptor family, which belongs to class A of G-protein coupled receptors (GPCRs), has been reported to play a critical role in neuropsychiatric disorders. Recently, the crystal structure of human dopamine D3 receptor was reported, which facilitates structure-based drug discovery of D3R significantly. We dock D3R-selective compounds into the crystal structure of D3R and homology structure of D2R. Then we perform 20 ns molecular dynamics (MD) of the receptor with selective compounds bound in explicit lipid and water. Our docking and MD results indicate the important residues related to the selectivity of D3R. Specifically, residue Thr7.39 in D3R may contribute to the high selectivity of R-22 with D3R. Meanwhile, the 4-carbon linker and phenylpiperazine of R-22 improve the binding affinity and the selectivity with D3R. We also dock the agonists, including dopamine, into D3R and perform MD. Our molecular dynamics results of D3R with agonist bound show strong conformational changes from TM5, TM6, and TM7, outward movement of intracellular part of TM6, fluctuation of “ionic lock” motif and conformational change of Tyr7.53, which is consistent with recent crystal structures of active GPCRs and illustrates the dynamical process during activation. Our results reveal the mechanism of selectivity and activation for D3R, which is important for developing high selective antagonists and agonists for D3R.
Co-reporter:Xiaohui Yu;Tingjun Hou;Xuhui Sun
ChemPhysChem 2012 Volume 13( Issue 6) pp:1514-1521
Publication Date(Web):
DOI:10.1002/cphc.201101012
Abstract
TiO2 doped with transition metals shows improved photocatalytic efficiency. Herein the electronic and optical properties of Mo-doped TiO2 with defects are investigated by DFT calculations. For both rutile and anatase phases of TiO2, the bandgap decreases continuously with increasing Mo doping level. The 4d electrons of Mo introduce localized states into the forbidden band of TiO2, and this shifts the absorption edge into the visible-light region and enhances the photocatalytic activity. Since defects are universally distributed in TiO2 or doped TiO2, the effect of oxygen deficiency due to oxygen vacancies or interstitial Mo atoms is systemically studied. Oxygen vacancies associated with the Mo dopant atoms or interstitial Mo will reduce the spin polarization and magnetic moment of Mo-doped TiO2. Moreover, oxygen deficiency has a negative impact on the improved photocatalytic activity of Mo-doped TiO2. The current results indicate that substitutional Mo, interstitial Mo, and oxygen vacancy have different impacts on the electronic/optical properties of TiO2 and are suited to different applications.
Co-reporter:Yang Li, Jing Gao, Qinliang Li, Mingfa Peng, Xuhui Sun, Youyong Li, Gang Yuan, Wen Wen and M. Meyyappan
Journal of Materials Chemistry A 2011 vol. 21(Issue 19) pp:6944-6947
Publication Date(Web):11 Apr 2011
DOI:10.1039/C1JM10419E
We report the preparation of α- and κ-phase In2Se3 nanowires by thermal evaporation and investigation of their phase transformations in situ by synchrotron radiation X-ray diffraction (XRD) during a thermal annealing process. The κ-phase transformed into the α-phase at 500 °C and eventually transformed to high temperature α-phase with a layered structure of 5 atoms-5 atoms at 700 °C irreversibly. Different atomistic structures of In2Se3 were modeled and optimized by DFT, which correlate well with the XRD results. The In2Se3 nanowires also exhibit a large difference in resistivity before and after annealing.
Co-reporter:Yi Wang ; Tingjun Hou ; Sheng Tian ; Shuit-Tong Lee
The Journal of Physical Chemistry C 2011 Volume 115(Issue 15) pp:7706-7716
Publication Date(Web):March 30, 2011
DOI:10.1021/jp111203e
Zinc oxide, ZnO, can be synthesized into a variety of morphologies including nanowires, nanorods, tetrapods, nanobelts, nanoflowers, nanoparticles, etc., and zinc oxide nanomaterials are promising candidates for nanoelectronics and photonics. Doping in ZnO is one of the most efficient approaches to improve its structure and properties. Here we use the density-functional theory (DFT) to investigate the doping effect of Mn or Co on ZnO. Our calculation results reveal that the transition pressure from the wurtzite (B4) phase to the rocksalt (B1) phase of ZnO decreases with Mn or Co doping, which is consistent with the in situ high-pressure X-ray powder diffraction results from synchrotron radiation. For both Mn and Co doping, the doping effect on different structures of ZnO increases in the order: B1 < intermediate tetragonal structure < intermediate hexagonal structure < B4. This causes the decrease of the transition barrier between B4 and B1, which accounts for the decreased transition pressure of Mn- or Co-doped ZnO. Our results provide a theoretical basis for the doping approach to control the structures and properties of ZnO and other similar materials.
Co-reporter:Xiaohui Yu, Tingjun Hou, Xuhui Sun, Youyong Li
Solid State Communications (May 2013) Volume 162() pp:28-33
Publication Date(Web):1 May 2013
DOI:10.1016/j.ssc.2013.03.012
•First time study the crystal structures/ phase transition of In2Se3 by DFT.•We identify the stable structures and phase transition mechanism of In2Se3.•Thermal annealing merges the layer structures and improves conductivity of In2Se3.In2Se3 has potential application in photovoltaic cell, solid-state batteries, phase change memories, and in the manufacture of detectors of ionizing radiation. Here we study the crystal structures and phase transition of In2Se3 by using DFT calculations. Crystal structures of In2Se3 include layered structures and vacancy-ordered-in-screw-form structures. Combining with SR-XRD techniques, our studies show that thermal annealing will merge the layer structures of In2Se3 and improves conductivity of In2Se3. Our results provide structural information for different phases and phase transition mechanism of In2Se3.Graphical abstractDownload high-res image (190KB)Download full-size image
Co-reporter:Huilong Dong, Lu Wang, Liujiang Zhou, Tingjun Hou, Youyong Li
Carbon (March 2017) Volume 113() pp:
