Co-reporter:Kangning Zhu, Zhiyuan Zhu, Haiou Zhou, Jingyan Zhang, Shiyong Liu
Chinese Chemical Letters 2017 Volume 28, Issue 6(Volume 28, Issue 6) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.cclet.2017.03.020
Two reduction-cleavable ABA triblock copolymers possessing two disulfide linkages, PMMA-ss-PMEO3MA-ss-PMMA and PDEA-ss-PEO-ss-PDEA were synthesized via facile substitution reactions from homopolymer precursors, where PMMA, PMEO3MA, PDEA, and PEO represent poly(methyl methacrylate), poly(tri(ethylene glycol) monomethyl ether methacrylate, poly(2-(diethylamino)ethyl methacrylate), and poly(ethylene oxide), respectively. Spherical micelles were obtained through supramolecular self-assembly of these two triblock copolymers in aqueous solutions. The resultant micelles with abundant disulfide bonds could serve as soft templates and precisely accommodate gold nanoparticles in the core/shell interface as a result of the formation of Au–S bonds.Download high-res image (152KB)Download full-size imageSupramolecular self-assembly of the triblock copolymers and precisely install the gold nanoparticles at the core/shell interface of the micelles.
Co-reporter:Dr. Guhuan Liu; Dr. Guohai Shi;Haoyue Sheng;Dr. Yanyan Jiang; Dr. Haojun Liang; Dr. Shiyong Liu
Angewandte Chemie International Edition 2017 Volume 56(Issue 30) pp:8686-8691
Publication Date(Web):2017/07/17
DOI:10.1002/anie.201702748
AbstractIn situ quantification of the conjugation efficiency of azide-terminated synthetic polymers/imaging probes and thiol-functionalized antibodies/proteins/peptides was enabled by a doubly caged profluorescent and heterodifunctional core molecule C1 as a self-sorting bridging unit. Orthogonal dual “click” coupling of C1 with azide- and thiol-functionalized precursors led to highly fluorescent bioconjugates, whereas single-click products remained essentially nonfluorescent. Integration with FRET processes was also possible. For the construction of antibody–probe conjugates from an anti-carcinoembryonic antigen and a quinone-caged profluorescent naphthalimide derivative, the dual “click” coupling process with C1 was monitored on the basis of the emission turn-on of C1, whereas prominent changes in FRET ratios occurred for antibody–imaging-probe conjugates when specifically triggered by quinone oxidoreductase (NQO1), which is overexpressed in various types of cancer cells.
Co-reporter:Dr. Guhuan Liu; Dr. Guohai Shi;Haoyue Sheng;Dr. Yanyan Jiang; Dr. Haojun Liang; Dr. Shiyong Liu
Angewandte Chemie 2017 Volume 129(Issue 30) pp:8812-8817
Publication Date(Web):2017/07/17
DOI:10.1002/ange.201702748
AbstractIn situ quantification of the conjugation efficiency of azide-terminated synthetic polymers/imaging probes and thiol-functionalized antibodies/proteins/peptides was enabled by a doubly caged profluorescent and heterodifunctional core molecule C1 as a self-sorting bridging unit. Orthogonal dual “click” coupling of C1 with azide- and thiol-functionalized precursors led to highly fluorescent bioconjugates, whereas single-click products remained essentially nonfluorescent. Integration with FRET processes was also possible. For the construction of antibody–probe conjugates from an anti-carcinoembryonic antigen and a quinone-caged profluorescent naphthalimide derivative, the dual “click” coupling process with C1 was monitored on the basis of the emission turn-on of C1, whereas prominent changes in FRET ratios occurred for antibody–imaging-probe conjugates when specifically triggered by quinone oxidoreductase (NQO1), which is overexpressed in various types of cancer cells.
Co-reporter:Jinming Hu
Science China Chemistry 2017 Volume 60( Issue 9) pp:1153-1161
Publication Date(Web):19 July 2017
DOI:10.1007/s11426-017-9083-1
Polymer chain architectures play a crucial role in the physical properties of polymers and this unique phenomenon has been recognized as the topological effects. As one of the most representative architectures, macrocyclic polymers characterized by the endless topology have received extensive attention due to their distinct physical properties as compared to the linear counterparts. To understand these differences and unravel the underlying mechanisms, there is a long pursuit to efficiently fabricate macrocyclic polymers. To date, both ring-closing and ring-expansion strategies have been developed, which drastically elevates the accessibility of macrocyclic polymers. The improved availability of macrocyclic polymers enables the further investigation of the biomedical applications and the preliminary results suggest that macrocyclic polymers outperform their linear analogs in terms of improving gene delivery efficiency, elevating blood circulation time, and enhancing colloidal stability of nanoparticles.
Co-reporter:Cheng Wang, Guoying Zhang, Guhuan Liu, Jinming Hu, Shiyong Liu
Journal of Controlled Release 2017 Volume 259(Volume 259) pp:
Publication Date(Web):10 August 2017
DOI:10.1016/j.jconrel.2016.11.007
Hydrogels have found promising applications in drug delivery due to their biocompatibility, high drug loading capability, and tunable release profiles. However, hydrogel-based carriers are primarily employed for delivering hydrophilic payloads while hydrophobic drugs cannot be efficiently delivered due to the lack of hydrophobic domains within conventional hydrogel matrices. Herein, we report that thermo- and photo-responsive hydrogels could be constructed from amphiphilic triblock copolymers, poly(N-isopropylacrylamide)-b-poly(4-acryloylmorpholine)-b-poly(2-((((2-nitrobenzyl)oxy)carbonyl) amino)ethyl methacrylate) (PNIPAM-b-PNAM-b-PNBOC), and the resulting hydrogels could be further engineered a new carrier for both hydrophilic gemcitabine (GCT) and hydrophobic doxorubicin (DOX). PNIPAM-b-PNAM-b-PNBOC triblock copolymers were first self-assembled into micelles with hydrophobic photosensitive PNBOC cores, hydrophilic PNAM inner shells, and thermoresponsive PNIPAM coronas below the lower critical solution temperature (LCST), while hydrogels of physically cross-linked micellar nanoparticles were achieved at elevated polymer concentrations and high temperatures above the critical gelation temperature (CGT). Rheological experiments revealed that the CGT was highly dependent on polymer compositions and concentrations, that is, a longer hydrophobic PNBOC block or a higher polymer concentration led to a decreased CGT. However, the CGT prior to UV irradiation (CGT0) could be drastically elevated after UV irradiation (CGTUV) as a result of UV irradiation-induced concurrently cross-linking and hydrophobic-to-hydrophilic transition within PNBOC cores. As such, gel-to-sol transition could be accomplished by either temperature decrease or exposure to UV irradiation at a fixed temperature lower than the CGTUV. Note that both GCT and DOX could be simultaneously encapsulated into the hydrogels due to the coexistence of extramicellar aqueous phase and hydrophobic micellar cores. Intriguingly, the subsequent co-release of GCT and DOX could be regulated by taking advantage of either temperature or UV irradiation-mediated gel-to-sol transitions.Download high-res image (318KB)Download full-size image
Co-reporter:Zhengyu Deng, Yinfeng Qian, Yongqiang Yu, Guhuan Liu, Jinming Hu, Guoying Zhang, and Shiyong Liu
Journal of the American Chemical Society 2016 Volume 138(Issue 33) pp:10452-10466
Publication Date(Web):August 3, 2016
DOI:10.1021/jacs.6b04115
Reactive oxygen species (ROS) and oxidative stress are implicated in various physiological and pathological processes, and this feature provides a vital biochemical basis for designing novel therapeutic and diagnostic nanomedicines. Among them, oxidation-responsive micelles and vesicles (polymersomes) of amphiphilic block copolymers have been extensively explored; however, in previous works, oxidation by ROS including H2O2 exclusively leads to microstructural destruction of polymeric assemblies. For oxidation-responsive polymersomes, fast release of encapsulated hydrophilic drugs and bioactive macromolecules will occur upon microstructural disintegration. Under certain application circumstances, this does not meet design requirements for sustained-release drug nanocarriers and long-acting in vivo nanoreactors. Also note that conventional polymersomes possess thick hydrophobic bilayers and compromised membrane permeability, rendering them as ineffective nanocarriers and nanoreactors. We herein report the fabrication of oxidation-responsive multifunctional polymersomes exhibiting intracellular milieu-triggered vesicle bilayer cross-linking, permeability switching, and enhanced imaging/drug release features. Mitochondria-targeted H2O2 reactive polymersomes were obtained through the self-assembly of amphiphilic block copolymers containing arylboronate ester-capped self-immolative side linkages in the hydrophobic block, followed by surface functionalization with targeting peptides. Upon cellular uptake, intracellular H2O2 triggers cascade decaging reactions and generates primary amine moieties; prominent amidation reaction then occurs within hydrophobic bilayer membranes, resulting in concurrent cross-linking and hydrophobic-to-hydrophilic transition of polymersome bilayers inside live cells. This process was further utilized to achieve integrated functions such as sustained drug release, (combination) chemotherapy monitored by fluorescence and magnetic resonance (MR) imaging turn-on, and to construct intracellular fluorogenic nanoreactors for cytosolic thiol-containing bioactive molecules.
Co-reporter:Sidan Tian, Guhuan Liu, Xiaorui Wang, Tao Wu, Jinxian Yang, Xiaodong Ye, Guoying Zhang, Jinming Hu, and Shiyong Liu
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 6) pp:3693
Publication Date(Web):November 19, 2015
DOI:10.1021/acsami.5b08970
The mimicking of biological supramolecular interactions and their mutual transitions to fabricate intelligent artificial systems has been of increasing interest. Herein, we report the fabrication of supramolecular micellar nanoparticles consisting of quaternized poly(ethylene oxide)-b-poly(2-dimethylaminoethyl methacrylate) (PEO-b-PQDMA) and tetrakis(4-carboxylmethoxyphenyl)ethene (TPE-4COOH), which was capable of reversible transition between polyion complexes (PIC) and hydrogen bonding complexes (HBC) with tunable aggregation-induced emission (AIE) mediated by solution pH. At pH 8, TPE-4COOH chromophores can be directly dissolved in aqueous milieu without evident fluorescence emission. However, upon mixing with PEO-b-PQDMA, polyion complexes were formed by taking advantage of electrostatic interaction between carboxylate anions and quaternary ammonium cations and the most compact PIC micelles were achieved at the isoelectric point (i.e., [QDMA+]/[COO–] = 1), as confirmed by dynamic light scattering (DLS) measurement. Simultaneously, fluorescence spectroscopy revealed an evident emission turn-on and the maximum fluorescence intensity was observed near the isoelectric point due to the restriction of intramolecular rotation of TPE moieties within the PIC cores. The kinetic study supported a micelle fusion/fission mechanism on the formation of PIC micelles at varying charge ratios, exhibiting a quick time constant (τ1) relating to the formation of quasi-equilibrium micelles and a slow time constant (τ2) corresponding to the formation of final equilibrium micelles. Upon deceasing the pH of PIC micelles from 8 to 2 at the [QDMA+]/[COO–] molar ratio of 1, TPE-4COOH chromophores became gradually protonated and hydrophobic. The size of micellar nanoparticles underwent a remarkable decrease, whereas the fluorescence intensity exhibited a further increase by approximately 7.35-fold, presumably because of the formation of HBC micelles comprising cationic PQDMA coronas and PEO/TPE-4COOH hydrogen-bonded cores, an inverted micellar structures compared to initial PIC micelles. Moreover, the pH-mediated schizophrenic micellar transition from PIC to HBC with tunable AIE characteristic was reversible.Keywords: aggregation-induced emission; hydrogen bonding complex; pH-responsive; polyion complex; schizophrenic micelle; supramolecular interaction
Co-reporter:Jingyan Zhang, Liangliang Jiang, Zhiyuan Zhu and Shiyong Liu
RSC Advances 2016 vol. 6(Issue 45) pp:39016-39023
Publication Date(Web):13 Apr 2016
DOI:10.1039/C6RA04593F
The formation of unilamellar vesicles was successfully established by the addition of hydrotropic salt p-toluidine hydrochloride (PTHC) to solutions of the anionic surfactant sodium dodecyl sulfate (SDS) at high salt concentrations ([PTHC]/[SDS], xPTHC > 0.6). Further studies on the existing vesicles were conducted in terms of changes of concentrations or temperatures of the aqueous solutions. Upon dilution or heating these vesicles can transform into long, flexible wormlike micelles (WLMs). In this process, the solutions switch from an aqueous solution of bluish and nearly Newtonian liquids with low viscosity to clear and viscoelastic solutions with an ability to trap bubbles, which was called “dilution-thickening” or “thermo-thickening”. It should be noted that a microscopic phase separation always emerges at a narrow range of concentrations or temperatures during the transition. Rheological techniques, laser light scattering (LLS), transmission electron microscopy (TEM), micro-differential scanning calorimetry (micro-DSC), and transmittance measurements were used to confirm the formation of vesicles and their reversible transformation with WLMs. Finally, a tentative mechanism for the reversible vesicle-to-WLM transition was proposed to explain the results.
Co-reporter:Yamin Li;Dr. Guhuan Liu;Xiaorui Wang;Dr. Jinming Hu ;Dr. Shiyong Liu
Angewandte Chemie 2016 Volume 128( Issue 5) pp:1792-1796
Publication Date(Web):
DOI:10.1002/ange.201509401
Abstract
Antimicrobial resistance poses serious public health concerns and antibiotic misuse/abuse further complicates the situation; thus, it remains a considerable challenge to optimize/improve the usage of currently available drugs. We report a general strategy to construct a bacterial strain-selective delivery system for antibiotics based on responsive polymeric vesicles. In response to enzymes including penicillin G amidase (PGA) and β-lactamase (Bla), which are closely associated with drug-resistant bacterial strains, antibiotic-loaded polymeric vesicles undergo self-immolative structural rearrangement and morphological transitions, leading to sustained release of antibiotics. Enhanced stability, reduced side effects, and bacterial strain-selective drug release were achieved. Considering that Bla is the main cause of bacterial resistance to β-lactam antibiotic drugs, as a further validation, we demonstrate methicillin-resistant S. aureus (MRSA)-triggered release of antibiotics from Bla-degradable polymeric vesicles, in vitro inhibition of MRSA growth, and enhanced wound healing in an in vivo murine model.
Co-reporter:Chenzhi Yao, Xiaorui Wang, Guhuan Liu, Jinming Hu, and Shiyong Liu
Macromolecules 2016 Volume 49(Issue 21) pp:8282-8295
Publication Date(Web):October 25, 2016
DOI:10.1021/acs.macromol.6b01374
The construction of intelligent vesicular nanocarriers and nanoreactors has received increasing interests due to their potential in mimicking natural counterparts such as cells and organelles. Herein, we report thermoresponsive and photoreactive vesicles could be fabricated from amphiphilic block copolymers (BCPs), poly(N-isopropylacrylamide)-b-poly(2-((((2-nitrobenzyl)oxy)carbonyl)amino)ethyl acrylate) (PNIPAM-b-PNBOCA), which were synthesized via consecutive reversible addition–fragmentation chain transfer (RAFT) polymerizations. The resulting BCPs self-assembled into vesicles when temperatures were lower than the lower critical solution temperature (LCST) of PNIPAM blocks (defined as LCST0). However, the resulting vesicles irreversibly formed collapsed vesicles upon temperature rise (T > LCST0), and a further temperature increase (T > Tagg,0) led to the formation of irregular aggregates of collapsed vesicles. On the other hand, upon UV irradiation, the initially hydrophobic PNBOCA bilayers underwent aminolysis-induced cross-linking and hydrophobic-to-hydrophilic transition, resulting in elevated LCST (defined as LCSTuv). Although the thermo-induced collapse of PNIPAM coronas (T > LCSTuv) and the formation of aggregates of cross-linked vesicles (T > Tagg,uv) were observed, the initially vesicular morphology could be restored when cooling to lower than LCSTuv, as opposed to irreversible morphological transition without UV irradiation. The vesicular assemblies were engineered as nanocarriers for both hydrophilic (doxorubicin hydrochloride, DOX) and hydrophobic (Nile red, NR) payloads. The corelease profiles could be delicately regulated by both temperature variations and UV irradiation. Interestingly, DOX release could be also regulated by thermo-induced vesicle collapse without recourse to UV irradiation or by near-infrared (NIR) irradiation-induced vesicle collapse in the presence of photothermal agents coloaded within vesicular interiors as a result of the relatively low glass transition temperature of PNBOCA blocks. Moreover, nanoreactors were constructed by loading glucose oxidase (GOx) into the aqueous interiors of the vesicles, allowing for activating fluorogenic reactions by UV irradiation and temperature change.
Co-reporter:Yamin Li;Dr. Guhuan Liu;Xiaorui Wang;Dr. Jinming Hu ;Dr. Shiyong Liu
Angewandte Chemie International Edition 2016 Volume 55( Issue 5) pp:1760-1764
Publication Date(Web):
DOI:10.1002/anie.201509401
Abstract
Antimicrobial resistance poses serious public health concerns and antibiotic misuse/abuse further complicates the situation; thus, it remains a considerable challenge to optimize/improve the usage of currently available drugs. We report a general strategy to construct a bacterial strain-selective delivery system for antibiotics based on responsive polymeric vesicles. In response to enzymes including penicillin G amidase (PGA) and β-lactamase (Bla), which are closely associated with drug-resistant bacterial strains, antibiotic-loaded polymeric vesicles undergo self-immolative structural rearrangement and morphological transitions, leading to sustained release of antibiotics. Enhanced stability, reduced side effects, and bacterial strain-selective drug release were achieved. Considering that Bla is the main cause of bacterial resistance to β-lactam antibiotic drugs, as a further validation, we demonstrate methicillin-resistant S. aureus (MRSA)-triggered release of antibiotics from Bla-degradable polymeric vesicles, in vitro inhibition of MRSA growth, and enhanced wound healing in an in vivo murine model.
Co-reporter:Xiaorui Wang; Jinming Hu; Guhuan Liu; Jie Tian; Huijuan Wang; Ming Gong
Journal of the American Chemical Society 2015 Volume 137(Issue 48) pp:15262-15275
Publication Date(Web):November 19, 2015
DOI:10.1021/jacs.5b10127
We report on the fabrication of photochromic polymersomes exhibiting photoswitchable and reversible bilayer permeability from newly designed poly(ethylene oxide)-b-PSPA (PEO-b-PSPA) diblock copolymers, where SPA is spiropyran (SP)-based monomer containing a unique carbamate linkage. Upon self-assembling into polymersomes, SP moieties within vesicle bilayers undergo reversible phototriggered isomerization between hydrophobic spiropyran (SP, λ2 > 450 nm irradiation) and zwitterionic merocyanine (MC, λ1 < 420 nm irradiation) states. For both SP and MC polymersomes, their microstructures are stabilized by multiple cooperative noncovalent interactions including hydrophobic, hydrogen bonding, π–π stacking, and paired electrostatic (zwitterionic) interactions, with the latter two types being exclusive for MC polymersomes. Control experiments using analogous block copolymers of hydrophobic SP monomer with a carbonate linkage (SPO) and conventional spiropyran methacrylate monomer (SPMA) containing a single ester functionality were then conducted, revealing that carbamate-incurred hydrogen bonding interactions in PEO-b-PSPA are crucial for polymersome stabilization in the zwitterionic MC state. Moreover, reversible phototriggered SP-to-MC polymersome transition is accompanied by membrane polarity and permeability switching from being nonimpermeable to selectively permeable toward noncharged, charged, and zwitterionic small molecule species below critical molar masses. Intriguingly, UV-actuated MC polymersomes possess two types of release modules: (1) sustained release upon short UV irradiation duration by taking advantage of the unexpectedly slow spontaneous MC-to-SP transition kinetics (t1/2 > 20 h) under dark conditions; (2) on-demand and switchable release under alternated UV–vis light irradiation. We further demonstrate photoswitchable spatiotemporal release of 4′,6-diamidino-2-phenylindole (DAPI, cell nuclei-staining dye) within living HeLa cells.
Co-reporter:Guhuan Liu; Guofeng Zhang; Jinming Hu; Xiaorui Wang; Mingqiang Zhu
Journal of the American Chemical Society 2015 Volume 137(Issue 36) pp:11645-11655
Publication Date(Web):September 1, 2015
DOI:10.1021/jacs.5b05060
Upon stimuli-triggered single cleavage of capping moieties at the focal point and chain terminal, self-immolative dendrimers (SIDs) and linear self-immolative polymers (l-SIPs) undergo spontaneous domino-like radial fragmentation and cascade head-to-tail depolymerization, respectively. The nature of response selectivity and signal amplification has rendered them a unique type of stimuli-responsive materials. Moreover, novel design principles are required for further advancement in the field of self-immolative polymers (SIPs). Herein, we report the facile fabrication of water-dispersible SIPs with a new chain topology, hyperbranched self-immolative polymers (hSIPs), by utilizing one-pot AB2 polycondensation methodology and sequential postfunctionalization. The modular engineering of three categories of branching scaffolds, three types of stimuli-cleavable capping moieties at the focal point, and seven different types of peripheral functional groups and polymeric building blocks affords both structurally and functionally diverse hSIPs with chemically tunable amplified-release features. On the basis of the hSIP platform, we explored myriad functions including visible light-triggered intracellular release of peripheral conjugated drugs in a targeted and spatiotemporally controlled fashion, intracellular delivery and cytoplasmic reductive milieu-triggered plasmid DNA release via on/off multivalency switching, mitochondria-targeted fluorescent sensing of H2O2 with a detection limit down to ∼20 nM, and colorimetric H2O2 assay via triggered dispersion of gold nanoparticle aggregates. To further demonstrate the potency and generality of the hSIP platform, we further configure it into biosensor design for the ultrasensitive detection of pathologically relevant antigens (e.g., human carcinoembryonic antigen) by integrating with enzyme-mediated cycle amplification with positive feedback and enzyme-linked immunosorbent assay (ELISA).
Co-reporter:Xianglong Hu, Yang Li, Tao Liu, Guoying Zhang, and Shiyong Liu
ACS Applied Materials & Interfaces 2015 Volume 7(Issue 28) pp:15551
Publication Date(Web):June 26, 2015
DOI:10.1021/acsami.5b04025
Intracellular temperature plays a prominent role in cellular functions and biochemical activities inside living cells, but effective intracellular temperature sensing and imaging is still in its infancy. Herein, thermoresponsive double hydrophilic block copolymers (DHBCs)-based fluorescent thermometers were fabricated to investigate their application in intracellular temperature imaging. Blue-emitting coumarin monomer, CMA, green-emitting 7-nitro-2,1,3-benzoxadiazole (NBD) monomer, NBDAE, and red-emitting rhodamine B monomer, RhBEA, were copolymerized separately with N-isopropylacrylamide (NIPAM) to afford dye-labeled PEG-b-P(NIPAM-co-CMA), PEG-b-P(NIPAM-co-NBDAE), and PEG-b-P(NIPAM-co-RhBEA). Because of the favorable fluorescence resonance energy transfer (FRET) potentials between CMA and NBDAE, NBDAE and RhBEA, as well as the slight tendency between CMA and RhBEA fluorophore pairs, three polymeric thermometers based on traditional one-step FRET were fabricated by facile mixing two of these three fluorescent DHBCs, whereas exhibiting limited advantages. Thus, two-step cascade FRET among three polymeric fluorophores was further interrogated, in which NBD acted as a bridging dye by transferring energy from CMA to RhBEA. Through the delicate optimization of the molar contents of three polymeric components, a ∼8.4-fold ratio change occurred in the temperature range of 20–44 °C, and the detection sensitivity improved significantly, reached as low as ∼0.4 °C, which definitely outperformed other one-step FRET thermometers in the intracellular temperature imaging of living cells. To our knowledge, this work represents the first example of polymeric ratiometric thermometer employing thermoresponsive polymer-based cascade FRET mechanism.Keywords: cascade FRET; intracellular temperature imaging; PNIPAM; polymeric ratiometric thermometers; thermoresponsive DHBCs;
Co-reporter:Guhuan Liu, Jinming Hu, Guoying Zhang, and Shiyong Liu
Bioconjugate Chemistry 2015 Volume 26(Issue 7) pp:1328
Publication Date(Web):December 16, 2014
DOI:10.1021/bc500548r
Spatiotemporal switching of respective phototherapy modes at the cellular level with minimum side effects and high therapeutic efficacy is a major challenge for cancer phototherapy. Herein we demonstrate how to address this issue by employing photosensitizer-conjugated pH-responsive block copolymers in combination with intracellular endocytic pH gradients. At neutral pH corresponding to extracellular and cytosol milieu, the copolymers self-assemble into micelles with prominently quenched fluorescence emission and low 1O2 generation capability, favoring a highly efficient photothermal module. Under mildly acidic pH associated with endolysosomes, protonation-triggered micelle-to-unimer transition results in recovered emission and enhanced photodynamic 1O2 efficiency, which synergistically actuates release of encapsulated drugs, endosomal escape, and photochemical internalization processes.
Co-reporter:Xianglong Hu and Shiyong Liu
Dalton Transactions 2015 vol. 44(Issue 9) pp:3904-3922
Publication Date(Web):23 Dec 2014
DOI:10.1039/C4DT03609C
Responsive polymeric assemblies and hybrid superstructures fabricated from stimuli-sensitive polymers and inorganic nanoparticles (NPs) have been the subject of extensive investigations during the past few decades due to their distinct advantages such as an improved water solubility, stimuli-responsiveness, excellent biocompatibility, and facile introduction of functional units. In addition, the chemical compositions of polymeric assemblies and corresponding hybrid superstructures can be modulated via the initial synthetic design to target desired functions, fabricate smart nanostructures, and explore morphology-dependent functional optimization. Promising applications in the field of imaging, sensing, drug/gene delivery, diagnostics, and nanoreactors are being extensively investigated. This perspective article focuses on recent developments, microstructural control, and biomedical applications of stimuli-responsive polymeric assemblies as well as responsive hybrid superstructures fabricated from responsive polymers and inorganic NP building blocks (gold NPs and magnetic iron oxide NPs), and highlights their current status and future developments with selected literature reports.
Co-reporter:Jinming Hu
Macromolecular Chemistry and Physics 2015 Volume 216( Issue 6) pp:591-604
Publication Date(Web):
DOI:10.1002/macp.201400578
Co-reporter:Yanyan Jiang, Guhuan Liu, Xiaorui Wang, Jinming Hu, Guoying Zhang, and Shiyong Liu
Macromolecules 2015 Volume 48(Issue 3) pp:764-774
Publication Date(Web):January 21, 2015
DOI:10.1021/ma502389w
Supramolecular aggregates of stimuli-responsive block copolymers are increasingly utilized as drug nanocarriers. Although in situ tracking their triggered disintegration and drug release processes at the cellular level is highly desirable, it remains a considerable challenge. We report the fabrication of double hydrophilic block copolymers covalently conjugated with α,β-unsaturated ketone-caged coumarin functionalities in the thermoresponsive block. Upon thermo-induced micellization and cellular uptake, Michael addition reaction of unsaturated ketone moieties with thiol compounds (GSH and Cys) in the reductive subcellular compartments leads to micelle-to-unimer transition. This is accompanied by concomitant fluorescence emission turn-on and triggered drug release, allowing for the process visualization.
Co-reporter:Jinming Hu, Xiao Wang, Yinfeng Qian, Yongqiang Yu, Yanyan Jiang, Guoying Zhang, and Shiyong Liu
Macromolecules 2015 Volume 48(Issue 16) pp:5959-5968
Publication Date(Web):August 4, 2015
DOI:10.1021/acs.macromol.5b01110
Nonviral gene delivery vectors need to overcome both extracellular and intracellular obstacles before releasing plasmid DNA in the transcriptionally active form. However, serum and transport stability desired for cationic polymer/pDNA polyplexes contradicts with the eventual plasmid release requirement; chain lengths of cationic polymer vectors render additional compromise between cytotoxicity and transfection efficiency. Although the introduction of stimuli-triggered degradable cationic polymers can partially solve these issues, the quest for novel design criteria and elucidation of elementary cellular transport pathways are highly desirable. Herein we report a supramolecular approach to construct fluorogenic gene delivery vectors via self-assembly of intracellular milieu-reactive cationic amphiphiles. This new type of micellar nanocarriers can effectively bind pDNA to form polyplexes due to multivalent cationic segments at micellar coronas. Upon cellular uptake and endosomal escape, hydrophobic micellar cores are subjected to fluorogenic Michael addition reactions with highly hydrophilic cytoplasmic thiols, leading to micellar disintegration, pDNA release, and emission turn-on for image-guided delivery.
Co-reporter:Lei Wang, Guhuan Liu, Xiaorui Wang, Jinming Hu, Guoying Zhang, and Shiyong Liu
Macromolecules 2015 Volume 48(Issue 19) pp:7262-7272
Publication Date(Web):October 1, 2015
DOI:10.1021/acs.macromol.5b01709
Supramolecular vesicles, also referred to as polymersomes, self-assembled from amphiphilic polymers capable of synchronically loading with both hydrophilic and hydrophobic payloads have shown promising potential in drug delivery application. Herein, we report the fabrication of pH-responsive polymersomes via supramolecular self-assembly of amphiphilic diblock copolymers, poly(ethylene oxide)-b-poly(2-((((5-methyl-2-(2,4,6-trimethoxyphenyl)-1,3-dioxan-5-yl)methoxy)carbonyl)amino)ethyl methacrylate) (PEO-b-PTTAMA), which were synthesized via reversible addition–fragmentation chain transfer (RAFT) polymerization of a pH-responsive monomer (i.e., TTAMA) using a PEO-based macroRAFT agent. The resultant amphiphilic diblock copolymer then self-assembled into vesicles consisting of hydrophilic PEO coronas and pH-responsive hydrophobic bilayers, as confirmed by TEM and DLS measurements. The polymersomes containing cyclic benzylidene acetals in the hydrophobic bilayers were relatively stable under neutral pH, whereas they underwent hydrolysis with the liberation of hydrophobic 2,4,6-trimethoxybenzaldehyde and the simultaneous generation of hydrophilic diol moieties upon exposure to acidic pH milieu, which could be monitored by UV/vis spectroscopy, SEM, and TEM observations. By loading hydrophobic model drug (Nile red) as well as hydrophilic chemotherapeutic drug (doxorubicin hydrochloride, DOX·HCl) into the bilayer and aqueous interior of the polymersomes, the subsequent release of Nile red and DOX·HCl payloads was remarkably regulated by the solution pH values, and a lower pH value led to a faster drug release profile. In vitro experiment, observed by a confocal laser scanning microscope (CLSM), revealed that the pH-responsive polymersomes were easily taken up by HeLa cells and were primarily located in the acidic organelles after internalization, where the pH-responsive cyclic acetal moieties were hydrolyzed and the embedded payloads were therefore released, allowing for on-demand release of the encapsulants mediated by intracellular pH. In addition to small molecule chemotherapeutic drugs, biomacromolecules (alkaline phosphatase, ALP) can also be encapsulated into the aqueous lumen of the polymersomes. Significantly, the pH-triggered degradation of polymersomes could also regulate the release of encapsulated ALP, as confirmed by ALP-activated fluorogenic reaction.
Co-reporter:Jinming Hu, Guoying Zhang, Zhishen Ge, Shiyong Liu
Progress in Polymer Science 2014 Volume 39(Issue 6) pp:1096-1143
Publication Date(Web):June 2014
DOI:10.1016/j.progpolymsci.2013.10.006
In the past decade, responsive polymers exhibiting reversible or irreversible changes in physical properties and/or chemical structures in response to external stimuli have been extensively investigated. Among them, tertiary amine methacrylate-based block copolymers represent a unique category considering their responsiveness to multiple external stimuli (e.g., pH, temperature and salts), which are essentially relevant to the biological milieu. These intriguing properties allow for their applications in a variety of fields ranging from drug or gene delivery, imaging, diagnostics, antibacterial coatings, catalysis, and bio-separations. This review article highlights tertiary amine methacrylate-based block copolymers, focusing on recent advances in the synthesis of tertiary amine methacrylate-based block copolymers with varying chemical structures and chain topologies, their supramolecular self-assembly in aqueous media as well as in the bulk state, and the emerging functional applications.
Co-reporter:Jinming Hu and Shiyong Liu
Accounts of Chemical Research 2014 Volume 47(Issue 7) pp:2084-2095
Publication Date(Web):April 17, 2014
DOI:10.1021/ar5001007
In this Account, we summarize recent progress in the field of responsive polymeric materials containing host–guest recognition motifs with selected examples and highlight their versatile functional applications, whereas small molecule-oriented host–guest supramolecular systems are excluded. We demonstrate how the introduction of host–guest chemistry into conventional polymer systems can modulate their responsive modes to external stimuli. Moreover, the responsive specificity and selectivity of polymeric systems can also be inherited from the host–guest recognition motifs, and these features provide extra advantages in terms of function integration. The following discussions are categorized in terms of design and functions, namely, host–guest chemistry toward the fabrication of responsive polymers and assemblies, optical sensing and imaging, drug and gene delivery, and self-healing materials. A concluding remark on future developments is also presented. We wish this prosperous field would incur more original and evolutionary ideas and benefit fundamental research and our daily life in a more convenient way.
Co-reporter:Yamin Li;Hansen Yu;Yinfeng Qian;Jinming Hu
Advanced Materials 2014 Volume 26( Issue 39) pp:6734-6741
Publication Date(Web):
DOI:10.1002/adma.201402797
Co-reporter:Xiaorui Wang ; Jinming Hu ; Guoying Zhang
Journal of the American Chemical Society 2014 Volume 136(Issue 28) pp:9890-9893
Publication Date(Web):July 1, 2014
DOI:10.1021/ja505278w
The development of a highly selective and fast responsive fluorogenic biosensor for diverse analytes ranging from bioactive small molecules to specific antigens is highly desirable but remains a considerable challenge. We herein propose a new approach by integrating substrate-selective enzymatic reactions with fluorogens exhibiting aggregation-induced emission feature. Tyrosine-functionalized tetraphenylethene, TPE-Tyr, molecularly dissolves in aqueous media with negligible fluorescence emission; upon addition of horseradish peroxidase (HRP) and H2O2, effective cross-linking occurs due to HRP-catalyzed oxidative coupling of tyrosine moieties in TPE-Tyr. This leads to fluorescence emission turn-on and fast detection of H2O2 with high sensitivity and selectivity. As a validation of the new strategy’s generality, we further configure it into the biosensor design for glucose through cascade enzymatic reactions and for pathologically relevant antigens (e.g., human carcinoembryonic antigen) by combining with the ELISA kit.
Co-reporter:Guhuan Liu ; Xiaorui Wang ; Jinming Hu ; Guoying Zhang
Journal of the American Chemical Society 2014 Volume 136(Issue 20) pp:7492-7497
Publication Date(Web):April 30, 2014
DOI:10.1021/ja5030832
Stimuli-triggered disassembly of block copolymer vesicles or polymersomes has been conventionally achieved via solubility switching of the bilayer-forming block, requiring cooperative changes of most of the repeating units. Herein we report an alternative approach by incorporating hydrophobic blocks exhibiting stimuli-triggered head-to-tail cascade depolymerization features. Amphiphilic block copolymers bearing this motif self-assemble into self-immolative polymersomes (SIPsomes). By modular design of terminal capping moieties, visible light, UV light, and reductive milieu can be utilized to actuate SIPsomes disintegration into water-soluble small molecules and hydrophilic blocks. The design of SIPsomes allows for triggered drug co-release and controllable access toward protons, oxygen, and enzymatic substrates. We also demonstrate programmed (OR-, AND-, and XOR-type logic) enzymatic reactions by integrating SIPsome encapsulation and trigger/capping moiety-selective cascade depolymerization events.
Co-reporter:Yang Li, Yinfeng Qian, Tao Liu, Guoying Zhang, Jinming Hu and Shiyong Liu
Polymer Chemistry 2014 vol. 5(Issue 5) pp:1743-1750
Publication Date(Web):29 Oct 2013
DOI:10.1039/C3PY01278F
