Co-reporter:Madduri Srinivasarao and Philip S. Low
Chemical Reviews October 11, 2017 Volume 117(Issue 19) pp:12133-12133
Publication Date(Web):September 12, 2017
DOI:10.1021/acs.chemrev.7b00013
Safety and efficacy constitute the major criteria governing regulatory approval of any new drug. The best method to maximize safety and efficacy is to deliver a proven therapeutic agent with a targeting ligand that exhibits little affinity for healthy cells but high affinity for pathologic cells. The probability of regulatory approval can conceivably be further enhanced by exploiting the same targeting ligand, conjugated to an imaging agent, to select patients whose diseased tissues display sufficient targeted receptors for therapeutic efficacy. The focus of this Review is to summarize criteria that must be met during design of ligand-targeted drugs (LTDs) to achieve the required therapeutic potency with minimal toxicity. Because most LTDs are composed of a targeting ligand (e.g., organic molecule, aptamer, protein scaffold, or antibody), spacer, cleavable linker, and therapeutic warhead, criteria for successful design of each component will be described. Moreover, because obstacles to successful drug design can differ among human pathologies, limitations to drug delivery imposed by the unique characteristics of different diseases will be considered. With the explosion of genomic and transcriptomic data providing an ever-expanding selection of disease-specific targets, and with tools for high-throughput chemistry offering an escalating diversity of warheads, opportunities for innovating safe and effective LTDs has never been greater.
Co-reporter:Ananda Kumar Kanduluru and Philip S. Low
Molecular Pharmaceutics November 6, 2017 Volume 14(Issue 11) pp:3859-3859
Publication Date(Web):October 2, 2017
DOI:10.1021/acs.molpharmaceut.7b00583
The neurokinin-1 receptor (NK1R) plays a significant role in the progression and metastasis of several neuroendocrine tumors. Due to its upregulation in these cancers, NK1R constitutes an attractive receptor for development of ligand-targeted imaging and therapeutic agents. In this report, we present the design and synthesis of an NK1R targeting ligand conjugated to the chemotherapeutic agent, tubulysin B hydrazide (TubBH), via a self-immolative linker. We then explore the ability of this low molecular weight tubulysin conjugate to kill NK1R overexpressing cancer cells both in vitro and in vivo without killing receptor negative healthy cells. Because similar studies in mice bearing NK1-negative tumors reveal no therapeutic impact, we conclude that our NK1R targeting ligand is specific for NK1R-expressing cells. Taken together, the data suggest a possible new approach for the treatment of NK1R-positive neuroendocrine cancers.Keywords: conjugation; drug targeting; ligand-targeted therapy; neuroendocrine cancer; neurokinin-1 receptor; receptors; substance P; tubulysin; tumor;
Co-reporter:Sakkarapalayam M. Mahalingam, Vadim Y. Dudkin, Shalom Goldberg, Donna Klein, Fang Yi, Sunil Singhal, Karyn T. O’Neil, and Philip S. Low
Bioconjugate Chemistry November 15, 2017 Volume 28(Issue 11) pp:2865-2865
Publication Date(Web):September 25, 2017
DOI:10.1021/acs.bioconjchem.7b00566
Tumor-targeted near-infrared fluorescent dyes have the potential to improve cancer surgery by enabling surgeons to locate and resect more malignant lesions where good visualization tools are required to ensure complete removal of malignant tissue. Although the tumor-targeted fluorescent dyes used in humans to date have been either small organic molecules or high molecular weight antibodies, low molecular weight protein scaffolds have attracted significant attention because they penetrate solid tumors almost as efficiently as small molecules, but can be infinitely mutated to bind almost any antigen. Here we describe the use of a 10 kDa protein scaffold, a Centyrin, to target a near-infrared fluorescent dye to tumors that overexpress the epidermal growth factor receptor (EGFR) for fluorescence-guided surgery (FGS). We have developed and optimized the dose and time required for imaging small tumor burdens with minimal background fluorescence in real-time fluorescence-guided surgery of EGFR-expressing tumor xenografts in murine models. We demonstrate that the Centyrin-near-infrared dye conjugate (CNDC) binds selectively to human EGFR+ cancer cells with an EC50 of 2 nM, localizes to EGFR+ tumor xenografts in athymic nude mice and that uptake of the dye in xenografts is significantly reduced when EGFR are blocked by preinjection of excess unlabeled Centyrin. Taken together, these data suggest that CNDCs can be used for intraoperative identification and surgical removal of EGFR-expressing lesions and that Centyrins targeted to other tumor-specific antigens should prove similarly useful in fluorescence guided surgery of cancer. In addition, we demonstrate that the CNDC is detected in the NIR region of the spectrum and can be utilized for fluorescence-guided surgery (FGS). In addition, we propose that with its eventual complete clearance from EGFR-negative tissues and its quantitative retention in the tumor mass for >24 h, a Centyrin-targeted NIR dye should provide excellent tumor contrast when injected at least 6–8 h before initiation of cancer surgery in human patients.
Co-reporter:Qingshou Chen, Xiangjun Meng, Paul McQuade, Daniel Rubins, Shu-An Lin, Zhizhen Zeng, Hyking Haley, Patricia Miller, Dinko González Trotter, and Philip S. Low
Molecular Pharmaceutics December 4, 2017 Volume 14(Issue 12) pp:4353-4353
Publication Date(Web):October 13, 2017
DOI:10.1021/acs.molpharmaceut.7b00415
The folate receptor (FR) has been established as a promising target for imaging and therapy of cancer (FR-α), inflammation, and autoimmune diseases (FR-β). Several folate based PET radiotracers have been reported in the literature, but an 18F-labeled folate-PET imaging agent with optimal properties for clinical translation is still lacking. In the present study, we report the design and preclinical evaluation of folate-PEG12-NOTA-Al18F (1), a new folate-PET agent with improved potential for clinical applications. Radiochemical synthesis of 1 was achieved via a one-pot labeling process by heating folate-PEG12-NOTA in the presence of in situ prepared Al18F for 15 min at 105 °C, followed by HPLC purification. Specific binding of 1 to FR was evaluated on homogenates of KB (FR-positive) and A549 (FR-deficient) tumor xenografts in the presence and absence of excess folate. In vivo tumor imaging with folate-PEG12-NOTA-Al18F was compared to imaging with 99mTc-EC20 using nu/nu mice bearing either KB or A549 tumor xenografts. Specific accumulation of 1 in tumor and other tissues was assessed by high-resolution micro-PET and ex vivo biodistribution in the presence and absence of excess folate. Radiosynthesis of 1 was accomplished within ∼35 min, affording pure radiotracer 1 in 8.4 ± 1.3% (decay corrected) radiochemical yield with ∼100% radiochemical purity after HPLC purification and a specific activity of 35.8 ± 15.3 GBq/mmol. Further in vitro and in vivo examination of 1 demonstrated highly specific FR-mediated uptake in FR+ tumor, with Kd of ∼0.4 nM (KB), and reduced accumulation in liver. Given its facile preparation and improved properties, the new radiotracer, folate-PEG12-NOTA-Al18F (1), constitutes a promising tool for identification and classification of patients with FR overexpressing cancers.Keywords: 18F-PET imaging; Al18F-NOTA; cancer imaging; folate conjugate; folate receptor;
Co-reporter:Peng-Cheng Lv, Karson S. Putt, and Philip S. Low
Bioconjugate Chemistry 2016 Volume 27(Issue 7) pp:1762
Publication Date(Web):June 30, 2016
DOI:10.1021/acs.bioconjchem.6b00271
As tumors grow, vasculature is often deficient or malformed, resulting in many localized areas of hypoxia. Cells located in these hypoxic regions exhibit an altered gene expression pattern that can significantly alter resistance to conventional anticancer treatments such as ionizing radiation and chemotherapeutic drugs. A priori knowledge of the level of hypoxia within a tumor may better guide clinical care. In an effort to create a hypoxia specific imaging agent, a ligand for the tissue hypoxia marker, carbonic anhydrase IX (CA IX), was synthesized and used as a targeting ligand to deliver an attached 99mTc-chelating agent. Binding of the resulting conjugates to hypoxic cancer cells was first characterized in vitro. Whole animal imaging and biodistribution studies then were performed to determine tumor specificity in vivo. Several conjugates were found to bind selectively to CA IX expressing tumors in a receptor-dependent manner. We suggest that such conjugates could prove useful in identifying hypoxic cancers and/or quantitating the level of hypoxia within a tumor.
Co-reporter:Ananda Kumar Kanduluru, Madduri Srinivasarao, and Philip S. Low
Bioconjugate Chemistry 2016 Volume 27(Issue 9) pp:2157
Publication Date(Web):August 16, 2016
DOI:10.1021/acs.bioconjchem.6b00374
The neurokinin-1 receptor (NK1R) is implicated in the growth and metastasis of many tumors, including cancers of the brain (e.g., gliomas, glioblastomas, and astrocytomas), skin (e.g., melanomas), and neuroendocrine tissues (cancers of the breast, stomach, pancreas, larynx, and colon). Because overexpression of NK1R has been reported in most of these malignancies, we have undertaken designing an NK1R-targeted near-infrared (NIR) fluorescent dye for fluorescence-guided surgeries of these cancers. We demonstrate here that an NK1R-binding ligand linked to the NIR dye LS288 selectively accumulates in NK1R-expressing tumor xenografts with high affinity (Kd = 13 nM), allowing intraoperative imaging of these cancers in live mice. Because tumor accumulation is nearly quantitatively blocked by excess unlabeled ligand, and because NK1R-negative tumors and normal tissues display virtually no uptake, we conclude that the observed tumor retention is NK1R-mediated. Results on the synthesis, in vitro characterization, and animal testing of NK1R-targeted NIR dye are presented.