Publication Date(Web):March 2017
DOI:10.1016/j.carbon.2016.11.029
The graphene-based gas sensors are limited by the weak physisorption to gas molecules, and the doping of silicon is an effective way to enhance its sensitivity. By density functional investigations, we reported a novel SiC5 siligraphene (g-SiC5) with great potential in detecting and sensing some specific air pollutants. The g-SiC5 is semimetallic as graphene is, possessing good structural and thermal stability. By comparing the adsorption performance of 12 common gas molecules, we have revealed that the nucleophilic gas molecules could form stable chemisorption with g-SiC5. Among the 12 gas molecules, NO, HCHO and SO2 exhibit moderate adsorption energies in the range of 0.4–0.6 eV when chemisorb on g-SiC5 and would open up a considerable band gap due to the orbital hybridization, thus changing the conductivity of g-SiC5 and making the sensing feasible. The further comparative research demonstrates that the g-SiC5 is superior to Si-doped graphene and other siligraphenes in gas sensing, enabling g-SiC5 to be a promising material as gas sensor for detecting NO, HCHO or SO2 from air mixture.The g-SiC5 is semimetallic and exhibits good selectivity and sensitivity for air pollutants due to the band gap opening caused by chemisorption, making it promising in gas sensing.
Co-reporter:Yujin Ji, Mingye Yang, Huilong Dong, Lu Wang, Tingjun Hou and Youyong Li
Journal of Materials Chemistry A 2017 - vol. 5(Issue 4) pp:NaN1741-1741
Publication Date(Web):2016/12/09
DOI:10.1039/C6TA08321H
Exploration of oxygen reduction reaction (ORR) catalysts based on non-precious metals is of great importance to the development of fuel cells. In this work, we theoretically investigate the practical feasibility and catalytic activity of two-dimensional (2D) group IVA monochalcogenides MXs (M = Ge/Sn and X = S/Se) as potential and efficient ORR catalysts. Through first-principles calculations, it is found that these 2D MXs are semiconductors with high adsorption affinity and dissociation activity for oxygen molecules. Furthermore, the free energy diagrams along the four-electron pathway are simulated to elucidate the whole ORR mechanism catalyzed by the monolayer MXs (GeS, GeSe, and SnSe here), with all the energetically favorable intermediates being exothermic in free energy. In the acidic environment, the GeS monolayer exhibits the best performance due to its considerably low overpotential (0.52 V). While in the alkaline environment, there is no overpotential for the ORR catalyzed by the 2D MXs (GeS, GeSe, and SnSe). Our work provides a novel insight into the electrochemical applications of IVA monochalcogenides, which is expected to realize experimentally.
Co-reporter:Yaguang Zhao, Huilong Dong, Youyong Li and Xuefeng Fu
Chemical Communications 2012 - vol. 48(Issue 29) pp:NaN3508-3508
Publication Date(Web):2012/02/17
DOI:10.1039/C2CC00114D
Living radical polymerization (LRP) of organic and water soluble acrylates and acrylamides was mediated by a versatile cobalt porphyrin complex (TMP-OH)Co. The capability of this cobalt complex to mediate LRP in both polar and non-polar media permits the direct synthesis of useful and previously difficult to prepare functional block copolymers.
Co-reporter:Xiaotian Sun, Yunxia Liu, Zhigang Song, Yongdan Li, Weizhou Wang, Haiping Lin, Lu Wang and Youyong Li
Journal of Materials Chemistry A 2017 - vol. 5(Issue 17) pp:NaN4166-4166
Publication Date(Web):2017/03/24
DOI:10.1039/C7TC00306D
Defects are unavoidable during the synthesis of materials, especially for two-dimensional (2D) nanomaterials. They are usually seen as detrimental to device properties, but sometimes bring about new beneficial effects. In order to clarify the influence of defects on the structural and electronic properties, we have performed first-principles calculations to systematically investigate the structural stability, mobility and electronic properties of typical point defects in 2D arsenene (h-As), antimonene (h-Sb) and antimony arsenide (h-AsSb), including the Stone–Wales defects, single vacancies (SVs), double vacancies (DVs) and adatoms. To provide visual guidance for experimental observations, scanning tunnelling microscopy (STM) images are simulated. Compared to defects in graphene and silicene, these defects form more easily with lower formation energies, and SVs can diffuse very quickly to the edges with a lower diffusion barrier of less than 1 eV. Monolayer arsenene, antimonene and antimony arsenide are indirect band gap semiconductors, and the defective structures significantly reduce the band gaps. Most of the SV and adatom defects carry magnetic moments due to the dangling bonds resulting from the absent or extra atom. Our present results have demonstrated that the point defects induce significant effects on the electronic properties of pristine arsenene, antimonene and the antimony arsenide alloy, which should be considered in their future applications.
Co-reporter:Ningning Yu, Lu Wang, Min Li, Xiaotian Sun, Tingjun Hou and Youyong Li
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 17) pp:NaN11704-11704
Publication Date(Web):2015/03/25
DOI:10.1039/C5CP00161G
Molybdenum disulfide (MoS2), a kind of graphene-like, two-dimensional material, has attracted great interest because of its unique properties and potential applications in electronics and sensors. In this paper, first-principle calculations and grand canonical Monte Carlo (GCMC) simulations are performed and used to show that the MoS2 layer is efficient at absorbing non-polar gases. Compared with the popular gas sorbents (metal organic frameworks and carbon-based materials), MoS2 has additional advantages, including large surface to volume ratio and tunable properties. The non-polar gas [carbon dioxide (CO2) and methane (CH4)] adsorption on the MoS2 layer with and without vacancies has been systematically studied. The perfect MoS2 shows little or no adsorption for CO2 and CH4 molecules, but the MoS2 with a single S vacancy and double S vacancies exhibits an excellent adsorption ability for CO2 and CH4 gases. The adsorption energies were 65 kJ mol−1 for CO2 and 47 kJ mol−1 for CH4 (van der Waals-D2), respectively. An orbital coupling between the p orbital of the CO2 (or CH4) molecule and the d orbital of the Mo atom was observed. GCMC simulation results show that MoS2 with a single S vacancy could absorb 42.1 wt% of CO2 and 37.6 wt% of CH4 under a pressure of 80 bar at room temperature. The results given in this paper indicate that monolayer MoS2 with defects is a highly efficient absorbent for non-polar gases.