We report on the synthesis of star copolymers possessing dual functions of gene delivery vectors and magnetic resonance (MR) imaging contrast enhancement. Starting from asymmetrically functionalized β-cyclodextrin (β-CD) comprising 7 azide moieties and 14 α-bromopropionate functionalities at the upper and lower rim of a rigid toroidal β-CD core, (DOTA-Gd)7-CD-(PDMA)14 star copolymers were synthesized via atom transfer radical polymerization (ATRP) of N,N-dimethylaminoethyl methacrylate (DMA) and subsequent click reaction with an alkynyl-functionalized gadolinium (Gd3+) complex, DOTA-Gd, where DOTA is 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid. The obtained Janus-type star copolymers, (DOTA-Gd)7-CD-(PDMA)14, could completely complex with anionic plasmid DNA (pDNA) via electrostatic interactions at N/P ratios equal to or higher than 2 and exhibit optimal in vitro transfection efficiency at an N/P ratio of 8. In addition, in vitro MR imaging experiments demonstrated considerably enhanced T1 relaxivity (r1 ∼ 10.9 s−1 mM−1) for the star copolymer compared to that of commercially available small molecular MR imaging contrast agents (2.4–3.2 s−1 mM−1). The star-type topology of asymmetrically functionalized β-CD based copolymers in combination with the integrated design of diagnostic and therapeutic functions augurs well for their potential applications in the field of image-guided gene therapy.
Co-reporter:Yang Li;Tao Liu;Guoying Zhang;Zhishen Ge
Macromolecular Rapid Communications 2014 Volume 35( Issue 4) pp:466-473
Publication Date(Web):
DOI:10.1002/marc.201300719
Co-reporter:Jingyan Zhang, Sangui Chen, Zhiyuan Zhu and Shiyong Liu
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 1) pp:117-127
Publication Date(Web):15 Oct 2013
DOI:10.1039/C3CP53608D
The formation of soluble polyion complexes (PICs) from anionic block copolymers, poly(ethylene oxide)-b-poly(sodium 4-styrene sulfonate) (PEO-b-PSSNa) and cationic block copolymers, poly(ethylene oxide)-b-poly(quaternized 2-(dimethyl amino)ethyl methacrylate) (PEO-b-PQDMA) was investigated by fluorescence spectroscopy, laser light scattering (LLS), and stopped-flow light scattering. Colloidally stabilized dispersions could be obtained upon direct mixing of the aqueous solutions of these two block copolymers, which indicated the formation of core–shell nanostructures with the core consisting of interpolymer electrostatic complexes between PSSNa and PQDMA blocks and the corona of PEO block. Both LLS and fluorescence results revealed that the most compact complex micelles formed at the equal molar ratio of oppositely charged SSNa and QDMA residues. The kinetics of the assembly process was studied via stopped-flow upon direct mixing of the two polymer solutions. The complexation process between PEO-b-PQDMA and PEO-b-PSSNa was fast and could finish within seconds. Moreover, the relaxation process can only be detected at near equal SSNa to QDMA molar ratios. The relaxation curves can be well fitted by a double-exponential function, leading to a fast relaxation process related to the initial quasi-equilibrium complex formation and a slow process related to the pre-complex structure rearrangements to the final equilibrium complexes. Both stages are determined as second-order reactions and processed through a micelle fusion–fission mechanism. Fluorescence kinetic studies revealed that the neutralization of an oppositely charged polyion was too fast to be detected and should be completed within the stopped-flow dead-time. Thermodynamic studies revealed that spontaneous complexation is entropy driven. Upon increasing the ionic strength of the solutions, the complexation processes became slower due to the decrease of entropy driving force. The PIC dissociation process was further studied and considered to consist of two competing processes: a second-order process depending on PIC concentration and a first-order process independent of the PIC concentration.
Co-reporter:Dr. Xianglong Hu;Yang Li;Dr. Tao Liu; Guoying Zhang; Shiyong Liu
Chemistry – An Asian Journal 2014 Volume 9( Issue 8) pp:2148-2155
Publication Date(Web):
DOI:10.1002/asia.201402171
Abstract
We report on the fabrication of a photodegradable gene-delivery vector based on PEO-b-P(GMA-g-PDMAEMA) neutral–cationic brush block copolymers that possess cationic poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA) brushes for DNA compaction, poly(ethylene oxide) (PEO) as a hydrophilic block, and poly(glycidyl methacrylate) (PGMA) as the backbone. The PEO-b-P(GMA-g-PDMAEMA) copolymers were synthesized through the combination of reversible addition–fragmentation transfer (RAFT) polymerization and postmodification. A photocleavable PEO-based macroRAFT agent was first synthesized; next, the PEO-b-PGMA block copolymer was prepared by RAFT polymerization of GMA; this was followed by a click reaction to introduce the RAFT initiators on the side chains of the PGMA block; then, RAFT polymerization of DMAEMA afforded the PEO-b-P(GMA-g-PDMAEMA) copolymer. The obtained neutral–cationic brush block copolymer could effectively complex plasmid DNA (pDNA) into nanoparticles at an N/P ratio (i.e., the number of nitrogen residues per DNA phosphate) of 4. Upon UV irradiation, pDNA could be released owing to cleavage of the pDNA-binding cationic PDMAEMA side chains as well as the nitrobenzyl ester linkages at the diblock junction point. In addition, in vitro gene transfection results demonstrated that the polyplexes could be effectively internalized by cells with good transfection efficiency, and the UV irradiation protocol could considerably enhance the efficiency of gene transfection.
Co-reporter:Jinming Hu, Guhuan Liu, Cheng Wang, Tao Liu, Guoying Zhang, and Shiyong Liu
Biomacromolecules 2014 Volume 15(Issue 11) pp:
Publication Date(Web):October 15, 2014
DOI:10.1021/bm501296d
Endosomal escape is of crucial importance to increase the therapeutic efficacy for nanoparticle-based drug and gene delivery. It has been long presumed that pH-responsive polymeric nanocarriers are potent in aiding endosomal escape due to the “proton sponge” effect; however, the intracellular pH (pHi) gradients subjected by pH-responsive nanocarriers during endocytic and endosomal escaping processes remain to be quantified and elucidated. We herein report the fabrication of ultrasensitive ratiometric fluorescent pHi imaging probes with robust endosomal escaping capability derived from dual dye-labeled pH-responsive block copolymers, which can directly monitor endosomal escape in living cells and quantitatively measure pHi variations during the entire endocytic and endosomolytic processes. Micellar nanoparticle-based pHi sensors could be efficiently internalized into cells via endocytosis where micelle-to-unimer transition occurs, followed by endosomal escape into the cytosol. This process is accompanied by deactivation of blue coumarin emission within acidic organelles and restored blue/red dual emissions within the neutral cytosolic milieu, allowing for ratiometric fluorescent imaging of entire pHi gradients subjected by micellar nanoparticles following the endocytic transport pathway.
Co-reporter:Yamin Li, Xianglong Hu, Sidan Tian, Yang Li, Guoqing Zhang, Guoying Zhang, Shiyong Liu
Biomaterials 2014 35(5) pp: 1618-1626
Publication Date(Web):
DOI:10.1016/j.biomaterials.2013.10.077
Co-reporter:Xiaorui Wang;Guhuan Liu;Dr. Jinming Hu;Dr. Guoying Zhang ;Dr. Shiyong Liu
Angewandte Chemie International Edition 2014 Volume 53( Issue 12) pp:3138-3142
Publication Date(Web):
DOI:10.1002/anie.201310589
Abstract
The fabrication of block copolymer (BCP) vesicles (polymersomes) exhibiting synchronized covalent crosslinking and bilayer permeabilization remains a considerable challenge as crosslinking typically leads to compromised membrane permeability. Herein it is demonstrated how to solve this dilemma by employing a stimuli-triggered crosslinking strategy with amphiphilic BCPs containing photolabile carbamate-caged primary amines. Upon self-assembling into polymersomes, light-triggered self-immolative decaging reactions release primary amine moieties and extensive amidation reactions then occur due to suppressed amine pKa within hydrophobic milieu. This leads to serendipitous vesicle crosslinking and the process is associated with bilayer hydrophobicity-to-hydrophilicity transition and membrane permeabilization.
Co-reporter:Xiaorui Wang;Guhuan Liu;Dr. Jinming Hu;Dr. Guoying Zhang ;Dr. Shiyong Liu
Angewandte Chemie 2014 Volume 126( Issue 12) pp:3202-3206
Publication Date(Web):
DOI:10.1002/ange.201310589
Abstract
The fabrication of block copolymer (BCP) vesicles (polymersomes) exhibiting synchronized covalent crosslinking and bilayer permeabilization remains a considerable challenge as crosslinking typically leads to compromised membrane permeability. Herein it is demonstrated how to solve this dilemma by employing a stimuli-triggered crosslinking strategy with amphiphilic BCPs containing photolabile carbamate-caged primary amines. Upon self-assembling into polymersomes, light-triggered self-immolative decaging reactions release primary amine moieties and extensive amidation reactions then occur due to suppressed amine pKa within hydrophobic milieu. This leads to serendipitous vesicle crosslinking and the process is associated with bilayer hydrophobicity-to-hydrophilicity transition and membrane permeabilization.
Co-reporter:Ming-Qiang Zhu, Guo-Feng Zhang, Zhe Hu, Matthew P. Aldred, Chong Li, Wen-Liang Gong, Tao Chen, Zhen-Li Huang, and Shiyong Liu
Macromolecules 2014 Volume 47(Issue 5) pp:1543-1552
Publication Date(Web):February 18, 2014
DOI:10.1021/ma5001157
We report on the reversible fluorescence switching of biodegradable nanoparticles of spiropyran-terminated poly(ε-caprolactone) (SP-PCL) for super-resolution fluorescence imaging. SP-PCL was synthesized via ring-opening polymerization using hydroxyl-containing SP derivative as the initiator. SP-PCL solution in THF or dioxane exhibits fast photochromism from colorless to blue upon UV irradiation due to the transformation of SPs in SP-PCL into merocyanines (MCs). Although both SP-PCL solution and MC-PCL solution do not fluoresce, SP-PCL nanoparticle dispersion fabricated via nanoprecipitation in aqueous media, in which SP molecules were embedded into the hydrophobic PCL matrix, displays considerable green emission at 530 nm at an excitation wavelength of 420 nm. Upon <420 nm irradiation, the resulting MC-PCL nanoparticles show strong red emission at 650 nm when excited at 420 nm. SP-PCL nanoparticles display green–red dual-color intrinsic fluorescence switching upon alternated UV/vis illumination. Green emission from SP in SP-PCL nanoparticles is observed before UV irradiation while red emission from MCs in MC-PCL nanoparticles after UV irradiation. For both SP-PCL and MC-PCL nanoparticles, the critical excitation wavelength is determined at 420 nm, at which the photoinduced interconversion of MC- and SP-forms are found to be at equilibrium. Positive and inverse photoisomerizations monitored using time-dependent fluorescence spectra show that blue light excitation above 420 nm yields green emission of SPs in SP-PCL nanoparticles while light irradiation below 420 nm imparts photoisomerization (SP to MC) and thus red emission of MCs in MC-PCL nanoparticles. Green and red fluorescence can be optically switched and imaged under fluorescent microscopy. Biodegradable SP-PCL nanoparticles are demonstrated to be promising photoswitchable fluorophores for localization-based super-resolution microscopy, evidencing by resolving nanostructures with sub-50 nm resolution in poly(vinyl alcohol) (PVA) film and live cells.
Co-reporter:XiangLong Hu;Yang Li;Tao Liu;GuoYing Zhang
Science China Chemistry 2014 Volume 57( Issue 4) pp:615-623
Publication Date(Web):2014 April
DOI:10.1007/s11426-014-5077-z
We report on the fabrication of fluorescent and multicolor probes for Zn2+ ions and temperature from a mixture of three types of fluorophore-labeled responsive block copolymers in aqueous media. Quinoline-based Zn2+-recognizing fluorescent monomer ZQMA, red-emitting rhodamine B-based monomer RhBEA, and blue-emitting coumarin derivative Coum-OH, were synthesized first. A ZQMA-labeled well-defined double hydrophilic block copolymer (DHBC), PEG-b-P(MEO2MA-co-ZQMA), was synthesized via reversible addition-fragmentation chain-transfer (RAFT) polymerization of 2-(2-methoxyethoxy)ethyl methacrylate (MEO2MA) and ZQMA by utilizing a PEG-based macroRAFT agent. Following similar procedures, PEG-b-P(St-co-RhBEA) amphiphilic diblock copolymer and PEG-b-P(MEO2MA-co-Coum) DHBC were also synthesized, where P(St-co-RhBEA) was a RhBEA-labeled polystyrene (PS) block. At room temperature in aqueous solution, almost nonfluorescent PEG-b-P(MEO2MA-co-ZQMA) can effectively bind Zn2+ ions, leading to prominent green fluorescence enhancement due to the coordination of ZQMA with Zn2+ ions. However, by mixing red-emitting PEG-b-P(St-co-RhBEA) and blue-emitting PEG-b-P(MEO2MA-co-Coum) with PEG-b-P(MEO2MA-co-ZQMA) at an appropriate ratio, three color transitions could be observed. In the absence of Zn2+ ions, a mixed pink fluorescent originating from Coum and RhBEA was observed; upon the addition of a certain amount of Zn2+ ions, the green fluorescence enhanced dramatically, leading to a white fluorescence readout. By further increasing the amount of Zn2+ ions, the green fluorescence further enhanced and overwhelmed the blue and red emissions, leading to a green-dominant mixed-fluorescence emission. In addition, upon increasing the temperature, the fluorescence of Coum decreased considerably due to the fluorescence-resonance energy transfer (FRET) between Coum and ZQMA moieties. In this way, a ratiometric fluorescent thermometer can be constructed.
Co-reporter:Jun Yin, Shengyu Shi, Jinming Hu, and Shiyong Liu
Langmuir 2014 Volume 30(Issue 31) pp:9551-9559
Publication Date(Web):2017-2-22
DOI:10.1021/la501918s
We report on the construction of a polyelectrolyte-responsive system evolved from sterically stabilized protonated poly(2-vinylpyridine) (P2VPH+) microgels. Negatively charged sodium dodecylbenzenesulfonate (SDBS) surfactants could be readily internalized into the cationic microgels by means of electrostatic interactions, resulting in microgel collapse and concomitant formation of surfactant micellar domains (P2VPH+/SDBS)-contained electrostatic complexes. These internal hydrophobic domains conferred the opportunity of fluorescent dyes to be loaded. The obtained fluorescent microgel complexes could be further disintegrated in the presence of anionic polyelectrolyte, poly(sodium 4-styrenesulfonate) (PNaStS). The stronger electrostatic attraction between multivalent P2VPH+ microgels and PNaStS polyelectrolyte than single-charged surfactant led to triggered release of the encapsulated pyrene dyes from the hydrophobic interiors into microgel dispersion. The process was confirmed by laser light scattering (LLS) and fluorescence measurements. Furthermore, the entire dynamic process of PNaStS adsorption into P2VPH+ microgel interior was further studied by stopped-flow equipment as a function of polyelectrolyte concentration and degree of polymerization. The whole adsorption process could be well fitted with a double-exponential function, suggesting a fast (τ1) and a slow (τ2) relaxation time, respectively. The fast process (τ1) was correlated well with the approaching of PNaStS with P2VPH+ microgel to form a nonequilibrium complex within the microgel shell, while the slow process (τ2) was consistent with the formation of equilibrium complexes in the microgel deeper inside. This simple yet feasible design augurs well for the promising applications in controlled release fields.
Co-reporter:Jun Yin, Jinming Hu, Guoying Zhang, and Shiyong Liu
Langmuir 2014 Volume 30(Issue 9) pp:2551-2558
Publication Date(Web):2017-2-22
DOI:10.1021/la500133y
A variety of slightly cross-linked poly(2-vinylpyridine)–poly(N-isopropylacrylamide) (P2VP–PNIPAM) core–shell microgels with pH- and temperature-responsive characteristic were prepared via seeded emulsion polymerization. Negatively charged sodium 2,6-naphthalenedisulfonate (2,6-NDS) could be internalized into the inner core, followed by formation of (P2VPH+/SO32–) supramolecular complex through the electrostatic attractive interaction in acid condition. The thermoresponsive characteristic feature of the (P2VPH+/SO32–)–PNIPAM core–shell microgels was investigated by laser light scattering and UV–vis measurement, revealing an integration of upper critical solution temperature (UCST) and lower critical solution temperature (LCST) behaviors in the temperature range of 20–55 °C. The UCST performance arised from the compromised electrostatic attractive interaction between P2VPH+ and 2,6-NDS at elevated temperatures, while the subsequent LCST transition is correlated to the thermo-induced collapse of PNIPAM shells. The controlled release of 2,6-NDS was monitored by static fluorescence spectra as a function of temperature change. Moreover, stopped-flow equipped with a temperature-jump accessory was then employed to assess the dynamic process, suggesting a millisecond characteristic relaxation time of the 2,6-NDS diffusion process. Interestingly, the characteristic relaxation time is independent of the shell cross-link density, whereas it was significantly affected by shell thickness. We believe that these dual thermoresponsive core–shell microgels with thermotunable volume phase transition may augur promising applications in the fields of polymer science and materials, particularly for temperature-triggered release.
Co-reporter:Zhishen Ge and Shiyong Liu
Chemical Society Reviews 2013 vol. 42(Issue 17) pp:7289-7325
Publication Date(Web):03 Apr 2013
DOI:10.1039/C3CS60048C
Self-assembled nanostructures of amphiphilic and double hydrophilic block copolymers have been increasingly utilized as potent polymeric nanocarriers of therapeutic drugs, genes, bioactive molecules, and imaging/contrast agents due to improved water solubility, bioavailability, and extended blood circulation duration. Though passive and active targeted drug delivery strategies have long been proposed to promote desirable drug accumulation specifically at the disease sites, the introduction of stimuli-responsiveness into self-assembled block copolymer nanocarriers can additionally lead to controlled/triggered release of therapeutic/imaging agents into target pathological tissues and cells, with concomitant advantages of enhanced delivery efficiency and therapeutic efficacy. Appropriately designed stimuli-responsive block copolymer assemblies can exhibit chemical structure transformation, microstructural rearrangement and inversion, or even disassembly into unimers or smaller ones under external stimuli such as pH, temperature, ion strength, redox potential, light, electric, and magnetic fields, and specific bioactive molecules and metabolites. Compared to normal tissues, pathological sites such as tumor tissues typically exhibit vascular abnormalities, weak acidity (∼pH 6.8), abnormal temperatures, over-expressed proteins and enzymes, hypoxia, high levels of metabolites and reactive small molecule species, etc. Moreover, upon cellular uptake, drug-loaded polymeric nanocarriers will be subjected to intracellular pH gradients (pH 5.9–6.2 in early endosomes and pH 5.0–5.5 in late endosomes and lysosomes) and redox and H2O2 gradients within different cell organelles and the cytosol. Thus, block copolymer nanocarriers responsive to the above described bio-relevant stimuli or biochemical signals characteristic of pathologic tissues and cells will provide an alternative type of “active targeting” strategy, which can be utilized to further boost therapeutic efficacy and imaging sensitivity via disease site-specific delivery and controlled release. A variety of extracellular or intracellular stimuli innate to disease sites, such as mildly acidic pH, temperature, enzymes (matrix metalloproteinase, β-glucuronidase, and phosphatase), oxidative/reductive microenvironments, and abnormal levels of bioactive molecules or metabolites, have been utilized for this purpose. In this review, we summarize recent advances in stimuli-responsive block copolymer assemblies which are responsive to tumor and intracellular microenvironments and their applications in anticancer drug delivery and enhanced imaging sensitivity.
Co-reporter:Xianglong Hu ; Jinming Hu ; Jie Tian ; Zhishen Ge ; Guoying Zhang ; Kaifu Luo
Journal of the American Chemical Society 2013 Volume 135(Issue 46) pp:17617-17629
Publication Date(Web):October 25, 2013
DOI:10.1021/ja409686x
Solution self-assembly of block copolymers (BCPs) typically generates spheres, rods, and vesicles. The reproducible bottom-up fabrication of stable planar nanostructures remains elusive due to their tendency to bend into closed bilayers. This morphological vacancy renders the study of shape effects on BCP nanocarrier-cell interactions incomplete. Furthermore, the fabrication of single BCP assemblies with built-in drug delivery functions and geometry-optimized performance remains a major challenge. We demonstrate that PEG-b-PCPTM polyprodrug amphiphiles, where PEG is poly(ethylene glycol) and PCPTM is polymerized block of reduction-cleavable camptothecin (CPT) prodrug monomer, with >50 wt % CPT loading content can self-assemble into four types of uniform nanostructures including spheres, large compound vesicles, smooth disks, and unprecedented staggered lamellae with spiked periphery. Staggered lamellae outperform the other three nanostructure types, exhibiting extended blood circulation duration, the fastest cellular uptake, and unique internalization pathways. We also explore shape-modulated CPT release kinetics, nanostructure degradation, and in vitro cytotoxicities. The controlled hierarchical organization of polyprodrug amphiphiles and shape-tunable biological performance opens up new horizons for exploring next-generation BCP-based drug delivery systems with improved efficacy.
Co-reporter:Junjie Li, Zhishen Ge and Shiyong Liu
Chemical Communications 2013 vol. 49(Issue 62) pp:6974-6976
Publication Date(Web):12 Jun 2013
DOI:10.1039/C3CC43576H
A matrix metalloproteinase-cleavable peptide-linked block copolymer was fabricated and utilized to construct PEG-sheddable polyplex micelles as smart gene delivery vectors, which were demonstrated to exhibit higher cellular uptake, improved endosomal escape, and high-efficiency gene transfection in the presence of matrix metalloproteinase-2.
Co-reporter:Tao Liu;Jinming Hu;Zhenyu Jin;Fan Jin
Advanced Healthcare Materials 2013 Volume 2( Issue 12) pp:1576-1581
Publication Date(Web):
DOI:10.1002/adhm.201200436
Co-reporter:Xianglong Hu, Hui Li, Shizhong Luo, Tao Liu, Yanyan Jiang and Shiyong Liu
Polymer Chemistry 2013 vol. 4(Issue 3) pp:695-706
Publication Date(Web):18 Sep 2012
DOI:10.1039/C2PY20701J
We report on the fabrication of dynamic covalent shell cross-linked (SCL) micelles of amphiphilic diblock copolymers functionalized with aldehyde moieties in the hydrophilic block by utilizing difunctional crosslinkers cleavable in response to pH and thiols. Well-defined amphiphilic diblock copolymer, PCL-b-P(OEGMA-co-MAEBA), was synthesized via ring opening polymerization (ROP) of ε-caprolactone (CL) and atom transfer radical polymerization (ATRP) of oligo(ethylene glycol) monomethyl ether methacrylate (OEGMA) and p-(methacryloxyethoxy)benzaldehyde (MAEBA) comonomers. In aqueous solution, the diblock copolymer self-assembles into micelles consisting of hydrophobic PCL cores and hydrophilic P(OEGMA-co-MAEBA) coronas covalently anchored with aldehyde groups. The subsequent shell cross-linking reaction was conducted at pH 6.2 upon addition of difunctional dithiolbis(propanoic dihydrazide) (DTP). The formation of dynamic acylhydrazone cross-linking linkages was facilitated under the catalysis of aniline. The obtained SCL micelles can be de-crosslinked via two biologically relevant modes, namely, acidic pH-triggered cleavage of acylhydrazone bonds into aldehyde and hydrazide and thiol-triggered cleavage of disulfide linkages, which have been utilized for triggered release of physically encapsulated chemotherapeutic drugs. The dual-responsive dynamic covalent SCL micelles were examined by dynamic laser light scattering (LLS), 1H NMR, Ellman's assay, and enzymatic degradation tests. In addition, camptothecin (CPT)-loaded SCL micelles were used to investigate thiol and pH-modulated CPT release profiles. Compared with CPT-loaded non-cross-linked (NCL) micelles, CPT-loaded SCL micelles can largely minimize drug leakage under physiological conditions, whilst exhibiting accelerated drug release under mildly acidic or thiol-rich microenvironments, which are relevant to those of acidic organelles (endosomes and lysosomes) or cytosol within tumor cells. Cell cytotoxicity studies revealed that drug-free SCL micelles are almost nontoxic, whereas CPT-loaded SCL micelles can efficiently deliver chemotherapeutic drug (CPT) into HepG2 cells, leading to considerable nucleic accumulation at extended incubation duration. The reported dynamic covalent shell cross-linking strategy can exert intricate control concerning the micellar stability and the release profile of encapsulated drugs in response to biological microenvironments, which augurs well for their potential use as novel smart nanocarriers for drug delivery in cancer chemotherapy.
Co-reporter:Xuejuan Wan;Tao Liu;Jinming Hu
Macromolecular Rapid Communications 2013 Volume 34( Issue 4) pp:341-347
Publication Date(Web):
DOI:10.1002/marc.201200673
Abstract
The fabrication of photo-degradable, protein–polyelectrolyte complex (PPC)-coated, mesoporous silica nanoparticles (MSNs) and their controlled co-release of protein and model drugs is reported. Random copolymers composed of oligo(ethylene glycol) monomethyl ether methacrylate (OEGMA), and a photolabile o-nitrobenzyl-containing monomer, 5-(2′-(dimethylamino)ethoxy)-2-nitrobenzyl methacrylate (DENBMA), are first anchored onto the MSNs and then quaternary aminated, to obtain positively charged P(OEGMA-co-TENBMA) which exhibits photo-induced charge conversion characteristics. PPCs consisting of P(OEGMA-co-TENBMA) and the protein bovine serum albumin (BSA) are utilized as capping agents for the nanopores of the MSNs. Upon UV irradiation, charge conversion of P(OEGMA-co-TENBMA) can lead to the disruption of PPCs on MSNs and co-release of BSA and rhodamine B by electrostatic repulsion.
Co-reporter:Jinming Hu;Tao Liu;Guoying Zhang;Fan Jin
Macromolecular Rapid Communications 2013 Volume 34( Issue 9) pp:749-758
Publication Date(Web):
DOI:10.1002/marc.201200613
Co-reporter:Zhishen Ge
Macromolecular Rapid Communications 2013 Volume 34( Issue 11) pp:922-930
Publication Date(Web):
DOI:10.1002/marc.201300072
Co-reporter:Yonghao Wu, Huamin Hu, Jinming Hu, Tao Liu, Guoying Zhang, and Shiyong Liu
Langmuir 2013 Volume 29(Issue 11) pp:3711-3720
Publication Date(Web):February 20, 2013
DOI:10.1021/la400145f
We report on thermo- and light-regulated formation and disintegration of double hydrophilic block copolymer (DHBC) micelles associated with tunable fluorescence emissions by employing two types of DHBCs covalently labeled with fluorescence resonance energy transfer (FRET) donor and acceptor moieties, respectively, within the light and temperature dually responsive block. Both DHBCs are molecularly soluble at room temperature in their aqueous mixture, whereas, upon heating to above the critical micellization temperature (CMT, ∼31 °C), they coassemble into mixed micelles possessing hydrophilic coronas and mixed cores containing FRET donors and acceptors. Accordingly, the closer spatial proximity between the FRET pair (NBDAE and RhBEA moieties) within micellar cores leads to substantially enhanced FRET efficiency, compared to that in the non-aggregated unimer state. Moreover, upon UV irradiation, the light-reactive moieties undergo light-cleavage reaction and transform into negatively charged carboxylate residues, leading to elevated CMT (∼46 °C). Thus, thermo-induced mixed micelles in the intermediate temperature range (31 °C < T < 46 °C) undergo light-triggered disintegration into unimers, accompanied with the decrease of FRET efficiency. Overall, the coassembly and disassembly occurring in the mixed DHBC solution can be dually regulated by temperature and UV irradiation, and most importantly, these processes can be facilely monitored via changes in FRET efficiency and distinct emission colors.
Co-reporter:Tao Liu;Yan-feng Zhang;Shi-yong Liu 刘世勇
Chinese Journal of Polymer Science 2013 Volume 31( Issue 6) pp:924-937
Publication Date(Web):2013 June
DOI:10.1007/s10118-013-1281-0
We report on the fabrication of self-assembled micelles from ABC-type miktoarm star polypeptide hybrid copolymers consisting of poly(ethylene oxide), poly(L-lysine), and poly(ɛ-caprolactone) arms, PEO(-b-PLL)-b-PCL, and their functional applications as co-delivery nanocarriers of chemotherapeutic drugs and plasmid DNA. Miktoarm star copolymer precursors, PEO(-b-PZLL)-b-PCL, were synthesized at first via the combination of consecutive “click” reactions and ring-opening polymerizations (ROP), where PZLL is poly(ɛ-benzyloxycarbonyl-L-lysine). Subsequently, the deprotection of PZLL arm afforded amphiphilic miktoarm star copolymers, PEO(-b-PLL)-b-PCL. In aqueous media at pH 7.4, PEO(-b-PLL)-b-PCL self-assembles into micelles consisting of PCL cores and hydrophilic PEO/PLL hybrid coronas. The hydrophobic micellar cores can effectively encapsulate model hydrophobic anticancer drug, paclitaxel; whereas positively charged PLL arms within mixed micellar corona are capable of forming electrostatic polyplexes with negatively charged plasmid DNA (pDNA) at N/P ratios higher than ca. 2. Thus, PEO(-b-PLL)-b-PCL micelles can act as co-delivery nanovehicles for both chemotherapeutic drugs and genes. Furthermore, polyplexes of pDNA with paclitaxel-loaded PEO(-b-PLL)-b-PCL micelles exhibited improved transfection efficiency compared to that of pDNA/blank micelles. We expect that the reported strategy of varying chain topologies for the fabrication of co-delivery polymeric nanocarriers can be further applied to integrate with other advantageous functions such as targeting, imaging, and diagnostics.
Co-reporter:Xianglong Hu, Jie Tian, Tao Liu, Guoying Zhang, and Shiyong Liu
Macromolecules 2013 Volume 46(Issue 15) pp:6243-6256
Publication Date(Web):July 22, 2013
DOI:10.1021/ma400691j
We report on the fabrication of dynamic covalent shell cross-linked (SCL) micelles with hydrophobic cores conjugated with photocaged chemotherapeutic drugs and coronas functionalized with ligands for tumor cell targeting. Two types of amphiphilic diblock copolymers, P(CL-g-CPT)-b-P(OEGMA-co-MAEBA)-CPT and PCL-b-P(OEGMA-co-MAEBA-co-FA), were synthesized via the combination of ring-opening copolymerization (ROP) of ε-caprolactone (CL) and 2-bromo-ε-caprolactone (CL-Br), atom transfer radical polymerization (ATRP) of oligo(ethylene glycol) monomethyl ether methacrylate (OEGMA) and p-(methacryloxyethoxy) benzaldehyde (MAEBA) comonomers, and “click” post-functionalization with photocaged camptothecin (CPT) prodrug and alkynyl-functionalized folic acid (FA) moieties, respectively. Mixed micelles coassembled from PCL-b-P(OEGMA-co-MAEBA-co-FA) and P(CL-g-CPT)-b-P(OEGMA-co-MAEBA)-CPT possess hydrophobic cores conjugated with photocaged CPT prodrugs and hydrophilic outer coronas covalently attached with aldehyde groups and FA moieties for subsequent shell cross-linking and cancer cell targeting. Shell cross-linking was performed at pH 6.2 upon addition of difunctional cross-linker, dithiol bis(propanoic dihydrazide) (DTP), under the catalysis of aniline. The obtained FA-decorated SCL micelles contain acylhydrazone and disulfide linkages in the outer coronas, which can be de-cross-linked under two biologically relevant conditions, mildly acidic or reductive microenvironments, that is, endosomal/lysosomal pH or high GSH level in the cytosol. The cleavage of caged CPT drug within the cores of SCL micelles can be effectively actuated under photo irradiation, whereas its diffusion out of micellar nanocarriers can be further modulated by pH and thiol levels due to the dually responsive nature of DTP cross-linker. Compared with the control, FA-decorated SCL micelles can more efficiently enter folate-receptor expressing cancer cells than folate-receptor deficient ones. Cell viability assays revealed that SCL micelles displayed at least ∼9.7-fold enhanced cytotoxicity upon light irradiation. The reported targeting ligand decorated and prodrug-conjugated dynamic covalent SCL micelles exert intricate control concerning micellar stability, cancer cell targeting, photo-triggered parent drug release with photoactivated cytotoxicity, and tunable drug release profiles. All of these augur well for their potential application as a novel integrated platform for targeted drug delivery in cancer chemotherapy.
Co-reporter:Jinming Hu, Guoqing Zhang and Shiyong Liu
Chemical Society Reviews 2012 vol. 41(Issue 18) pp:5933-5949
Publication Date(Web):13 Jun 2012
DOI:10.1039/C2CS35103J
Being responsive and adaptive to external stimuli is an intrinsic feature characteristic of all living organisms and soft matter. Specifically, responsive polymers can exhibit reversible or irreversible changes in chemical structures and/or physical properties in response to a specific signal input such as pH, temperature, ionic strength, light irradiation, mechanical force, electric and magnetic fields, and analyte of interest (e.g., ions, bioactive molecules, etc.) or an integration of them. The past decade has evidenced tremendous growth in the fundamental research of responsive polymers, and accordingly, diverse applications in fields ranging from drug or gene nanocarriers, imaging, diagnostics, smart actuators, adaptive coatings, to self-healing materials have been explored and suggested. Among a variety of external stimuli that have been utilized for the design of novel responsive polymers, enzymes have recently emerged to be a promising triggering motif. Enzyme-catalyzed reactions are highly selective and efficient toward specific substrates under mild conditions. They are involved in all biological and metabolic processes, serving as the prime protagonists in the chemistry of living organisms at a molecular level. The integration of enzyme-catalyzed reactions with responsive polymers can further broaden the design flexibility and scope of applications by endowing the latter with enhanced triggering specificity and selectivity. In this tutorial review, we describe recent developments concerning enzyme-responsive polymeric assemblies, nanoparticles, and hydrogels by highlighting this research area with selected literature reports. Three different types of systems, namely, enzyme-triggered self-assembly and aggregation of synthetic polymers, enzyme-driven disintegration and structural reorganization of polymeric assemblies and nanoparticles, and enzyme-triggered sol-to-gel and gel-to-sol transitions, are described. Their promising applications in drug controlled release, biocatalysis, imaging, sensing, and diagnostics are also discussed.
Co-reporter:Jinming Hu ; Tao Wu ; Guoying Zhang
Journal of the American Chemical Society 2012 Volume 134(Issue 18) pp:7624-7627
Publication Date(Web):April 23, 2012
DOI:10.1021/ja302019q
We report on a robust approach to the size-selective and template-free synthesis of asymmetrically functionalized ultrasmall (<4 nm) gold nanoparticles (AuNPs) stably anchored with a single amphiphilic triblock copolymer chain per NP. Directed NP self-assembly in aqueous solution can be facilely accomplished to afford organic/inorganic hybrid micelles, vesicles, rods, and large compound micelles by taking advantage of the rich microphase separation behavior of the as-synthesized AuNP hybrid amphiphilic triblock copolymers, PEO–AuNP–PS, which act as the polymer–metal–polymer analogue of conventional amphiphilic triblock copolymers. Factors affecting the size-selective fabrication and self-assembly characteristics and the time-dependent morphological evolution of NP assemblies were thoroughly explored.
Co-reporter:Guocan Yu ; Chengyou Han ; Zibin Zhang ; Jianzhuang Chen ; Xuzhou Yan ; Bo Zheng ; Shiyong Liu ;Feihe Huang
Journal of the American Chemical Society 2012 Volume 134(Issue 20) pp:8711-8717
Publication Date(Web):April 27, 2012
DOI:10.1021/ja302998q
The trans form of an azobenzene-containing guest can complex with a pillar[6]arene, while it cannot complex with pillar[5]arenes due to the different cavity sizes of the pillar[6]arene and the pillar[5]arenes. The spontaneous aggregation of its host–guest complex with the pillar[6]arene can be reversibly photocontrolled by irradiation with UV and visible light, leading to a switch between irregular aggregates and vesicle-like aggregates. This new pillar[6]arene-based photoresponsive host–guest recognition motif can work in organic solvents and is a good supplement to the existing widely used cyclodextrin/azobenzene recognition motif.
Co-reporter:Xiaorui Wang, Jinming Hu, Tao Liu, Guoying Zhang and Shiyong Liu
Journal of Materials Chemistry A 2012 vol. 22(Issue 17) pp:8622-8628
Publication Date(Web):22 Mar 2012
DOI:10.1039/C2JM16510D
In this work, we integrated the concept of aggregation-induced emission (AIE) with the specific supramolecular recognition between K+ ions and crown ether moieties to develop more effective fluorometric K+ probes. We synthesized a novel crown ether-functionalized tetraphenylethene (TPE) derivative, TPE-(B15C5)4, via the thiol-ene click reaction of thiol-derivatized TPE, TPE-(SH)4, with maleimide-functionalized benzo-15-crown-5 (B15C5). In TPE-(B15C5)4, the TPE core and four outer B15C5 moieties serve as the AIE-active motif and supramolecular K+-recognizing functionalities, respectively. TPE- (B15C5)4 molecularly dissolves in THF with negligible fluorescence emission. As we have envisaged, upon K+ addition, TPE-(B15C5)4 can be effectively induced to aggregate due to K+-mediated cross-linking via the formation of K+/B15C5 (1/2 molar ratio) molecular recognition complex in a sandwiched manner. This process is concomitantly accompanied with the turn-on of fluorescence emission via the AIE mechanism. Thus, TPE-(B15C5)4 can serve as highly sensitive and selective fluorometric off–on K+ probes.