Co-reporter:Peng-Cheng Lv; Jyoti Roy; Karson S. Putt
Molecular Pharmaceutics 2016 Volume 13(Issue 5) pp:1618-1625
Publication Date(Web):April 4, 2016
DOI:10.1021/acs.molpharmaceut.6b00065
Proof-of-principle studies in ovarian, lung, and brain cancer patients have shown that fluorescence-guided surgery can enable removal of otherwise undetectable malignant lesions, decrease the number of cancer-positive margins, and permit identification of disease-containing lymph nodes that would have normally evaded resection. Unfortunately, the current arsenal of tumor-targeted fluorescent dyes does not permit identification of all cancers, raising the need to design new tumor-specific fluorescent dyes to illuminate the currently undetectable cancers. In an effort to design a more universal fluorescent cancer imaging agent, we have undertaken to synthesize a fluorophore that could label all hypoxic regions of tumors. We report here the synthesis, in vitro binding, and in vivo imaging of a near-infrared (NIR) fluorescent dye that is targeted to carbonic anhydrase IX (CA IX), i.e., a widely accepted marker of hypoxic tissues. The low molecular weight NIR probe, named Hypoxyfluor, is shown to bind CA IX with high affinity and accumulate rapidly and selectively in CA IX positive tumors. Because nearly all human cancers contain hypoxic regions that express CA IX abundantly, this NIR probe should facilitate surgical resection of a wide variety of solid tumors.
Co-reporter:Qingshou Chen; Xiangjun Meng; Paul McQuade; Daniel Rubins; Shu-An Lin; Zhizhen Zeng; Hyking Haley; Patricia Miller; Dinko González Trotter
Molecular Pharmaceutics 2016 Volume 13(Issue 5) pp:1520-1527
Publication Date(Web):April 7, 2016
DOI:10.1021/acs.molpharmaceut.5b00989
Folate-receptor-targeted PET radiotracers can potentially serve as versatile imaging agents for the diagnosis, staging, and prediction of response to therapy of patients with folate-receptor (FR)-expressing cancers. Because current FR-targeted PET reagents can be compromised by complex labeling procedures, low specific activities, poor radiochemical yields, or unwanted accumulation in FR negative tissues, we have undertaken to design an improved folate-PET agent that might be more amenable for clinical development. For this purpose, we have synthesized a folate-NOTA-Al18F radiotracer and examined its properties both in vitro and in vivo. Methods: Radiochemical synthesis of folate-NOTA-Al18F was achieved by incubating 18F– with AlCl3 for 2 min followed by heating in the presence of folate-NOTA for 15 min at 100 °C. Binding of folate-NOTA-Al18F to FR was quantitated in homogenates of KB and Cal51 tumor xenografts in the presence and absence of excess folic acid as a competitor. In vivo imaging was performed on nu/nu mice bearing either FR+ve (KB cell) or FR-ve (A549 cell) tumor xenografts, and specific accumulation of the radiotracer in tumor and other tissues was assessed by high-resolution micro-PET and ex vivo biodistribution in the presence and absence of excess folic acid. Image quality of folate-NOTA-Al18F was compared with that of 99mTc-EC20, a clinically established folate-targeted SPECT imaging agent. Results: Total radiochemical synthesis and purification of folate-NOTA-Al18F was completed within 37 min, yielding a specific activity of 68.82 ± 18.5 GBq/μmol, radiochemical yield of 18.6 ± 4.5%, and radiochemical purity of 98.3 ± 2.9%. Analysis of FR binding revealed a Kd of ∼1.0 nM, and micro-PET imaging together with ex vivo biodistribution analyses demonstrated high FR-mediated uptake in an FR+ tumor and the kidneys. Conclusions: Folate-NOTA-Al18F constitutes an easily prepared FR-targeted PET imaging agent with improved radiopharmaceutical properties and high specificity for folate receptor expressing tumors. Given its improved properties over 99mTc-EC20 (i.e., higher resolution, shorter image acquisition time, etc.), we conclude that folate-NOTA-Al18F constitutes a viable alternative to 99mTc-EC20 for use in identification, diagnosis, and staging of patients with FR-expressing cancers.
Co-reporter:Estela Puchulu-Campanella;Francesco M. Turrini;Yen-Hsing Li
PNAS 2016 Volume 113 (Issue 48 ) pp:13732-13737
Publication Date(Web):2016-11-29
DOI:10.1073/pnas.1611904113
Src homology 2 (SH2) domains are composed of weakly conserved sequences of ∼100 aa that bind phosphotyrosines in signaling
proteins and thereby mediate intra- and intermolecular protein–protein interactions. In exploring the mechanism whereby tyrosine
phosphorylation of the erythrocyte anion transporter, band 3, triggers membrane destabilization, vesiculation, and fragmentation,
we discovered a SH2 signature motif positioned between membrane-spanning helices 4 and 5. Evidence that this exposed cytoplasmic
sequence contributes to a functional SH2-like domain is provided by observations that: (i) it contains the most conserved sequence of SH2 domains, GSFLVR; (ii) it binds the tyrosine phosphorylated cytoplasmic domain of band 3 (cdb3-PO4) with Kd = 14 nM; (iii) binding of cdb3-PO4 to erythrocyte membranes is inhibited both by antibodies against the SH2 signature sequence and dephosphorylation of cdb3-PO4; (iv) label transfer experiments demonstrate the covalent transfer of photoactivatable biotin from isolated cdb3-PO4 (but not cdb3) to band 3 in erythrocyte membranes; and (v) phosphorylation-induced binding of cdb3-PO4 to the membrane-spanning domain of band 3 in intact cells causes global changes in membrane properties, including (i) displacement of a glycolytic enzyme complex from the membrane, (ii) inhibition of anion transport, and (iii) rupture of the band 3–ankyrin bridge connecting the spectrin-based cytoskeleton to the membrane. Because SH2-like motifs
are not retrieved by normal homology searches for SH2 domains, but can be found in many tyrosine kinase-regulated transport
proteins using modified search programs, we suggest that related cases of membrane transport proteins containing similar motifs
are widespread in nature where they participate in regulation of cell properties.
Co-reporter:Lindsay E. Kelderhouse;Sakkarapalayam Mahalingam;Philip S. Low
Molecular Imaging and Biology 2016 Volume 18( Issue 2) pp:
Publication Date(Web):2016/04/01
DOI:10.1007/s11307-015-0876-y
Although current therapies for many inflammatory/autoimmune diseases are effective, a significant number of patients still exhibit only partial or negligible responses to therapeutic intervention. Since prolonged use of an inadequate therapy can result in both progressive tissue damage and unnecessary expense, methods to identify nonresponding patients are necessary.Four murine models of inflammatory disease (rheumatoid arthritis, ulcerative colitis, pulmonary fibrosis, and atherosclerosis) were induced, treated with anti-inflammatory agents, and evaluated for inflammatory response. The mice were also injected intraperitoneally with OTL0038, a folate receptor-targeted near-infrared dye that accumulates in activated macrophages at sites of inflammation. Uptake of OTL0038 in inflamed lesions was then correlated with clinical measurements of disease severity.OTL0038 accumulated at sites of inflammation in all four animal models. More importantly, changes in lesion-associated OTL0038 preceded changes in clinical symptoms in mice treated with all anti-inflammatory drugs examined.OTL0038 has the ability to predict responses to multiple therapies in four murine models of inflammation.
Co-reporter:Bindu Varghese;Chrystal Paulos
Inflammation 2016 Volume 39( Issue 4) pp:1345-1353
Publication Date(Web):2016 August
DOI:10.1007/s10753-016-0366-7
Folate-targeted immunotherapy constitutes a powerful method for the treatment of established arthritis in multiple animal models of the disease. The therapy involves immunization of the animal against a hapten to induce anti-hapten antibodies, followed by injection with a folate-hapten conjugate to decorate the surface of folate receptor-positive (activated) macrophages with the antigenic hapten. The hapten-marked macrophages are then recognized by the anti-hapten antibodies and eliminated by immune mechanisms, leading to attenuation of disease symptoms. In the following paper, we optimize the therapy for elimination of inflammatory macrophages and suppression of rheumatoid arthritis symptoms. We also demonstrate a tight correlation between folate receptor-positive macrophage abundance in the liver and inflammation of affected joints. The results suggest that therapies that reduce folate receptor-positive macrophage populations in the body should constitute effective treatments for rheumatoid arthritis.
Co-reporter:Charity Wayua; Jyoti Roy; Karson S. Putt
Molecular Pharmaceutics 2015 Volume 12(Issue 7) pp:2477-2483
Publication Date(Web):June 4, 2015
DOI:10.1021/acs.molpharmaceut.5b00218
As the delivery of selectively targeted cytotoxic agents via antibodies or small molecule ligands to malignancies has begun to show promise in the clinic, the need to identify and validate additional cellular targets for specific therapeutic delivery is critical. Although a multitude of cancers have been targeted using the folate receptor, PSMA, bombesin receptor, somatostatin receptor, LHRH, and αvβ3, there is a notable lack of specific small molecule ligand/receptor pairs to cellular targets found within cancers of the GI tract. Because of the selective GI tract expression of the cholecystokinin 2 receptor (CCK2R), we undertook the creation of conjugates that would deliver microtubule-disrupting drugs to malignancies through the specific targeting of CCK2R via a high affinity small molecule ligand. The cytotoxic activity of these conjugates were shown to be receptor mediated in vitro and in vivo with xenograft mouse models exhibiting delayed growth or regression of tumors that expressed CCK2R. Overall, this work demonstrates that ligands to CCK2R can be used to create selectively targeted therapeutic conjugates.
Co-reporter:Lindsay E. Kelderhouse; Meridith T. Robins; Katelyn E. Rosenbalm; Emily K. Hoylman; Sakkarapalayam Mahalingam
Molecular Pharmaceutics 2015 Volume 12(Issue 10) pp:3547-3555
Publication Date(Web):September 2, 2015
DOI:10.1021/acs.molpharmaceut.5b00134
The ability to select patients who will respond to therapy is especially acute for autoimmune/inflammatory diseases, where the costs of therapies can be high and the progressive damage associated with ineffective treatments can be irreversible. In this article we describe a clinical test that will rapidly predict the response of patients with an autoimmune/inflammatory disease to many commonly employed therapies. This test involves quantitative assessment of uptake of a folate receptor-targeted radioimaging agent (99mTc-EC20) by a subset of inflammatory macrophages that accumulate at sites of inflammation. Murine models of four representative inflammatory diseases (rheumatoid arthritis, inflammatory bowel disease, pulmonary fibrosis, and atherosclerosis) show markedly decreased uptake of 99mTc-EC20 in inflamed lesions upon initiation of successful therapies, but no decrease in uptake upon administration of ineffective therapies, in both cases long before changes in clinical symptoms can be detected. This predictive capability should reduce costs and minimize morbidities associated with failed autoimmune/inflammatory disease therapies.