Co-reporter:Xiaotian Kong, Peichen Pan, Dan Li, Sheng Tian, Youyong Li and Tingjun Hou
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 8) pp:NaN6113-6113
Publication Date(Web):2015/01/27
DOI:10.1039/C4CP05440G
Anaplastic lymphoma kinase (ALK) has gained increased attention as an attractive therapeutic target for the treatment of various cancers, especially non-small-cell lung cancer (NSCLC). Recently, piperidine carboxamides were reported as Type I1/2 inhibitors of ALK, which occupy both the ATP binding site and the back ATP hydrophobic cavity in DFG-in conformation. Due to the dynamic behavior of ALK in the binding of Type I1/2 inhibitors, the accurate predictions of the binding structures and relative binding potencies of these inhibitors are quite challenging. In this study, different modeling techniques, including molecular docking, ensemble docking based on multiple receptor conformations, molecular dynamics simulations and free energy calculations, were utilized to explore the binding mechanisms of piperidine carboxamides. Our predictions show that the conventional docking protocols are not sufficient to predict the relative binding potencies of the studied inhibitors with high accuracy, but incorporating protein flexibility before or after docking is quite effective to improve the prediction accuracy. Notably, the binding free energies predicted by MM/GBSA or MM/PBSA based on the MD simulations for the docked poses give the highest correlation with the experimental data, highlighting the importance of the inclusion of receptor flexibility for the accurate predictions of the binding potencies for Type I1/2 inhibitors of ALK. Furthermore, the comprehensive analysis of several pairs of representative inhibitors demonstrates the importance of hydrophobic interactions in improving the binding affinities of the inhibitors with the hot-spot residues surrounding the binding pocket. This work is expected to provide valuable clues for further rational design of novel and potent Type I1/2 ALK inhibitors.
Co-reporter:Yang Li, Jing Gao, Qinliang Li, Mingfa Peng, Xuhui Sun, Youyong Li, Gang Yuan, Wen Wen and M. Meyyappan
Journal of Materials Chemistry A 2011 - vol. 21(Issue 19) pp:NaN6947-6947
Publication Date(Web):2011/04/11
DOI:10.1039/C1JM10419E
We report the preparation of α- and κ-phase In2Se3 nanowires by thermal evaporation and investigation of their phase transformations in situ by synchrotron radiation X-ray diffraction (XRD) during a thermal annealing process. The κ-phase transformed into the α-phase at 500 °C and eventually transformed to high temperature α-phase with a layered structure of 5 atoms-5 atoms at 700 °C irreversibly. Different atomistic structures of In2Se3 were modeled and optimized by DFT, which correlate well with the XRD results. The In2Se3 nanowires also exhibit a large difference in resistivity before and after annealing.
Co-reporter:Min Li, Lu Wang, Ningning Yu, Xiaotian Sun, Tingjun Hou and Youyong Li
Journal of Materials Chemistry A 2015 - vol. 3(Issue 15) pp:NaN3649-3649
Publication Date(Web):2015/02/25
DOI:10.1039/C5TC00209E
Based on density functional theory (DFT) calculations, we have constructed and investigated different types of single-side hydrogenated graphene (SSHG) structures from their structural motifs. The structural stability and electronic properties of these SSHG structures are extensively analyzed and compared with the reported structures. The single-side hydrogenation causes a severe bending in graphene at high H coverage, which leads to a greater formation energy with increasing H coverage. Among the SSHG structures that we have considered, the configurations with H attached along the armchair direction show the lowest formation energies due to a relatively small buckling compared to other configurations. Moreover, only the armchair hydrogenated graphene opens a band gap near the Fermi level, and the band gap can be modulated from zero to 1.44 eV by varying the H coverage from zero to 50%. Our results suggest an efficient way to prepare graphene-based materials and devices with suitable band gaps.
Co-reporter:Yunxia Liu, Yao-Jun Dong, Zeyuan Tang, Xue-Feng Wang, Lu Wang, Tingjun Hou, Haiping Lin and Youyong Li
Journal of Materials Chemistry A 2016 - vol. 4(Issue 26) pp:NaN6385-6385
Publication Date(Web):2016/06/07
DOI:10.1039/C6TC01328G
Recently, borophene was reported to be produced on silver surfaces. We employed density functional theory and electronic transport calculations to investigate the stabilities, electronic structures and transport properties of borophene nanoribbons. The stability of a borophene nanoribbon increases with its width and only the lined-edged borophene nanoribbons are stable in the free-standing form. Such anisotropic stability dependence is ascribed to the large scale delocalization of π electrons along the boron rows. In particular, all line-edge borophene nanoribbons undergo edge reconstructions, in which the out-of-plane bulking edge atoms are reconstructed to form quasi planar edge structures. Such edge reconstructions have not yet been reported, which is critical for the formation of boron nanostructrues. Subsequent electronic transport calculations based on a non-equilibrium Green’s function indicated that line-edge borophene nanoribbons exhibit low-resistivity Ohmic conductance. Our results indicated that the line-edge borophene nanoribbons present remarkable properties (high thermal stabilities, Ohmic conductance with low electrical resistivity and good rigidities) and are promising for applications as one-dimensional electrical connections in compact nanoscale circuits.