Co-reporter:Tao Wu, Qianqian Zhang, Jinming Hu, Guoying Zhang and Shiyong Liu
Journal of Materials Chemistry A 2012 vol. 22(Issue 11) pp:5155-5163
Publication Date(Web):03 Feb 2012
DOI:10.1039/C2JM15530C
We report on the fabrication of water-dispersible composite silica nanospheres covalently anchored with gold nanoparticles (AuNPs) possessing thermo-tunable spatial distributions at the outer periphery of thermoresponsive poly(N-isopropylacrylamide) (PNIPAM) brushes. Starting from initiator-functionalized silica nanoparticles, surface-initiated atom transfer radical polymerization (ATRP) of N-isopropylacrylamide (NIPAM) afforded hybrid silica nanoparticles coated with PNIPAM brushes. The substitution reaction of halogen terminal groups of grafted PNIPAM chains with sodium azide and subsequent click reaction with 1,2-dithiolane-3-pentanoic acid-N-propargylamide afforded hybrid silica nanoparticles coated with 1,2-dithiolane end-capped PNIPAM brushes. AuNPs were then covalently anchored to the outer periphery of hybrid silica nanoparticles by utilizing strong chemisorption of surface-attached dithiolane moieties to AuNPs. Dynamic laser light scattering (LLS) measurements revealed that thermosensitive PNIPAM brushes at the surface of hybrid silica nanoparticles exhibit reversible thermo-induced collapse/swelling transitions, leading to the facile thermo-modulation of spatial distributions of AuNPs covalently attached at the periphery of composite silica nanospheres and thermo-reversible surface plasmon absorption band shift. The reported strategy of covalent assembly of AuNPs into well-defined composite nanospheres possessing thermo-tunable characteristics might be further exploited for colorimetric temperature sensing and responsive SERS detection purposes.
Co-reporter:Tao Liu, Yinfeng Qian, Xianglong Hu, Zhishen Ge and Shiyong Liu
Journal of Materials Chemistry A 2012 vol. 22(Issue 11) pp:5020-5030
Publication Date(Web):02 Feb 2012
DOI:10.1039/C2JM15092A
We report on the utilization of mixed diblock copolymer micelles as an integrated multifunctional platform for the cancer cell-targeted delivery of chemotherapeutic drugs and magnetic resonance (MR) imaging contrast enhancement under in vitro and in vivo conditions. Two types of amphiphilic diblock copolymers, PCL-b-P(OEGMA-FA) and PCL-b-P(OEGMA-Gd), consisting of a hydrophobic poly(ε-caprolactone) (PCL) block and a hydrophilic poly(oligo(ethylene glycol) monomethyl ether methacrylate) (POEGMA) block, covalently attached with folic acid (FA) and DOTA-Gd (Gd) moieties, respectively, were synthesized via the combination of atom transfer radical polymerization (ATRP), ring-opening polymerization (ROP), and “click” post-functionalization. Mixed micelles co-assembled from PCL-b-P(OEGMA-FA) and PCL-b-P(OEGMA-Gd) possess hydrophobic PCL cores for loading chemotherapeutic drugs and hydrophilic POEGMA outer coronas functionalized with FA and Gd complexes for synergistic functions of targeted delivery and MR imaging contrast enhancement. As-prepared nanosized mixed micelles are capable of physically encapsulating paclitaxel, a well-known hydrophobic anticancer drug, with a loading content of ∼5.0 w/w%, exhibiting controlled release of up to ∼60% loaded drugs over a duration of ∼130 h. In vitrocell viability assays revealed that drug-free mixed micelles are almost non-cytotoxic up to a concentration of 0.2 g L−1, whereas paclitaxel-loaded ones can effectively kill HeLa cells at the same concentration. In vitro MR imaging experiments indicated dramatically increased T1 relaxivity (26.29 s−1mM−1) for mixed micelles compared to that of small molecule counterpart, alkynyl-DOTA-Gd (3.12 s−1mM−1). Further in vivo MR imaging experiments in rabbits revealed considerably enhanced signal intensity, prominent positive contrast enhancement, improved accumulation and retention, and extended blood circulation duration for FA-labeled mixed micellar nanoparticles within the rabbit liver, as compared to those for FA-free mixed micelles and small molecule alkynyl-DOTA-Gd complex. These preliminary results indicate that the reported mixed micellar nanocarriers possess synergistically integrated functions of cancer-targeted drug delivery and controlled release, and MR imaging contrast enhancement, which augurs well for their potential application as a novel type of theranostic platform.
Co-reporter:Xingxing Sun, Xuepeng Zhang, Xinyang Li, Shiyong Liu and Guoqing Zhang
Journal of Materials Chemistry A 2012 vol. 22(Issue 33) pp:17332-17339
Publication Date(Web):04 Jul 2012
DOI:10.1039/C2JM32809G
Mechanochromic luminescence (ML) refers to the luminescence color and/or intensity change of solid-state materials induced by mechanical perturbations. For organic molecular solids, this phenomenon is related to the specific packing modes and orientations of individual fluorophores, which could give rise to different excited-state interactions. The molecular solids of difluoroboron dibenzoylmethane (BF2dbm) derivatives were previously found to exhibit reversible ML at room temperature and are promising as self-healing optical materials. In this report, we aim to shed some light on the mechanism of BF2dbm ML by trying to understand the excited-state interactions among solid-state BF2dbm molecules and elucidate how these interactions change upon mechanical stimulation. We first investigated the optical properties of monomeric, dimeric, and polymeric BF2dbm derivatives in optically dilute solutions and demonstrated unambiguously that BF2dbm moieties have a propensity to form H-aggregates. Next, we studied the physical properties of these boron complexes in the solid state including their crystal structures, fluorescence emissions, and mechanochromic luminescence. By correlating solution data with the solid-state characterization results, it was concluded that two coupled processes, force-induced emissive H-aggregate formation and energy transfer to the emissive H-aggregates, are responsible for the observed BF2dbm ML in the solid state.
Co-reporter:Changhua Li and Shiyong Liu
Chemical Communications 2012 vol. 48(Issue 27) pp:3262-3278
Publication Date(Web):09 Feb 2012
DOI:10.1039/C2CC17695E
Fluorescent polymeric assemblies and nanoparticles (NPs) of nanoscale dimensions have become a focus of intensive investigations during the past few decades due to combined advantages such as improved biocompatibility, water dispersibility, stimuli-responsiveness, facile integration into optical detection devices, and the ability of further functionalization. In addition, the chemical composition and morphology of polymeric assemblies and NPs can be modulated via synthetic approaches, leading to the precise spatial organization of multiple fluorophores. Thus, polymeric assemblies and NPs have been utilized to optimize the photoluminescent properties of covalently or physically attached fluorophores and facilely modulate the fluorescence resonance energy transfer (FRET) processes when the polymeric matrix is endowed with stimuli-responsiveness. These fascinating fluorescent polymeric assemblies and NPs offer unique and versatile platforms for the construction of novel detection, imaging, biolabeling, and optoelectronic systems. This feature article focuses on the recent developments of polymeric assemblies and NPs-based stimuli-tunable fluorescent systems and highlights their future practical applications with selected literature reports.
Co-reporter:Yonghao Wu;Huamin Hu;Jinming Hu
Macromolecular Rapid Communications 2012 Volume 33( Issue 21) pp:1852-1860
Publication Date(Web):
DOI:10.1002/marc.201200411
Abstract
The fabrication of a novel type of positively charged acid-disintegrable microgel loaded with insulin by electrostatic interactions and covalently immobilized with glucose oxidase (GOx) and catalase by inverse emulsion polymerization is reported, aiming for glucose-regulated insulin release by utilizing GOx/catalase cascade enzymatic reactions to trigger local pH decrease and acid-cleavage of crosslinking moieties. At the same time, a local pH decrease within the microgels also leads to the diminishment of net surface negative charges of encapsulated insulin. The above two factors both synergistically contribute to the prominently enhanced insulin release at high glucose levels (∼10–20 mM) compared to that in the absence of glucose.
Co-reporter:Jean-François Lutz;Brent Sumerlin
Macromolecular Rapid Communications 2012 Volume 33( Issue 9) pp:
Publication Date(Web):
DOI:10.1002/marc.201200171
No abstract is available for this article.
Co-reporter:Changhua Li, Jinming Hu and Shiyong Liu
Soft Matter 2012 vol. 8(Issue 27) pp:7096-7102
Publication Date(Web):09 May 2012
DOI:10.1039/C2SM25582K
Polymeric micelles and polymersomes self-assembled from amphiphilic and double hydrophilic block copolymers (DHBCs) offer great promise as smart nanocarriers of chemotherapeutic drugs, genes, and imaging or contrast agents. Specifically, fluorescent polymeric assemblies and nanoparticles render possible the in situ tracking of intracellular transport, in vivo circulation characteristics, and biodistribution of drug-loaded or conjugated nanocarriers. The introduction of fluorescence resonance energy transfer (FRET) processes into polymeric assemblies and nanoparticles can further enhance relevant functions due to the fact that the tracking can be conducted in a ratiometric manner to effectively exclude background interference and enhance spatiotemporal detection resolution. Moreover, if the FRET efficiency is responsive to the variation of solution conditions such as pH value, temperature, metal ions, glucose, and tissue-specific enzymes by taking advantage of the responsiveness of polymeric matrix, sensing of the external microenvironment will also be achieved. This article highlights recent developments of fluorescent polymers, polymeric assemblies and nanoparticles involving FRET events, in which the modulation of FRET efficiency can be achieved by tuning the spatial distribution of fluorescent donors and acceptors.
Co-reporter:Changhua Li;Tao Wu;Dr. Chunyan Hong;Dr. Guoqing Zhang;Dr. Shiyong Liu
Angewandte Chemie 2012 Volume 124( Issue 2) pp:470-474
Publication Date(Web):
DOI:10.1002/ange.201105735
Co-reporter:Yamin Li, Yinfeng Qian, Tao Liu, Guoying Zhang, and Shiyong Liu
Biomacromolecules 2012 Volume 13(Issue 11) pp:
Publication Date(Web):September 27, 2012
DOI:10.1021/bm301425j
Polymeric drug nanocarriers integrated with diagnostic and sensing functions are capable of in situ monitoring the biodistribution of chemotherapeutic drugs and imaging/contrasting agents, which enables the establishment of image-guided personalized cancer therapeutic protocols. Responsive multifunctional theranostic nanocarriers possessing external stimuli-tunable drug release rates and imaging signal intensities represent another promising direction in this field. In this work, we fabricated responsive amphiphilic diblock copolymer micelles exhibiting light-triggered hydrophobic–hydrophilic transition within micellar cores and the concomitant enhancement of magnetic resonance (MR) imaging contrast performance and release rate of physically encapsulated hydrophobic drugs. POEGMA-b-P(NIPAM-co-NBA-co-Gd) diblock copolymer covalently labeled with Gd3+ complex (Gd) in the light-responsive block was synthesized at first, where OEGMA, NIPAM, and NBA are oligo(ethylene glycol) monomethyl ether methacrylate, N-isopropylacrylamide, and o-nitrobenzyl acrylate, respectively. The amphiphilic diblock copolymer spontaneously self-assembles in aqueous solution into micellar nanoparticles possessing hydrophobic P(NIPAM-co-NBA-co-Gd) cores and hydrophilic POEGMA coronas, which can physically encapsulate doxorubicin (Dox) as a model chemotherapeutic drug. Upon UV irradiation, hydrophobic NBA moieties within micellar cores transform into hydrophilic carboxyl derivatives, triggering micelle microstructural changes and core swelling. During this process, the microenvironment surrounding Gd3+ complexes was subjected to a transition from being hydrophobic to hydrophilic, leading to the enhancement of MR imaging contrast performance, that is, ∼1.9-fold increase in longitudinal relaxivity (r1). In addition, the release rate of encapsulated Dox was also enhanced (∼65% of Dox release in 12 h upon UV irradiation versus ∼47% Dox release in 25 h for the control). The reported strategy of light-triggered coenhancement of MR imaging contrast performance and drug release profiles represents a general route to the construction of next generation smart polymeric theranostic nanocarriers.
Co-reporter:Changhua Li;Tao Wu;Dr. Chunyan Hong;Dr. Guoqing Zhang;Dr. Shiyong Liu
Angewandte Chemie International Edition 2012 Volume 51( Issue 2) pp:455-459
Publication Date(Web):
DOI:10.1002/anie.201105735
Co-reporter:Tao Liu, Xiaojie Li, Yinfeng Qian, Xianglong Hu, Shiyong Liu
Biomaterials 2012 33(8) pp: 2521-2531
Publication Date(Web):
DOI:10.1016/j.biomaterials.2011.12.013
Co-reporter:Jinming Hu, Yinfeng Qian, Xiaofeng Wang, Tao Liu, and Shiyong Liu
Langmuir 2012 Volume 28(Issue 4) pp:2073-2082
Publication Date(Web):November 2, 2011
DOI:10.1021/la203992q
We report on the fabrication of organic/inorganic hybrid micelles of amphiphilic block copolymers physically encapsulated with hydrophobic drugs within micellar cores and stably embedded with superparamagnetic iron oxide (SPIO) nanoparticles within hydrophilic coronas, which possess integrated functions of chemotherapeutic drug delivery and magnetic resonance (MR) imaging contrast enhancement. Poly(ε-caprolactone)-b-poly(glycerol monomethacrylate), PCL-b-PGMA, and PCL-b-P(OEGMA-co-FA) amphiphilic block copolymers were synthesized at first by combining ring-opening polymerization (ROP), atom transfer radical polymerization (ATRP), and post- modification techniques, where OEGMA and FA are oligo(ethylene glycol) monomethyl ether methacrylate and folic acid-bearing moieties, respectively. A model hydrophobic anticancer drug, paclitaxel (PTX), and 4 nm SPIO nanoparticles were then loaded into micellar cores and hydrophilic coronas, respectively, of mixed micelles fabricated from PCL-b-PGMA and PCL-b-P(OEGMA-co-FA) diblock copolymers by taking advantage of the hydrophobicity of micellar cores and strong affinity between 1,2-diol moieties in PGMA and Fe atoms at the surface of SPIO nanoparticles. The controlled and sustained release of PTX from hybrid micelles was achieved, exhibiting a cumulative release of ∼61% encapsulated drugs (loading content, 8.5 w/w%) over ∼130 h. Compared to that of surfactant-stabilized single SPIO nanoparticles (r2 = 28.3 s–1 mM–1 Fe), the clustering of SPIO nanoparticles within micellar coronas led to considerably enhanced T2 relaxivity (r2 = 121.1 s–1 mM–1 Fe), suggesting that hybrid micelles can serve as a T2-weighted MR imaging contrast enhancer with improved performance. Moreover, preliminary experiments of in vivo MR imaging were also conducted. These results indicate that amphiphilic block copolymer micelles surface embedded with SPIO nanoparticles at the hydrophilic corona can act as a new generation of nanoplatform integrating targeted drug delivery, controlled release, and disease diagnostic functions.
Co-reporter:Jinming Hu, Tao Wu, Guoqing Zhang, and Shiyong Liu
Macromolecules 2012 Volume 45(Issue 9) pp:3939-3947
Publication Date(Web):April 25, 2012
DOI:10.1021/ma3006558
We report on the fabrication of a new type of polymeric fluorescent Hg2+ probe covering a broad effective concentration range from nanomolar to micromolar levels and exhibiting considerably enhanced detection selectivity. Two amphiphilic diblock copolymers colabeled with Hg2+-reactive caged dye (RhBHA) and Hg2+-catalyzed caged fluorophore (HCMA) in the hydrophilic segments, PS-b-P(DMA-co-HCMA) and PS-b-P(DMA-co-RhBHA), were synthesized via sequential reversible addition–fragmentation chain transfer (RAFT) polymerization, where PS, DMA, HCMA, and RhBHA are polystyrene, N,N-dimethylacrylamide, hydrazone-caged coumarin, and rhodamine B (RhB) derivatives, respectively. The two amphiphilic diblock copolymers can spontaneously self-assemble into mixed micelles in aqueous solution possessing hydrophobic PS cores and HCMA and RhBHA moieties colabeled hydrophilic PDMA coronas. Fluorescence emissions of caged RhBHA and HCMA moieties can effectively turn on in the presence of low and high Hg2+ concentrations via Hg2+-induced ring-opening reaction and Hg2+-catalyzed hydrolysis mechanisms, respectively. The drastically different, but self-complementary reaction kinetics and optimum working concentration ranges of RhBHA and HCMA moieties endow the sensing system with high selectivity and broad sensing concentration range (from nanomolar to micromolar). In addition, the Hg2+-sensing platform can be further employed for the fluorescent ratiometric detection of Cu2+ ion via its selective quenching of the emission of acyclic RhBHA moieties. This work presents a new example of ensembling two partially selective chemical reaction-based fluorometric sensing motifs to achieve enhanced metal ion sensing selectivity and broadened working concentration ranges, which can be further generalized for the construction of other highly selective and broad dynamic range sensing systems.
Co-reporter:Jun Yin, Tao Wu, Jibin Song, Qian Zhang, Shiyong Liu, Rong Xu, and Hongwei Duan
Chemistry of Materials 2011 Volume 23(Issue 21) pp:4756
Publication Date(Web):October 13, 2011
DOI:10.1021/cm201791r
We report a new class of turn-on surface enhanced Raman scattering (SERS) sensors for the sensitive and selective detection of cadmium ion (Cd2+) by taking advantage of the interparticle plasmonic coupling generated in the process of Cd2+-selective nanoparticle self-aggregation. The SERS-active nanoparticles consist of 41-nm gold nanoparticles, encoded with a Raman-active dye through a disulfide anchoring group, and a layer of Cd2+-chelating polymer brush coating grafted on the nanoparticle via surface-initiated atom transfer radical polymerization. These SERS nanoparticles are optimized to remain spectrally silent when staying as single particles. Addition of Cd2+ leads to interparticle self-aggregation and immediately turns on the SERS fingerprint signal with up to 90-fold of signal enhancement. The selectivity of the SERS nanoparticle for Cd2+ was also examined, showing that various common metal ions cannot induce interparticle self-aggregation and the turn-on of SERS signal. In contrast to nanoparticle-based colorimetric assays, the SERS probe is also capable of detecting Cd2+ in heavily colored samples.Keywords: cadmium ion; metal nanoparticles; nanosensors; SERS; SI-ATRP;
Co-reporter:Tao Wu, Zhishen Ge, and Shiyong Liu
Chemistry of Materials 2011 Volume 23(Issue 9) pp:2370
Publication Date(Web):April 13, 2011
DOI:10.1021/cm200102g
We report on the fabrication of thermoresponsive cross-linked hollow poly(N- isopropylacrylamide) (PNIPAM) nanocapsules and silver nanoparticle-embedded hybrid PNIPAM nanocapsules with controlled shell thickness via the combination of surface-initiated atom transfer radical polymerization (ATRP) and “click” cross-linking. Starting from initiator- functionalized silica nanoparticles, the surface-initiated ATRP of N-isopropylacrylamide (NIPAM) and 3-azidopropylacrylamide (AzPAM) afforded hybrid silica nanoparticles surface coated with P(NIPAM-co-AzPAM) brushes. Hybrid PNIPAM nanocapsules were then fabricated by the “click” cross-linking of PNIPAM shell layer with a trifunctional molecule, 1,1,1-tris(4-(2-propynyloxy)phenyl)ethane, followed by the subsequent removal of silica cores via HF etching. Shell cross-linked hybrid silica nanoparticles can further serve as templates for the in situ preparation of silver nanoparticles within the cross-linked PNIPAM layer. After HF etching, silver nanoparticle-embedded hybrid PNIPAM nanocapsules were obtained. Due to the thermoresponsiveness of PNIPAM, cross-linked PNIPAM nanocapsules and silver nanoparticle-embedded hybrid PNIPAM nanocapsules exhibit thermo-induced collapse/swelling transitions. In the latter case, the spatial distribution of Ag nanoparticles within the hybrid PNIPAM nanocapsules can be facilely modulated by temperature variations, as revealed by the thermo-induced red shift of surface plasmon absorption band. Dynamic laser light scattering (LLS) measurements revealed that PNIPAM nanocapsules and Ag nanoparticle- embedded hybrid PNIPAM nanocapsules exhibit more prominent thermo-induced dimensional changes, as compared to shell cross-linked hybrid silica/PNIPAM nanoparticles loaded with or without Ag nanoparticles, respectively. Due to that the surface-initiated ATRP can be conducted in a controlled manner, the current strategy employed for the fabrication of structurally stable cross-linked PNIPAM nanocapsules and Ag nanoparticle-embedded hybrid PNIPAM nanocapsules can be further applied to the preparation of other functional hollow hybrid nanostructures with controlled dimensions.Keywords: hybrid capsules; poly(N-isopropylacrylamide); silica nanoparticles; silver nanoparticles; spatial distribution; surface-initiated ATRP; thermoresponsive nanocapsules;
Co-reporter:Jinming Hu, Xiaozheng Zhang, Di Wang, Xianglong Hu, Tao Liu, Guoying Zhang and Shiyong Liu
Journal of Materials Chemistry A 2011 vol. 21(Issue 47) pp:19030-19038
Publication Date(Web):31 Oct 2011
DOI:10.1039/C1JM13575A
We report on the fabrication of highly sensitive ratiometric fluorescent pH and temperature probes based on thermoresponsive double hydrophilic block copolymers (DHBCs) with the two blocks labeled with two types of dyes possessing different pH-switchable emission characteristics. P(NIPAM-co-FITC)-b-P(OEGMA-co-RhBAM) DHBCs were synthesized via consecutive reversible addition–fragmentation chain transfer (RAFT) polymerizations in combination with post-modifications, where NIPAM, OEGMA, FITC, and RhBAM are N-isopropylacrylamide, oligo(ethylene glycol) monomethyl ether methacrylate, fluorescein isothiocyanate, and rhodamine B-based derivatives, respectively. Due to that FITC and RhBAM moieties exhibit prominent decrease and increase in emission intensities with decreasing solution pH, respectively, intensity ratios of characteristic RhBAM and FITC emission bands, I582/I522, of P(NIPAM-co-FITC)-b-P(OEGMA-co-RhBAM) unimers at 25 °C exhibit ∼39-fold changes in the range of pH 2–10. At elevated temperatures, thermo-induced formation of PNIPAM-core micelles enables effective fluorescence resonance energy transfer (FRET) between FITC and RhBAM moieties respectively located within micellar cores and coronas, and I582/I522 exhibits ∼52.5-fold changes in the same pH range. The reported dually modulated multicolor-emitting P(NIPAM-co-FITC)-b-P(OEGMA-co-RhBAM) DHBCs are capable of ultrasensitive fluorometric detection of solution pH and temperature in a ratiometric manner, which augurs well for their practical applications in sensing, imaging, and the fabrication of new generation of theranostic systems.
Co-reporter:Xuejuan Wan and Shiyong Liu
Journal of Materials Chemistry A 2011 vol. 21(Issue 28) pp:10321-10329
Publication Date(Web):01 Jun 2011
DOI:10.1039/C1JM10332F
We reported on the synthesis of well-defined thermoresponsive polymers labeled with fluorescence resonance energy transfer (FRET) pairs at chain middle and terminals, which can act as single chain-based dual ratiometric fluorescent probes for pH and temperature under extremely dilute conditions. Starting from difunctional initiator containing a 7-nitro-2,1,3-benzoxadiazole (NBD) moiety, the atom transfer radical polymerization (ATRP) of oligo(ethylene glycol) monomethyl ether methacrylate (OEGMA) and di(ethylene glycol) monomethyl ether methacrylate (DEGMA), and the subsequent terminal group functionalization with Rhodamine B (RhB)-ethylenediamine derivative afforded thermoresponsive NBD-P(OEGMA-co-DEGMA)-RhB2, which were labeled with FRET donor (NBD) and acceptor moieties (RhB) at the chain middle and terminals. The fluorescence emission of terminal RhB functionalities is highly pH-dependent, i.e, non-fluorescent in neutral or alkaline media (spirolactam form) and highly fluorescent in acidic media (ring-opened acyclic form), thus the off/on switching of FRET process can be facilely modulated by solution pH. Moreover, at acidic pH and highly dilute conditions, the thermo-induced chain collapse and extension of NBD-P(OEGMA-co-DEGMA)-RhB2 can effectively modulate the spatial distance between FRET donor and acceptor moieties, leading to prominent changes in FRET efficiencies. The site-specific incorporation of one FRET donor and two pH-switchable acceptors at the chain middle and terminals of thermoresponsive polymers allows for the effective off/on switching and the modulation of efficiency of FRET processes by dually playing with solution pH and temperatures. This work represents the first report of single thermoresponsive polymer chains acting as dual ratiometric fluorescent probes under highly dilute conditions.
Co-reporter:Jun Yin, Haibo Hu, Yonghao Wu and Shiyong Liu
Polymer Chemistry 2011 vol. 2(Issue 2) pp:363-371
Publication Date(Web):28 Sep 2010
DOI:10.1039/C0PY00254B
We report on the fabrication of thermo- and light-responsive P(NIPAM-DMNA-NBDAE-RhBEA) microgels consisting of N-isopropylacrylamide (NIPAM), photocleavable moieties, 5-(2′-(dimethylamino)ethoxy)-2-nitrobenzyl acrylate (DMNA), fluorescence resonance energy transfer (FRET) donors, 4-(2-acryloyloxyethylamino)-7-nitro-2,1,3-benzoxadiazole (NBDAE), and rhodamine B-based FRET acceptors (RhBEA) via the free radical emulsion polymerization technique. Thermo-induced collapse and swelling of responsive microgels above and below the lower critical solution temperatures (LCSTs), respectively, can finely tune the spatial proximity between NBDAE and RhBEA dyes, leading to the facile modulation of FRET efficiencies. Upon UV irradiation (pH 8.5), DMNA moieties within microgels undergo photolysis reactions and the formation of sodium carboxylate residues on microgel scaffolds results in elevated LCST. Thus, UV irradiation of microgel dispersions in the intermediate temperature range (between the LCST of original microgels and that of UV-irradiated microgels) can directly lead to the re-swelling of initially collapsed microgels. The incorporation of FRET pairs (NBDAE and RhBEA dyes) allow for the in situ monitoring of thermo- and UV irradiation-induced volume phase transitions (VPTs) of the reported dually responsive microgels. This work represents the first report of thermoresponsive microgels with VPTs tunable by photolabile 2-nitrobenzyl ester moieties.
Co-reporter:Tao Liu and Shiyong Liu
Analytical Chemistry 2011 Volume 83(Issue 7) pp:2775
Publication Date(Web):March 2, 2011
DOI:10.1021/ac200095f
We report on the fabrication of responsive double hydrophilic block copolymers (DHBCs)-based dual fluorescent chemosensors for Zn2+ ions and temperatures and investigate the effects of thermo-induced micellization and detection conditions on the probing sensitivity and binding reversibility of Zn2+ ions. A novel quinoline-based polarity-sensitive and Zn2+-recognizing fluorescent monomer (ZQMA, 6) was synthesized at first. Well-defined DHBCs bearing quinoline-based Zn2+-recognizing moieties (ZQMA) in the thermoresponsive block, PEG-b-P(MEO2MA-co-OEGMA-co-ZQMA), were synthesized via reversible addition−fragmentation chain transfer (RAFT) polymerization of 2-(2-methoxyethoxy)ethyl methacrylate (MEO2MA), oligo(ethylene glycol) monomethyl ether methacrylate (OEGMA), and ZQMA in the presence of PEG-based macroRAFT agent. The OEGMA contents in the thermoresponsive block varied in the range of 0−12.0 mol % to tune their lower critical solution temperatures (LCSTs). At 20 °C, almost nonfluorescent PEG-b-P(MEO2MA-co-ZQMA) molecularly dissolved in water and can selectively bind with Zn2+ ions over other common metal ions, leading to prominent fluorescence enhancement due to the coordination of ZQMA with Zn2+. At a polymer concentration of 0.2 g/L, the Zn2+ detection limit can be down to ∼3.0 nM. PEG-b-P(MEO2MA-co-ZQMA) self-assembles into micelles possessing P(MEO2MA-co-ZQMA) cores and well-solvated PEG coronas upon heating to above the LCST, and the fluorescence intensity exhibit ∼6.0-fold increase due to the fact that ZQMA moieties are now located in a more hydrophobic microenvironment. Compared to the unimer state at 20 °C, although PEG-b-P(MEO2MA-co-ZQMA) micelles possess a slightly decreased detection limit for Zn2+ (∼14 nM), reversible binding between ZQMA moieties and Zn2+ ions at 37 °C can be achieved, as evidenced by the on/off switching of fluorescence emission via the sequential addition of Zn2+ and EDTA. In vitro fluorescence imaging studies suggested that the micelles can effectively enter into living cells and sensitively respond to Zn2+ ions. This work represents the first example of a purely aqueous-based polymeric Zn2+ sensing system by integrating the well-developed small molecule Zn2+-sensing moieties with stimuli-responsive DHBCs.
Co-reporter:Zhishen Ge;Hao Liu;Yanfeng Zhang
Macromolecular Rapid Communications 2011 Volume 32( Issue 1) pp:68-73
Publication Date(Web):
DOI:10.1002/marc.201000367
Co-reporter:Jinming Hu;Changhua Li;Yue Cui
Macromolecular Rapid Communications 2011 Volume 32( Issue 7) pp:610-615
Publication Date(Web):
DOI:10.1002/marc.201100024
Co-reporter:Xuejuan Wan;Guoying Zhang
Macromolecular Rapid Communications 2011 Volume 32( Issue 14) pp:1082-1089
Publication Date(Web):
DOI:10.1002/marc.201100198
Co-reporter:Jingyan Zhang and Shiyong Liu
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 27) pp:12545-12553
Publication Date(Web):13 Jun 2011
DOI:10.1039/C0CP02856H
The kinetics of thermo-induced micelle-to-vesicle transitions in a catanionic surfactant system consisting of sodium dodecyl sulfate (SDS) and dodecyltriethylammonium bromide (DEAB) were investigated by the stopped-flow temperature jump technique, which can achieve T-jumps within ∼2–3 ms. SDS/DEAB aqueous mixtures ([SDS]/[DEAB] = 2/1, 10 mM) undergo microstructural transitions from cylindrical micelles to vesicles when heated above 33 °C. Upon T-jumps from 20 °C to final temperatures in the range of 25–31 °C, relaxation processes associated with negative amplitudes can be ascribed to the dilution-induced structural rearrangement of cylindrical micelles and to the dissolution of non-equilibrium mixed aggregates. In the final temperature range of 33–43 °C the obtained dynamic traces can be fitted by single exponential functions, revealing one relaxation time (τ) in the range of 82–440 s, which decreases with increasing temperature. This may be ascribed to the transformation of floppy bilayer structures into precursor vesicles followed by further growth into final equilibrium vesiclesvia the exchange and insertion/expulsion of surfactant monomers. In the final temperature range of 45–55 °C, vesicles are predominant. Here T-jump relaxations revealed a distinctly different kinetic behavior. All dynamic traces can only be fitted with double exponential functions, yielding two relaxation times (τ1 and τ2), exhibiting a considerable decrease with increasing final temperatures. The fast process (τ1 ∼ 5.2–28.5 s) should be assigned to the formation of non-equilibrium precursor vesicles, and the slow process (τ2 ∼ 188–694 s) should be ascribed to their further growth into final equilibrium vesiclesvia the fusion/fission of precursor vesicles. In contrast, the reverse vesicle-to-micelle transition process induced by a negative T-jump from elevated temperatures to 20 °C occurs quite fast and almost completes within the stopped-flow dead time (∼2–3 ms).
Co-reporter:Xuejuan Wan, Tao Liu, and Shiyong Liu
Langmuir 2011 Volume 27(Issue 7) pp:4082-4090
Publication Date(Web):March 2, 2011
DOI:10.1021/la104911r
We report on the fabrication of core cross-linked (CCL) micelles possessing thermoresponsive cores and their application as sensitive and selective ratiometric Hg2+ probes with thermo-tunable detection efficiency. Well-defined double hydrophilic block copolymer (DHBC) bearing naphthalimide-based Hg2+-reactive moieties (NUMA, 4), PEO-b-P(NIPAM-co-NAS-co-NUMA), was synthesized via reversible addition−fragmentation chain transfer (RAFT) polymerization, where PEO, NIPAM, and NAS represent poly(ethylene oxide), N-isopropylacrylamide, and N-acryloxysuccinimide. At 25 °C, PEO-b-P(NIPAM-co-NAS-co-NUMA) unimers in aqueous solution can act as ratiometric Hg2+ probes with a detection limit of ∼10.1 nM. After core cross-linking of the micellar nanoparticles formed at elevated temperatures, structurally stable CCL micelles with well-solvated PEO coronas and thermoresponsive cores embedded with Hg2+-reactive NUMA moieties were obtained. Upon Hg2+ addition, the aqueous dispersion of CCL micelles exhibit a colorimetric transition from yellowish to colorless and a fluorometric emission transition from green to bright blue. Moreover, Hg2+ detection limits of CCL micelles were considerably enhanced to 3.0 and 1.8 nM at 25 and 40 °C, when the thermoresponsive cores are at their swollen and collapsed state, respectively. The high selectivity of CCL micelles to Hg2+ over other common cations was also demonstrated. Furthermore, in vitro studies revealed that CCL micelles can effectively enter into living cells and sensitively respond to the presence of Hg2+ ions via the change of fluorescence emission color. This work represents the first example of DHBC-based CCL micelle acting as highly selective and sensitive ratiometric metal ion probes. The structural stability, water dispersibility, biocompatibility, and most importantly the thermo-tunable detection sensitivity of this novel type of CCL micelle-based sensing systems augur well for their future applications as multifunctional nanocarriers for drug delivery, sensing, imaging, and diagnosis.
Co-reporter:Jinming Hu, Guoying Zhang, Yanhou Geng, and Shiyong Liu
Macromolecules 2011 Volume 44(Issue 20) pp:8207-8214
Publication Date(Web):September 19, 2011
DOI:10.1021/ma201777p
We report on the fabrication of a novel type of ratiometric fluorescent polymeric probes for fluoride ions (F–) based on self-assembled micellar nanoparticles of P(MMA-co-NBDAE)-b-PF-b-P(MMA-co-NBDAE) coil–rod–coil triblock copolymer, where MMA, NBDAE, and PF are methyl methacrylate, 4-(2-acryloyloxyethylamino)-7-nitro-2,1,3-benzoxadiazole, and polyfluorene, respectively. Blue-emitting conjugated PF block and green-emitting NBDAE moieties with F– turn-off characteristics within the PMMA block serve as fluorescence resonance energy transfer (FRET) donors and switchable acceptors, respectively. For coil–rod–coil triblock copolymer in a good solvent such as THF, the blue emission of PF block dominates due to unimolecularly dissolved state associated with ineffective FRET process. The addition of F– ions only leads to ∼2.92-fold decrease of fluorescence intensity ratio, I515/I417, of characteristic NBDAE and PF emission bands. In acetone, the triblock copolymer spontaneously self-assembles into micelles possessing PF cores and NBDAE-labeled PMMA coronas. In the absence of F– ions, effective FRET processes between micellar cores and coronas occurs, resulting in prominently enhanced NBDAE emission. Upon addition of F– ions, the quenching of NBDAE emission bands leads to ∼8.75-fold decrease in the emission intensity ratio, I515/I417, which is also accompanied by naked eye-discernible fluorometric transition from cyan to blue emissions and colorimetric transition from green to yellowish. At a micellar concentration of 0.1 g/L in acetone at 25 °C, the detection limit of F– ions can be down to ∼4.78 μM (∼0.09 ppm). This work presents a new example of polymeric micelles-based optical F– probes and manifests that, upon proper structural design and optimization of spatial distribution of FRET donors and acceptors, self-assembled micelles of coil–rod–coil triblock copolymers serve as better ratiometric fluorescent F– ion sensors possessing visual detection capability, as compared to that of molecularly dissolved chains.
Co-reporter:Xuejuan Wan, Tao Liu, and Shiyong Liu
Biomacromolecules 2011 Volume 12(Issue 4) pp:
Publication Date(Web):February 18, 2011
DOI:10.1021/bm101463d
We report on the facile synthesis of well-defined amphiphilic and thermoresponsive tadpole-shaped linear-cyclic diblock copolymers via ring-opening polymerization (ROP) directly initiating from cyclic precursors, their self-assembling behavior in aqueous solution, and the application of micellar assemblies as controlled release drug nanocarriers. Starting from a trifunctional core molecule containing alkynyl, hydroxyl, and bromine moieties, alkynyl-(OH)-Br, macrocyclic poly(N-isopropylacrylamide) (c-PNIPAM) bearing a single hydroxyl functionality was prepared by atom transfer radical polymerization (ATRP), the subsequent end group transformation into azide functionality, and finally the intramacromolecular ring closure reaction via click chemistry. The target amphiphilic tadpole-shaped linear-cyclic diblock copolymer, (c-PNIPAM)-b-PCL, was then synthesized via the ROP of ε-caprolactone (CL) by directly initiating from the cyclic precursor. In aqueous solution at 20 °C, (c-PNIPAM)-b-PCL self-assembles into spherical micelles consisting of hydrophobic PCL cores and well-solvated coronas of cyclic PNIPAM segments. For comparison, linear diblock copolymer with comparable molecular weight and composition, (l-PNIPAM)-b-PCL, was also synthesized. It was found that the thermoresponsive coronas of micelles self-assembled from (c-PNIPAM)-b-PCL exhibit thermoinduced collapse and aggregation at a lower critical thermal phase transition temperature (Tc) compared with those of (l-PNIPAM)-b-PCL. Temperature-dependent drug release profiles from the two types of micelles of (c-PNIPAM)-b-PCL and (l-PNIPAM)-b-PCL loaded with doxorubicin (Dox) were measured, and the underlying mechanism for the observed difference in releasing properties was proposed. Moreover, MTT assays revealed that micelles of (c-PNIPAM)-b-PCL are almost noncytotoxic up to a concentration of 1.0 g/L, whereas at the same polymer concentration, micelles loaded with Dox lead to ∼60% cell death. Overall, chain topologies of thermoresponsive block copolymers, that is, (c-PNIPAM)-b-PCL versus (l-PNIPAM)-b-PCL, play considerable effects on the self-assembling and thermal phase transition properties and their functions as controlled release drug nanocarriers.