Co-reporter:Guo Li, Philip S. Low
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 8) pp:1792-1798
Publication Date(Web):15 April 2015
DOI:10.1016/j.bmcl.2015.02.033
Over-expression of the somatostatin-2 (SST2) receptor on plasma membranes of neuroendocrine cancer cells renders it attractive for use in targeting both imaging and therapeutic agents to neuroendocrine tumors. Peptide analogs of somatostatin have dominated this approach to date, however, many peptide analogs are either unstable in vivo or exhibit unwanted non-specific uptake in the liver and kidneys. The purpose of this Letter is to describe the preparation and evaluation of a non-peptide SST2 agonist for use in targeting drugs to neuroendocrine cancers.A non-peptide ligand for the SST2 receptor was identified from the literature as a candidate for development of targeted pharmaceuticals for neuroendocrine tumors, based on its SST2 binding affinity and selectivity for SST2 over other somatostatin receptors. It also offered a multiplicity of possible conjugation sites. Rhodamine conjugates in two positions were used for optical imaging and two compounds were internalized in an SST2 receptor transduced cell line (C6-SST2) via SST2 receptor-mediated endocytosis. Radionuclide conjugates were prepared for in vivo imaging and biodistribution studies in mice. The in vitro binding affinity of 99mTc conjugates ranged from a Kd of 37–494. Of these, one 99mTc conjugate was selected and dosed by IV injection into mice bearing C6-SST2 tumor xenografts. The highest uptake was into tumor, intestine and skin four hours after IV injection. Competition studies with octreotide, a synthetic peptide and SST2 agonist, confirmed that uptake was SST2 receptor mediated. While relatively high uptake in intestine, liver, kidneys and skin discouraged further development of the conjugate for delivery of chemotherapeutic agents, the conjugate may still be worthy of further development for neuroendocrine tumor imaging.
Co-reporter:Michael J. Hansen;N. Achini Bandara
Inflammation Research 2015 Volume 64( Issue 9) pp:697-706
Publication Date(Web):2015 September
DOI:10.1007/s00011-015-0849-2
Adipose tissue macrophages (ATMs) have been implicated in a number of obesity-related diseases. Because the activated macrophages associated with many types of autoimmune and inflammatory diseases express a folate receptor (FR) that can be exploited for FR-targeted drug delivery, we examined the visceral adipose tissue of obese mice and humans to determine whether ATMs also express FR that are accessible by folate conjugates.C57BL/6 or FATSO mice fed on either a low- or high-fat diet were used in murine studies. Human adipose tissue were obtained from healthy volunteers during adipose reduction surgery.Visceral adipose tissue was collected from both obese mice and humans, collagenase digested, and stained with folate-Oregon Green and antibodies for macrophage markers including F4/80, mannose receptor (CD206), CD11b, and CD11c. Cells were then examined for expression of the above markers by flow cytometry. Furthermore, the ability of folate conjugates to target the FR-expressing ATMs in obese mice was evaluated in vivo.A subset of the ATMs harvested from obese mice were found to express FR. Subpopulations of ATMs also simultaneously express both pro- and anti-inflammatory markers, and FR is expressed on both subsets. We then demonstrate that FR-expressing ATMs can be targeted with folate-linked fluorescent dyes in vivo.FR are expressed on multiple subsets of ATMs and these subsets can be targeted with folate-linked drugs, allowing for the possible development of FR-targeted therapies for obesity-related inflammatory diseases.
Co-reporter:Bindu Varghese, Erina Vlashi, Wei Xia, Wilfredo Ayala Lopez, Chrystal M. Paulos, Joseph Reddy, Le-Cun Xu, and Philip S. Low
Molecular Pharmaceutics 2014 Volume 11(Issue 10) pp:3609-3616
Publication Date(Web):August 28, 2014
DOI:10.1021/mp500348e
Activated macrophages overexpress a receptor for the vitamin folic acid termed the folate receptor β (FR-β). Because conjugation of folate to low molecular weight drugs, genes, liposomes, nanoparticles, and imaging agents has minor effects on FR binding, the vitamin can be exploited to target both therapeutic and imaging agents to activated macrophages without promoting their uptake by other healthy cells. In this paper, we characterize the binding, internalization, and recycling kinetics of FR-β on activated macrophages in inflamed tissues of rats with adjuvant-induced arthritis. Our results demonstrate that saturation of macrophage FR is achieved at injection doses of ∼150–300 nmol/kg, with more rapidly perfused tissues saturating at lower doses than inflamed appendages. After binding, FR-β internalizes and recycles back to the cell surface every ∼10–20 min, providing empty receptors for additional folate conjugate uptake. Because the half-life of low molecular weight folate conjugates in the vasculature is usually <1 h, these data suggest that targeting of folate conjugates to activated macrophages in vivo can be maximized by frequent dosing at conjugate concentrations that barely saturate FR (∼150 nmol/kg), thereby minimizing nonspecific binding to receptor-negative tissues and maximizing the probability that unoccupied cell surface receptors will be exposed to folate-drug conjugate.Keywords: arthritis; endocytosis; folate; internalization; receptor recycling;
Co-reporter:Rajesh K. Pandey, Gregory G. Jarvis and Philip S. Low
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 11) pp:1707-1710
Publication Date(Web):22 Jan 2014
DOI:10.1039/C3OB41230J
The chemical synthesis of staphyloferrin A, a siderophore used by Staphylococcus bacteria for ferric iron retrieval, has been achieved with 79% yield via solid phase peptide synthesis (SPPS). Biological activity of synthetic staphyloferrin A has been confirmed by demonstrating its capture and uptake by live S. aureus.
Co-reporter:Charity Wayua and Philip S. Low
Molecular Pharmaceutics 2014 Volume 11(Issue 2) pp:468-476
Publication Date(Web):December 10, 2013
DOI:10.1021/mp400429h
Surgical resection of malignant disease remains one of the most effective tools for treating cancer. Tumor-targeted near-infrared dyes have the potential to improve contrast between normal and malignant tissues, thereby enabling surgeons to more quantitatively resect malignant disease. Because the cholecystokinin 2 receptor (CCK2R and its tumor-specific splice variant CCK2i4svR) is overexpressed in cancers of the lungs, colon, thyroid, pancreas, and stomach, but absent or inaccessible to parenterally administered drugs in most normal tissues, we have undertaken to design a targeting ligand that can deliver attached near-infrared dyes to CCK2R+ tumors. We report here the synthesis and biological characterization of a CCK2R-targeted conjugate of the near-infrared dye, LS-288 (CRL-LS288). We demonstrate that CRL-LS288 binds selectively to CCK2R+ cancer cells with low nanomolar affinity (Kd = 7 × 10–9 M). We further show that CRL-LS288 localizes primarily to CCK2R-expressing HEK 293 murine tumor xenografts and that dye uptake in these xenografts is significantly reduced when CCK2R are blocked by preinjection of excess ligand (CRL) or when mice are implanted with CCK2R-negative tumors. Because CRL-LS288 is also found to reveal the locations of distant tumor metastases, we suggest that CRL-LS288 has the potential to facilitate intraoperative identification of malignant disease during a variety of cancer debulking surgeries.Keywords: cholecystokinin 2 receptor; colorectal cancer; fluorescence-guided surgery; gastrin receptor; gastrointestinal cancer; intraoperative imaging; medullary thyroid cancer; near-infrared dye; pancreatic cancer; small cell lung cancer;
Co-reporter:N. Achini Bandara, Michael J. Hansen, and Philip S. Low
Molecular Pharmaceutics 2014 Volume 11(Issue 3) pp:1007-1013
Publication Date(Web):January 21, 2014
DOI:10.1021/mp400659t
The folate receptor (FR) is a GPI anchored cell surface glycoprotein that functions to facilitate folic acid uptake and mediate signal transduction. With the introduction of multiple folate-targeted drugs into the clinic, the question has arisen regarding how frequently a patient can be dosed with a FR-targeted drug or antibody and whether dosing frequency exerts any impact on the availability of FR for subsequent rounds of FR-mediated drug uptake. Although the rate of FR recycling has been examined in murine tumor models, little or no information exists on the impact of FR occupancy on its rate of endocytosis. The present study quantitates the number of cell surface FR-α and FR-β following exposure to saturating concentrations of a variety of folate-linked molecules and anti-FR antibodies, including the unmodified vitamin, folate-linked drug mimetics, multifolate derivatized nanoparticles, and monoclonal antibodies to FR. We report here that FR occupancy has no impact on the rate of FR internalization. We also demonstrate that multivalent conjugates that bind and cross-link FRs at the cell surface internalize at the same rate as monovalent folate conjugates that have no impact on FR clustering, even though the multivalent conjugates traffic through a different endocytic pathway.Keywords: antibodies to folate receptors; folate receptor endocytosis; ligand targeted drugs; ligand valency on nanomedicines; receptor recycling;
Co-reporter:Lindsay E. Kelderhouse, Venkatesh Chelvam, Charity Wayua, Sakkarapalayam Mahalingam, Scott Poh, Sumith A. Kularatne, and Philip S. Low
Bioconjugate Chemistry 2013 Volume 24(Issue 6) pp:1075
Publication Date(Web):May 5, 2013
DOI:10.1021/bc400131a
Complete surgical resection of malignant disease is the only reliable method to cure cancer. Unfortunately, quantitative tumor resection is often limited by a surgeon’s ability to locate all malignant disease and distinguish it from healthy tissue. Fluorescence-guided surgery has emerged as a tool to aid surgeons in the identification and removal of malignant lesions. While nontargeted fluorescent dyes have been shown to passively accumulate in some tumors, the resulting tumor-to-background ratios are often poor, and the boundaries between malignant and healthy tissues can be difficult to define. To circumvent these problems, our laboratory has developed high affinity tumor targeting ligands that bind to receptors that are overexpressed on cancer cells and deliver attached molecules selectively into these cells. In this study, we explore the use of two tumor-specific targeting ligands (i.e., folic acid that targets the folate receptor (FR) and DUPA that targets prostate specific membrane antigen (PSMA)) to deliver near-infrared (NIR) fluorescent dyes specifically to FR and PSMA expressing cancers, thereby rendering only the malignant cells highly fluorescent. We report here that all FR- and PSMA-targeted NIR probes examined bind cultured cancer cells in the low nanomolar range. Moreover, upon intravenous injection into tumor-bearing mice with metastatic disease, these same ligand–NIR dye conjugates render receptor-expressing tumor tissues fluorescent, enabling their facile resection with minimal contamination from healthy tissues.