Co-reporter:Chunmiao Du, Haiping Lin, Bin Lin, Zeyao Ma, Tingjun Hou, Jianxin Tang and Youyong Li
Journal of Materials Chemistry A 2015 - vol. 3(Issue 46) pp:NaN23119-23119
Publication Date(Web):2015/10/29
DOI:10.1039/C5TA05084G
Late transition metals, such as Rh, Ir, Pd and Pt, have a strong tendency to form a square-planar 16-electron complex. Although this feature has been widely used in organometallics to develop homogeneous catalysts, a single-atom heterogeneous analogue has not yet been reported. In this work, we show that a 16-electron complex may act as an important transition state in the CO oxidation over a single Pt atom supported by a MoS2 monolayer (Pt/MoS2). The catalytic oxidation reaction prefers to start with the Langmuir–Hinshelwood (L–H) reaction, where the CO and O2 molecules are first co-adsorbed on the Pt atom, then cross a small barrier of 0.40 eV to form a square-planar 16-electron intermediate state, and subsequently the first CO2 is released. The activation barrier of the following Eley–Rideal (E–R) reaction is only 0.23 eV. The superior catalytic reactivity of the Pt/MoS2 surface can be explained by the quantum confinement effect of the Pt-5d orbitals and the stability of the square-planar 16-electron transition state. In addition, MoS2 may serve as a defect-free two dimensional anchoring substrate for Pt atomic adsorption. It provides not only a very large surface-to-volume ratio, but also a well-defined structure with a uniform distribution of anchoring points. The square-planar 16-electron intermediate state of the L–H reaction, together with the new MoS2 anchoring substrate, may provide a new opportunity for the design of single-atom catalysts on two-dimensional surfaces.
Co-reporter:Xiao Yuan, Mingye Yang, Lu Wang and Youyong Li
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 21) pp:NaN13854-13854
Publication Date(Web):2017/05/02
DOI:10.1039/C7CP01727H
Based on the first-principles calculations, we systematically studied the structures and electronic properties of two-dimensional (2D) transition metal dichalcogenide (TMD) alloys with half-to-half mixing of S and Se. Using the chemical potentials of S and Se, the energetic phase diagrams for both single phases and mixed phases of TMD were constructed. A new heterolayer structure (for Sc and Ti) and alternating structure (for Cr, Mn, Fe, Zr, Mo, and W) were proposed for the first time, which were thermodynamically stable for MSSe alloys under the S-poor (relatively low chemical potential of S) and Se-rich (relatively high chemical potential of Se) conditions, and further compared with the disordered structures. Moreover, band gaps, carrier effective mass, and work functions were calculated for these stable mixed phases. Compared to the single phases of MS2 and MSe2, MSSe alloys showed superior electronic properties including tunable band gaps and work functions. Importantly, the significantly reduced effective mass of the carriers in the MSSe alloys may induce higher carrier mobility, providing better performance of TMD materials in electronic devices.
Co-reporter:Mingye Yang, Lu Wang, Tingjun Hou and Youyong Li
Journal of Materials Chemistry A 2017 - vol. 5(Issue 1) pp:NaN207-207
Publication Date(Web):2016/11/29
DOI:10.1039/C6TC04487E
Two-dimensional nanomaterials have attracted extensive interest and their heterostructures possess excellent electronic and optical properties suitable for various applications. Based on first-principles calculations, we investigated the structural stability and electronic properties of WS2 and graphene oxide (GO) heterostructures. We considered three types of GO, including epoxy only, hydroxyl only and both epoxy and hydroxyl on the GO surface. Our results show that the interlayer binding energy per WS2 unit increases from 0.117 eV to 0.214 eV as the surface oxygen concentration of GO increases. The band gap of the WS2/GO heterostructures can be efficiently tuned in a wide range from 0.13 eV to 1.91 eV by changing the oxygen functionalities and the concentration. The spatial separation of the conduction band minimum and valence band maximum is observed, which are distributed in different layers. In addition, the work function of WS2 can also be modulated by GO in the range of 4.09 eV to 6.34 eV, which potentially increases the carrier concentration and broadens the applications of WS2 and other transition metal dichalcogenide materials in optoelectronic devices.
Co-reporter:Xiaotian Kong, Huiyong Sun, Peichen Pan, Sheng Tian, Dan Li, Youyong Li and Tingjun Hou
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 3) pp:NaN2046-2046
Publication Date(Web):2015/12/02
DOI:10.1039/C5CP05622E
Due to the high sequence identity of the binding pockets of cyclin-dependent kinases (CDKs), designing highly selective inhibitors towards a specific CDK member remains a big challenge. 4-(thiazol-5-yl)-2-(phenylamino) pyrimidine derivatives are effective inhibitors of CDKs, among which the most promising inhibitor 12u demonstrates high binding affinity to CDK9 and attenuated binding affinity to other homologous kinases, such as CDK2. In this study, in order to rationalize the principle of the binding preference towards CDK9 over CDK2 and to explore crucial information that may aid the design of selective CDK9 inhibitors, MM/GBSA calculations based on conventional molecular dynamics (MD) simulations and enhanced sampling simulations (umbrella sampling and steered MD simulations) were carried out on two representative derivatives (12u and 4). The calculation results show that the binding specificity of 12u to CDK9 is primarily controlled by conformational change of the G-loop and variation of the van der Waals interactions. Furthermore, the enhanced sampling simulations revealed the different reaction coordinates and transient interactions of inhibitors 12u and 4 as they dissociate from the binding pockets of CDK9 and CDK2. The physical principles obtained from this study may facilitate the discovery and rational design of novel and specific inhibitors of CDK9.