Co-reporter:Changhua Li, Jinming Hu, Tao Liu, and Shiyong Liu
Macromolecules 2011 Volume 44(Issue 3) pp:429-431
Publication Date(Web):January 12, 2011
DOI:10.1021/ma102608a
Co-reporter:Xiaojie Li, Yinfeng Qian, Tao Liu, Xianglong Hu, Guoying Zhang, Yezi You, Shiyong Liu
Biomaterials 2011 32(27) pp: 6595-6605
Publication Date(Web):
DOI:10.1016/j.biomaterials.2011.05.049
Co-reporter:Yanyan Jiang, Xianglong Hu, Jinming Hu, Hao Liu, Hui Zhong, and Shiyong Liu
Macromolecules 2011 Volume 44(Issue 22) pp:8780-8790
Publication Date(Web):October 25, 2011
DOI:10.1021/ma2018588
We report on the fabrication of a novel type of responsive double hydrophilic block copolymer (DHBC)-based highly selective and sensitive fluorescence “turn-on” reactive probes for fluoride ions (F–) working in purely aqueous media by exploiting F–-induced cyclization reaction of nonfluorescent moieties to induce the formation of fluorescent coumarin moieties, as inspired by the previous work of the Swager research group ( Angew. Chem. Int. Ed. 2003, 42, 4803). Diblock copolymers bearing F–-reactive moieties (SiCouMA) in the thermoresponsive block, PEO-b-P(MEO2MA-co-OEGMA-co-SiCouMA), were synthesized at first via reversible addition–fragmentation chain transfer (RAFT) technique followed by postmodification, where PEO, MEO2MA, and OEGMA are poly(ethylene glycol), di(ethylene glycol) monomethyl ether methacrylate, and oligo(ethylene glycol) monomethyl ether methacrylate, respectively. As-synthesized diblock copolymers molecularly dissolve in water at room temperature and self-assemble into micellar nanoparticles above the critical micellization temperature (33 °C). In the presence of F– ions, deprotection of nonfluorescent SiCouMA moieties followed by spontaneous cyclization reaction leads to the formation of highly fluorescent coumarin residues (CouMA). Thus, PEO-b-P(MEO2MA-co-OEGMA-co- SiCouMA) diblock copolymers can serve as highly efficient and selective fluorescence “turn-on” reaction probes for F– ions in aqueous media. In the range of 0–1600 equiv of F– ions, diblock unimers and micellar solutions at 20 and 40 °C exhibit ∼88-fold and ∼30-fold increase in fluorescence emission intensity (20 min incubation time), respectively. The detection limits were determined to be 0.065 and 0.05 ppm for diblock unimers and micelles, respectively. Most importantly, in the low F– concentration range, excellent linear correlation between F– concentration and emission intensity was observed (0–15 ppm for unimers at 20 °C and 0–8 ppm for micelles at 40 °C). Interestingly, upon complete transformation of nonfluorescent SiCouMA moieties into fluorescent CouMA, the emission intensity of diblock copolymer solution decreases linearly with temperatures in the range of 20–60 °C, suggesting its further application as fluorometric temperature sensors. To the best of our knowledge, this work represents the first example of F–-reactive polymeric probes working in purely aqueous media, which are capable of highly sensitive and selective fluorescent F– sensing in the form of both unimers and micellar nanoparticles.
Co-reporter:Zhishen Ge, Yueming Zhou, Zhen Tong, and Shiyong Liu
Langmuir 2011 Volume 27(Issue 3) pp:1143-1151
Publication Date(Web):January 10, 2011
DOI:10.1021/la1048166
A series of thermoresponsive double hydrophilic (AB)n multiblock and ABA triblock copolymers of N,N-dimethylacrylamide (DMA) and N-isopropylacrylamide (NIPAM) with varying sequence lengths were synthesized via successive reversible addition−fragmentation chain transfer (RAFT) polymerizations by employing polytrithiocarbonate as the chain transfer agent. Previously, we reported that multiblock copolymers in dilute aqueous solutions can form either unimolecular or multimolecular micelles at elevated temperatures depending on the relative chain lengths of PDMA and PNIPAM sequences (Zhou et al. Langmuir 2007, 23, 13076−13084). In this follow-up work, we further explored and compared the chain architectural (multiblock vs triblock) and Hofmeister effects (addition of various sodium salts) on the gelation behavior of multiblock and ABA triblock copolymers at high concentrations and attempted to establish a correlation between the aggregation behavior and gelation properties of multiblock copolymers at low and high polymer concentrations, respectively. It was found that only m-PDMAp−PNIPAMq multiblock copolymers with PDMA and PNIPAM sequence lengths located within a specific range can form physical gels at elevated temperatures. Rheology measurements revealed that multiblock copolymers possess considerably lower critical gelation temperatures (CGT) and higher gel storage modulus, G′gel, as compared to those of PNIPAM-b-PDMA-b-PNIPAM triblock copolymers possessing comparable sequence lengths. The addition of inorganic sodium salts can effectively facilitate thermogelling for multiblock and triblock copolymers, resulting in decreasing CGTs and critical gelation concentrations (CGCs) in the order of Hofmeister series with increasing hydration capabilities. The unique thermogelling behavior of aqueous multiblock copolymer solutions in the absence and presence of inorganic salts, as compared to that of ABA triblock copolymers, augurs well for their potential applications in various fields such as biomaterials and biomedicines.
Co-reporter:Di Wang, Tao Liu, Jun Yin, and Shiyong Liu
Macromolecules 2011 Volume 44(Issue 7) pp:2282-2290
Publication Date(Web):March 11, 2011
DOI:10.1021/ma200053a
We report on the fabrication of multifunctional ratiometric probes for glucose and temperatures based on thermoresponsive poly(N-isopropylacrylamide) (PNIPAM) microgels covalently incorporated with glucose-recognizing moieties, N-acryloyl-3-aminophenylboronic acid (APBA), fluorescence resonance energy transfer (FRET) donor dyes, 4-(2-acryloyloxyethylamino)-7-nitro-2,1,3-benzoxadiazole (NBDAE), and rhodamine B-based FRET acceptors (RhBEA). P(NIPAM-APBA-NBDAE-RhBEA) microgels containing FRET pairs and APBA were synthesized via free radical emulsion copolymerization. The spatial proximity of FRET donors and acceptors within microgels can be tuned via thermo-induced microgel collapse or glucose-induced microgel swelling at appropriate pH and temperatures, leading to the facile modulation of FRET efficiencies. APBA moieties within P(NIPAM-APBA-NBDAE-RhBEA) microgels can bind with glucose at appropriate pH to form cyclic boronate moieties, which can decrease the pKa of APBA residues and increase the volume phase transition (VPT) temperature of microgels. The gradual addition of glucose into fluorescent microgel dispersions at intermediate temperatures, i.e., between microgel VPT temperatures in the absence and presence of glucose, respectively, can lead to the reswelling of initially collapsed microgels. Thus, P(NIPAM-APBA-NBDAE-RhBEA) microgels can serve as dual ratiometric fluorescent probes for glucose and temperatures by monitoring the changes in fluorescence emission intensity ratios. Moreover, P(NIPAM-APBA-NBDAE-RhBEA) microgels at pH 8 and 37 °C can serve as a ratiometric fluorescent glucose sensor with improved detection sensitivity as compared to that at 25 °C. MTT assays further revealed that thermoresponsive microgels are almost noncytotoxic up to a concentration of 1.6 g/L. These results augur well for the application of P(NIPAM-APBA-NBDAE-RhBEA) microgels for multifunctional purposes such as sensing, imaging, and triggered-release nanocarriers under in vivo conditions.
Co-reporter:Jinming Hu, Lu Dai, and Shiyong Liu
Macromolecules 2011 Volume 44(Issue 12) pp:4699-4710
Publication Date(Web):May 31, 2011
DOI:10.1021/ma2001146
We report on the fabrication of amphiphilic thermoresponsive diblock copolymer micelle-based multifunctional ratiometric fluorescent chemosensors for metal ions (Hg2+ and Cu2+), pH, and temperatures. A fluorescence resonance energy transfer (FRET) pair consisting of 4-(2-acryloyloxyethylamino)-7-nitro-2,1,3-benzoxadiazole (NBDAE) donor and rhodamine B-based potential acceptor (RhBHA) in the spirolactam form with pH and Hg2+(Cu2+)-reactive characteristics were respectively copolymerized into the hydrophobic PS and thermoresponsive PNIPAM block of P(St-co-NBDAE)-b-P(NIPAM-co-RhBHA) amphiphilic diblock copolymers, where PS and PNIPAM represent polystyrene and poly(N-isopropylacrylamide). In aqueous solution, the FRET pair-labeled diblock copolymer self-assembles into nanosized micelles with NBDAE moieties located in the micellar cores and RhBHA in the thermoresponsive coronas. Because of that Hg2+ ions and acidic pH can induce the transformation of RhBHA from the nonfluorescent spirolactam form to highly fluorescent acyclic form, and the FRET process between NBDAE and RhBHA moieties, located respectively within micellar cores and coronas, can be effectively switched on. Thus, these nanosized micelles can serve as excellent ratiometric fluorescent probes for Hg2+ ions and pH, accompanied by fluorometric transition from green to orange and colorimetric change from almost colorless to pink. At a micellar concentration of 0.05 g/L and 25 °C, the detection limit of Hg2+ ions can be down to ∼14.8 ppb. On the other hand, Cu2+ ions can quantitatively induce the ring-opening of RhBHA moieties and afford nonfluorescent residues, which can effectively quench the NBDAE emission. On the basis of the relative changes in NBDAE emission intensities, the Cu2+ detection limit can be down to ∼4.3 ppb. Most importantly, the spatial distance of the FRET pair can be facilely tuned via thermo-induced collapse of PNIPAM micellar coronas, which dramatically increase the FRET efficiency and enhance the pH detection sensitivity. This work represents a proof-of-concept example of amphiphilic block copolymer micelles-based multifunctional ratiometric fluorescent probes for two types of metal ions (Hg2+ and Cu2+), pH, and temperatures, which augurs well for their potential applications as nanocarriers with integrated functions such as imaging, sensing, and controlled-release of therapeutics.
Co-reporter:Dr. Xuejuan Wan; Guoying Zhang; Zhishen Ge; Ravin Narain; Shiyong Liu
Chemistry – An Asian Journal 2011 Volume 6( Issue 10) pp:2835-2845
Publication Date(Web):
DOI:10.1002/asia.201100489
Abstract
We report on the fabrication of well-defined polymer–protein bioconjugates with varying chain architectures, including star polymers, star block copolymers, and heteroarm star copolymers through the specific noncovalent interaction between avidin and biotinylated synthetic polymer precursors. Homopolymer and diblock precursors site-specifically labeled with a single biotin moiety at the chain terminal, chain middle, or diblock junction point were synthesized by a combination of atom-transfer radical polymerization (ATRP) and click reactions. By taking advantage of molecular recognition between avidin and biotin moieties, supramolecular star polymers, star block copolymers, and heteroarm star copolymers were successfully fabricated. This specific binding process was also assessed by using the diffraction optic technology (DOT) technique. We further investigated the effects of polymer molecular weights, location of biotin functionality within the polymer chain, and polymer chain conformations, that is, steric hindrance effects, on the binding numbers of biotinylated polymer chains per avidin within the polymer–protein bioconjugates, which were determined by the standard avidin/2-(4-hydroxyazobenzene)benzoic acid (HABA) assay. The binding numbers vary in the range of 1.9–3.3, depending on the molecular weights, locations of biotin functionality within synthetic polymer precursors, and polymer chain conformations.
Co-reporter:Changhua Li and Shiyong Liu
Journal of Materials Chemistry A 2010 vol. 20(Issue 47) pp:10716-10723
Publication Date(Web):24 Sep 2010
DOI:10.1039/C0JM01828G
We report on the fabrication of thermoresponsive poly(N-isopropylacrylamide) nanogel-based dual fluorescent sensors for temperature and Hg2+ ions, and the effects of thermo-induced nanogel collapse on the detection sensitivity of Hg2+ ions. Near-monodisperse thermoresponsive nanogels were prepared via emulsion polymerization of N-isopropylacrylamide (NIPAM) and a novel 1,8-naphthalimide-based polarity-sensitive and Hg2+-reactive fluorescent monomer (NPTUA, 3). At room temperature, PNIPAM nanogels labeled with a single type of naphthalimide-based dye (NPTUA) can act as ratiometric Hg2+ probes at the nanomolar level. Upon heating above the phase transition temperature, the fluorescence intensity of NPTUA-labeled nanogels in the absence of Hg2+ exhibit ∼3.4-fold increase due to that NPTUA moieties are now located in a more hydrophobic microenvironment. Moreover, it was observed that the detection sensitivity to Hg2+ can be further improved above the nanogel phase transition temperature. At a nanogel concentration of 0.05 g L−1 and in the same Hg2+ concentration range (0–3.0 equiv.), ∼10 fold and ∼57 fold increase in fluorescence emission intensity ratio changes can be achieved at 25 and 40 °C, respectively.
Co-reporter:Cong Liu, Kaka Zhang, Daoyong Chen, Ming Jiang and Shiyong Liu
Chemical Communications 2010 vol. 46(Issue 33) pp:6135-6137
Publication Date(Web):27 Jul 2010
DOI:10.1039/C0CC00902D
The deliberately prepared one ssDNA/one micelle complex has an unstable toroidal DNA-bound region and stable upper and lower hemispheres, and thus can self-assemble along the plane of the unstable toroidal region into free-suspending films.
Co-reporter:Xuejuan Wan
Macromolecular Rapid Communications 2010 Volume 31( Issue 23) pp:2070-2076
Publication Date(Web):
DOI:10.1002/marc.201000340
Co-reporter:Jinming Hu;Di Wang;Jian Xu;Zhiyuan Zhu
Macromolecular Chemistry and Physics 2010 Volume 211( Issue 24) pp:2573-2584
Publication Date(Web):
DOI:10.1002/macp.201000476
Co-reporter:Jinming Hu and Shiyong Liu
Macromolecules 2010 Volume 43(Issue 20) pp:8315-8330
Publication Date(Web):September 17, 2010
DOI:10.1021/ma1005815
The past two decades have evidenced a tremendous growth in the field of responsive polymers, which can exhibit reversible or irreversible changes in physical properties and/or chemical structures to an external stimulus such as pH, temperature, ionic strength, light irradiation, mechanical forces, electric and magnetic fields, specific analytes, external additives (ions, bioactive molecules, etc.), or a combination of them. Responsive polymers can exist in the form of solutions, gels, self-assembled nanoparticles, (multilayer) films, and bulk solids. The field of responsive polymers has nowadays evolved well beyond the demonstration of novel and interesting properties. Currently, the exploitation of useful and advanced functions, e.g., drug or gene carriers with triggered release properties, catalysis, detection and imaging, environmentally adaptive coatings, and self-healing materials, has emerged to be a more relevant subject. In this Perspective, we focus on recent developments of responsive polymer-based chemo- and biosensors, highlighting this concept with selected literature reports. Such functional polymeric materials show prominent advantages such as tunable detection sensitivity, structural stability, aqueous dispersibility, biocompatibility, processability, and facile integration into detection devices, as compared to their small molecule analogues.
Co-reporter:XueJuan Wan;Jian Xu
Science China Chemistry 2010 Volume 53( Issue 12) pp:2520-2527
Publication Date(Web):2010 December
DOI:10.1007/s11426-010-4135-4
We report a facile synthesis method of dendrimer-like star-branched poly(N-isopropylacrylamide) (PNIPAM) via the combination of click chemistry and atom transfer radical polymerization (ATRP) by employing the arm-first approach. First, the α-azido-ω-chloro-heterodifunctionalized building block, N3-PNIPAM-Cl (G0-Cl), was synthesized via ATRP by 3-azidopropyl 2-chloropropionate as the initiator. Taking advantage of click chemistry, the first generation (G1) of dendrimer-like star-branched PNIPAM, G1-(Cl)3, was facilely prepared via the click coupling reaction between G0-Cl and tripropargylamine. For the construction of second generation (G2) dendrimer-like star-branched PNIPAM, G2-(Cl)6, terminal chloride moieties of G1-(Cl)3 were first converted to azide, and then reacted with excess tripropargylamine to give G1-(alkynyl)6; G2-(Cl)6 was subsequently prepared via click reaction between G1-(alkynyl)6 and G0-Cl. Gel permeation chromatography (GPC) and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry were employed to confirm the successful construction of dendrimer-like star-branched polymers. The unique thermal phase transition behavior of this dendrimer-like star-branched polymer in aqueous solutions was further investigated by turbidimetry and micro-differential scanning calorimetry (Micro-DSC).
Co-reporter:Jinming Hu, Changhua Li and Shiyong Liu
Langmuir 2010 Volume 26(Issue 2) pp:724-729
Publication Date(Web):September 11, 2009
DOI:10.1021/la9024102
We report on novel type of responsive double hydrophilic block copolymer (DHBC)-based multifunctional chemosensors to Hg2+ ions, pH, and temperatures and investigate the effects of thermo-induced micellization on the detection sensitivity. Well-defined DHBCs bearing rhodamine B-based Hg2+-reactive moieties (RhBHA) in the thermo-responsive block, poly(ethylene oxide)-b-poly(N-isopropylacrylamide-co-RhBHA) (PEO-b-P(NIPAM-co-RhBHA)), were synthesized via reversible addition−fragmentation chain transfer (RAFT) polymerization. Nonfluorescent RhBHA moieties are subjected to selective ring-opening reaction upon addition of Hg2+ ions or lowering solution pH, producing highly fluorescent acyclic species. Thus, at room temperature PEO-b-P(NIPAM-co-RhBHA) DHBCs can serve as water-soluble multifunctional and efficient fluorescent chemosensors to Hg2+ ions and pH. Upon heating above the lower critical solution temperature (∼36 °C) of the PNIPAM block, they self-assemble into micelles possessing P(NIPAM-co-RhBHA) cores and well-solvated PEO coronas, which were fully characterized by dynamic and static laser light scattering. It was found that the detection sensitivity to Hg2+ ions and pH could be dramatically improved at elevated temperatures due to fluorescence enhancement of RhBHA residues in the acyclic form, which were embedded within hydrophobic cores of thermo-induced micellar aggregates. This work represents a proof-of-concept example of responsive DHBC-based multifunctional fluorescent chemosensors for the highly efficient detection of Hg2+ ions, pH, and temperatures with tunable detection sensitivity. Compared to reaction-based small molecule Hg2+ probes in previous literature reports, the integration of stimuli-responsive block copolymers with well-developed small molecule-based selective sensing moieties in the current study are expected to exhibit preferred advantages including enhanced detection sensitivity, water dispersibility, biocompatibility, facile incorporation into devices, and the ability of further functionalization for targeted imaging and detection.
Co-reporter:Xuejuan Wan, Di Wang, and Shiyong Liu
Langmuir 2010 Volume 26(Issue 19) pp:15574-15579
Publication Date(Web):September 14, 2010
DOI:10.1021/la102148x
We report on the fabrication of fluorescent pH-sensing organic/inorganic hybrid mesoporous silica nanoparticles (MSN) capable of tunable redox-responsive release of embedded guest molecules. The reversible addition−fragmentation chain transfer (RAFT) copolymerization of N-(acryloxy)succinimide (NAS), oligo(ethylene glycol) monomethyl ether methacrylate (OEGMA), and 1,8-naphthalimide-based pH-sensing monomer (NaphMA) at the surface of MSN led to fluorescent organic/inorganic hybrid MSN. The obtained hybrid MSN exhibits excellent water dispersibility and acts as sensitive fluorescent pH probes in the range pH 4−8 due to the presence of NaphMA moieties. After loading with rhodamine B (RhB) as a model drug molecule, P(NAS-co-OEGMA-co- NaphMA) brushes at the surface of hybrid MSN were cross-linked with cystamine to block nanopore entrances for the effective retention of guest molecules. Taking advantage of disulfide-containing cross-linkers, the release rate of RhB can be easily adjusted by adding varying concentrations of dithiothreitol (DTT), which can cleave the disulfide linkage to open blocked nanopores. The increase of DTT concentration from 0 to 20 mM led to 20−30 times enhancement of RhB release rate. The reported multifunctional hybrid MSN augurs well for applications in controlled-release nanocarriers, cell and tissue imaging, and clinical diagnosis.
Co-reporter:Jun Yin, Changhua Li, Di Wang, and Shiyong Liu
The Journal of Physical Chemistry B 2010 Volume 114(Issue 38) pp:12213-12220
Publication Date(Web):September 8, 2010
DOI:10.1021/jp1052369
We report on the fabrication of ratiometric fluorescent K+ sensors based on thermoresponsive poly(N-isopropylacrylamide) (PNIPAM) microgels covalently incorporated with K+-recognizing 4-acrylamidobenzo-18-crown-6 residues (B18C6Am), fluorescence resonance energy transfer (FRET) donor dyes, 4-(2-acryloyloxyethylamino)-7-nitro-2,1,3-benzoxadiazole (NBDAE), and rhodamine-B-based FRET acceptors (RhBEA) by utilizing K+-induced changes in microgel volume phase transition (VPT) temperatures. P(NIPAM-B18C6Am-NBDAE-RhBEA) microgels were synthesized via the free radical emulsion copolymerization technique. The spatial proximity between FRET pairs (NBDAE and RhBEA dyes) within microgels can be tuned via thermoinduced collapse and swelling of thermoresponsive microgels above and below VPT temperatures, leading to the facile modulation of FRET efficiencies. B18C6Am moieties within P(NIPAM-B18C6Am-NBDAE-RhBEA) microgels can preferentially capture K+ via the formation of 1:1 molecular recognition complexes, resulting in the enhancement of microgel hydrophilicity and elevated VPT temperatures. Thus, the gradual addition of K+ into microgel dispersions at intermediate temperatures, i.e., between VPT temperatures of P(NIPAM-B18C6Am-NBDAE-RhBEA) microgels in the absence and presence of K+ ions, respectively, can directly lead to the reswelling of initially collapsed microgels. This process can be monitored by changes in fluorescence intensity ratios, i.e., FRET efficiencies. The presence of FRET pairs within P(NIPAM-B18C6Am-NBDAE-RhBEA) microgels allows for the facile in situ monitoring of thermoinduced and K+-induced VPTs of dually responsive microgels. The response time for fluorescent K+-sensing was further investigated via the stopped-flow technique, which reveals that the process completes within ∼4 s. This work represents the first report of thermoresponsive microgel-based ratiometric fluorescent sensors for both K+ ions and temperatures.
Co-reporter:Jinming Hu, Zhishen Ge, Yueming Zhou, Yanfeng Zhang and Shiyong Liu
Macromolecules 2010 Volume 43(Issue 12) pp:5184-5187
Publication Date(Web):May 25, 2010
DOI:10.1021/ma100813m
Co-reporter:Changhua Li;Yanxi Zhang;Jinming Hu;Jianjun Cheng Dr. Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 30) pp:5120-5124
Publication Date(Web):
DOI:10.1002/anie.201002203
Co-reporter:Tao Liu, Jinming Hu, Jun Yin, Yanfeng Zhang, Changhua Li and Shiyong Liu
Chemistry of Materials 2009 Volume 21(Issue 14) pp:3439
Publication Date(Web):July 1, 2009
DOI:10.1021/cm901070a
We report on the fabrication of responsive microgel-based Cu2+ chemosensors possessing tunable detection sensitivity via thermo-induced microgel collapse/swelling. A novel phenanthroline-containing fluorescent monomer capable of Cu2+-binding and fluorescence sensing, PhenUMA (4), was synthesized at first by reacting 5-amino-1,10-phenanthroline (2) with 2-(3-isocyanato-propionyloxy)ethyl methacrylate (3). Near-monodisperse Cu2+-sensing microgels were synthesized via emulsion polymerization of N-isopropylacrylamide (NIPAM) in the presence of N,N′-methylenebis(acrylamide) (BIS), an anionic surfactant, and PhenUMA (4) at around neutral pH and 70 °C. At 20 °C, PhenUMA-labeled microgels in their swollen state can selectively bind Cu2+ over other metal ions (Al3+, Mg2+, Zn2+, Fe3+, Mn2+, Ni2+, Ag+, Cd2+, Hg2+, and Pb2+), leading to prominent quenching of fluorescence emission intensity. At a microgel concentration of 0.25 g/L, the Cu2+ detection limit can be down to ∼125 nM. When heated above 32 °C, fluorescence intensity of PhenUMA-labeled microgels in the absence of Cu2+ exhibits an approximately 33% increase due to their volume phase transition, which is reasonable considering that fluorescent PhenUMA moieties are now located in a nonpolar environment. Furthermore, Cu2+ detection sensitivity of PhenUMA-labeled microgels can be dramatically enhanced via thermo-induced microgel collapse at elevated temperatures. At a microgel concentration of 0.083 g/L, detection limits of Cu2+ ions can be drastically improved from ∼28 nM at 20 °C to ∼8 nM at 40 °C. A plausible mechanism for the thermo-induced enhancement of Cu2+ detection sensitivity has been proposed. To the best of our knowledge, this proof-of-concept work represents the first example of responsive microgel-based metal ion chemosensor with thermo-tunable detection sensitivity, which simultaneously combining advantageous properties of small molecule sensing moieties and stimuli-responsive soft matter entities.
Co-reporter:Tao Wu, Gang Zou, Jinming Hu and Shiyong Liu
Chemistry of Materials 2009 Volume 21(Issue 16) pp:3788
Publication Date(Web):July 29, 2009
DOI:10.1021/cm901072g
We report on the fabrication of hybrid silica nanoparticles densely grafted with thermoresponsive poly(N-isopropylacrylamide) (PNIPAM) brushes with inner and outer layers selectively labeled with fluorescence resonance energy transfer (FRET) donors, 4-(2-acryloyloxyethylamino)-7-nitro-2,1,3-benzoxadiazole (NBDAE), and photoswitchable acceptors, 1′-(2-methacryloxyethyl)-3′,3′-dimethyl-6-nitro-spiro(2H-1-benzo-pyran-2,2′-indoline) (SPMA), respectively, via surface-initiated sequential atom transfer radical polymerization (ATRP). P(NIPAM-co-NBDAE)-b-P(NIPAM-co-SPMA) brushes at the surface of silica core exhibit collapse in the broad temperature range of 20−37 °C. UV irradiation of the aqueous dispersion of hybrid silica nanoparticles induces the transformation of SPMA moieties in the outer layer of polymer brushes from nonfluorescent spiropyran (SP) form to fluorescent merocyanine (MC) form, leading to occurrence of the FRET process between NBDAE and SPMA residues. Most importantly, the FRET efficiency can be facilely tuned via thermo-induced collapse/swelling of P(NIPAM-co-NBDAE)-b-P(NIPAM-co-SPMA) brushes by changing the relative distance between donor and acceptor species located within the inner and outer layers of polymer brushes, respectively. Thus, hybrid silica nanoparticles coated with P(NIPAM-co-NBDAE)-b-P(NIPAM-co-SPMA) brushes can serve as a sensitive ratiometric fluorescent thermometer. On the other hand, when the hybrid nanoparticle dispersion was irradiated with visible light again after UV irradiation, the MC form of SPMA moieties reverts back to the nonfluorescent SP form, leading to the turn-off of FRET process. Overall, aqueous dispersion of this novel type of hybrid silica nanoparticle is capable of emitting multicolor fluorescence, which can be facilely tuned by UV irradiation, visible light, and temperatures or a proper combination of these factors.
Co-reporter:Yanfeng Zhang;Hao Liu;Jinming Hu;Changhua Li
Macromolecular Rapid Communications 2009 Volume 30( Issue 11) pp:941-947
Publication Date(Web):
DOI:10.1002/marc.200800820
Co-reporter:Zhishen Ge
Macromolecular Rapid Communications 2009 Volume 30( Issue 18) pp:1523-1532
Publication Date(Web):
DOI:10.1002/marc.200900182
Co-reporter:Zhishen Ge, Jian Xu, Jinming Hu, Yanfeng Zhang and Shiyong Liu
Soft Matter 2009 vol. 5(Issue 20) pp:3932-3939
Publication Date(Web):31 Jul 2009
DOI:10.1039/B907906H
We report on the synthesis and stimuli-responsive self-assembly of novel double hydrophilic Janus-type A7B14 heteroarm star copolymers with two types of water-soluble polymer arms emanating from the two opposing sides of the rigid toroidal β-CD core. Janus-type A7B14 star copolymers of N-isopropylacrylamide (NIPAM) and 2-(diethylamino)ethyl methacrylate (DEA), (PDEA)7-CD-(PNIPAM)14, were synthesized by coupling atom transfer radical polymerization (ATRP) and click chemistry techniques, starting from a well-defined β-CD derivative. At first, β-CD-(I)7 was obtained by reacting β-CD with I2 in the presence of PPh3 at 70 °C, which can selectively transform seven primary hydroxyl groups of β-CD into iodine moieties. After converting to β-CD-(N3)7viaazidation of β-CD-(I)7, the subsequent esterification reaction of β-CD-(N3)7 with 2-bromopropionic bromide afforded (N3)7-CD-(Br)14. The ATRP of NIPAM monomer in 2-propanol/DMF mixture at 25 °C using (N3)7-CD-(Br)14 as the multifunctional initiator led to azide-containing 14-arm star polymers, (N3)7-CD-(PNIPAM)14. Well-defined Janus-type double hydrophilic star copolymers were then prepared by the click reaction of (N3)7-CD-(PNIPAM)14 with an excess of monoalkynyl-terminated PDEA. Upon adjusting solution pH and temperatures, (PDEA30)7-CD-(PNIPAM25)14 can reversibly self-assemble into two distinct types of polymeric vesicles with “inverted” nanostructures in aqueous solution.
Co-reporter:Xiaoze Jiang, Guoying Zhang, Ravin Narain and Shiyong Liu
Soft Matter 2009 vol. 5(Issue 7) pp:1530-1538
Publication Date(Web):13 Feb 2009
DOI:10.1039/B819396G
Alkynyl-terminated double hydrophilic ABC triblock copolymer, poly(oligo(ethylene glycol) monomethyl ether methacrylate)-b-poly(2-(dimethylamino) ethyl methacrylate)-b-poly(2-(diethylamino) ethyl methacrylate) (alkynyl-POEGMA-b-PDMA-b-PDEA), was synthesized via atom transfer radical polymerization (ATRP) by sequential monomer addition using propargyl 2-bromoisobutyrate (PgBiB) as the initiator. The obtained triblock copolymer dissolves molecularly in acidic media and self-assembles into alkynyl surface-functionalized three-layer “onion-like” micelles consisting of a PDEA core, a PDMA inner shell, and a POEGMA corona at alkaline pH. Selective cross-linking of the PDMA inner shell with 2-bis(2-iodoethoxy)ethane (BIEE) results in structurally stable and surface ‘clickable’ shell cross-linked (SCL) micelles with pH-responsive PDEA cores. This new kind of SCL micelles could be further surface functionalized or conjugated with other azido- terminated polymer chains, functional groups, or biomolecules via Click chemistry. Four layer nanoparticles (SCL-PNIPAM) which have pH-responsive PDEA cores and temperature responsive PNIPAM outer coronas were fabricated from surface ‘clickable’ shell cross-linked (SCL) micelles and azide-terminated poly(N-isopropylacrylamide) (PNIPAM-N3) using Click chemistry. These novel four layer nanoparticles might act as suitable nano-sized drug delivery vehicles for the encapsulation and release of hydrophobic drugs as a function of either temperature or pH of the environment
Co-reporter:Hao Liu;Yanfeng Zhang;Jinming Hu;Changhua Li
Macromolecular Chemistry and Physics 2009 Volume 210( Issue 24) pp:2125-2137
Publication Date(Web):
DOI:10.1002/macp.200900279
Co-reporter:Zhishen Ge, Di Wang, Yueming Zhou, Hewen Liu and Shiyong Liu
Macromolecules 2009 Volume 42(Issue 8) pp:2903-2910
Publication Date(Web):March 31, 2009
DOI:10.1021/ma802585k
We report the synthesis of quatrefoil-shaped star-cyclic polystyrene, star-cyclic PS, containing a polyhedral oligomeric silsesquioxane (POSS) core via the combination of atom transfer radical polymerization (ATRP) and click chemistry techniques. The obtained star-cyclic PS represents a new chain topology in the category of nonlinear-shaped polymers. Using octa(3-chloropropyl) polyhedral oligomeric silsesquioxane, POSS-(Cl)8, as the starting material, its azidation and subsequent click reaction with a slight excess of propargyl 2-bromobutyrate afforded octafunctional initiator, POSS-(Br)8. 8-arm star-linear PS-N3 was obtained by the azidation of star-linear PS-Br, which was synthesized by the ATRP of styrene using POSS-(Br)8 as the initiator. Model reaction between α,ω-diazido-terminated PS (N3-PS-N3) and difunctional propargyl ether confirmed that bimolecular click cyclization reaction can effectively occur under highly dilute conditions. Next, intramolecular click ring closure of star-linear PS-N3 was conducted under highly dilute conditions, using propargyl ether as the difunctional linker and CuBr/PMDETA as the catalyst, affording quatrefoil-shaped star-cyclic PS. Gel permeation chromatography (GPC), 1H NMR, and FT-IR analysis confirmed the complete consumption of azide moieties in star-linear PS-N3 and that the coupling reaction proceeded via the intramolecular manner. Differential scanning calorimetry (DSC) results revealed that star-cyclic PS possesses higher glass transition temperature (Tg) than that of star-linear PS, possibly due to the ring topology of PS arms in the former.
Co-reporter:Changhua Li, Jinming Hu, Jun Yin and Shiyong Liu
Macromolecules 2009 Volume 42(Issue 14) pp:5007-5016
Publication Date(Web):May 21, 2009
DOI:10.1021/ma900788k
We report on the synthesis of well-defined thermoresponsive water-soluble diblock copolymer and homopolymers functionalized with controlled numbers of C60 moieties at predetermined positions via the combination of atom transfer radical polymerization (ATRP) and click chemistry. Azide-containing polymer precursors including monoazide-terminated and α,α-diazide-terminated poly(N-isopropylacrylamide), N3- PNIPAM and (N3)2-PNIPAM, as well as poly(ethylene glycol)-b-PNIPAM with one azide moiety at the diblock junction, PEG(-N3)-b-PNIPAM, were synthesized via ATRP using specific azide-functionalized small molecule and polymeric initiators. On the other hand, the reaction of 4-prop-2-ynyloxybenzaldehyde with pristine C60 in the presence of glycine afforded alkynyl-modified C60, alkynyl-C60. Subsequently, the click reaction of N3-PNIPAM, (N3)2-PNIPAM, and PEG(-N3)-b-PNIPAM led to the facile preparation of thermoresponsive diblock copolymer and homopolymers functionalized with controlled numbers of C60 at designed positions, including C60-PNIPAM, (C60)2-PNIPAM, and PEG(-C60)-b-PNIPAM. All the intermediate and final products were characterized by 1H NMR, Fourier transform infrared spectroscopy (FT-IR), UV−vis spectroscopy, thermogravimetric analysis (TGA), and gel permeation chromatograph (GPC) equipped with UV/RI dual detectors. C60-containing hybrid nanoparticles were then fabricated via supramolecular self-assembly of C60-PNIPAM, (C60)2-PNIPAM, and PEG(-C60)-b-PNIPAM in aqueous solution, which were characterized by dynamic and static laser light scattering (LLS) and transmission electron microscopy (TEM). These novel fullerenated polymers retain the thermoresponsiveness of PNIPAM-based precursors, and self-assembled hybrid nanoparticles exhibit thermo-induced collapse/aggregation behavior due to the lower critical solution temperature (LCST) phase transition of PNIPAM chains.
Co-reporter:Jian Xu
Journal of Polymer Science Part A: Polymer Chemistry 2009 Volume 47( Issue 2) pp:404-419
Publication Date(Web):
DOI:10.1002/pola.23157
Abstract
The syntheses of well-defined 7-arm and 21-arm poly(N-isopropylacrylamide) (PNIPAM) star polymers possessing β-cyclodextrin (β-CD) cores were achieved via the combination of atom transfer radical polymerization (ATRP) and click reactions. Heptakis(6-deoxy-6-azido)-β-cyclodextrin and heptakis[2,3,6-tri-O-(2-azidopropionyl)]-β-cyclodextrin, β-CD-(N3)7 and β-CD-(N3)21, precursors were prepared and thoroughly characterized by nuclear magnetic resonance and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. A series of alkynyl terminally functionalized PNIPAM (alkyne-PNIPAM) linear precursors with varying degrees of polymerization (DP) were synthesized via atom transfer radical polymerization (ATRP) of N-isopropylacrylamide using propargyl 2-chloropropionate as the initiator. The subsequent click reactions of alkyne-PNIPAM with β-CD-(N3)7 and β-CD-(N3)21 led to the facile preparation of well-defined 7-arm and 21-arm star polymers, namely β-CD-(PNIPAM)7 and β-CD-(PNIPAM)21. The thermal phase transition behavior of 7-arm and 21-arm star polymers with varying molecular weights were examined by temperature-dependent turbidity and micro-differential scanning calorimetry, and the results were compared to those of linear PNIPAM precursors. The anchoring of PNIPAM chain terminal to β-CD cores and high local chain density for star polymers contributed to their considerably lower critical phase separation temperatures (Tc) and enthalpy changes during phase transition as compared with that of linear precursors. © 2008 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 47: 404–419, 2009
Co-reporter:Yanfeng Zhang;Changhua Li
Journal of Polymer Science Part A: Polymer Chemistry 2009 Volume 47( Issue 12) pp:3066-3077
Publication Date(Web):
DOI:10.1002/pola.23388
Abstract