Co-reporter:Jiayin Shen, Venkatesh Chelvam, Gregory Cresswell, and Philip S. Low
Molecular Pharmaceutics 2013 Volume 10(Issue 5) pp:1918-1927
Publication Date(Web):April 23, 2013
DOI:10.1021/mp3006962
Pro-inflammatory macrophages play a prominent role in such autoimmune diseases as rheumatoid arthritis, Crohn’s disease, psoriasis, sarcoidosis, and atherosclerosis. Because pro-inflammatory macrophages have also been shown to overexpress a receptor for the vitamin folic acid (i.e., folate receptor beta; FR-β), folate-linked drugs have been explored for use in imaging and treatment of these same diseases. To determine whether allergic inflammatory disorders might be similarly targeted with folate-linked drugs, we have examined the characteristics of macrophages that are prominent in the pathogenesis of asthma. We report here that macrophages from the lungs of mice with experimental allergic asthma express FR-β. We further document that these FR-β+ macrophages coexpress markers of alternatively activated (M2-type) macrophages, including the mannose receptor and arginase-1. Finally, we demonstrate that folate-conjugated fluorescent dyes and radioimaging agents can be specifically targeted to these asthmatic lung macrophages, with little uptake by macrophages present in healthy lung tissue. These data suggest strategies for the development of novel diagnostic agents for the imaging of asthma and other diseases involving alternatively activated macrophages.Keywords: alternatively activated macrophages; EC20; folate receptor-β; folate targeting; radioimaging of asthma; technetium 99m imaging;
Co-reporter:Erina Vlashi, Lindsay E. Kelderhouse, Jennifer E. Sturgis, and Philip S. Low
ACS Nano 2013 Volume 7(Issue 10) pp:8573
Publication Date(Web):September 10, 2013
DOI:10.1021/nn402644g
Targeted therapies are emerging as a preferred strategy for the treatment of cancer and other diseases. To evaluate the impact of a high affinity targeting ligand on the rate and extent of tumor penetration of different sized nanomedicines, we have used intravital multiphoton microscopy to quantitate the kinetics of tumor accumulation of a homologous series of folate-PEG-rhodamine conjugates prepared with polyethylene glycols (PEG) of different molecular weights. We demonstrate that increasing the size of the folate-PEG-rhodamine conjugates results in both longer circulation times and slower tumor penetration rates. Although a “binding site barrier” is observed with the folate-linked polymers in folate receptor expressing tumors, ligand targeting eventually leads to increased tumor accumulation, with endocytosis of the targeted nanocarriers contributing to their enhanced tumor retention. Because the effects of nanocarrier size, shape, chemistry, and targeting ligand are interconnected and complex, we suggest that these parameters must be carefully optimized for each nanocarrier to ensure optimal drug delivery in vivo.Keywords: binding site barrier; folate receptor; intravital multiphoton microscopy; nanomedicines size; tumor accumulation
Co-reporter:Youngsoon Kim, David P. Lyvers, Alexander Wei, Ronald G. Reifenberger and Philip S. Low
Lab on a Chip 2012 vol. 12(Issue 5) pp:971-976
Publication Date(Web):25 Jan 2012
DOI:10.1039/C2LC20904G
Pathogenic bacteria obtain the iron necessary for survival by releasing an iron chelator, termed a siderophore, and retrieving the iron-siderophore complex via a cell surface siderophore receptor. We have exploited the high affinity of Yersinia enterocolitica for its siderophore, deferoxamine, to develop a rapid method for capture and identification of Yersinia. In this methodology, a deferoxamine-bovine serum albumin conjugate is printed onto a gold-plated chip in a parallel line pattern. After flowing a suspension of Yersinia across the siderophore-derivatized chip, any Yersinia that binds to the chip is detected by dark-field microscopy analysis of the scattered light, followed by Fourier transform analysis of the scattering pattern. Since peak intensities are found to correlate with pathogen concentration, pathogen titers as low as 103 cfu/ml can be readily detected. Moreover, immobilized deferoxamine can distinguish Y. enterocolitica, which binds ferrioxamine (deferoxamine-Fe), from Staphylococcus aureus, Mycobacterium smegmatis and Pseudomonas aeruginosa, which don't. Because human pathogens cannot easily mutate their iron retrieval systems without loss of viability, we suggest that few if any mutant Yersinia will emerge that can avoid detection. Together with previous results demonstrating selective capture of Pseudomonas aeruginosa by its immobilized siderophore (pyoverdin), these data suggest that pathogen-specific siderophores may constitute effective and immutable capture ligands for rapid detection and identification of their cognate pathogens.
Co-reporter:Jun J. Yang, Sumith A. Kularatne, Xianming Chen, Philip S. Low, and Exing Wang
Molecular Pharmaceutics 2012 Volume 9(Issue 2) pp:310-317
Publication Date(Web):December 15, 2011
DOI:10.1021/mp200483t
Due to the overexpression of a folate receptor (FR) on many malignant cells, folate-targeted drugs have been developed to improve the cancer specificity of chemotherapeutic agents. Therapeutic index is further enhanced with the use of self-immolative linkers that efficiently release the attached drug upon cellular internalization of the folate–drug conjugate. Because FR is also abundant in normal kidney proximal tubule (PT) cells, we sought to examine in real time the trafficking and release of folate-targeted drugs in the kidney in vivo. Thus, we conducted two-photon kidney imaging studies in mice utilizing a Förster resonance energy transfer (FRET) based folate conjugate that undergoes a color shift from red to green upon reduction of the disulfide bond linking folate to a surrogate drug molecule. Following infusion via intravenous injection, folate–FRET reached the kidney in its intact unreduced form. The folate–FRET conjugate was then filtered into the lumen of PT, where it was efficiently captured by FR. As FR transcytosed across PT, some disulfide reduction occurred, with reduced folate–FRET detectable in PT vesicles 30 min postinjection. Prolonged monitoring of folate–FRET in mice showed modest progression of reduction in PT cells over time. Moreover, inhibition of FR trafficking in PT cells by colchicine did not significantly affect the rate or extent of folate–FRET reduction. Finally, the lack of cytosolic accumulation of released drug surrogate in the PT suggests that drug release via disulfide bond reduction should cause little kidney toxicity.Keywords: disulfide reduction; drug release; FRET (Förster resonance energy transfer); intravital microscopy; kidney toxicity; two-photon;
Co-reporter:Walter A. Henne, Ryan Rothenbuhler, Wilfredo Ayala-Lopez, Wei Xia, Bindu Varghese, and Philip S. Low
Molecular Pharmaceutics 2012 Volume 9(Issue 5) pp:1435-1440
Publication Date(Web):April 2, 2012
DOI:10.1021/mp3000138
EC20, a folate-targeted 99mTc based radioimaging agent with a high folate receptor (FR) binding affinity, has been used for both the diagnosis and the staging of FR positive malignancies (currently in phase III trials) and also for the localization of inflamed lesions characterized by the accumulation of FR+ macrophages. Because recent evidence has suggested that FR+ macrophages might accumulate at sites of infectious disease, this study evaluated whether EC20 might prove similarly useful for imaging bacterial infection foci. Using gamma scintigraphic imaging, it was demonstrated that EC20 accumulated at sites of Staphylococcus aureus infection with a significant difference (P < 0.0001, n = 12) in enrichment noted between infected and noninfected limbs. Confirmation that the elevated uptake of EC20 in infected limbs was FR-mediated was supported by suppression of EC20 accumulation in the presence of a 200-fold excess of free folic acid (P < 0.0001, n = 12). This study establishes for the first time the use of EC20 to image and localize sites of infectious disease.Keywords: EC20; folate; folic acid; gamma scintigraphy; infection imaging; macrophage; technetium;
Co-reporter:Martiana F. Sega, Haiyan Chu, John Christian, and Philip S. Low
Biochemistry 2012 Volume 51(Issue 15) pp:
Publication Date(Web):March 27, 2012
DOI:10.1021/bi201623v
The partial pressure of oxygen constitutes an important factor in the regulation of human erythrocyte physiology, including control of cell volume, membrane structure, and glucose metabolism. Because band 3 is thought to be involved in all three processes and because binding of hemoglobin (Hb) to the cytoplasmic domain of band 3 (cdb3) is strongly oxygen-dependent, the possibility that the reversible association of deoxyhemoglobin (deoxyHb) with cdb3 might constitute an O2-dependent sensor that mediates O2-regulated changes in erythrocyte properties arises. While several lines of evidence support this hypothesis, a major opposing argument lies in the fact that the deoxyHb binding sequence on human cdb3 is not conserved. Moreover, no effect of O2 pressure on Hb–band 3 interactions has ever been demonstrated in another species. To explore whether band 3–Hb interactions might be widely involved in O2-dependent regulation of erythrocyte physiology, we undertook characterization of the effect of O2 on band 3–Hb interactions in the mouse. We report here that murine band 3 binds deoxyHb with significantly greater affinity than oxyHb, despite the lack of significant homology within the deoxyHb binding sequence. We further map the deoxyHb binding site on murine band 3 and show that deletion of the site eliminates deoxyHb binding. Finally, we identify mutations in murine cdb3 that either enhance or eliminate its affinity for murine deoxyHb. These data demonstrate that despite a lack of homology in the sequences of both murine band 3 and murine Hb, a strong oxygen-dependent association of the two proteins has been conserved.
Co-reporter:Jesse L. Grey, Gayani C. Kodippili, Katya Simon, and Philip S. Low
Biochemistry 2012 Volume 51(Issue 34) pp:
Publication Date(Web):August 3, 2012
DOI:10.1021/bi300693k
The red cell membrane is stabilized by a spectrin/actin-based cortical cytoskeleton connected to the phospholipid bilayer via multiple protein bridges. By virtue of its interaction with ankyrin and adducin, the anion transporter, band 3 (AE1), contributes prominently to these bridges. In a previous study, we demonstrated that an exposed loop comprising residues 175–185 of the cytoplasmic domain of band 3 (cdB3) constitutes a critical docking site for ankyrin on band 3. In this paper, we demonstrate that an adjacent loop, comprising residues 63–73 of cdB3, is also essential for ankyrin binding. Data that support this hypothesis include the following. (1) Deletion or mutation of residues within the latter loop abrogates ankyrin binding without affecting cdB3 structure or its other functions. (2) Association of cdB3 with ankyrin is inhibited by competition with the loop peptide. (3) Resealing of the loop peptide into erythrocyte ghosts alters membrane morphology and stability. To characterize cdB3–ankyrin interaction further, we identified their interfacial contact sites using molecular docking software and the crystal structures of D3D4-ankyrin and cdB3. The best fit for the interaction reveals multiple salt bridges and hydrophobic contacts between the two proteins. The most important ion pair interactions are (i) cdB3 K69–ankyrin E645, (ii) cdB3 E72–ankyrin K611, and (iii) cdB3 D183–ankyrin N601 and Q634. Mutation of these four residues on ankyrin yielded an ankyrin with a native CD spectrum but little or no affinity for cdB3. These data define the docking interface between cdB3 and ankyrin in greater detail.