![2,1,3-Benzothiadiazole, 4,4'-[4,4-bis(2-ethylhexyl)-4H-silolo[3,2-b:4,5-b']dithiophene-2,6-diyl]bis[7-bromo-](/data/chemimg/164900/1401032-28-7.png)
![2,1,3-Benzothiadiazole, 4,4'-[4,4-bis(2-ethylhexyl)-4H-silolo[3,2-b:4,5-b']dithiophene-2,6-diyl]bis[7-bromo-](/data/chemimg/164900/1401032-28-7_b.png)
![1H-Indole-6-carboxylic acid, 2,3-dihydro-3-[[[4-[methyl[2-(4-methyl-1-piperazinyl)acetyl]amino]phenyl]amino]phenylmethylene]-2-oxo-, methyl ester](http://img.cochemist.com/ccimg/1160300/1160294-26-7.png)
![1H-Indole-6-carboxylic acid, 2,3-dihydro-3-[[[4-[methyl[2-(4-methyl-1-piperazinyl)acetyl]amino]phenyl]amino]phenylmethylene]-2-oxo-, methyl ester](http://img.cochemist.com/ccimg/1160300/1160294-26-7_b.png)











![5-((3-Chlorophenyl)amino)benzo[c][2,6]naphthyridine-8-carboxylic acid](http://img.cochemist.com/ccimg/1009900/1009820-21-6.png)
![5-((3-Chlorophenyl)amino)benzo[c][2,6]naphthyridine-8-carboxylic acid](http://img.cochemist.com/ccimg/1009900/1009820-21-6_b.png)
![Thieno[3,2-d]pyrimidine, 2-(1H-indazol-4-yl)-6-[[4-(methylsulfonyl)-1-piperazinyl]methyl]-4-(4-morpholinyl)-](http://img.cochemist.com/ccimg/957100/957054-30-7.png)
![Thieno[3,2-d]pyrimidine, 2-(1H-indazol-4-yl)-6-[[4-(methylsulfonyl)-1-piperazinyl]methyl]-4-(4-morpholinyl)-](http://img.cochemist.com/ccimg/957100/957054-30-7_b.png)
![1,3,2-Dioxaborolane, 2-[4-(hexyloxy)phenyl]-4,4,5,5-tetramethyl-](http://img.cochemist.com/ccimg/922000/921937-76-0.png)
![1,3,2-Dioxaborolane, 2-[4-(hexyloxy)phenyl]-4,4,5,5-tetramethyl-](http://img.cochemist.com/ccimg/922000/921937-76-0_b.png)