We report on the one-pot synthesis of well-defined ABC miktoarm star terpolymers consisting of poly(2-(dimethylamino)ethyl methacrylate), poly(ε-caprolactone), and polystyrene or poly(ethylene oxide) arms, PS(-b-PCL)-b-PDMA and PEO (-b-PCL)-b-PDMA, taking advantage of the compatibility and mutual tolerability of reaction conditions (catalysts and monomers) employed for atom transfer radical polymerization (ATRP), ring-opening polymerization (ROP), and click reactions. At first, a novel trifunctional core molecule bearing alkynyl, hydroxyl group, and bromine moieties, alkynyl(OH)Br, was synthesized via the esterification reaction of 5-ethyl-5-hydroxymethyl-2,2-dimethyl-1,3-dioxane with 4-oxo-4-(prop-2-ynyloxy)butanoic acid, followed by deprotection and monoesterification of alkynyl(OH)2 with 2-bromoisobutyryl bromide. In the presence of trifunctional core molecule, alkynyl(OH)Br, and CuBr/PMDETA/Sn(Oct)2 catalytic mixtures, target ABC miktoarm star terpolymers, PS(-b-PCL)-b-PDMA and PEO(-b-PCL)-b-PDMA, were successfully synthesized in a one-pot manner by simultaneously conducting the ATRP of 2-(dimethylamino)ethyl methacrylate (DMA), ROP of ε-caprolactone (ε-CL), and the click reaction with azido-terminated PS (PS-N3) or azido-terminated PEO (PEO-N3). Considering the excellent tolerability of ATRP to a variety of monomers and the fast expansion of click chemistry in the design and synthesis of polymeric and biorelated materials, it is quite anticipated that the one-pot concept can be applied to the preparation of well-defined polymeric materials with more complex chain architectures. © 2009 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 47: 3066–3077, 2009
Co-reporter:Jun Yin;Zhishen Ge;Hao Liu
Journal of Polymer Science Part A: Polymer Chemistry 2009 Volume 47( Issue 10) pp:2608-2619
Publication Date(Web):
DOI:10.1002/pola.23346
Abstract
We report on the synthesis of well-defined amphiphilic copolymer brushes possessing alternating poly(methyl methacrylate) and poly(N-isopropylacrylamide) grafts, poly(PMMA-alt-PNIPAM), via a combination of atom transfer radical polymerization (ATRP) and click reaction (Scheme 1). Firstly, the alternating copolymerization of N-[2-(2-bromoisobutyryloxy)ethyl]maleimide (BIBEMI) with 4-vinylbenzyl azide (VBA) affords poly(BIBEMI-alt-VBA). Bearing bromine and azide moieties arranged in an alternating manner, multifunctional poly(BIBEMI-alt-VBA) is capable of initiating ATRP and participating in click reaction. The subsequent ATRP of methyl methacrylate (MMA) using poly(BIBEMI-alt-VBA) as the macroinitiator leads to poly(PMMA-alt-VBA) copolymer brush. Finally, amphiphilic poly(PMMA-alt-PNIPAM) copolymer brush bearing alternating PMMA and PNIPAM grafts is synthesized via the click reaction of poly(PMMA-alt-VBA) with an excess of alkynyl-terminated PNIPAM (alkynyl-PNIPAM). The click coupling efficiency of PNIPAM grafts is determined to be ∼80%. Differential scanning calorimetry (DSC) analysis of poly(PMMA-alt-PNIPAM) reveals two glass transition temperatures (Tg). In aqueous solution, poly(PMMA-alt-PNIPAM) supramolecularly self-assembles into spherical micelles consisting of PMMA cores and thermoresponsive PNIPAM coronas, which were characterized via a combination of temperature-dependent optical transmittance, micro-differential scanning calorimetry (micro-DSC), dynamic and static laser light scattering (LLS), and transmission electron microscopy (TEM). © 2009 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 47: 2608–2619, 2009
Co-reporter:Zhishen Ge;Jinming Hu;Feihe Huang Dr. Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 10) pp:1798-1802
Publication Date(Web):
DOI:10.1002/anie.200805712
Co-reporter:Changhua Li, Zhishen Ge, Jin Fang and Shiyong Liu
Macromolecules 2009 Volume 42(Issue 8) pp:2916-2924
Publication Date(Web):March 25, 2009
DOI:10.1021/ma900165z
We report on the synthesis and self-assembly of well-defined coil−rod double hydrophilic diblock copolymer with pH- and thermo-responsive asymmetric centipede-shaped polymer brush as the rod segment via a combination of atom transfer radical polymerization (ATRP) and click chemistry (Schemes 1 and 2). At first, poly(ethylene oxide)-b-poly(glycidyl methacrylate), PEO-b-PGMA, was prepared by ATRP using PEO-based macroinitiator. The ring-opening of pendent epoxide moieties in PEO-b-PGMA with NaN3 followed by esterification with 2-bromoisobutyryl bromide afforded multifunctional PEO-b-[PGMA-(N3)(Br)] bearing one azide and one bromine moieties on each monomer repeating unit of PGMA. The subsequent ATRP of 2-(2-methoxyethoxy)ethyl methacrylate (MEO2MA) using PEO-b-[PGMA-(N3)(Br)] as the macroinitiator yielded PEO-b-[PGMA-g-(N3)(PMEO2MA)] coil−brush diblock copolymer possessing one residual azide moiety at each grafting site. Finally, the target coil−rod diblock copolymer with asymmetric centipede-shaped polymer brush as the rod segment, PEO-b-[PGMA-g-(PDEA)(PMEO2MA)], was obtained via the click reaction of PEO-b-[PGMA-g-(N3)(PMEO2MA)] with an excess of alkynyl-terminated poly(2-(diethylamino)ethyl methacrylate) (alkynyl-PDEA). All the intermediate and final products were characterized by 1H NMR, Fourier transform infrared spectroscopy (FT-IR), and gel permeation chromatography (GPC). Atomic force microscopy (AFM) analysis revealed that PEO-b-[PGMA-g-(PDEA)(PMEO2MA)] coil−rod diblock unimer chains adopt a wormlike conformation in aqueous solution at pH 4 and room temperature. Possessing pH-responsive PDEA and thermo-responsive PMEO2MA grafts arranged in an asymmetric centipede manner within the rod segment, PEO-b-[PGMA-g-(PDEA)(PMEO2MA)] self-assembles into two types spherical aggregates in aqueous solution, depending on solution pH and temperatures. The multiresponsive switching between wormlike unimers and two types of micellar aggregates were characterized by temperature-dependent optical transmittance, dynamic laser light scattering (LLS), AFM, and transmission electron microscopy (TEM).
Co-reporter:Changhua Li;Zhishen Ge;Hewen Liu
Journal of Polymer Science Part A: Polymer Chemistry 2009 Volume 47( Issue 16) pp:4001-4013
Publication Date(Web):
DOI:10.1002/pola.23461
Abstract
Well-defined amphiphilic and thermoresponsive ABC miktoarm star terpolymer consisting of poly(ethylene glycol), poly(tert-butyl methacrylate), and poly(N-isopropylacrylamide) arms, PEG(-b-PtBMA)-b-PNIPAM, was synthesized via a combination of consecutive click reactions and atom transfer radical polymerization (ATRP). Click reaction of monoalkynyl-terminated PEG with a trifunctional core molecule bis(2-azidoethyl)amine, (N3)2NH, afforded difunctional PEG possessing an azido and a secondary amine moiety at the chain end, PEG-NHN3. Next, the amidation of PEG-NHN3 with 2-chloropropionyl chloride led to PEG-based ATRP macroinitiator, PEG(N3)Cl. The subsequent ATRP of N-isopropylacrylamide (NIPAM) using PEG(N3)Cl as the macroinitiator led to PEG(N3)-b-PNIPAM bearing an azido moiety at the diblock junction point. Finally, well-defined ABC miktoarm star terpolymer, PEG(-b-PtBMA)-b-PNIPAM, was prepared via the click reaction of PEG(N3)-b-PNIPAM with monoalkynyl-terminated PtBMA. In aqueous solution, the obtained ABC miktoarm star terpolymer self-assembles into micelles consisting of PtBMA cores and hybrid PEG/PNIPAM coronas, which are characterized by dynamic and static laser light scattering, and transmission electron microscopy. On heating above the phase transition temperature of PNIPAM in the hybrid corona, micelles initially formed at lower temperatures undergo further structural rearrangement and fuse into much larger aggregates solely stabilized by PEG coronas. © 2009 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 47: 4001–4013, 2009
Co-reporter:Yanfeng Zhang;Hao Liu;Hefei Dong;Changhua Li
Journal of Polymer Science Part A: Polymer Chemistry 2009 Volume 47( Issue 6) pp:1636-1650
Publication Date(Web):
DOI:10.1002/pola.23273
Abstract
Amphiphilic ABC miktoarm star terpolymers consisting of polystyrene, poly(ε-caprolactone), and poly(N-isopropylacrylamide) arms, PS(-b-PNIPAM)-b-PCL, were synthesized via a combination of atom transfer radical polymerization, ring-opening polymerization (ROP), and click chemistry. Difunctional PS bearing an alkynyl and a primary hydroxyl moiety at the chain end, PS-alknyl-OH, was prepared by reacting azido-terminated PS with an excess of 3,5-bis(propargyloxy)benzyl alcohol (BPBA) under click conditions. The subsequent ROP of ε-caprolactone using PS-alknyl-OH macroinitiator afforded PS(-alkynyl)-b-PCL copolymer bearing an alkynyl moiety at the diblock junction point. Target PS(-b-PNIPAM)-b-PCL amphiphilic ABC miktoarm star terpolymers were then prepared via click reaction between PS(-alkynyl)-b-PCL and an excess of azido-terminated PNIPAM (PNIPAM-N3). The removal of excess PNIPAM-N3 was accomplished by “clicking” onto alkynyl-functionalized Wang resin. All the intermediate and final products were characterized by gel permeation chromatography, 1H NMR, and FTIR. In aqueous solution, the obtained amphiphilic ABC miktoarm star terpolymer self-assembles into micelles possessing mixed PS/PCL cores and thermoresponsive shells, which were further characterized by dynamic laser light scattering and transmission electron microscopy. © 2009 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 47: 1636–1650, 2009
Co-reporter:Jun Yin, Xuefeng Guan, Di Wang and Shiyong Liu
Langmuir 2009 Volume 25(Issue 19) pp:11367-11374
Publication Date(Web):August 26, 2009
DOI:10.1021/la901377h
We report on the fabrication of Cu2+-sensing thermoresponsive poly(N-isopropylacrylamide) (PNIPAM) microgels labeled with metal-chelating acceptor and fluorescent reporter moieties. Cu2+ detection sensitivity can be considerably enhanced via thermo-induced collapse of the sensing matrix, which can easily optimize the relative spatial distribution of Cu2+-binding sites and fluorescence readout functionalities. A novel picolinamine-containing monomer with Cu2+-binding capability, N-(2-(2-oxo-2-(pyridine 2-yl-methylamino)ethylamino)ethyl)acrylamide (PyAM, 3), was synthesized at first. Nearly monodisperse Cu2+-sensing microgels were prepared via emulsion polymerization of N-isopropylacrylamide (NIPAM) in the presence of a nonionic surfactant, N,N′-Methylene-bis(acrylamide) (BIS), PyAM (3), and fluorescent dansylaminoethyl- acrylamide (DAEAM, 5) monomers at around neutral pH and 70 °C. At 20 °C, as-synthesized microgels in their swollen state can selectively bind Cu2+ over other metal ions (Hg2+, Mg2+, Zn2+, Pb2+, Ag+, and Al3+), leading to prominent quenching of fluorescence emission intensity. Above the volume phase transition temperature, P(NIPAM-co-PyAM-co-DAEAM) microgels exhibit increased fluorescence intensity. It was observed that Cu2+ detection sensitivity can be dramatically enhanced via thermo-induced microgel collapse at elevated temperatures. At a microgel concentration of 3.0 × 10−6 g/mL, the detection limit drastically improved from ∼46 nM at 20 °C to ∼8 nM at 45 °C. The underlying mechanism for this novel type of sensor with thermotunable detection sensitivity was tentatively proposed.
Co-reporter:Xiaoze Jiang, Shiyong Liu and Ravin Narain
Langmuir 2009 Volume 25(Issue 23) pp:13344-13350
Publication Date(Web):October 30, 2009
DOI:10.1021/la9034276
We report on the fabrication of core cross-linked (CCL) micelles possessing thermoresponsive and degradable cores and biocompatible coronas cofunctionalized with carbohydrate and biotin moieties. Well-defined poly(2-aminoethylmethacrylamide) (PAEMA) homopolymer was first synthesized in a controlled fashion via the reversible addition−fragmentation chain transfer (RAFT) process. CCL micelles comprising of well-solvated PAEMA coronas and thermoresponsive cores were then obtained in a one-pot manner via RAFT copolymerization of N-isopropylacrylamide (NIPAM) and bis(2-methacryloyloxyethyl) disulfide (DSDMA) difunctional monomers by employing PAEMA as the macro-RAFT agent. In the presence of dithiothreitol (DTT), the obtained CCL micelles can be disintegrated into unimers due to the cleavage of disulfide cross-linkers, whereas deswelling of micellar cores can be achieved via heating above the phase transition temperature of PNIPAM. Thus, the release profiles of this type of nanocarriers are expected to be triggered by temperature and thiols or a combination of both. Furthermore, primary amine residues located within coronas of CCL micelles have been further exploited for surface functionalization with biotin and carbohydrate moieties, rendering them biocompatible and bioactive. The availability of biotin within the coronas of CCL micelles was confirmed by HABA/avidin binding assay and Diffractive Optics Technology (DOT) biosensing instrument. After the micelles were immobilized on the surface of avidin-sensor chip, specific biorecognition of the available biotins and carbohydrate moieties on the CCL micelles was further confirmed. We expect that this novel type of bioactive and potentially biocompatible CCL micelles can be employed as smart nanocarriers for targeted drug delivery and controlled release.
Co-reporter:JingYi Rao;ZhiYuan Zhu
Science Bulletin 2009 Volume 54( Issue 11) pp:1912-1917
Publication Date(Web):2009 June
DOI:10.1007/s11434-009-0244-x
Polypeptide hybrid triblock copolymer, poly(L-glutamic acid)-b-poly(propylene oxide)-b-poly (L-glutamic acid) (PLGA-b-PPO-b-PLGA), was synthesized by the ring-opening polymerization of benzyl-L-glutamic N-carboxyanhydride (BLG-NCA) using poly(propylene glycol) bis(2-aminopropyl ether) as initiator, followed by the subsequent deprotection step. The obtained double hydrophilic triblock copolymer exhibits “schizophrenic” micellization behavior in aqueous solution upon dually playing with solution pH and temperature. The multi-responsive micellization behavior of this polypeptide hybrid triblock copolymer has been thoroughly investigated by 1H NMR, laser light scattering (LLS), temperature-dependent optical transmittance, and circular dichroism spectroscopy (CD).
Co-reporter:Tao Wu, Yanfeng Zhang, Xiaofeng Wang and Shiyong Liu
Chemistry of Materials 2008 Volume 20(Issue 1) pp:101
Publication Date(Web):December 12, 2007
DOI:10.1021/cm702073f
This article reports on the fabrication of hybrid silica nanoparticles densely grafted with thermoresponsive poly(N-isopropylacrylamide) (PNIPAM) brushes and their thermal phase transition behavior. Surface-initiated atom transfer radical polymerization (ATRP) of N-isopropylacrylamide (NIPAM) was conducted in 2-propanol at ambient temperature using CuCl/CuCl2/tris(2-(dimethylamino)ethyl)amine (Me6TREN) as the catalytic system, starting from the surface of silica nanoparticles derivatized with ATRP initiators (0.35 nm2/initiator). The surface-initiated ATRP can be conducted in a well-controlled manner, as revealed by the linear kinetic plot, linear evolution of number-average molecular weights (Mn) versus monomer conversions, and the relatively narrow molecular weight distributions (Mw/Mn < 1.25) of the grafted PNIPAM chains. The grafting density of PNIPAM chains at the surface of silica nanoparticls was estimated to be 2.2 nm2/chain based on transmission electron microscopy (TEM) and thermogravimetric analysis (TGA) characterization results. Laser light scattering (LLS) and optical transmittance were then employed to study the thermal phase transitions of PNIPAM brushes at the surface of silica nanoparticles. Both the intensity-average hydrodynamic radius, ⟨Rh⟩, and average radius of gyration, ⟨Rg⟩, exhibit a two-stage decrease upon heating over the broad temperature range of 20–37 °C, which is in contrast to the fact that free PNIPAM homopolymer in aqueous solution exhibits a phase transition at ca. 32 °C within a narrow temperature range. The first phase transition takes place in the temperature range of 20–30 °C, which can be tentatively ascribed to the n-cluster-induced collapse of the inner region of PNIPAM brushes close to the silica core; the second phase transition occurs above 30 °C, which can be ascribed to the outer region of PNIPAM brushes, possessing much lower chain density compared to that of the inner part. We tentatively expect that the observed unique double phase transition behavior of polymer brushes coated at the surface of inorganic nanoparticle cores can be further utilized to fabricate novel nanostructured devices with more complex functions.
Co-reporter:Xiaofeng Wang;Yanfeng Zhang;Zhiyuan Zhu
Macromolecular Rapid Communications 2008 Volume 29( Issue 4) pp:340-346
Publication Date(Web):
DOI:10.1002/marc.200700811
Co-reporter:Jian Xu and Shiyong Liu
Soft Matter 2008 vol. 4(Issue 9) pp:1745-1749
Publication Date(Web):25 Jul 2008
DOI:10.1039/B807696K
Polymeric micelles self-assembled from amphiphilic copolymers in aqueous solution have emerged as versatile drug nanocarriers in the past few decades. To enhance the bioavailability of drugs at the target disease site and upon cellular internalization, the use of stimuli-responsive nanocarriers with triggered release characteristics is highly desirable. This article highlights the recent developments in the field of polymeric nanocarriers possessing thermoresponsive coronas, focusing on the fabrication of structurally stable multi-responsive micelles via core or shell cross-linking, the phase transition behavior of thermoresponsive polymer brushes at the corona of unimolecular micelles, and their application as vehicles for targeted drug delivery and stimuli-responsive on-demand release.
Co-reporter:Zhishen Ge;Jian Xu;Danlu Wu;Ravin Narain
Macromolecular Chemistry and Physics 2008 Volume 209( Issue 7) pp:754-763
Publication Date(Web):
DOI:10.1002/macp.200700520
Co-reporter:Jing Ye, Jian Xu, Jinming Hu, Xiaofeng Wang, Guangzhao Zhang, Shiyong Liu and Chi Wu
Macromolecules 2008 Volume 41(Issue 12) pp:4416-4422
Publication Date(Web):May 20, 2008
DOI:10.1021/ma702196g
We have comparatively studied the association of cyclic- and linear-poly(N-isopropylacrylamide) (c-PNIPAM and l-PNIPAM) chains with varying chain lengths and end groups in dilute aqueous solutions by laser light scattering (LLS) and stopped-flow temperature-jump measurements. Dynamic and static LLS results reveal that the heating leads to a microphase transition. The resultant structures of interchain aggregates depend on the heating rate and the chain topologies. In comparison with l-PNIPAM chains, a slow heating of c-PNIPAM chains in the solution results in stable mesoglobules with a lower average aggregation number, a looser structure, and a smaller average size (∼290 nm). The temperature-jump-induced association of c-PNIPAM chains in the stopped-flow measurement reveals two kinetic growth stages, which were tentatively ascribed to the loose packing of contracted c-PNIPAM chains and further contraction-induced fragmentation of initially packed c-PNIPAM chains due to the lack of interchain entanglements. On the other hand, for l-PNIPAM chains, the intrachain contraction and interchain penetration/entanglement simultaneously occur as the temperature increases, leading to larger and more compact aggregates whose size increases with the solution temperature.
Co-reporter:Yueming Zhou;Kunqiang Jiang;Yaqiong Chen
Journal of Polymer Science Part A: Polymer Chemistry 2008 Volume 46( Issue 19) pp:6518-6531
Publication Date(Web):
DOI:10.1002/pola.22961
Abstract
A novel primary amine-containing monomer, 1-(3′-aminopropyl)-4-acrylamido-1,2,3-triazole hydrochloride (APAT), was prepared from N-propargylacrylamide and 3-azidopropylamine hydrochloride via copper-catalyzed Huisgen 1,3-dipolar cycloaddition (click reaction). Poly(N-isopropylacrylamide)-b-poly(1-(3′-aminopropyl)-4-acrylamido-1,2,3-triazole hydrochloride), PNIPAM-b-PAPAT, was then synthesized via consecutive reversible addition-fragmentation chain transfer polymerizations of N-isopropylacrylamide and APAT. In aqueous solution, the obtained thermoresponsive double hydrophilic block copolymer dissolves molecularly at room temperature and self-assembles into micelles with PNIPAM cores and PAPAT shells at elevated temperature. Because of the presence of highly reactive primary amine moieties in PAPAT block, two types of covalently stabilized nanoparticles namely core crosslinked and shell crosslinked micelles with ‘inverted’ core-shell nanostructures were facilely prepared upon the addition of glutaric dialdehyde at 25 and 50 °C, respectively. In addition, the obtained structure-fixed micelles were incorporated with gold nanoparticles via in situ reduction of preferentially loaded HAuCl4. High resolution transmission electron microscopy revealed that gold nanoparticles can be selectively loaded into the crosslinked cores or shells, depending on the micelle templates employed. © 2008 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 46: 6518–6531, 2008
Co-reporter:Jingyi Rao, Yanfeng Zhang, Jingyan Zhang and Shiyong Liu
Biomacromolecules 2008 Volume 9(Issue 10) pp:
Publication Date(Web):July 9, 2008
DOI:10.1021/bm800462q
Well-defined AB2 Y-shaped miktoarm star polypeptide copolymer, PZLL-b-(PBLG)2, was synthesized via a combination of ring-opening polymerization (ROP) of α-amino acid N-carboxyanhydride (NCA) and click chemistry, where PZLL is poly(ϵ-benzyloxycarbonyl-l-lysine) and PBLG is poly(γ-benzyl-l-glutamate). First, two types of primary-amine-containing initiators, N-aminoethyl 3,5-bis(propargyloxyl)-benzamide and 3-azidopropylamine, were synthesized and employed for the ROP of NCA, leading to the formation of dialkynyl-terminated PZLL and azide-terminated PBLG, dialkynyl-PZLL and PBLG-N3, respectively. The subsequent copper(I)-catalyzed cycloaddition reaction between dialkynyl-PZLL and slightly excess PBLG-N3 led to facile preparation of PZLL-b-(PBLG)2 Y-shaped miktoarm star polypeptide copolymer. The excess PBLG-N3 was scavenged off by reacting with alkynyl-functionalized Wang resin. The obtained Y-shaped miktoarm star polypeptide copolymer was characterized by gel permeation chromatograph (GPC), Fourier transform-infrared spectroscopy (FT-IR), and1H NMR. Moreover, after the hydrolysis of protecting benzyl and benzyloxycarbonyl groups of PZLL-b-(PBLG)2, water-soluble pH-responsive Y-shaped miktoarm star polypeptide copolymer, PLL-b-(PLGA)2, was obtained, where PLL is poly(l-lysine) and PLGA is poly(l-glutamic acid). It can self-assemble into PLGA-core micelles at acidic pH and PLL-core micelles at alkaline pH, accompanied with the coil-to-helix transition of PLGA and PLL sequences, respectively. The spontaneous pH-responsive supramolecular assembly of PLL-b-(PLGA)2 miktoarm star polypeptide copolymer has been investigated via a combination of 1H NMR, laser light scattering (LLS), transmission electron microscopy (TEM), and circular dichroism (CD) spectroscopy.
Co-reporter:Yanfeng Zhang;Weiyin Gu;Hangxun Xu
Journal of Polymer Science Part A: Polymer Chemistry 2008 Volume 46( Issue 7) pp:2379-2389
Publication Date(Web):
DOI:10.1002/pola.22572
Abstract
Poly(2-(dimethylamino)ethyl methacrylate)-b-poly(γ-methacryloxypropyl-trimethoxysilane) (PDMA-b-PMPS) was synthesized via consecutive reversible addition-fragmentation chain transfer (RAFT) polymerizations in 1,4-dioxane. Subsequent micellization of the obtained amphiphilic diblock polymer in aqueous solution led to the formation of nanoparticles consisting of hydrophobic PMPS cores and well-solvated PDMA shells. Containing tertiary amine residues, PDMA blocks in micelle coronas can spontaneously catalyze the sol–gel reactions of trimethoxysilyl groups within PMPS cores, leading to the formation of hybrid nanoparticles coated with PDMA brushes. Transmission electron microscopy (TEM) and laser light scattering (LLS) revealed the presence of monodisperse spherical hybrid nanoparticles, and the grafting density of PDMA chains at the surface of nanoparticle cores was estimated to be ∼5.8 nm2/chain. PDMA brushes exhibit dual stimuli-responsiveness, and the swelling/collapse of them can be finely tuned with solution pH and temperatures. The obtained multi-responsive hybrid nanoparticles might find potential applications in fields such as smart devices, recyclable catalysts, and intelligent nanocarriers for drug delivery or gene transfection. © 2008 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 46: 2379–2389, 2008
Co-reporter:Xiaoze Jiang;Jingyan Zhang;Yueming Zhou;Jian Xu
Journal of Polymer Science Part A: Polymer Chemistry 2008 Volume 46( Issue 3) pp:860-871
Publication Date(Web):
DOI:10.1002/pola.22430
Abstract
Double hydrophilic diblock copolymer, poly(N,N-dimethylacrylamide)-b-poly(N-isopropylacrylamide-co-3-azidopropylacrylamide) (PDMA-b-P(NIPAM-co-AzPAM), containing azide moieties in one of the blocks was synthesized via consecutive reversible addition-fragmentation chain transfer polymerization. The obtained diblock copolymer molecularly dissolves in aqueous solution at room temperature, and can further supramolecularly self-assemble into core-shell nanoparticles consisting of thermoresponsive P(NIPAM-co-AzPAM) cores and water-soluble PDMA coronas above the lower critical solution temperature of P(NIPAM-co-AzPAM) block. As the micelle cores contain reactive azide residues, core crosslinking can be facilely achieved upon addition of difunctional propargyl ether via click chemistry. In an alternate approach in which the PDMA-b-P(NIPAM-co-AzPAM) diblock copolymer was dissolved in a common organic solvent (DMF), the core-crosslinked (CCL) micelles can be fabricated via “click” crosslinking upon addition of propargyl ether and subsequent dialysis against water. CCL micelles prepared by the latter approach typically possess larger sizes and broader size distributions, compared with that obtained by the former one. In both cases, the obtained (CCL) micelles possess thermoresponsive cores, and the swelling/shrinking of which can be finely tuned with temperature, rendering them as excellent candidates as intelligent drug nanocarriers. Because of the high efficiency and quite mild conditions of click reactions, we expect that this strategy can be generalized for the structural fixation of other self-assembled nanostructures. © 2007 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 46: 860–871, 2008
Co-reporter:Jian Xu;Xiaoze Jiang
Journal of Polymer Science Part A: Polymer Chemistry 2008 Volume 46( Issue 1) pp:60-69
Publication Date(Web):
DOI:10.1002/pola.22358
Abstract
Controlled radical polymerizations of N-ethylmethylacrylamide (EMA) by atom transfer radical polymerization and reversible addition-fragmentation chain transfer processes were investigated in detail for the first time, employing complementary characterization techniques including gel permeation chromatography, 1H NMR spectroscopy, and matrix-assisted laser desorption ionization time-of-flight mass spectrometry. In both cases, relatively good control of the polymerization of EMA was achieved, as revealed by the linear evolution of molecular weights with monomer conversions and the low polydispersity of poly(N-ethylmethylacrylamide) (PEMA). The thermal phase transitions of well-defined PEMA homopolymers with polydispersities less than 1.2 and degrees of polymerization up to 320 in aqueous solution were determined by temperature-dependent turbidity measurements. The obtained cloud points (CPs) vary in the range of 58–68 °C, exhibiting inverse molecular weight and polymer concentration dependences. Moreover, the presence of a carboxyl group instead of an alkyl one at the PEMA chain end can elevate its CP by ∼3–4 °C. © 2007 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 46: 60–69, 2008
Co-reporter:Jun Yin, Damien Dupin, Junfang Li, Steven P. Armes and Shiyong Liu
Langmuir 2008 Volume 24(Issue 17) pp:9334-9340
Publication Date(Web):July 22, 2008
DOI:10.1021/la8014282
Near-monodisperse, sterically stabilized poly(2-vinylpyridine) (P2VP) microgels were synthesized by emulsion polymerization. These particles exhibited completely reversible pH-responsive swelling/deswelling behavior in aqueous solution. Stopped-flow light scattering was employed to investigate the kinetics of pH-induced deswelling in highly dilute dispersions. Upon a pH jump from 2 to various final solution pH values (≥5.4), the scattered light intensity of an aqueous dispersion of a 1960 nm microgel exhibited an abrupt initial increase, followed by a gradual decrease to the final equilibrium value. The whole microgel-to-latex deswelling process occurred over time scales of ∼0.5−1.0 s, which is much slower than the kinetics for latex-to-microgel swelling. The microgel deswelling kinetics depends on the final pH, with a higher final pH leading to a faster rate of shrinkage. Close inspection of the deswelling kinetics during the early stages (<0.2 s) revealed that initial microgel collapse occurred within ∼50 ms, with more rapid transitions being observed when higher final pH values were targeted. Addition of external salt significantly accelerates the kinetics of deswelling. Systematic studies of the microgel-to-latex transition for a series of six near-monodisperse P2VP particles (with swollen microgel diameters ranging from 1270 to 4230 nm) has also been investigated. The characteristic deswelling time for initial microgel collapse, τdeswell, correlated fairly well with the initial swollen microgel radius, R, in agreement with the Tanaka equation. Moreover, the collective diffusion coefficient of the gel network, D, calculated from the slope of the τdeswell−R2 curve, was of the order of 10−7 cm2 s−1.
Co-reporter:Lei Shen, Jianzhong Du, Steven P. Armes and Shiyong Liu
Langmuir 2008 Volume 24(Issue 18) pp:10019-10025
Publication Date(Web):August 6, 2008
DOI:10.1021/la801190z
The kinetics of pH-induced formation and dissociation of vesicles self-assembled from a biocompatible zwitterionic diblock copolymer, poly(2-(methacryloyloxy)ethyl phosphorylcholine)-b-poly(2-(diisopropylamino)ethyl methacrylate) (PMPC-b-PDPA), was investigated in detail via a combination of stopped-flow light scattering and laser light scattering (LLS). Upon jumping from pH 2 to 10, stopped-flow light scattering reveals three distinct relaxation processes for the early stages of vesicle self-assembly (0−40 s). Kinetic sequences associated with the obtained three characteristic relaxation times have been tentatively proposed. Moreover, the kinetics of vesicle formation in the later stage (from 3 min onward) was investigated by dynamic LLS. It was found that both the intensity-averaged hydrodynamic radius, ⟨Rh⟩, and the polydispersity, μ2/Γ2, decrease exponentially, yielding a characteristic relaxation time of ∼350 s. To our knowledge, this is the first report on the kinetics of the unimer-to-vesicle transition of a stimulus-responsive diblock copolymer. The kinetics of vesicle dissociation for a pH jump from 12 to 2 was also investigated. The breakdown of polymeric vesicles is extremely fast and is independent of polymer concentration; it is complete within ∼5 ms and is in marked contrast to the much slower rate of vesicle formation.
Co-reporter:Jingyan Zhang, Jian Xu and Shiyong Liu
The Journal of Physical Chemistry B 2008 Volume 112(Issue 36) pp:11284-11291
Publication Date(Web):August 15, 2008
DOI:10.1021/jp803700n
A series of well-defined poly(ethylene oxide)-b-poly(2-(diethylamino)ethyl methacrylate) (PEO-b-PDEA) diblock copolymers containing PEO block of identical chain length and PDEA block with varying degrees of polymerization (DP, in the range of 32−154) were prepared via atom transfer radical polymerization (ATRP) employing a PEO-based macroinitiator (DP = 113). Upon a pH-jump from 3 to 12 under highly efficient stopped-flow mixing conditions, PEO-b-PDEA copolymers spontaneously form spherical micelles of increasing sizes and aggregation numbers (Nagg) with increasing PDEA chain lengths. Stopped-flow light scattering technique was used to probe the pH-induced micellization kinetics of PEO-b-PDEA copolymers, aiming to elucidate the PDEA chain-length effects on the unimer-to-micelle transition process. Upon a stopped-flow pH-jump from 3 to 12, the obtained dynamic traces can be well-fitted with double exponential functions. The calculated fast and slow characteristic relaxation times (τ1 and τ2) can be ascribed to the formation of quasi-equilibrium micelles (fast process) and subsequent relaxation into final equilibrium micelles (slow process), respectively. For PEO113-b-PDEA32 and PEO113-b-PDEA61, τ2 is almost independent of polymer concentrations, suggesting that the relaxation from quasi-equilibrium micelles into final equilibrium micelles mainly proceeds via insertion/expulsion of unimer chains. Upon increasing the DP of pH-responsive PDEA block to 89, 117, and 154, the obtained slow relaxation time, τ2, tends to decrease with increasing polymer concentrations, suggesting that the slow process is dominated by the micelle fusion/fission mechanism. The apparent activation energy (Ea) associated with τ2 has also been determined from temperature-dependent micellization kinetics for five PEO-b-PDEA copolymers. It was found that during micellization, copolymers with longer PDEA blocks exhibit much lower Ea compared to those with shorter blocks. Thus, we observed experimentally for the first time that increasing the hydrophobic block length in double hydrophilic block copolymers (DHBCs) can transform the mechanism of the slow process from unimer insertion/expulsion to micelle fusion/fission.
Co-reporter:Jingyan Zhang, Zhishen Ge, Xiaoze Jiang, P.A. Hassan, Shiyong Liu
Journal of Colloid and Interface Science 2007 Volume 316(Issue 2) pp:796-802
Publication Date(Web):15 December 2007
DOI:10.1016/j.jcis.2007.08.067
The kinetics and mechanism of sphere-to-rod transitions of sodium alkyl sulfate micelles induced by hydrotropic salt, p -toluidine hydrochloride (PTHC), were investigated by stopped-flow with light scattering detection. Spherical sodium dodecyl sulfate (SDS) micelles transform into short ellipsoidal shapes at low salt concentrations ([PTHC]/[SDS], χPTHC=0.3χPTHC=0.3 and 0.4). Upon stopped-flow mixing aqueous solutions of spherical SDS micelles with PTHC, the scattered light intensity gradually increases with time. Single exponential fitting of the dynamic traces leads to characteristic relaxation time, τgτg, for the growth process from spherical to ellipsoidal micelles, and it increases with increasing SDS concentrations. This suggests that ellipsoidal micelles might be produced by successive insertion of unimers into spherical micelles, similar to the case of formation of spherical micelles as suggested by Aniansson–Wall (A–W) theory. At χPTHC⩾0.5χPTHC⩾0.5, rod-like micelles with much higher axial ratio form. The scattered light intensity exhibits an initially abrupt increase and then levels off. The dynamic curves can be well fitted with single exponential functions, and the obtained τgτg decreases with increasing SDS concentration. Thus, the growth from spherical to rod-like micelles might proceed via fusion of spherical micelles, in agreement with mechanism proposed by Ikeda et al. At χPTHC=0.3χPTHC=0.3 and 0.6, the apparent activation energies obtained from temperature dependent kinetic studies for the micellar growth are 40.4 and 3.6 kJ/mol, respectively. The large differences between activation energies for the growth from spherical to ellipsoidal micelles at low χPTHCχPTHC and the sphere-to-rod transition at high χPTHCχPTHC further indicate that they should follow different mechanisms. Moreover, the sphere-to-rod transition kinetics of sodium alkyl sulfate with varying hydrophobic chain lengths (n=10n=10, 12, 14, and 16) are also studied. The longer the carbon chain lengths, the slower the sphere-to-rod transition.
Co-reporter:Shiyong Liu;Tao Wu;Yanfeng Zhang
Macromolecular Chemistry and Physics 2007 Volume 208(Issue 23) pp:2492-2501
Publication Date(Web):26 SEP 2007
DOI:10.1002/macp.200700293
Double hydrophilic diblock copolymer, poly(N-isopropylacrylamide)-block-poly(2-diethylamino ethyl methacrylate) (PNIPAM-b-PDEA), was synthesized via reversible addition-fragmentation chain transfer (RAFT) polymerization. Containing the well-known thermo-responsive PNIPAM block and pH-responsive PDEA block, this novel diblock copolymer exhibits intriguing “schizophrenic” micellization behavior in aqueous solution, forming PDEA-core micelles at alkaline pH and room temperature, and PNIPAM-core micelles at acidic pH and elevated temperatures. The kinetics of the pH- and thermo-responsive micellization processes were studied in detail using a stopped-flow apparatus equipped with a newly developed millisecond temperature jump (mT-jump) accessory. Upon a pH jump from 4 to 12 at 25 °C, the early stages of relaxation curves monitoring the formation PDEA-core micelles can be well-fitted using a double-exponential function, leading to two characteristic relaxation time constants, τ1 and τ2. As τ2 decreases with increasing polymer concentration, the slow process is thus expected to proceed via micelle fusion/fission mechanism, approaching the final equilibrium state. Upon a temperature jump from 20 to 45 °C at pH 4, the relaxation curves monitoring the formation PNIPAM-core micelles can also be well-fitted using a double-exponential function. The fast process (τ1) is associated with the quick association of unimers into a large amount of small micelles and the formation of quasi-equilibrium micelles. τ2 is almost independent of polymer concentration, suggesting that unimer insertion/expulsion is the main mechanism for the slow process. The protonated PDEA corona of quasi-equilibrium micelles renders the micelle fusion/fission mechanism less favorable due to electrostatic repulsion.
Co-reporter:Jingyi Rao, Zhaofeng Luo, Zhishen Ge, Hao Liu and Shiyong Liu
Biomacromolecules 2007 Volume 8(Issue 12) pp:
Publication Date(Web):November 3, 2007
DOI:10.1021/bm700830b