Co-reporter:Rajesh K. Pandey, Gregory G. Jarvis, Philip S. Low
Tetrahedron Letters 2012 Volume 53(Issue 13) pp:1627-1629
Publication Date(Web):28 March 2012
DOI:10.1016/j.tetlet.2012.01.074
Chemical synthesis of petrobactin, a siderophore for Bacillus anthracis, has been achieved via Sb(OEt)3-mediated ester–amide exchange.
Co-reporter:Jacob A. Galan;Haiyan Chu;Estela Puchulu-Campanella;Joseph F. Hoffman;W. Andy Tao
PNAS 2012 Volume 109 (Issue 31 ) pp:12794-12799
Publication Date(Web):2012-07-31
DOI:10.1073/pnas.1209014109
The type of metabolic compartmentalization that occurs in red blood cells differs from the types that exist in most eukaryotic
cells, such as intracellular organelles. In red blood cells (ghosts), ATP is sequestered within the cytoskeletal–membrane
complex. These pools of ATP are known to directly fuel both the Na+/K+ and Ca2+ pumps. ATP can be entrapped within these pools either by incubation with bulk ATP or by operation of the phosphoglycerate
kinase and pyruvate kinase reactions to enzymatically generate ATP. When the pool is filled with nascent ATP, metabolic labeling
of the Na+/K+ or Ca2+ pump phosphoproteins (ENa-P and ECa-P, respectively) from bulk [γ-32P]-ATP is prevented until the pool is emptied by various means. Importantly, the pool also can be filled with the fluorescent
ATP analog trinitrophenol ATP, as well as with a photoactivatable ATP analog, 8-azido-ATP (N3-ATP). Using the fluorescent ATP, we show that ATP accumulates and then disappears from the membrane as the ATP pools are
filled and subsequently emptied, respectively. By loading N3-ATP into the membrane pool, we demonstrate that membrane proteins that contribute to the pool’s architecture can be photolabeled.
With the aid of an antibody to N3-ATP, we identify these labeled proteins by immunoblotting and characterize their derived peptides by mass spectrometry. These
analyses show that the specific peptides that corral the entrapped ATP derive from sequences within β-spectrin, ankyrin, band
3, and GAPDH.
Co-reporter:Wei Xia
Journal of Medicinal Chemistry 2010 Volume 53(Issue 19) pp:6811-6824
Publication Date(Web):July 28, 2010
DOI:10.1021/jm100509v
Co-reporter:Sumith A. Kularatne ; Chelvam Venkatesh ; Hari-Krishna R. Santhapuram ; Kevin Wang ; Balasubramanian Vaitilingam ; Walter A. Henne
Journal of Medicinal Chemistry 2010 Volume 53(Issue 21) pp:7767-7777
Publication Date(Web):October 11, 2010
DOI:10.1021/jm100729b
Ligand-targeted therapeutics have increased in prominence because of their potential for improved potency and reduced toxicity. However, with the advent of personalized medicine, a need for greater versatility in ligand-targeted drug design has emerged, where each tumor-targeting ligand should be capable of delivering a variety of therapeutic agents to the same tumor, each therapeutic agent being selected for its activity on a specific patient's cancer. In this report, we describe the use of a prostate-specific membrane antigen (PSMA)-targeting ligand to deliver multiple unrelated cytotoxic drugs to human prostate cancer (LNCaP) cells. We demonstrate that the PSMA-specific ligand, 2-[3-(1, 3-dicarboxy propyl)ureido] pentanedioic acid, is capable of mediating the targeted killing of LNCaP cells with many different therapeutic warheads. These results suggest that flexibility can be designed into ligand-targeted therapeutics, enabling adaptation of a single targeting ligand for the treatment of patients with different sensitivities to different chemotherapies.
Co-reporter:Derek D. Doorneweerd, Walter A. Henne, Ronald G. Reifenberger, and Philip S. Low
Langmuir 2010 Volume 26(Issue 19) pp:15424-15429
Publication Date(Web):August 12, 2010
DOI:10.1021/la101962w
Rapid identification of infectious pathogens constitutes an important step toward limiting the spread of contagious diseases. Whereas antibody-based detection strategies are often selected because of their speed, mutation of the pathogen can render such tests obsolete. In an effort to develop a rapid yet mutation-proof method for pathogen identification, we have explored the use of “immutable ligands” to capture the desired microbe on a detection device. In this “proof-of-principle” study, we immobilize pyoverdine, a siderophore that Pseudomonas aeruginosa must bind to obtain iron, onto gold-plated glass chips and then examine the siderophore’s ability to capture P. aeruginosa for its subsequent identification. We demonstrate that exposure of pyoverdine-coated chips to increasing dilutions of P. aeruginosa allows detection of the bacterium down to concentrations as low as 102/mL. We further demonstrate that printing of the siderophore in a periodic pattern on the detection chip enables a sensitive method of detecting the bound pathogen by a Fourier transform analysis of light scattered by the patterned chip. Because unrelated bacteria are not captured on the pyoverdine chip, we conclude that pyoverdine can be exploited for the specific binding and identification of P. aeruginosa. It follows that the utilization of other microbe-specific “immutable ligands” may allow the specific identification of their cognate pathogens.
Co-reporter:Young-Su Yi, Wilfredo Ayala-López, Sumith A. Kularatne and Philip S. Low
Molecular Pharmaceutics 2009 Volume 6(Issue 4) pp:1228-1236
Publication Date(Web):April 17, 2009
DOI:10.1021/mp900070b
We have previously reported that disease symptoms can be greatly ameliorated in rodents with adjuvant-induced arthritis (AIA) by first immunizing the rodents against fluorescein and then treating the animals with folate−fluorescein. In this targeted hapten therapy, folate−fluorescein was shown to decorate folate receptor (FR)-expressing activated macrophages with fluorescein (an immunogenic hapten), leading to binding of antifluorescein antibodies and the consequent elimination of the activated macrophages by Fc receptor-expressing immune cells. In the current study, we compare the therapeutic potencies of a variety of FR-targeted haptens in treating the symptoms of AIA in rats. Rats were immunized with either dinitrophenyl (DNP) or trinitrophenyl (TNP) conjugated to keyhole limpet hemocyanin followed by induction of AIA with heat-inactivated Mycobacterium butyricum. Following development of arthritis, rats were treated with one of five folate−hapten conjugates (folate−DNP1, folate−DNP2, folate−DNP3, folate−FITC, or folate−TNP) at two different doses (30 nmol/kg or 200 nmol/kg) 5×/week for 25 days. Symptoms of AIA in treated rats, including paw swelling, arthritis score, splenomegaly, bone erosion, and FR+ activated macrophage density in inflamed tissues, were quantitated over the course of therapy. Although all folate−hapten conjugates promoted a reduction in disease symptoms, folate−TNP and folate−FITC proved to be more potent than any of the 3 folate−DNP conjugates. We conclude that both folate−TNP and folate−FITC constitute promising haptens for use in FR-targeted immunotherapy of arthritis.Keywords: Activated macrophages; arthritis; dinitrophenyl; drug-targeting; fluorescein; folate receptor; immunotherapy; trinitrophenyl;
Co-reporter:Sumith A. Kularatne, Kevin Wang, Hari-Krishna R. Santhapuram and Philip S. Low
Molecular Pharmaceutics 2009 Volume 6(Issue 3) pp:780-789
Publication Date(Web):April 12, 2009
DOI:10.1021/mp900069d
Prostate cancer (PCa) is a major cause of mortality and morbidity in Western society today. Current methods for detecting PCa are limited, leaving most early malignancies undiagnosed and sites of metastasis in advanced disease undetected. Major deficiencies also exist in the treatment of PCa, especially metastatic disease. In an effort to improve both detection and therapy of PCa, we have developed a PSMA-targeted ligand that delivers attached imaging and therapeutic agents selectively to PCa cells without targeting normal cells. The PSMA-targeted radioimaging agent (DUPA−99mTc) was found to bind PSMA-positive human PCa cells (LNCaP cell line) with nanomolar affinity (KD = 14 nM). Imaging and biodistribution studies revealed that DUPA−99mTc localizes primarily to LNCaP cell tumor xenografts in nu/nu mice (% injected dose/gram = 11.3 at 4 h postinjection; tumor-to-muscle ratio = 75:1). Two PSMA-targeted optical imaging agents (DUPA−FITC and DUPA−rhodamine B) were also shown to efficiently label PCa cells and to internalize and traffic to intracellular endosomes. A PSMA-targeted chemotherapeutic agent (DUPA−TubH) was demonstrated to kill PSMA-positive LNCaP cells in culture (IC50 = 3 nM) and to eliminate established tumor xenografts in nu/nu mice with no detectable weight loss. Blockade of tumor targeting upon administration of excess PSMA inhibitor (PMPA) and the absence of targeting to PSMA-negative tumors confirmed the specificity of each of the above targeted reagents for PSMA. Tandem use of the imaging and therapeutic agents targeted to the same receptor could allow detection, staging, monitoring, and treatment of PCa with improved accuracy and efficacy.Keywords: chemotherapy for prostate cancer; diagnosis of prostate cancer; Prostate-specific membrane antigen; PSMA-targeted imaging and therapy; radioimaging and optical imaging of prostate cancer; tubulysin prodrug;
Co-reporter:Sumith A. Kularatne, Zhigang Zhou, Jun Yang, Carol B. Post and Philip S. Low
Molecular Pharmaceutics 2009 Volume 6(Issue 3) pp:790-800
Publication Date(Web):April 11, 2009
DOI:10.1021/mp9000712
The high mortality and financial burden associated with prostate cancer can be partly attributed to a lack of sensitive screening methods for detection and staging of the disease. Guided by in silico docking studies using the crystal structure of PSMA, we designed and synthesized a series of PSMA-targeted 99mTc−chelate complexes for imaging PSMA-expressing human prostate cancer cells (LNCaP cell line). Of the six targeted radioimaging agents synthesized, three were found to bind LNCaP cells with low nanomolar affinity. Moreover, the same three PSMA-targeted imaging agents were shown to localize primarily to LNCaP tumor xenografts in nu/nu mice, with an average of 9.8 ± 2.4% injected dose/g tissue accumulating in the tumor and only 0.11% injected dose/g tissue retained in the muscle at 4 h postinjection. Collectively, these high affinity, PSMA-specific radioimaging agents demonstrate significant potential for use in localizing prostate cancer masses, monitoring response to therapy, detecting prostate cancer recurrence following surgery, and selecting patients for subsequent PSMA-targeted chemotherapy.Keywords: 99mTc-radioimaging agents; diagnosis of prostate cancer; Prostate-specific membrane antigen; PSMA-targeted radioimaging;