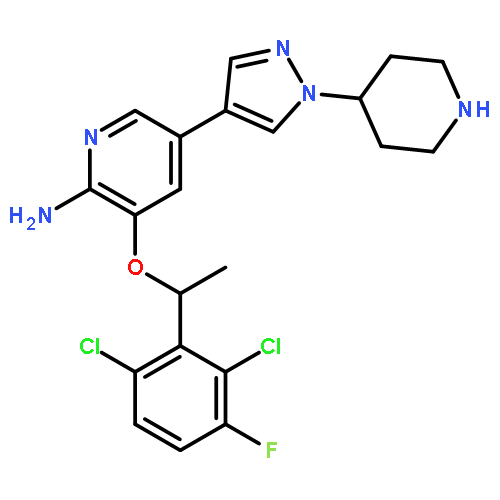


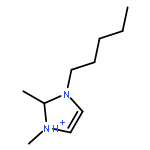
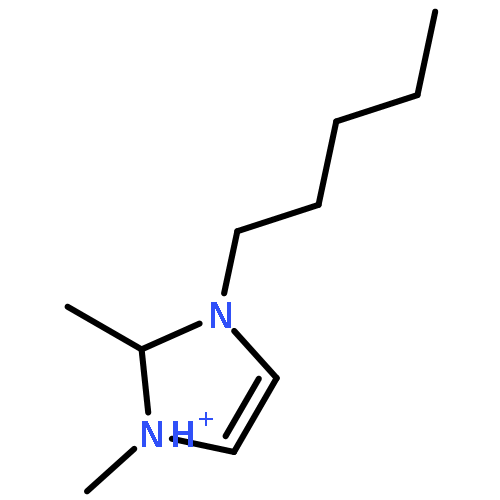


![(S)-N1-((1H-Benzo[d]imidazol-2-yl)methyl)-N1-(5,6,7,8-tetrahydroquinolin-8-yl)butane-1,4-diamine](http://img.cochemist.com/ccimg/558500/558447-26-0.png)
![(S)-N1-((1H-Benzo[d]imidazol-2-yl)methyl)-N1-(5,6,7,8-tetrahydroquinolin-8-yl)butane-1,4-diamine](http://img.cochemist.com/ccimg/558500/558447-26-0_b.png)


![3-({[(3-chlorobenzoyl)amino]carbonothioyl}amino)benzoic acid](http://img.cochemist.com/ccimg/434000/433942-71-3.png)
![3-({[(3-chlorobenzoyl)amino]carbonothioyl}amino)benzoic acid](http://img.cochemist.com/ccimg/434000/433942-71-3_b.png)




![3',5'-Difluoro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/350700/350682-84-7.png)
![3',5'-Difluoro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/350700/350682-84-7_b.png)






![3',5'-Dichloro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/191000/190911-79-6.png)
![3',5'-Dichloro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/191000/190911-79-6_b.png)
![2-[[9-(1-Methylethyl)-6-[(phenylmethyl)amino]-9H-purin-2-yl]amino]-1-butanol](http://img.cochemist.com/ccimg/186700/186692-44-4.png)
![2-[[9-(1-Methylethyl)-6-[(phenylmethyl)amino]-9H-purin-2-yl]amino]-1-butanol](http://img.cochemist.com/ccimg/186700/186692-44-4_b.png)











![1,1'-[1,4-Phenylenebis(methylene)]bis[1,4,8,11-tetraazacyclotetradecane]](http://img.cochemist.com/ccimg/155200/155148-31-5.png)
![1,1'-[1,4-Phenylenebis(methylene)]bis[1,4,8,11-tetraazacyclotetradecane]](http://img.cochemist.com/ccimg/155200/155148-31-5_b.png)








