Co-reporter:Bartosz Bieszczad
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 31) pp:6483-6492
Publication Date(Web):2017/08/09
DOI:10.1039/C7OB00751E
Tertiary alcohol precursors of both C2 diastereoisomers of α-tocopherol were prepared in three ways by our recently reported asymmetric Grignard synthesis. The versatility of Grignard chemistry inherent in its three-way disconnection was exploited to allow the synthesis of three product grades: 77 : 23 dr (5 steps), 81 : 19 dr (5 steps) and 96 : 4 dr (7 steps, one gram scale) from readily available and abundant starting materials. The products were converted to their respective α-tocopherols in 3 steps, which allowed a definitive re-assignment of their absolute configurations.
Co-reporter:Kamalraj V. Rajendran; Kirill V. Nikitin
Journal of the American Chemical Society 2015 Volume 137(Issue 29) pp:9375-9381
Publication Date(Web):July 17, 2015
DOI:10.1021/jacs.5b04415
The dynamic resolution of tertiary phosphines and phosphine oxides was monitored by NMR spectroscopy. It was found that the stereoselectivity is set during the formation of the diastereomeric alkoxyphosphonium salts (DAPS), such that their initial diastereomeric excess (de) limits the final enantiomeric excess (ee) of any phosphorus products derived from them. However, 31P NMR monitoring of the spontaneous thermal decomposition of the DAPS shows consistent diastereomeric self-enrichment, indicating a higher rate constant for decomposition of the minor diastereomer. This crucial observation was confirmed by reductive trapping of the unreacted enriched DAPS with lithium tri-sec-butylborohydride (commercially distributed as L-Selectride reagent) at different time intervals after the start of reaction, which gives progressively higher ee of the phosphine product with time. It is proposed that the Hammond postulate operates for both formation and decomposition of DAPS intermediate so that the lower rate of formation and faster subsequent collapse of the minor isomer are thermodynamically linked. This kinetic enhancement of kinetic resolution furnishes up to 97% ee product.
Co-reporter:Niall P. Kenny, Kamalraj V. Rajendran and Declan G. Gilheany
Chemical Communications 2015 vol. 51(Issue 92) pp:16561-16564
Publication Date(Web):28 Sep 2015
DOI:10.1039/C5CC06389B
A method is reported for the phosphoryl bond cleavage of O-alkyl phosphinates, phosphinothioates and certain phosphonamidates to furnish the corresponding P(III) borane adducts. The two-step procedure relies upon initial activation of the phosphoryl bond with an alkyl triflate, followed by reduction of the resulting intermediate using lithium borohydride.
Co-reporter:Sulaiman S. Al Sulaimi;Kamalraj V. Rajendran
European Journal of Organic Chemistry 2015 Volume 2015( Issue 27) pp:5959-5965
Publication Date(Web):
DOI:10.1002/ejoc.201500521
Abstract
We report LiBH4 as a preferred, simple and effective reagent for reductive boronation of achiral and racemic chlorophosphonium salts (CPS) and for diastereomeric alkoxyphosphonium salts (DAPS), both of which are, in turn, easily generated from either the corresponding phosphane or, more conveniently, the phosphane oxide. Further, we have shown that the DAPS reduction/boronation could be achieved with complete stereocontrol to give scalemic phosphane–borane directly in excellent yield and enantiomeric excess (ee). This new methodology was employed to investigate the effects of aryl substitution on the outcome of dynamic kinetic resolution of arylmethylphenylphosphanes and phosphane oxides via DAPS. It was found that substitution at the ortho position strongly affects the degree of stereoselection. However, surprisingly, we confirmed that there was no variation of stereoselectivity seen with the electronic effect of substituents on the para position.