Co-reporter:Erina Vlashi, Jennifer E. Sturgis, Mini Thomas and Philip S. Low
Molecular Pharmaceutics 2009 Volume 6(Issue 6) pp:1868-1875
Publication Date(Web):September 15, 2009
DOI:10.1021/mp900158d
Targeted therapies are emerging as a preferred strategy for treatment of cancer and other diseases. To evaluate the effect of high affinity receptors on the rate and extent of tumor penetration of receptor-targeted drugs, we have characterized the kinetics of folate−rhodamine uptake by folate receptor (FR)-expressing tumors in live mice. Folate−rhodamine was selected to model receptor-targeted drugs, because (i) it has high affinity (Kd = 10−9 M) for FR-rich tumors, (ii) its uptake can be monitored in vivo by multiphoton microscopy, and (iii) five folate-targeted drugs of similar size are currently undergoing clinical trials. We demonstrate that (1) folate−rhodamine saturates tumor FR in <5 min, <30 min, and <100 min following intravenous, paraorbital, and intraperitoneal injection, respectively; (2) complete clearance of folate−rhodamine from receptor-negative tissues requires ≥50 min, and (3) a “binding site barrier” may retard, but does not prevent, penetration of the ligand-targeted drug. We conclude that low molecular weight ligand-targeted drugs have appropriate pharmacokinetic properties for tumor-selective delivery.Keywords: folic acid; real time imaging; Targeted cancer therapy; tumor uptake;
Co-reporter:Ian A. Lewis;M. Estela Campanella;John L. Markley
PNAS 2009 Volume 106 (Issue 44 ) pp:18515-18520
Publication Date(Web):2009-11-03
DOI:10.1073/pnas.0905999106
Deoxygenation elevates glycolytic flux and lowers pentose phosphate pathway (PPP) activity in mammalian erythrocytes. The
membrane anion transport protein (band 3 or AE1) is thought to facilitate this process by binding glycolytic enzymes (GEs)
and inhibiting their activity in an oxygen-dependent manner. However, this regulatory mechanism has not been demonstrated
under physiological conditions. In this study, we introduce a 1H-13C NMR technique for measuring metabolic fluxes in intact cells. The role of band 3 in mediating the oxygenated/deoxygenated
metabolic transition was examined by treating cells with pervanadate, a reagent that prevents the GE–band 3 complex from forming.
We report that pervanadate suppresses oxygen-dependent changes in glycolytic and PPP fluxes. Moreover, these metabolic alterations
were not attributable to modulation of bisphosphoglycerate mutase, direct inhibition of GEs by pervanadate, or oxidation,
which are the major side effects of pervanadate treatment. These data provide direct evidence supporting the role of band
3 in mediating oxygen-regulated metabolic transitions.
Co-reporter:Philip S. Low, Walter A. Henne and Derek D. Doorneweerd
Accounts of Chemical Research 2008 Volume 41(Issue 1) pp:120
Publication Date(Web):July 27, 2007
DOI:10.1021/ar7000815
In order to avoid the toxicities associated with prescription drug use today, we have explored novel methods for delivering drugs selectively to pathologic cells, thereby avoiding the collateral damage that accompanies their uptake by healthy cells. In this Account, we describe our quest for the ideal targeted therapeutic agent. This effort began with a search for ligands that would bind selectively to pathologic cells, displaying no affinity for healthy cells. After identification of an optimal targeting ligand, effort was focused on construction of linkers that would carry the attached drug to pathologic cells with receptors for the selected ligand. In the case of cancer, we exploited the well-characterized up-regulation of folate receptors on malignant cells to target folate-linked pharmaceuticals to cancer tissues in vivo. Drugs that have been linked to folic acid for tumor-selective drug delivery to date include (i) protein toxins, (ii) chemotherapeutic agents, (iii) gene therapy vectors, (iv) oligonucleotides (including small interfering RNA (siRNA)), (v) radioimaging agents, (vi) magnetic resonance imaging (MRI) contrast agents, (vii) liposomes with entrapped drugs, (viii) radiotherapeutic agents, (ix) immunotherapeutic agents, and (x) enzyme constructs for prodrug therapy. Current clinical trials of four folate-linked drugs demonstrate that folate receptor-targeting holds great promise for increasing the potency while reducing toxicity of many cancer therapies. In the course of developing folate-conjugated drugs for cancer, we discovered that folate receptors are also overexpressed on activated (but not resting or quiescent) macrophages. Recognizing that activated macrophages either cause or contribute to such diseases as rheumatoid arthritis, Crohnʼs disease, atherosclerosis, lupus, inflammatory osteoarthritis, diabetes, ischemia reperfusion injury, glomerulonephritis, sarcoidosis, psoriasis, Sjogrenʼs disease, and vasculitis, we initiated studies aimed at developing folate-conjugated imaging and therapeutic agents for the diagnosis and treatment of such diseases. In very brief time, significant progress has been made towards identification of clinical candidates for targeted treatment of several inflammatory and autoimmune diseases. This Account summarizes the discovery and development of a variety of folate-targeted drugs for the diagnosis and therapy of cancers and inflammatory/autoimmune diseases.
Co-reporter:Emanuela I. Sega
Cancer and Metastasis Reviews 2008 Volume 27( Issue 4) pp:
Publication Date(Web):2008 December
DOI:10.1007/s10555-008-9155-6
Folate receptors are up-regulated on a variety of human cancers, including cancers of the breast, ovaries, endometrium, lungs, kidneys, colon, brain, and myeloid cells of hematopoietic origin. This over-expression of folate receptors (FR) on cancer tissues can be exploited to target folate-linked imaging and therapeutic agents specifically to FR-expressing tumors, thereby avoiding uptake by most healthy tissues that express few if any FR. Four folate-targeted therapeutic drugs are currently undergoing clinical trials, and several folate-linked chemotherapeutic agents are in late stage preclinical development. However, because not all cancers express FR, and because only FR-expressing cancers respond to FR-targeted therapies, FR-targeted imaging agents have been required to select patients with FR-expressing tumors likely to respond to folate-targeted therapies. This review focuses on recent advances in the use of the vitamin folic acid to target PET agents, γ-emitters, MRI contrast agents and fluorescent dyes to FR+ cancers for the purpose of diagnosing and imaging malignant masses with improved specificity and sensitivity.
Co-reporter:Bindu Varghese, Nicholas Haase and Philip S. Low
Molecular Pharmaceutics 2007 Volume 4(Issue 5) pp:679-685
Publication Date(Web):September 12, 2007
DOI:10.1021/mp0700615
Systemic lupus erythematosus (SLE) is an autoimmune disease involving deposition of immune complexes in normal tissues and the consequent accumulation of immune cells and tissue injury. Activated macrophages are thought to contribute to disease pathogenesis by releasing inflammatory mediators that both cause direct tissue damage and attract other immune cells that augment inflammation. Previous studies in animal models of rheumatoid arthritis have shown that activated macrophages express a folate receptor that can be targeted with folate-linked haptens, leading to (1) marking of the activated macrophages with highly immunogenic haptens, (2) recognition of the marked cells by Fc receptor-expressing immune cells, and (3) destruction of the antibody-coated macrophages by the body’s own immune system. Here we demonstrate that the same folate–hapten-targeted immunotherapy can greatly suppress symptoms of SLE in two animal models of the disease, resulting in reduced immune complex deposition, diminished damage to normal tissues, and prolonged animal survival.Keywords: Folate; folate receptor; immunotherapy; lupus; macrophage; SLE;
Co-reporter:Wei He;Haifeng Wang;Lynn C. Hartmann;Ji-Xin Cheng
PNAS 2007 Volume 104 (Issue 28 ) pp:11760-11765
Publication Date(Web):2007-07-10
DOI:10.1073/pnas.0703875104
Quantitation of circulating tumor cells (CTCs) constitutes an emerging tool for the diagnosis and staging of cancer, assessment
of response to therapy, and evaluation of residual disease after surgery. Unfortunately, no existing technology has the sensitivity
to measure the low numbers of tumor cells (<1 CTC per ml of whole blood) that characterize minimal levels of disease. We present
a method, intravital flow cytometry, that noninvasively counts rare CTCs in vivo as they flow through the peripheral vasculature. The method involves i.v. injection of a tumor-specific fluorescent ligand
followed by multiphoton fluorescence imaging of superficial blood vessels to quantitate the flowing CTCs. Studies in mice
with metastatic tumors demonstrate that CTCs can be quantitated weeks before metastatic disease is detected by other means.
Analysis of whole blood samples from cancer patients further establishes that human CTCs can be selectively labeled and quantitated
when present at ≈2 CTCs per ml, opening opportunities for earlier assessment of metastatic disease.
Co-reporter:Jun Yang;Hongtao Chen;Iontcho R. Vlahov;Ji-Xin Cheng
PNAS 2006 Volume 103 (Issue 37 ) pp:13872-13877
Publication Date(Web):2006-09-12
DOI:10.1073/pnas.0601455103
Despite functional evidence for disulfide bond-reducing activity in endosomal compartments, the mechanistic details pertaining
to such process (e.g., kinetics and sites of disulfide reduction) remain largely controversial. To address these questions
directly, we have synthesized a previously uncharacterized fluorescent folate conjugate, folate-(BODIPY FL)-SS-rhodamine (folate-FRET),
that changes fluorescence from red to green upon disulfide bond reduction. Using this construct, we have observed that disulfide
reduction: (i) occurs with a half-time of 6 h after folate-FRET endocytosis, (ii) begins in endosomes and does not depend significantly on redox machinery located on the cell surface or within the lysosome
or the Golgi apparatus, (iii) occurs independently of endocytic vesicle trafficking along microtubules, and (iv) yields products that are subsequently sorted into distinct endosomes and trafficked in different directions. Finally, colocalization
of folate and transferrin receptors suggest that conclusions derived from this study may apply to other endocytic pathways.