![Propanedinitrile,[2-[2-[4-(diethylamino)phenyl]ethenyl]-6-methyl-4H-pyran-4-ylidene]-](http://img.cochemist.com/ccimg/140700/140623-32-1.png)
![Propanedinitrile,[2-[2-[4-(diethylamino)phenyl]ethenyl]-6-methyl-4H-pyran-4-ylidene]-](http://img.cochemist.com/ccimg/140700/140623-32-1_b.png)

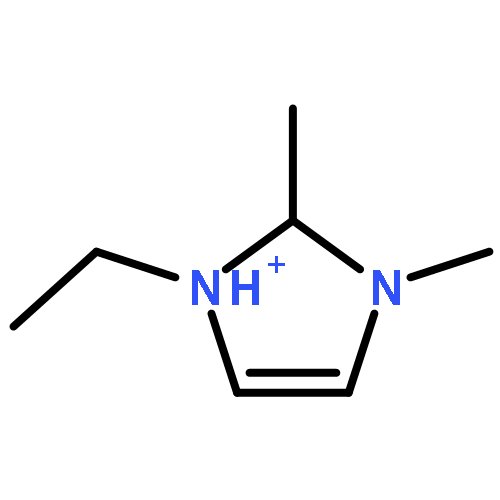


![[1,1':3',1''-Terphenyl]-4,4''-dicarboxaldehyde, 5'-(4-formylphenyl)-](/data/chemimg/113700/118688-53-2.png)
![[1,1':3',1''-Terphenyl]-4,4''-dicarboxaldehyde, 5'-(4-formylphenyl)-](/data/chemimg/113700/118688-53-2_b.png)











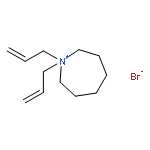
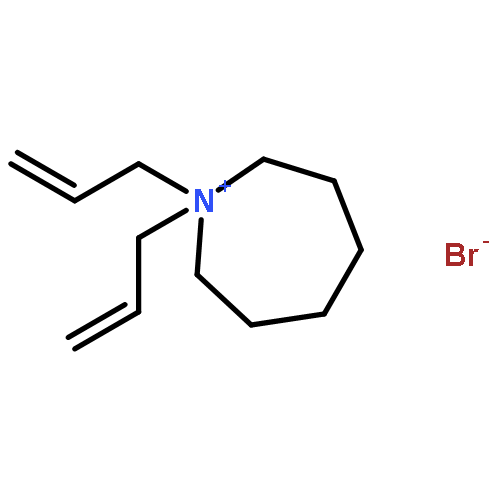


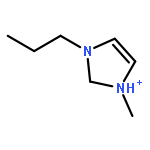
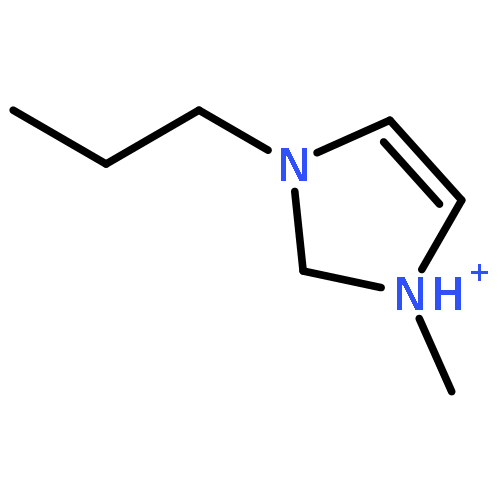
![1-[(4-ETHENYLPHENYL)METHYL]-2-METHYLIMIDAZOLE](http://img.cochemist.com/ccimg/78500/78430-93-0.png)
![1-[(4-ETHENYLPHENYL)METHYL]-2-METHYLIMIDAZOLE](http://img.cochemist.com/ccimg/78500/78430-93-0_b.png)




![[4-(phenylsulphonyl)phenyl]hydrazine](http://img.cochemist.com/ccimg/70800/70714-83-9.png)
![[4-(phenylsulphonyl)phenyl]hydrazine](http://img.cochemist.com/ccimg/70800/70714-83-9_b.png)
![Benzoic acid,4-[[(benzoylamino)thioxomethyl]amino]-, ethyl ester](http://img.cochemist.com/ccimg/69200/69165-46-4.png)
![Benzoic acid,4-[[(benzoylamino)thioxomethyl]amino]-, ethyl ester](http://img.cochemist.com/ccimg/69200/69165-46-4_b.png)