A polypeptide hybrid double hydrophilic diblock copolymer (DHBC), poly(N-isopropylacrylamide)-b-poly(l-glutamic acid) (PNIPAM-b-PLGA), was synthesized via the ring-opening polymerization of γ-benzyl-l-glutamate N-carboxyanhydride (BLG-NCA) using monoamino-terminated PNIPAM as the macroinitiator, followed by deprotection of benzyl groups under alkaline conditions. Containing a thermoresponsive PNIPAM block and a pH-responsive PLGA block, the obtained polypeptide hybrid diblock copolymer molecularly dissolves in aqueous solution at alkaline pH and room temperature but supramolecularly self-assembles into PNIPAM–core micelles at alkaline pH and elevated temperatures and PLGA–core micelles at acidic pH and room temperature accompanied with coil-to-helix transition of the PLGA sequence. The pH- and thermoresponsive “schizophrenic” micellization behavior of PNIPAM-b-PLGA diblock copolymer has been investigated by 1H NMR, optical transmittance, fluorescence probe measurement, transmission electron microscopy (TEM), dynamic and static laser light scattering (LLS), and circular dichroism (CD) spectroscopy. Moreover, the micellization process was investigated employing stopped-flow light scattering technique. The pH-induced micelle growth of PNIPAM-b-PLGA in aqueous solution exhibits drastically different kinetics compared to that of conventional pH-responsive DHBCs, probably due to the stabilization effects exerted by the formed α-helix secondary structures within the PLGA core at low pH. Exhibiting “schizophrenic” micellization, the polypeptide sequence of PNIPAM-b-PLGA can either locate within micelle cores or stabilizing coronas. The incorporation of polypeptide block into DHBCs can endow them with structural versatility, tunable spatial arrangement of chain segments within self-assembled nanostructures, and broader applications in the field of biomedicines.
Co-reporter:Hao Liu;Jian Xu;Jiali Jiang;Jun Yin;Ravin Narain;Yuanli Cai
Journal of Polymer Science Part A: Polymer Chemistry 2007 Volume 45(Issue 8) pp:1446-1462
Publication Date(Web):5 MAR 2007
DOI:10.1002/pola.21915
Well-defined amphiphilic PCL-b-(PDMA)2 and (PCL)2-b-PDMA Y-shaped miktoarm star copolymers and PCL-b-PDMA linear diblock copolymer were synthesized via a combination of ring-opening polymerization (ROP) and atom transfer radical polymerization (ATRP), where PCL is poly (ε-caprolactone) and PDMA is poly(2-(dimethylamino)ethyl methacrylate). All of these three types of copolymers have comparable PCL contents and overall molecular weights. The PCL block is hydrophobic while the PDMA block is hydrophilic, and they behave like polymeric surfactants and self-assemble into PCL-core micelles in aqueous media. The chain architectural effects on the micellization properties, including the aggregation number, size, polydispersity, and micelle densities of (PCL29)2-b-PDMA45, PCL61-b-(PDMA24)2, and PCL56-b-PDMA49 in dilute aqueous solution, were then explored by dynamic and static laser light scattering (LLS). The intensity–average hydrodynamic radius, 〈Rh〉, the aggregation number per micelle, Nagg, and the core radius, Rcore, of the PCL-core micelles all increased in the order PCL61-b-(PDMA24)2 < (PCL29)2-b-PDMA45 < PCL56-b-PDMA49. The surface area occupied per soluble PDMA block at the core/corona interface increased in the order PCL61-b-(PDMA24)2 < PCL56-b-PDMA49 < (PCL29)2-b-PDMA45. PCL61-b-(PDMA24)2 micelles had the largest overall micelle density, possibly because of that the presence of two soluble PDMA arms at the junction point favors the bending of the core–corona interface and thus the formation of densely-packed core-shell nanostructures. © 2006 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 45: 1446–1462, 2007
Co-reporter:Zhishen Ge;Daoyong Chen;Jingyan Zhang;Jingyi Rao;Jun Yin;Di Wang;Xuejuan Wan;Wenfang Shi
Journal of Polymer Science Part A: Polymer Chemistry 2007 Volume 45(Issue 8) pp:1432-1445
Publication Date(Web):5 MAR 2007
DOI:10.1002/pola.21914
We report the first instance of facile synthesis of dumbbell-shaped dendritic-linear-dendritic triblock copolymer, [G-3]-PNIPAM-[G-3], consisting of third generation poly(benzyl ether) monodendrons ([G-3]) and linear poly(N-isopropylacrylamide) (PNIPAM), via reversible addition-fragmentation chain transfer (RAFT) polymerization. The key step was the preparation of novel [G-3]-based RAFT agent, [G-3]-CH2SCSSCH2-[G-3] (1), from third-generation dendritic poly(benzyl ether) bromide, [G-3]-CH2Br. Due to the bulky nature of [G-3]-CH2Br, its transformation into trithiocarbonate 1 cannot go to completion, a mixture containing ∼80 mol % of 1 and 20 mol % [G-3]-CH2Br was obtained. Dumbbell-shaped [G-3]-PNIPAM310-[G-3] triblock copolymer was then successfully obtained by the RAFT polymerization of N-isopropylacylamide (NIPAM) using 1 as the mediating agent, and trace amount of unreacted [G-3]-CH2Br was conveniently removed during purification by precipitating the polymer into diethyl ether. The dendritic-linear-dendritic triblock structure was further confirmed by aminolysis, and fully characterized by gel permeation chromatography (GPC) and 1H-NMR. The amphiphilic dumbbell-shaped triblock copolymer contains a thermoresponsive PNIPAM middle block, in aqueous solution it self-assembles into spherical nanoparticles with the core consisting of hydrophobic [G-3] dendritic block and stabilized by the PNIPAM central block, forming loops surrounding the insoluble core. The micellar properties of [G-3]-PNIPAM310-[G-3] were then fully characterized. © 2006 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 45: 1432–1445, 2007
Co-reporter:Zhishen Ge;Shizhong Luo
Journal of Polymer Science Part A: Polymer Chemistry 2006 Volume 44(Issue 4) pp:1357-1371
Publication Date(Web):3 JAN 2006
DOI:10.1002/pola.21261
This article describes the syntheses and solution behavior of model amphiphilic dendritic–linear diblock copolymers that self-assemble in aqueous solutions into micelles with thermoresponsive shells. The investigated materials are constructed of poly(benzyl ether) monodendrons of the second generation ([G-2]) or third generation ([G-3]) and linear poly(N-isopropylacrylamide) (PNIPAM). [G-2]-PNIPAM and [G-3]-PNIPAM dendritic–linear diblock copolymers have been prepared by reversible addition–fragmentation transfer (RAFT) polymerizations of N-isopropylacrylamide with a [G-2]- or [G-3]-based RAFT agent, respectively. The critical micelle concentration (cmc) of [G-3]-PNIPAM220, determined by surface tensiometry, is 6.3 × 10−6 g/mL, whereas [G-2]-PNIPAM235 has a cmc of 1.0 × 10−5 g/mL. Transmission electron microscopy results indicate the presence of spherical micelles in aqueous solutions. The thermoresponsive conformational changes of PNIPAM chains located at the shell of the dendritic–linear diblock copolymer micelles have been thoroughly investigated with a combination of dynamic and static laser light scattering and excimer fluorescence. The thermoresponsive collapse of the PNIPAM shell is a two-stage process; the first one occurs gradually in the temperature range of 20–29 °C, which is much lower than the lower critical solution temperature of linear PNIPAM homopolymer, followed by the second process, in which the main collapse of PNIPAM chains takes place in the narrow temperature range of 29–31 °C. © 2006 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 44: 1357–1371, 2006
Co-reporter:Xianglong Hu; Guhuan Liu; Yang Li; Xiaorui Wang
Journal of the American Chemical Society () pp:
Publication Date(Web):December 12, 2014
DOI:10.1021/ja5105848
The rational design of theranostic nanoparticles exhibiting synergistic turn-on of therapeutic potency and enhanced diagnostic imaging in response to tumor milieu is critical for efficient personalized cancer chemotherapy. We herein fabricate self-reporting theranostic drug nanocarriers based on hyperbranched polyprodrug amphiphiles (hPAs) consisting of hyperbranched cores conjugated with reduction-activatable camptothecin prodrugs and magnetic resonance (MR) imaging contrast agent (Gd complex), and hydrophilic coronas functionalized with guanidine residues. Upon cellular internalization, reductive milieu-actuated release of anticancer drug in the active form, activation of therapeutic efficacy (>70-fold enhancement in cytotoxicity), and turn-on of MR imaging (∼9.6-fold increase in T1 relaxivity) were simultaneously achieved in the simulated cytosol milieu. In addition, guanidine-decorated hPAs exhibited extended blood circulation with a half-life up to ∼9.8 h and excellent tumor cell penetration potency. The hyperbranched chain topology thus provides a novel theranostic polyprodrug platform for synergistic imaging/chemotherapy and enhanced tumor uptake.
Co-reporter:Xiaoze Jiang ; Guoying Zhang ; Ravin Narain
Langmuir () pp:
Publication Date(Web):January 13, 2009
DOI:10.1021/la803616d
A well-defined ABC triblock copolymer, poly(2-(2-methoxyethoxy)ethyl methacrylate)-b-poly(2-(dimethylamino)ethyl methacrylate)-b-poly(2-(diethylamino)ethyl methacrylate) (PMEO2MA-b-PDMA-b-PDEA), was synthesized via sequential atom transfer radical polymerization using ethyl 2-bromoisobutyrate as the initiator. Reacting the triblock precursor with propargyl bromide in anhydrous tetrahydrofuran yielded PMEO2MA-b-P(DMA-co-QDMA)-b-PDEA triblock copolymer with “clickable” moieties, where QDMA was quaternized DMA residues. PMEO2MA-b-P(DMA-co-QDMA)-b-PDEA triblock copolymer exhibited “schizophrenic” micellization behavior in aqueous solution, forming three-layer onion-like PMEO2MA-core and PDEA-core micelles upon proper adjustment of the solution pH and temperature. For temperature-induced formation of PMEO2MA-core micelles at acidic pH, the critical micellization temperature can be tuned by incorporating oligo(ethylene glycol) methyl ether methacrylate (OEGMA; the mean degree of polymerization was 8−9) residues into the PMEO2MA block, shifting from 38 to 43 °C as the OEGMA contents varied in the range of 0−10 mol %. In both types of micelles, the inner shell layer consisted of the middle P(DMA-co-QDMA) segment. Subsequently, cross-linking with tetra(ethylene glycol) diazide via click chemistry in the presence of copper catalysts led to the facile preparation of two types of shell-cross-linked (SCL) micelles with “inverted” structures in purely aqueous solution. The cores and coronas of SCL micelles exhibited multiresponsive swelling/shrinking and collapse/aggregation behavior, respectively. To the best of our knowledge, this represents the first report of the fabrication of two types of SCL micelles with inverted structures from a single schizophrenic water-soluble triblock copolymer in purely aqueous solution.
Co-reporter:Hao Liu ; Changhua Li ; Hewen Liu
Langmuir () pp:
Publication Date(Web):February 24, 2009
DOI:10.1021/la803813r
We report the first example of the synthesis and pH-responsive supramolecular self-assembly of double hydrophilic ABC miktoarm star terpolymers. Well-defined ABC miktoarm star terpolymers consisting of poly(ethylene glycol), poly(tert-butyl methacrylate), and poly(2-(diethylamino)ethyl methacrylate) arms [PEG(-b-PtBMA)-b-PDEA] were synthesized via the combination of consecutive click reactions and atom transfer radical polymerization (ATRP), starting from a trifunctional core molecule, 1-azido-3-chloro-2-propanol (ACP). The click reaction of monoalkynyl-terminated PEG with an excess of ACP afforded difunctional PEG bearing a chlorine and a secondary hydroxyl moiety at the chain end, PEG113(-Cl)-OH (1). After azidation with NaN3, PEG-based macroinitiator PEG113(-N3)-Br (3) was prepared by the esterification of PEG113(-N3)-OH (2) with 2-bromoisobutyryl bromide and then employed in the ATRP of tert-butyl methacrylate (tBMA). The obtained PEG(-N3)-b-PtBMA copolymers (4) possessed an azido moiety at the diblock junction point. The preparation of PEG(-b-PtBMA)-b-PDEA miktoarm star terpolymers was then achieved via the click reaction of 4 with an excess of monoalkynyl-terminated PDEA. The obtained miktoarm star terpolymers were successfully converted into PEG(-b-PMAA)-b-PDEA, where PMAA is poly(methacrylic acid). In aqueous solution, PEG(-b-PMAA)-b-PDEA zwitterionic ABC miktoarm star terpolymers can self-assemble into three types of micellar aggregates by simply adjusting solution pH at room temperature. Above pH 8, PDEA-core micelles stabilized by PEG/ionized PMAA hybrid coronas were formed due to the insolubility of PDEA block. In the range of pH 5−7, micelles possessing polyion complex cores formed as a result of charge compensation between partially ionized PMAA and partially protonated PDEA sequences. At pH < 4, hydrogen bonding interactions between fully protonated PMAA and PEG led to the formation of another type of micellar aggregates possessing hydrogen-bonded complex cores stabilized by protonated PDEA coronas. The fully reversible pH-responsive formation of three types of aggregates were characterized by 1H NMR, dynamic and static laser light scattering (LLS), and transmission electron microscopy (TEM).
Co-reporter:Changhua Li and Shiyong Liu
Chemical Communications 2012 - vol. 48(Issue 27) pp:NaN3278-3278
Publication Date(Web):2012/02/09
DOI:10.1039/C2CC17695E
Fluorescent polymeric assemblies and nanoparticles (NPs) of nanoscale dimensions have become a focus of intensive investigations during the past few decades due to combined advantages such as improved biocompatibility, water dispersibility, stimuli-responsiveness, facile integration into optical detection devices, and the ability of further functionalization. In addition, the chemical composition and morphology of polymeric assemblies and NPs can be modulated via synthetic approaches, leading to the precise spatial organization of multiple fluorophores. Thus, polymeric assemblies and NPs have been utilized to optimize the photoluminescent properties of covalently or physically attached fluorophores and facilely modulate the fluorescence resonance energy transfer (FRET) processes when the polymeric matrix is endowed with stimuli-responsiveness. These fascinating fluorescent polymeric assemblies and NPs offer unique and versatile platforms for the construction of novel detection, imaging, biolabeling, and optoelectronic systems. This feature article focuses on the recent developments of polymeric assemblies and NPs-based stimuli-tunable fluorescent systems and highlights their future practical applications with selected literature reports.
Co-reporter:Xiaorui Wang, Jinming Hu, Tao Liu, Guoying Zhang and Shiyong Liu
Journal of Materials Chemistry A 2012 - vol. 22(Issue 17) pp:
Publication Date(Web):
DOI:10.1039/C2JM16510D
Co-reporter:Xingxing Sun, Xuepeng Zhang, Xinyang Li, Shiyong Liu and Guoqing Zhang
Journal of Materials Chemistry A 2012 - vol. 22(Issue 33) pp:NaN17339-17339
Publication Date(Web):2012/07/04
DOI:10.1039/C2JM32809G
Mechanochromic luminescence (ML) refers to the luminescence color and/or intensity change of solid-state materials induced by mechanical perturbations. For organic molecular solids, this phenomenon is related to the specific packing modes and orientations of individual fluorophores, which could give rise to different excited-state interactions. The molecular solids of difluoroboron dibenzoylmethane (BF2dbm) derivatives were previously found to exhibit reversible ML at room temperature and are promising as self-healing optical materials. In this report, we aim to shed some light on the mechanism of BF2dbm ML by trying to understand the excited-state interactions among solid-state BF2dbm molecules and elucidate how these interactions change upon mechanical stimulation. We first investigated the optical properties of monomeric, dimeric, and polymeric BF2dbm derivatives in optically dilute solutions and demonstrated unambiguously that BF2dbm moieties have a propensity to form H-aggregates. Next, we studied the physical properties of these boron complexes in the solid state including their crystal structures, fluorescence emissions, and mechanochromic luminescence. By correlating solution data with the solid-state characterization results, it was concluded that two coupled processes, force-induced emissive H-aggregate formation and energy transfer to the emissive H-aggregates, are responsible for the observed BF2dbm ML in the solid state.
Co-reporter:Jingyan Zhang and Shiyong Liu
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 27) pp:NaN12553-12553
Publication Date(Web):2011/06/13
DOI:10.1039/C0CP02856H
The kinetics of thermo-induced micelle-to-vesicle transitions in a catanionic surfactant system consisting of sodium dodecyl sulfate (SDS) and dodecyltriethylammonium bromide (DEAB) were investigated by the stopped-flow temperature jump technique, which can achieve T-jumps within ∼2–3 ms. SDS/DEAB aqueous mixtures ([SDS]/[DEAB] = 2/1, 10 mM) undergo microstructural transitions from cylindrical micelles to vesicles when heated above 33 °C. Upon T-jumps from 20 °C to final temperatures in the range of 25–31 °C, relaxation processes associated with negative amplitudes can be ascribed to the dilution-induced structural rearrangement of cylindrical micelles and to the dissolution of non-equilibrium mixed aggregates. In the final temperature range of 33–43 °C the obtained dynamic traces can be fitted by single exponential functions, revealing one relaxation time (τ) in the range of 82–440 s, which decreases with increasing temperature. This may be ascribed to the transformation of floppy bilayer structures into precursor vesicles followed by further growth into final equilibrium vesiclesvia the exchange and insertion/expulsion of surfactant monomers. In the final temperature range of 45–55 °C, vesicles are predominant. Here T-jump relaxations revealed a distinctly different kinetic behavior. All dynamic traces can only be fitted with double exponential functions, yielding two relaxation times (τ1 and τ2), exhibiting a considerable decrease with increasing final temperatures. The fast process (τ1 ∼ 5.2–28.5 s) should be assigned to the formation of non-equilibrium precursor vesicles, and the slow process (τ2 ∼ 188–694 s) should be ascribed to their further growth into final equilibrium vesiclesvia the fusion/fission of precursor vesicles. In contrast, the reverse vesicle-to-micelle transition process induced by a negative T-jump from elevated temperatures to 20 °C occurs quite fast and almost completes within the stopped-flow dead time (∼2–3 ms).
Co-reporter:Jingyan Zhang, Sangui Chen, Zhiyuan Zhu and Shiyong Liu
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 1) pp:NaN127-127
Publication Date(Web):2013/10/15
DOI:10.1039/C3CP53608D
The formation of soluble polyion complexes (PICs) from anionic block copolymers, poly(ethylene oxide)-b-poly(sodium 4-styrene sulfonate) (PEO-b-PSSNa) and cationic block copolymers, poly(ethylene oxide)-b-poly(quaternized 2-(dimethyl amino)ethyl methacrylate) (PEO-b-PQDMA) was investigated by fluorescence spectroscopy, laser light scattering (LLS), and stopped-flow light scattering. Colloidally stabilized dispersions could be obtained upon direct mixing of the aqueous solutions of these two block copolymers, which indicated the formation of core–shell nanostructures with the core consisting of interpolymer electrostatic complexes between PSSNa and PQDMA blocks and the corona of PEO block. Both LLS and fluorescence results revealed that the most compact complex micelles formed at the equal molar ratio of oppositely charged SSNa and QDMA residues. The kinetics of the assembly process was studied via stopped-flow upon direct mixing of the two polymer solutions. The complexation process between PEO-b-PQDMA and PEO-b-PSSNa was fast and could finish within seconds. Moreover, the relaxation process can only be detected at near equal SSNa to QDMA molar ratios. The relaxation curves can be well fitted by a double-exponential function, leading to a fast relaxation process related to the initial quasi-equilibrium complex formation and a slow process related to the pre-complex structure rearrangements to the final equilibrium complexes. Both stages are determined as second-order reactions and processed through a micelle fusion–fission mechanism. Fluorescence kinetic studies revealed that the neutralization of an oppositely charged polyion was too fast to be detected and should be completed within the stopped-flow dead-time. Thermodynamic studies revealed that spontaneous complexation is entropy driven. Upon increasing the ionic strength of the solutions, the complexation processes became slower due to the decrease of entropy driving force. The PIC dissociation process was further studied and considered to consist of two competing processes: a second-order process depending on PIC concentration and a first-order process independent of the PIC concentration.
Co-reporter:Jinming Hu, Xiaozheng Zhang, Di Wang, Xianglong Hu, Tao Liu, Guoying Zhang and Shiyong Liu
Journal of Materials Chemistry A 2011 - vol. 21(Issue 47) pp:NaN19038-19038
Publication Date(Web):2011/10/31
DOI:10.1039/C1JM13575A
We report on the fabrication of highly sensitive ratiometric fluorescent pH and temperature probes based on thermoresponsive double hydrophilic block copolymers (DHBCs) with the two blocks labeled with two types of dyes possessing different pH-switchable emission characteristics. P(NIPAM-co-FITC)-b-P(OEGMA-co-RhBAM) DHBCs were synthesized via consecutive reversible addition–fragmentation chain transfer (RAFT) polymerizations in combination with post-modifications, where NIPAM, OEGMA, FITC, and RhBAM are N-isopropylacrylamide, oligo(ethylene glycol) monomethyl ether methacrylate, fluorescein isothiocyanate, and rhodamine B-based derivatives, respectively. Due to that FITC and RhBAM moieties exhibit prominent decrease and increase in emission intensities with decreasing solution pH, respectively, intensity ratios of characteristic RhBAM and FITC emission bands, I582/I522, of P(NIPAM-co-FITC)-b-P(OEGMA-co-RhBAM) unimers at 25 °C exhibit ∼39-fold changes in the range of pH 2–10. At elevated temperatures, thermo-induced formation of PNIPAM-core micelles enables effective fluorescence resonance energy transfer (FRET) between FITC and RhBAM moieties respectively located within micellar cores and coronas, and I582/I522 exhibits ∼52.5-fold changes in the same pH range. The reported dually modulated multicolor-emitting P(NIPAM-co-FITC)-b-P(OEGMA-co-RhBAM) DHBCs are capable of ultrasensitive fluorometric detection of solution pH and temperature in a ratiometric manner, which augurs well for their practical applications in sensing, imaging, and the fabrication of new generation of theranostic systems.
Co-reporter:Changhua Li and Shiyong Liu
Journal of Materials Chemistry A 2010 - vol. 20(Issue 47) pp:NaN10723-10723
Publication Date(Web):2010/09/24
DOI:10.1039/C0JM01828G
We report on the fabrication of thermoresponsive poly(N-isopropylacrylamide) nanogel-based dual fluorescent sensors for temperature and Hg2+ ions, and the effects of thermo-induced nanogel collapse on the detection sensitivity of Hg2+ ions. Near-monodisperse thermoresponsive nanogels were prepared via emulsion polymerization of N-isopropylacrylamide (NIPAM) and a novel 1,8-naphthalimide-based polarity-sensitive and Hg2+-reactive fluorescent monomer (NPTUA, 3). At room temperature, PNIPAM nanogels labeled with a single type of naphthalimide-based dye (NPTUA) can act as ratiometric Hg2+ probes at the nanomolar level. Upon heating above the phase transition temperature, the fluorescence intensity of NPTUA-labeled nanogels in the absence of Hg2+ exhibit ∼3.4-fold increase due to that NPTUA moieties are now located in a more hydrophobic microenvironment. Moreover, it was observed that the detection sensitivity to Hg2+ can be further improved above the nanogel phase transition temperature. At a nanogel concentration of 0.05 g L−1 and in the same Hg2+ concentration range (0–3.0 equiv.), ∼10 fold and ∼57 fold increase in fluorescence emission intensity ratio changes can be achieved at 25 and 40 °C, respectively.
Co-reporter:Xuejuan Wan and Shiyong Liu
Journal of Materials Chemistry A 2011 - vol. 21(Issue 28) pp:NaN10329-10329
Publication Date(Web):2011/06/01
DOI:10.1039/C1JM10332F
We reported on the synthesis of well-defined thermoresponsive polymers labeled with fluorescence resonance energy transfer (FRET) pairs at chain middle and terminals, which can act as single chain-based dual ratiometric fluorescent probes for pH and temperature under extremely dilute conditions. Starting from difunctional initiator containing a 7-nitro-2,1,3-benzoxadiazole (NBD) moiety, the atom transfer radical polymerization (ATRP) of oligo(ethylene glycol) monomethyl ether methacrylate (OEGMA) and di(ethylene glycol) monomethyl ether methacrylate (DEGMA), and the subsequent terminal group functionalization with Rhodamine B (RhB)-ethylenediamine derivative afforded thermoresponsive NBD-P(OEGMA-co-DEGMA)-RhB2, which were labeled with FRET donor (NBD) and acceptor moieties (RhB) at the chain middle and terminals. The fluorescence emission of terminal RhB functionalities is highly pH-dependent, i.e, non-fluorescent in neutral or alkaline media (spirolactam form) and highly fluorescent in acidic media (ring-opened acyclic form), thus the off/on switching of FRET process can be facilely modulated by solution pH. Moreover, at acidic pH and highly dilute conditions, the thermo-induced chain collapse and extension of NBD-P(OEGMA-co-DEGMA)-RhB2 can effectively modulate the spatial distance between FRET donor and acceptor moieties, leading to prominent changes in FRET efficiencies. The site-specific incorporation of one FRET donor and two pH-switchable acceptors at the chain middle and terminals of thermoresponsive polymers allows for the effective off/on switching and the modulation of efficiency of FRET processes by dually playing with solution pH and temperatures. This work represents the first report of single thermoresponsive polymer chains acting as dual ratiometric fluorescent probes under highly dilute conditions.
Co-reporter:Tao Wu, Qianqian Zhang, Jinming Hu, Guoying Zhang and Shiyong Liu
Journal of Materials Chemistry A 2012 - vol. 22(Issue 11) pp:NaN5163-5163
Publication Date(Web):2012/02/03
DOI:10.1039/C2JM15530C
We report on the fabrication of water-dispersible composite silica nanospheres covalently anchored with gold nanoparticles (AuNPs) possessing thermo-tunable spatial distributions at the outer periphery of thermoresponsive poly(N-isopropylacrylamide) (PNIPAM) brushes. Starting from initiator-functionalized silica nanoparticles, surface-initiated atom transfer radical polymerization (ATRP) of N-isopropylacrylamide (NIPAM) afforded hybrid silica nanoparticles coated with PNIPAM brushes. The substitution reaction of halogen terminal groups of grafted PNIPAM chains with sodium azide and subsequent click reaction with 1,2-dithiolane-3-pentanoic acid-N-propargylamide afforded hybrid silica nanoparticles coated with 1,2-dithiolane end-capped PNIPAM brushes. AuNPs were then covalently anchored to the outer periphery of hybrid silica nanoparticles by utilizing strong chemisorption of surface-attached dithiolane moieties to AuNPs. Dynamic laser light scattering (LLS) measurements revealed that thermosensitive PNIPAM brushes at the surface of hybrid silica nanoparticles exhibit reversible thermo-induced collapse/swelling transitions, leading to the facile thermo-modulation of spatial distributions of AuNPs covalently attached at the periphery of composite silica nanospheres and thermo-reversible surface plasmon absorption band shift. The reported strategy of covalent assembly of AuNPs into well-defined composite nanospheres possessing thermo-tunable characteristics might be further exploited for colorimetric temperature sensing and responsive SERS detection purposes.
Co-reporter:Tao Liu, Yinfeng Qian, Xianglong Hu, Zhishen Ge and Shiyong Liu
Journal of Materials Chemistry A 2012 - vol. 22(Issue 11) pp:NaN5030-5030
Publication Date(Web):2012/02/02
DOI:10.1039/C2JM15092A
We report on the utilization of mixed diblock copolymer micelles as an integrated multifunctional platform for the cancer cell-targeted delivery of chemotherapeutic drugs and magnetic resonance (MR) imaging contrast enhancement under in vitro and in vivo conditions. Two types of amphiphilic diblock copolymers, PCL-b-P(OEGMA-FA) and PCL-b-P(OEGMA-Gd), consisting of a hydrophobic poly(ε-caprolactone) (PCL) block and a hydrophilic poly(oligo(ethylene glycol) monomethyl ether methacrylate) (POEGMA) block, covalently attached with folic acid (FA) and DOTA-Gd (Gd) moieties, respectively, were synthesized via the combination of atom transfer radical polymerization (ATRP), ring-opening polymerization (ROP), and “click” post-functionalization. Mixed micelles co-assembled from PCL-b-P(OEGMA-FA) and PCL-b-P(OEGMA-Gd) possess hydrophobic PCL cores for loading chemotherapeutic drugs and hydrophilic POEGMA outer coronas functionalized with FA and Gd complexes for synergistic functions of targeted delivery and MR imaging contrast enhancement. As-prepared nanosized mixed micelles are capable of physically encapsulating paclitaxel, a well-known hydrophobic anticancer drug, with a loading content of ∼5.0 w/w%, exhibiting controlled release of up to ∼60% loaded drugs over a duration of ∼130 h. In vitrocell viability assays revealed that drug-free mixed micelles are almost non-cytotoxic up to a concentration of 0.2 g L−1, whereas paclitaxel-loaded ones can effectively kill HeLa cells at the same concentration. In vitro MR imaging experiments indicated dramatically increased T1 relaxivity (26.29 s−1mM−1) for mixed micelles compared to that of small molecule counterpart, alkynyl-DOTA-Gd (3.12 s−1mM−1). Further in vivo MR imaging experiments in rabbits revealed considerably enhanced signal intensity, prominent positive contrast enhancement, improved accumulation and retention, and extended blood circulation duration for FA-labeled mixed micellar nanoparticles within the rabbit liver, as compared to those for FA-free mixed micelles and small molecule alkynyl-DOTA-Gd complex. These preliminary results indicate that the reported mixed micellar nanocarriers possess synergistically integrated functions of cancer-targeted drug delivery and controlled release, and MR imaging contrast enhancement, which augurs well for their potential application as a novel type of theranostic platform.
Co-reporter:Xianglong Hu and Shiyong Liu
Dalton Transactions 2015 - vol. 44(Issue 9) pp:NaN3922-3922
Publication Date(Web):2014/12/23
DOI:10.1039/C4DT03609C
Responsive polymeric assemblies and hybrid superstructures fabricated from stimuli-sensitive polymers and inorganic nanoparticles (NPs) have been the subject of extensive investigations during the past few decades due to their distinct advantages such as an improved water solubility, stimuli-responsiveness, excellent biocompatibility, and facile introduction of functional units. In addition, the chemical compositions of polymeric assemblies and corresponding hybrid superstructures can be modulated via the initial synthetic design to target desired functions, fabricate smart nanostructures, and explore morphology-dependent functional optimization. Promising applications in the field of imaging, sensing, drug/gene delivery, diagnostics, and nanoreactors are being extensively investigated. This perspective article focuses on recent developments, microstructural control, and biomedical applications of stimuli-responsive polymeric assemblies as well as responsive hybrid superstructures fabricated from responsive polymers and inorganic NP building blocks (gold NPs and magnetic iron oxide NPs), and highlights their current status and future developments with selected literature reports.
Co-reporter:Jinming Hu, Guoqing Zhang and Shiyong Liu
Chemical Society Reviews 2012 - vol. 41(Issue 18) pp:NaN5949-5949
Publication Date(Web):2012/06/13
DOI:10.1039/C2CS35103J
Being responsive and adaptive to external stimuli is an intrinsic feature characteristic of all living organisms and soft matter. Specifically, responsive polymers can exhibit reversible or irreversible changes in chemical structures and/or physical properties in response to a specific signal input such as pH, temperature, ionic strength, light irradiation, mechanical force, electric and magnetic fields, and analyte of interest (e.g., ions, bioactive molecules, etc.) or an integration of them. The past decade has evidenced tremendous growth in the fundamental research of responsive polymers, and accordingly, diverse applications in fields ranging from drug or gene nanocarriers, imaging, diagnostics, smart actuators, adaptive coatings, to self-healing materials have been explored and suggested. Among a variety of external stimuli that have been utilized for the design of novel responsive polymers, enzymes have recently emerged to be a promising triggering motif. Enzyme-catalyzed reactions are highly selective and efficient toward specific substrates under mild conditions. They are involved in all biological and metabolic processes, serving as the prime protagonists in the chemistry of living organisms at a molecular level. The integration of enzyme-catalyzed reactions with responsive polymers can further broaden the design flexibility and scope of applications by endowing the latter with enhanced triggering specificity and selectivity. In this tutorial review, we describe recent developments concerning enzyme-responsive polymeric assemblies, nanoparticles, and hydrogels by highlighting this research area with selected literature reports. Three different types of systems, namely, enzyme-triggered self-assembly and aggregation of synthetic polymers, enzyme-driven disintegration and structural reorganization of polymeric assemblies and nanoparticles, and enzyme-triggered sol-to-gel and gel-to-sol transitions, are described. Their promising applications in drug controlled release, biocatalysis, imaging, sensing, and diagnostics are also discussed.
Co-reporter:Zhishen Ge and Shiyong Liu
Chemical Society Reviews 2013 - vol. 42(Issue 17) pp:NaN7325-7325
Publication Date(Web):2013/04/03
DOI:10.1039/C3CS60048C
Self-assembled nanostructures of amphiphilic and double hydrophilic block copolymers have been increasingly utilized as potent polymeric nanocarriers of therapeutic drugs, genes, bioactive molecules, and imaging/contrast agents due to improved water solubility, bioavailability, and extended blood circulation duration. Though passive and active targeted drug delivery strategies have long been proposed to promote desirable drug accumulation specifically at the disease sites, the introduction of stimuli-responsiveness into self-assembled block copolymer nanocarriers can additionally lead to controlled/triggered release of therapeutic/imaging agents into target pathological tissues and cells, with concomitant advantages of enhanced delivery efficiency and therapeutic efficacy. Appropriately designed stimuli-responsive block copolymer assemblies can exhibit chemical structure transformation, microstructural rearrangement and inversion, or even disassembly into unimers or smaller ones under external stimuli such as pH, temperature, ion strength, redox potential, light, electric, and magnetic fields, and specific bioactive molecules and metabolites. Compared to normal tissues, pathological sites such as tumor tissues typically exhibit vascular abnormalities, weak acidity (∼pH 6.8), abnormal temperatures, over-expressed proteins and enzymes, hypoxia, high levels of metabolites and reactive small molecule species, etc. Moreover, upon cellular uptake, drug-loaded polymeric nanocarriers will be subjected to intracellular pH gradients (pH 5.9–6.2 in early endosomes and pH 5.0–5.5 in late endosomes and lysosomes) and redox and H2O2 gradients within different cell organelles and the cytosol. Thus, block copolymer nanocarriers responsive to the above described bio-relevant stimuli or biochemical signals characteristic of pathologic tissues and cells will provide an alternative type of “active targeting” strategy, which can be utilized to further boost therapeutic efficacy and imaging sensitivity via disease site-specific delivery and controlled release. A variety of extracellular or intracellular stimuli innate to disease sites, such as mildly acidic pH, temperature, enzymes (matrix metalloproteinase, β-glucuronidase, and phosphatase), oxidative/reductive microenvironments, and abnormal levels of bioactive molecules or metabolites, have been utilized for this purpose. In this review, we summarize recent advances in stimuli-responsive block copolymer assemblies which are responsive to tumor and intracellular microenvironments and their applications in anticancer drug delivery and enhanced imaging sensitivity.
Co-reporter:Cong Liu, Kaka Zhang, Daoyong Chen, Ming Jiang and Shiyong Liu
Chemical Communications 2010 - vol. 46(Issue 33) pp:NaN6137-6137
Publication Date(Web):2010/07/27
DOI:10.1039/C0CC00902D
The deliberately prepared one ssDNA/one micelle complex has an unstable toroidal DNA-bound region and stable upper and lower hemispheres, and thus can self-assemble along the plane of the unstable toroidal region into free-suspending films.
Co-reporter:Junjie Li, Zhishen Ge and Shiyong Liu
Chemical Communications 2013 - vol. 49(Issue 62) pp:NaN6976-6976
Publication Date(Web):2013/06/12
DOI:10.1039/C3CC43576H
A matrix metalloproteinase-cleavable peptide-linked block copolymer was fabricated and utilized to construct PEG-sheddable polyplex micelles as smart gene delivery vectors, which were demonstrated to exhibit higher cellular uptake, improved endosomal escape, and high-efficiency gene transfection in the presence of matrix metalloproteinase-2.
.jpg)
![Pentanoic acid, 4-cyano-4-[(phenylthioxomethyl)thio]-, 2-[[2-[(2-methyl-1-oxo-2-propen-1-yl)oxy]ethyl]dithio]ethyl ester](/data/chemimg/3784300/1173118-67-6.png)
![Pentanoic acid, 4-cyano-4-[(phenylthioxomethyl)thio]-, 2-[[2-[(2-methyl-1-oxo-2-propen-1-yl)oxy]ethyl]dithio]ethyl ester](/data/chemimg/3784300/1173118-67-6_b.png)
![2H-1-Benzopyran-2-one, 3-[1-oxo-3-(2-pyridinyl)-2-propen-1-yl]-7-(2-propyn-1-yloxy)-](/data/chemimg/3784300/1623450-83-8.png)
![2H-1-Benzopyran-2-one, 3-[1-oxo-3-(2-pyridinyl)-2-propen-1-yl]-7-(2-propyn-1-yloxy)-](/data/chemimg/3784300/1623450-83-8_b.png)
![2-Propenoic acid, 2-methyl-, 2-[[[2-(1',3'-dihydro-3',3'-dimethyl-6-nitrospiro[2H-1-benzopyran-2,2'-[2H]indol]-1'-yl)ethoxy]carbonyl]amino]ethyl ester](/data/chemimg/3784300/1836169-96-0.png)
![2-Propenoic acid, 2-methyl-, 2-[[[2-(1',3'-dihydro-3',3'-dimethyl-6-nitrospiro[2H-1-benzopyran-2,2'-[2H]indol]-1'-yl)ethoxy]carbonyl]amino]ethyl ester](/data/chemimg/3784300/1836169-96-0_b.png)