Co-reporter:Andrew R. Hilgenbrink
Journal of Pharmaceutical Sciences 2005 Volume 94(Issue 10) pp:2135-2146
Publication Date(Web):31 AUG 2005
DOI:10.1002/jps.20457
Folate targeted drug delivery has emerged as an alternative therapy for the treatment and imaging of many cancers and inflammatory diseases. Due to its small molecular size and high binding affinity for cell surface folate receptors (FR), folate conjugates have the ability to deliver a variety of molecular complexes to pathologic cells without causing harm to normal tissues. Complexes that have been successfully delivered to FR expressing cells, to date, include protein toxins, immune stimulants, chemotherapeutic agents, liposomes, nanoparticles, and imaging agents. This review will summarize the applications of folic acid as a targeting ligand and highlight the various methods being developed for delivery of therapeutic and imaging agents to FR-expressing cells. © 2005 Wiley-Liss, Inc. and the American Pharmacists Association J Pharm Sci 94:2135-2146, 2005
Co-reporter:M. Estela Campanella;Haiyan Chu
PNAS 2005 Volume 102 (Issue 7 ) pp:2402-2407
Publication Date(Web):2005-02-15
DOI:10.1073/pnas.0409741102
To characterize the location of glycolytic enzymes (GEs) in intact human erythrocytes, freshly drawn blood was fixed and stained
with Abs to GAPDH, aldolase, phosphofructokinase (PFK), pyruvate kinase (PK), lactate dehydrogenase (LDH), carbonic anhydrase
II, Hb, and band 3 (AE1). Confocal microscopy revealed that in cells where band 3 displays its expected membrane staining
and Hb is evenly distributed across the cytoplasm, GEs are largely limited to the membrane. Biochemical studies confirmed
that the membrane binding sites for GAPDH, aldolase, and PFK reside on band 3, but related analyses demonstrate that sites
for PK and LDH do not. Four lines of evidence demonstrate that the GEs are at least partially assembled into multimeric complexes
near the NH2 terminus of band 3. First, a mAb to residues 1–12 of band 3 displaces all of the above GEs from the membrane, including LDH
and PK, which do not bind band 3. Second, tyrosine phosphorylation of the NH2 terminus of band 3 (Y8 and Y21) reversibly releases all of the GEs from the membrane, including LDH and PK. Third, deoxygenation
of RBCs dislodges all GEs from the membrane, consistent with the established ability of deoxyHb but not oxyHb to bind the
NH2 terminus of band 3. Fourth, a large increase in the accessibility of enzyme epitopes is observed upon dissociation of GEs
from the membrane. We conclude, therefore, that GEs are organized into complexes on the membrane whose assembly is regulated
by oxygenation and phosphorylation.
Co-reporter:Yingjuan Lu, Philip S. Low
Journal of Controlled Release 2003 Volume 91(1–2) pp:17-29
Publication Date(Web):28 August 2003
DOI:10.1016/S0168-3659(03)00215-3
The cell surface receptor for the vitamin folic acid (termed the folate receptor), is often elevated in cancers of the ovary, kidney, lung, mammary gland, brain, endometrium, and myeloid cells of hematopoietic origin. Because the folate receptor (FR) is either absent from normal tissues or localized to the apical surfaces of polarized epithelia, where it is inaccessible to circulating drugs, folate-linked drugs do not normally accumulate in healthy tissues. However, since the same receptor is fully accessible on cancer cells, it has frequently been exploited as a target for receptor-directed cancer therapies, including chemotherapies and immunotherapies. In fact, most strategies for the immunotherapy of cancer have at some time been adapted to treat FR-expressing tumors. In this article, recent progress in the retargeting of the immune system to folate receptor-expressing cancers is summarized and future strategies for redirecting natural killer cells, antibodies and cytotoxic T lymphocytes to this large class of malignancies are proposed.
Co-reporter:J.A. Reddy, D.W. Clapp, P.S. Low
Journal of Controlled Release 2001 Volume 74(1–3) pp:77-82
Publication Date(Web):6 July 2001
DOI:10.1016/S0168-3659(01)00316-9
Viral vectors with high transfection efficiencies are not always those with optimal target cell binding specificities. As a consequence, virus pseudotyping has been developed to endow transfection competent viruses with improved cell binding specificities and affinities. We have hypothesized that chemical conjugation of a virus to a cell specific ligand might also alter its target cell specificity and produce a virus that would transfect only the desired cell type. To test this concept, an ecotropic replication-defective myeloproliferative sarcoma retrovirus and an amphotropic murine adenovirus containing the gene for β-galactosidase were chemically derivatized with folic acid. As expected from its strong ecotropism, the unmodified retrovirus did not induce β-galactosidase expression in nonhost KB cells, while the amphotropic adenovirus yielded high levels of gene expression in the same cell line. Surprisingly, although folate derivatization enabled avid binding of both viruses to folate receptor expressing KB cells, the folate conjugation did not promote retroviral gene expression and actually prevented the normal β-galactosidase expression seen with the adenoviral vector. The fact that co-administration of excess free folic acid to block uptake by folate receptor-mediated endocytosis restored adenoviral gene expression to the level obtained with unmodified virus suggests that folate derivatization per se does not hamper viral activity. We, therefore, conclude that neither retroviral nor adenoviral delivery via the folate endocytosis pathway is compatible with viral gene expression in KB cells.
Co-reporter:T. Franco, P.S. Low
Transfusion Clinique et Biologique (September 2010) Volume 17(Issue 3) pp:87-94
Publication Date(Web):1 September 2010
DOI:10.1016/j.tracli.2010.05.008
Adducin is an α, β heterotetramer that performs multiple important functions in the human erythrocyte membrane. First, adducin forms a bridge that connects the spectrin–actin junctional complex to band 3, the major membrane-spanning protein in the bilayer. Rupture of this bridge leads to membrane instability and spontaneous fragmentation. Second, adducin caps the fast growing (barbed) end of actin filaments, preventing the tetradecameric protofilaments from elongating into macroscopic F-actin microfilaments. Third, adducin stabilizes the association between actin and spectrin, assuring that the junctional complex remains intact during the mechanical distortions experienced by the circulating cell. And finally, adducin responds to stimuli that may be important in regulating the global properties of the cell, possibly including cation transport, cell morphology and membrane deformability. The text below summarizes the structural properties of adducin, its multiple functions in erythrocytes, and the consequences of engineered deletions of each of adducin subunits in transgenic mice.L’adducine est un hétérotétramère (α, β) qui a de multiples fonctions, importantes pour la membrane du globule rouge. Premièrement, l’adducine connecte le complexe spectrine–actine à la bande 3, la protéine transmembranaire majeure de la bicouche lipidique. La rupture de cette connection conduit à une instabilité membranaire et à une fragmentation spontanée. Deuxièmement, l’adducine en coiffant l’extrémité à croissance rapide des protofilaments d’actine les protège ainsi d’une élongation en microfilaments d’actine (F-actin). Troisièmement, l’adducine stabilise l’interaction entre spectrine et actine, permettant un maintien des complexes jonctionnels lors des distorsions mécaniques subies par le globule rouge au cours de sa circulation. Enfin, l’adducine répond à des stimuli qui pourraient être importants dans la régulation des propriétés globales de la cellule, notamment le transport cationique, la morphologie et la déformabilité du globule rouge. Cette revue rassemble les données sur les propriétés structurales de l’adducine, ses multiples fonctions dans le globule rouge et sur les conséquences de la délétion de chacune de ses sous-unités dans des modèles de souris transgéniques.
Co-reporter:Martiana F. Sega, Haiyan Chu, John A. Christian, Philip S. Low
Blood Cells, Molecules, and Diseases (October 2015) Volume 55(Issue 3) pp:
Publication Date(Web):1 October 2015
DOI:10.1016/j.bcmd.2015.07.004
Oxygen tension has emerged as a potent regulator of multiple erythrocyte properties, including glucose metabolism, cell volume, ATP release, and cytoskeletal organization. Because hemoglobin (Hb)1 binds to the cytoplasmic domain of band 3 (cdb3) in an oxygen dependent manner, with deoxyHb exhibiting significantly greater affinity for cdb3 than oxyHb, the deoxyHb-cdb3 interaction has been hypothesized to constitute the molecular switch for all O2-controlled erythrocyte processes. In this study, we describe a rapid and accurate method for quantitating the interaction of deoxyHb binding to cdb3. For this purpose, enhanced green fluorescent protein (eGFP) is fused to the COOH-terminus of cdb3, and the binding of Hb to the NH2-terminus of cdb3-eGFP is quantitated by Hb-mediated quenching of cdb3-eGFP fluorescence. As expected, the intensity of cdb3-eGFP fluorescence decreases only slightly following addition of oxyHb. However, upon deoxygenation of the same Hb-cdb3 solution, the fluorescence decreases dramatically (i.e. confirming that deoxyHb exhibits much greater affinity for cdb3 than oxyHb). Using this fluorescence quenching method, we not only confirm previously established characteristics of the Hb-cdb3 interaction, but also establish an assay that can be exploited to screen for inhibitors of the sickle Hb-cdb3 interaction that accelerates sickle Hb polymerization.