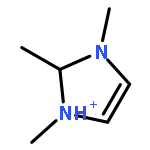
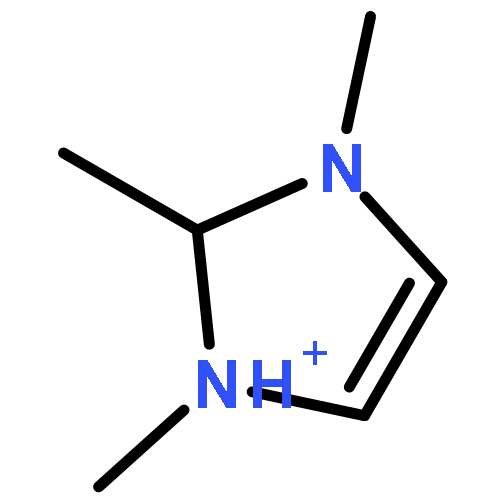
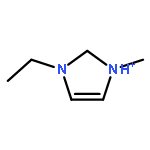

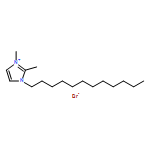
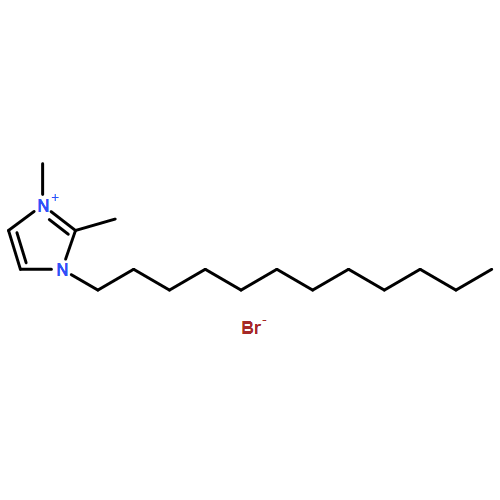
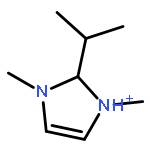








![Benzoic acid, 4-[[(benzoylamino)thioxomethyl]amino]-](http://img.cochemist.com/ccimg/40700/40611-30-1.png)
![Benzoic acid, 4-[[(benzoylamino)thioxomethyl]amino]-](http://img.cochemist.com/ccimg/40700/40611-30-1_b.png)


![4-Aza-1-azoniabicyclo[2.2.2]octane,1-methyl-, iodide (1:1)](http://img.cochemist.com/ccimg/15000/14968-74-2.png)
![4-Aza-1-azoniabicyclo[2.2.2]octane,1-methyl-, iodide (1:1)](http://img.cochemist.com/ccimg/15000/14968-74-2_b.png)

![[4-(4-hydrazinylphenyl)sulfonylphenyl]hydrazine](http://img.cochemist.com/ccimg/14100/14052-65-4.png)
![[4-(4-hydrazinylphenyl)sulfonylphenyl]hydrazine](http://img.cochemist.com/ccimg/14100/14052-65-4_b.png)





















![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3.png)
![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3_b.png)





![Benzo[1,2-b:4,5-b']dithiophene](/data/chemimg/477000/267-65-2.png)
![Benzo[1,2-b:4,5-b']dithiophene](/data/chemimg/477000/267-65-2_b.png)








![2H-Benzo[a]quinolizine-3-carboxamide,2-(acetyloxy)-N,N-diethyl-1,3,4,6,7,11b-hexahydro-9,10-dimethoxy-](http://img.cochemist.com/ccimg/100/63-12-7.png)
![2H-Benzo[a]quinolizine-3-carboxamide,2-(acetyloxy)-N,N-diethyl-1,3,4,6,7,11b-hexahydro-9,10-dimethoxy-](http://img.cochemist.com/ccimg/100/63-12-7_b.png)



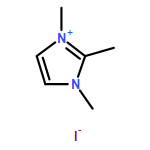
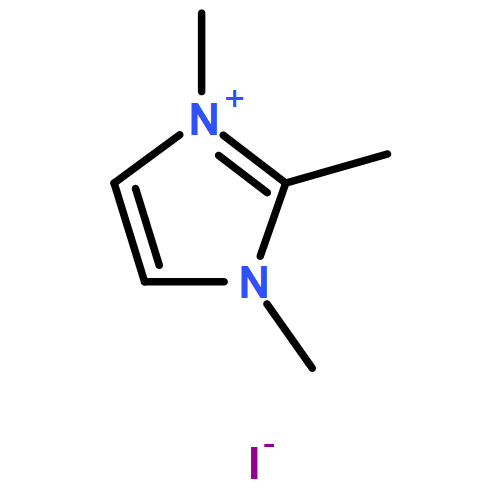
![[1,1':3',1''-Terphenyl]-4,4''-diol, 5'-(4-hydroxyphenyl)-](/data/chemimg/525300/15797-52-1.png)
![[1,1':3',1''-Terphenyl]-4,4''-diol, 5'-(4-hydroxyphenyl)-](/data/chemimg/525300/15797-52-1_b.png)