![2-Propenoic acid, 2-methyl-, 2-[[[2-(1',3'-dihydro-3',3'-dimethyl-6-nitrospiro[2H-1-benzopyran-2,2'-[2H]indol]-1'-yl)ethoxy]carbonyl]oxy]ethyl ester](/data/chemimg/3784300/1836170-01-4.png)
![2-Propenoic acid, 2-methyl-, 2-[[[2-(1',3'-dihydro-3',3'-dimethyl-6-nitrospiro[2H-1-benzopyran-2,2'-[2H]indol]-1'-yl)ethoxy]carbonyl]oxy]ethyl ester](/data/chemimg/3784300/1836170-01-4_b.png)
![Pentanoic acid, 4-cyano-4-[(phenylthioxomethyl)thio]-, 1,1'-(1,2-ethanediyl) ester](/data/chemimg/3785100/1563174-47-9.png)
![Pentanoic acid, 4-cyano-4-[(phenylthioxomethyl)thio]-, 1,1'-(1,2-ethanediyl) ester](/data/chemimg/3785100/1563174-47-9_b.png)


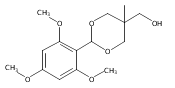
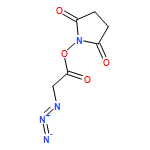
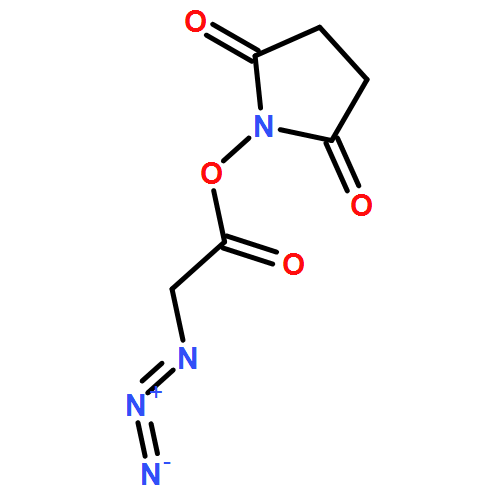