Co-reporter:Katie Giger, Ibrahim Habib, Ken Ritchie, Philip S. Low
Biochimica et Biophysica Acta (BBA) - Biomembranes (November 2016) Volume 1858(Issue 11) pp:
Publication Date(Web):1 November 2016
DOI:10.1016/j.bbamem.2016.08.012
•Glycophorin A and band 3 are abundant integral proteins in the erythrocyte membrane.•Evidence of a glycophorin A - band 3 interaction in erythrocytes remains equivocal.•Diffusion of glycophorin A appears to be restricted and locally confined.•Glycophorin A likely has an anchor to the cytoskeleton independent of band 3.Several lines of evidence suggest that glycophorin A (GPA) interacts with band 3 in human erythrocyte membranes including: i) the existence of an epitope shared between band 3 and GPA in the Wright b blood group antigen, ii) the fact that antibodies to GPA inhibit the diffusion of band 3, iii) the observation that expression of GPA facilitates trafficking of band 3 from the endoplasmic reticulum to the plasma membrane, and iv) the observation that GPA is diminished in band 3 null erythrocytes. Surprisingly, there is also evidence that GPA does not interact with band 3, including data showing that: i) band 3 diffusion increases upon erythrocyte deoxygenation whereas GPA diffusion does not, ii) band 3 diffusion is greatly restricted in erythrocytes containing the Southeast Asian Ovalocytosis mutation whereas GPA diffusion is not, and iii) most anti-GPA or anti-band 3 antibodies do not co-immunoprecipitate both proteins. To try to resolve these apparently conflicting observations, we have selectively labeled band 3 and GPA with fluorescent quantum dots in intact erythrocytes and followed their diffusion by single particle tracking. We report here that band 3 and GPA display somewhat similar macroscopic and microscopic diffusion coefficients in unmodified cells, however perturbations of band 3 diffusion do not cause perturbations of GPA diffusion. Taken together the collective data to date suggest that while weak interactions between GPA and band 3 undoubtedly exist, GPA and band 3 must have separate interactions in the membrane that control their lateral mobility.Download high-res image (114KB)Download full-size image
Co-reporter:Yingjuan Lu, Philip S. Low
Advanced Drug Delivery Reviews (December 2012) Volume 64(Supplement) pp:342-352
Publication Date(Web):1 December 2012
DOI:10.1016/j.addr.2012.09.020
The receptor for folic acid constitutes a useful target for tumor-specific drug delivery, primarily because: (1) it is upregulated in many human cancers, including malignancies of the ovary, brain, kidney, breast, myeloid cells and lung, (2) access to the folate receptor in those normal tissues that express it can be severely limited due to its location on the apical (externally-facing) membrane of polarized epithelia, and (3) folate receptor density appears to increase as the stage/grade of the cancer worsens. Thus, cancers that are most difficult to treat by classical methods may be most easily targeted with folate-linked therapeutics. To exploit these peculiarities of folate receptor expression, folic acid has been linked to both low molecular weight drugs and macromolecular complexes as a means of targeting the attached molecules to malignant cells. Conjugation of folic acid to macromolecules has been shown to enhance their delivery to folate receptor-expressing cancer cells in vitro in almost all situations tested. Folate-mediated macromolecular targeting in vivo has, however, yielded only mixed results, largely because of problems with macromolecule penetration of solid tumors. Nevertheless, prominent examples do exist where folate targeting has significantly improved the outcome of a macromolecule-based therapy, leading to complete cures of established tumors in many cases. This review presents a brief mechanistic background of folate-targeted macromolecular therapeutics and then summarizes the successes and failures observed with each major application of the technology.
Co-reporter:Rajesh K. Pandey, Gregory G. Jarvis and Philip S. Low
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 11) pp:NaN1710-1710
Publication Date(Web):2014/01/22
DOI:10.1039/C3OB41230J
The chemical synthesis of staphyloferrin A, a siderophore used by Staphylococcus bacteria for ferric iron retrieval, has been achieved with 79% yield via solid phase peptide synthesis (SPPS). Biological activity of synthetic staphyloferrin A has been confirmed by demonstrating its capture and uptake by live S. aureus.

![(S)-N-(3-([5,5'-Bipyrimidin]-2-ylamino)-4-methylphenyl)-4-((3-(dimethylamino)pyrrolidin-1-yl)methyl)-3-(trifluoromethyl)benzamide](http://img.cochemist.com/ccimg/859300/859212-16-1.png)
![(S)-N-(3-([5,5'-Bipyrimidin]-2-ylamino)-4-methylphenyl)-4-((3-(dimethylamino)pyrrolidin-1-yl)methyl)-3-(trifluoromethyl)benzamide](http://img.cochemist.com/ccimg/859300/859212-16-1_b.png)
![Benzamide,N-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]phenyl]-4-(1-piperazinylmethyl)-](http://img.cochemist.com/ccimg/404900/404844-02-6.png)
![Benzamide,N-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]phenyl]-4-(1-piperazinylmethyl)-](http://img.cochemist.com/ccimg/404900/404844-02-6_b.png)


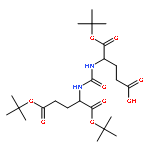
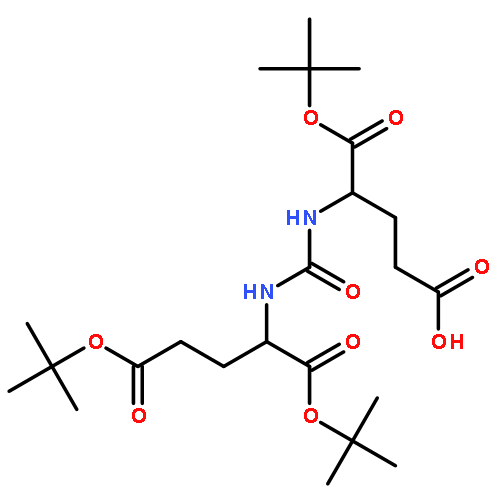


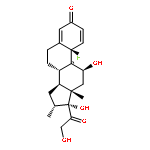
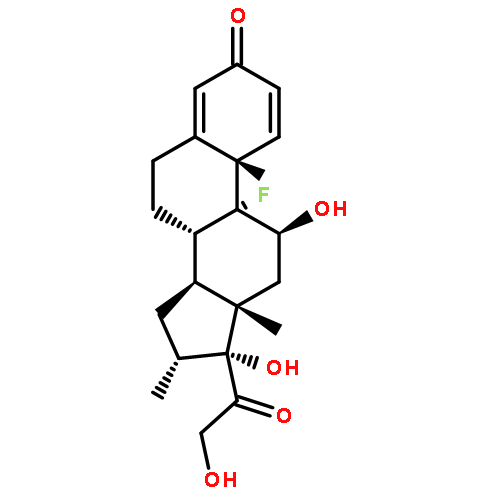
![1H-BENZOTRIAZOLE, 1-[[[2-(2-PYRIDINYLDITHIO)ETHOXY]CARBONYL]OXY]-](http://img.cochemist.com/ccimg/913200/913168-10-2.png)
![1H-BENZOTRIAZOLE, 1-[[[2-(2-PYRIDINYLDITHIO)ETHOXY]CARBONYL]OXY]-](http://img.cochemist.com/ccimg/913200/913168-10-2_b.png)









![Piperidine,3-[[3,5-bis(trifluoromethyl)phenyl]methoxy]-2-phenyl-, (2S,3S)-](http://img.cochemist.com/ccimg/148800/148700-85-0.png)
![Piperidine,3-[[3,5-bis(trifluoromethyl)phenyl]methoxy]-2-phenyl-, (2S,3S)-](http://img.cochemist.com/ccimg/148800/148700-85-0_b.png)




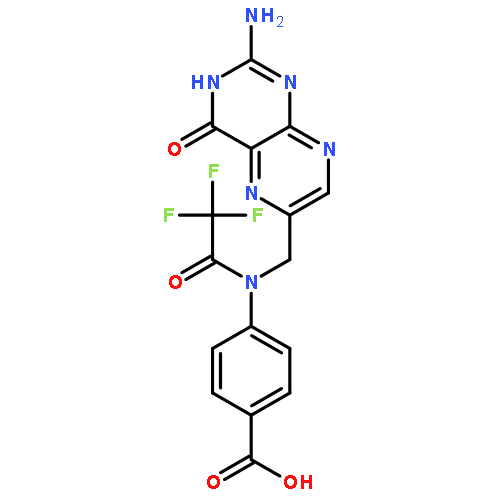




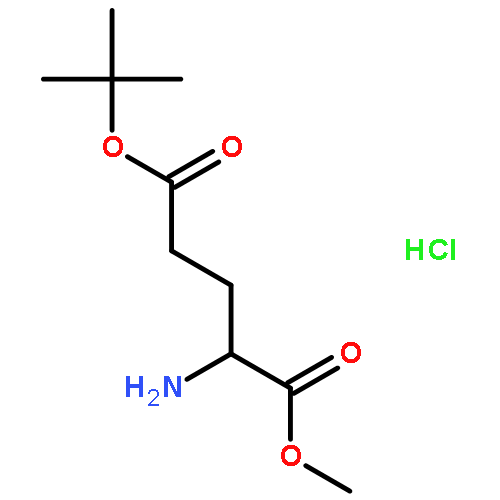
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)








![Butanedioic acid,2-[2-[[(1R)-1-carboxy-4-[[(3S)-3,4-dicarboxy-3-hydroxy-1-oxobutyl]amino]butyl]amino]-2-oxoethyl]-2-hydroxy-,(2S)-](http://img.cochemist.com/ccimg/128000/127902-98-1.png)
![Butanedioic acid,2-[2-[[(1R)-1-carboxy-4-[[(3S)-3,4-dicarboxy-3-hydroxy-1-oxobutyl]amino]butyl]amino]-2-oxoethyl]-2-hydroxy-,(2S)-](http://img.cochemist.com/ccimg/128000/127902-98-1_b.png)
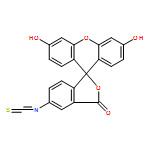


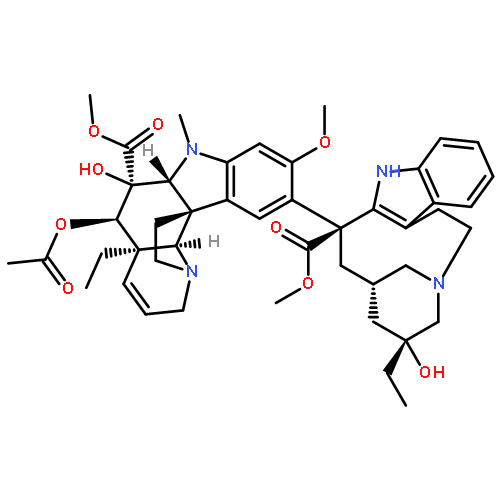
![2-ETHYL-1-(PHENYLSULFONYL)-1H-PYRROLO[2,3-B]PYRIDINE](http://img.cochemist.com/ccimg/343400/343326-69-2.png)
![2-ETHYL-1-(PHENYLSULFONYL)-1H-PYRROLO[2,3-B]PYRIDINE](http://img.cochemist.com/ccimg/343400/343326-69-2_b.png)