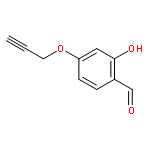






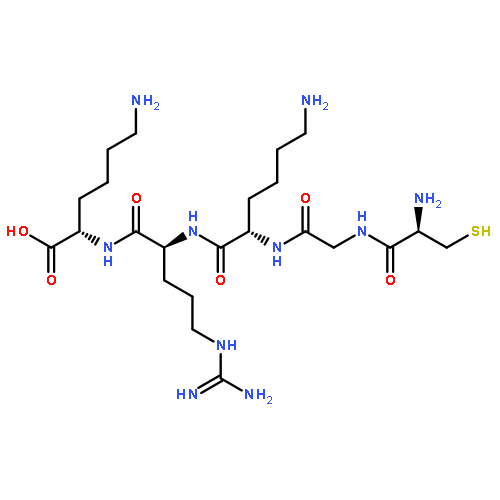
![2-[4-(aminoiminomethyl)phenyl]-1H-Indole-6-carboximidamide](http://img.cochemist.com/ccimg/47200/47165-04-8.png)
![2-[4-(aminoiminomethyl)phenyl]-1H-Indole-6-carboximidamide](http://img.cochemist.com/ccimg/47200/47165-04-8_b.png)
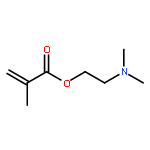
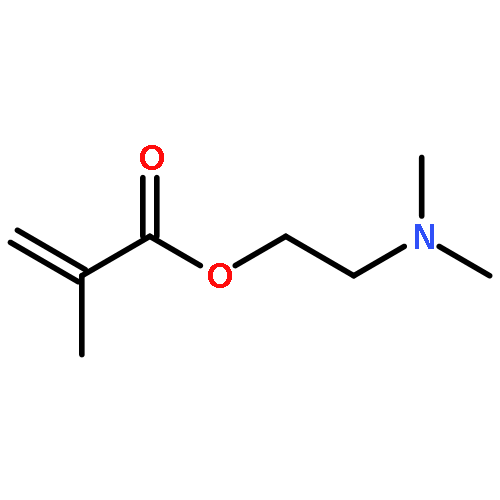


![2-Propenoic acid, 2-methyl-, 2-[(3-acetyl-2-oxo-2H-1-benzopyran-7-yl)oxy]ethyl ester, polymer with 2-[bis(1-methylethyl)amino]ethyl 2-methyl-2-](http://img.cochemist.com/ccimg/1683600/1683569-22-3.png)
![2-Propenoic acid, 2-methyl-, 2-[(3-acetyl-2-oxo-2H-1-benzopyran-7-yl)oxy]ethyl ester, polymer with 2-[bis(1-methylethyl)amino]ethyl 2-methyl-2-](http://img.cochemist.com/ccimg/1683600/1683569-22-3_b.png)
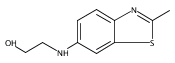












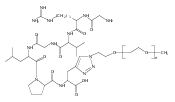












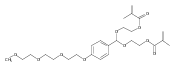
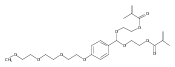




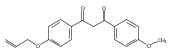















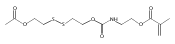
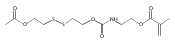






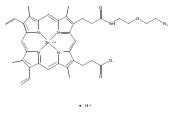
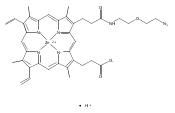




























































































































![2-Propenoic acid, 2-methyl-, 2-[(2-hydroxyethyl)dithio]ethyl ester](/data/chemimg/147300/1205532-50-8.png)
![2-Propenoic acid, 2-methyl-, 2-[(2-hydroxyethyl)dithio]ethyl ester](/data/chemimg/147300/1205532-50-8_b.png)







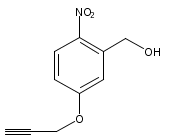


![Pentanoic acid, 4-cyano-4-[(phenylthioxomethyl)thio]-, 2-propyn-1-yl ester](/data/chemimg/190000/1075252-15-1.png)
![Pentanoic acid, 4-cyano-4-[(phenylthioxomethyl)thio]-, 2-propyn-1-yl ester](/data/chemimg/190000/1075252-15-1_b.png)


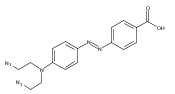



![Spiro[1H-isoindole-1,9'-[9H]xanthen]-3(2H)-one, 2-(2-aminoethyl)-3',6'-bis(diethylamino)-](/data/chemimg/106200/950846-89-6.png)
![Spiro[1H-isoindole-1,9'-[9H]xanthen]-3(2H)-one, 2-(2-aminoethyl)-3',6'-bis(diethylamino)-](/data/chemimg/106200/950846-89-6_b.png)









![Propanoic acid, 2-[[(ethylthio)thioxomethyl]thio]-2-methyl-](/data/chemimg/113500/881037-62-3.png)
![Propanoic acid, 2-[[(ethylthio)thioxomethyl]thio]-2-methyl-](/data/chemimg/113500/881037-62-3_b.png)
![Ethanol, 2-[[2-(acetyloxy)ethyl]dithio]-](http://img.cochemist.com/ccimg/877900/877864-03-4.png)
![Ethanol, 2-[[2-(acetyloxy)ethyl]dithio]-](http://img.cochemist.com/ccimg/877900/877864-03-4_b.png)





























![Benzaldehyde, 4-[2-[2-(2-methoxyethoxy)ethoxy]ethoxy]-](http://img.cochemist.com/ccimg/153400/153364-63-7.png)
![Benzaldehyde, 4-[2-[2-(2-methoxyethoxy)ethoxy]ethoxy]-](http://img.cochemist.com/ccimg/153400/153364-63-7_b.png)







![Gadolinate(1-),[1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetato(4-)-kN1,kN4,kN7,kN10,kO1,kO4,kO7,kO10]-, sodium (9CI)](http://img.cochemist.com/ccimg/93000/92923-44-9.png)
![Gadolinate(1-),[1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetato(4-)-kN1,kN4,kN7,kN10,kO1,kO4,kO7,kO10]-, sodium (9CI)](http://img.cochemist.com/ccimg/93000/92923-44-9_b.png)


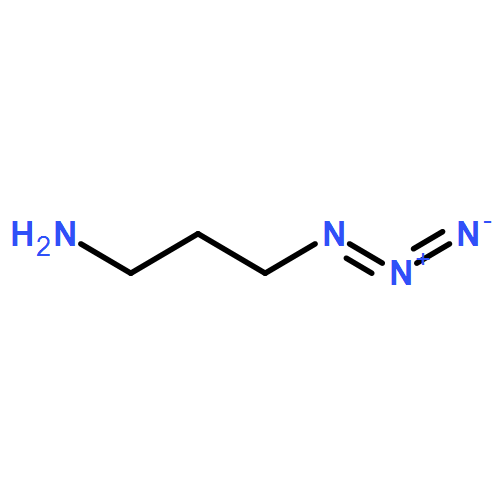

![Spiro[1H-isoindole-1,9'-[9H]xanthen]-3(2H)-one, 2-amino-3',6'-bis(diethylamino)-](/data/chemimg/659200/74317-53-6.png)
![Spiro[1H-isoindole-1,9'-[9H]xanthen]-3(2H)-one, 2-amino-3',6'-bis(diethylamino)-](/data/chemimg/659200/74317-53-6_b.png)

![2-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]ethanol](/data/chemimg/1305800/65703-47-1.png)
![2-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]ethanol](/data/chemimg/1305800/65703-47-1_b.png)


![1H-Benz[de]isoquinoline-1,3(2H)-dione, 6-bromo-2-(2-hydroxyethyl)-](/data/chemimg/3232400/52559-37-2.png)
![1H-Benz[de]isoquinoline-1,3(2H)-dione, 6-bromo-2-(2-hydroxyethyl)-](/data/chemimg/3232400/52559-37-2_b.png)
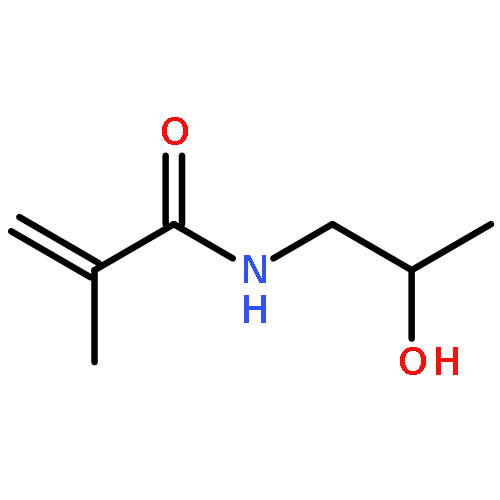







![2-Propenoic acid, 2-methyl-, 2-(3',3'-dimethyl-6-nitrospiro[2H-1-benzopyran-2,2'-[2H]indol]-1'(3'H)-yl)ethyl ester](/data/chemimg/3778900/25952-50-5.png)
![2-Propenoic acid, 2-methyl-, 2-(3',3'-dimethyl-6-nitrospiro[2H-1-benzopyran-2,2'-[2H]indol]-1'(3'H)-yl)ethyl ester](/data/chemimg/3778900/25952-50-5_b.png)



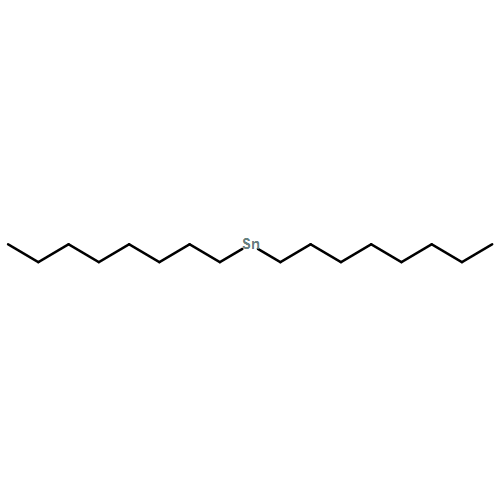




![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3.png)
![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3_b.png)







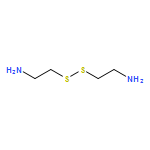
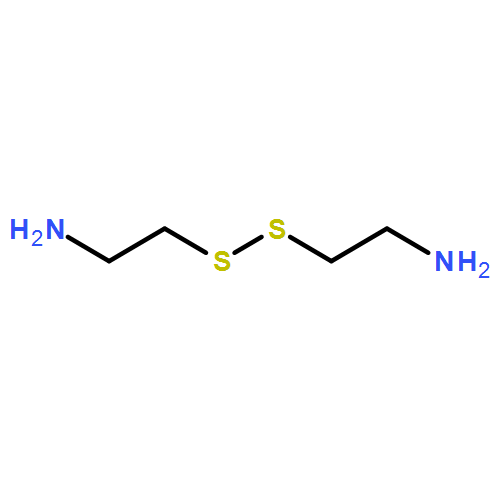


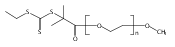

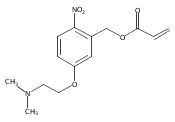
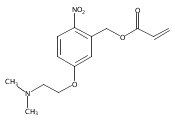
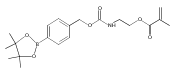



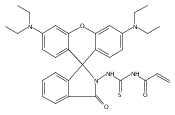
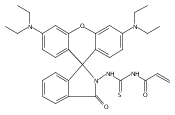








![2-Propenoic acid, 2-methyl-, 2-[(3-acetyl-2-oxo-2H-1-benzopyran-7-yl)oxy]ethyl ester](/data/chemimg/117300/841223-05-0.png)
![2-Propenoic acid, 2-methyl-, 2-[(3-acetyl-2-oxo-2H-1-benzopyran-7-yl)oxy]ethyl ester](/data/chemimg/117300/841223-05-0_b.png)







![Xanthylium, 3,6-bis(diethylamino)-9-[2-[[2-[(1-oxo-2-propen-1-yl)oxy]ethoxy]carbonyl]phenyl]-, chloride (1:1)](http://img.cochemist.com/data/images/1156509-18-0.png)
![Xanthylium, 3,6-bis(diethylamino)-9-[2-[[2-[(1-oxo-2-propen-1-yl)oxy]ethoxy]carbonyl]phenyl]-, chloride (1:1)](http://img.cochemist.com/data/images/1156509-18-0_b.png)


![Propanoic acid, 2-methyl-2-[[(propylthio)thioxomethyl]thio]-](/data/chemimg/190000/1254954-46-5.png)
![Propanoic acid, 2-methyl-2-[[(propylthio)thioxomethyl]thio]-](/data/chemimg/190000/1254954-46-5_b.png)