Co-reporter:Peter A. Byrne;Jimmy Muldoon;Yannick Ortin;Helge Müller-Bunz
European Journal of Organic Chemistry 2014 Volume 2014( Issue 1) pp:86-98
Publication Date(Web):
DOI:10.1002/ejoc.201301103
Abstract
Within the currently accepted mechanism of the Li-salt-free Wittig reaction, the phenomenon of stereochemical drift remains the one remaining “loose end” in an otherwise internally consistent explanation of a large body of diverse observations. The term describes the nonstereospecific decomposition of the oxaphosphetane (OPA) intermediate in reactions of certain alkylides with certain aldehydes. In this paper, it is shown that the previous examples in which drift occurs are not merely isolated aberrations from the observed norm, but rather that there is a general phenomenon in reactions of ethylides with benzaldehydes. Variable-temperature NMR (VTNMR) spectroscopy was used to establish that the amount and diastereomeric ratio of the OPA intermediates do not change below a certain temperature. At and above the temperature at which OPA decomposition to alkene and phosphine oxide begins to occur, the alkene shows a different diastereomeric ratio to the OPA, which indicates the occurrence of stereochemical drift. In one example, owing to an apparent remarkable coincidence of rates, the diastereomeric ratio of the OPA does not change above the decomposition temperature, even though stereochemical drift occurs in the formation of the alkene product. An alternative mechanism for drift involving its catalysis by aldehyde was not confirmed. Drift was also shown not to occur in similar Wittig reactions of structurally related longer-chain alkylides by stereospecific decomposition of OPA intermediates generated from β-hydroxyphosphonium salts (β-HPSs). The extremely useful (and generally applicable) NMR techniques, 1H–31P HMBC and selective 1H{31P}, which we have utilised to establish kinetic diastereomeric ratios, are described in full for the first time. Details of the determination of the relative stereochemistry of two β-HPSs (derived from acid quenching of OPAs) by X-ray crystallography are also given.
Co-reporter:Peter A. Byrne and Declan G. Gilheany
Chemical Society Reviews 2013 vol. 42(Issue 16) pp:6670-6696
Publication Date(Web):14 May 2013
DOI:10.1039/C3CS60105F
The mechanism of the Wittig reaction has long been a contentious issue in organic chemistry. Even now, more than 50 years after its announcement, its presentation in many modern undergraduate textbooks is either overly simplified or entirely inaccurate. In this review, we gather together the huge body of evidence that has been amassed to show that the Li salt-free Wittig reactions of non-stabilised, semi-stabilised and stabilised ylides all occur under kinetic control by a common mechanism in which oxaphosphetane (OPA) is the first-formed and only intermediate. The numerous recent significant additions to the subject – including computational studies and experimental material pertinent to both steps of the reaction (OPA formation and its decomposition) are discussed in detail, and the currently accepted explanations for the source of the stereoselectivity in Wittig reactions are given. We also present the other mechanistic proposals that have been made during the history of the Wittig reaction, and show how they are unable to account for all of the experimental evidence that is now available. Details of certain experimental facts to do with Wittig reactions in the presence of Li cation are also included, although the precise mechanistic details of such reactions are yet to be established conclusively. We make the case that a clear distinction should henceforth be made between the unknown “Li-present” and the now well-established “Li salt-free” Wittig mechanisms.
Co-reporter:Kirill Nikitin, Helge Müller-Bunz and Declan Gilheany
Chemical Communications 2013 vol. 49(Issue 14) pp:1434-1436
Publication Date(Web):21 Dec 2012
DOI:10.1039/C2CC38363B
Triphenylhalophosphonium halides, Ph3PX2, form crystals comprising bridged linear cations [Ph3P–X–X–X–PPh3]+ where the X3 bridge is shortened from 6.56 Å in Cl–Cl–Cl to 6.37 Å in the Br–Br–Br system. It is proposed that this structure is stabilised by five-centre/six-electron (5c–6e) hypervalent interactions.
Co-reporter:Kamalraj V. Rajendran, Lorna Kennedy, Cormac T. O’Connor, Enda Bergin, Declan G. Gilheany
Tetrahedron Letters 2013 Volume 54(Issue 51) pp:7009-7012
Publication Date(Web):18 December 2013
DOI:10.1016/j.tetlet.2013.10.044
A wide selection of phosphine activators has been screened to improve the selection process in the asymmetric Appel reaction. Of the activators screened, hexachloroacetone (HCA) gave the highest selectivity with excellent yield, but at least one of its by-products, pentachloroacetone (PCA), can become involved in the selection process. In addressing this, a new reaction of phosphines with oxalyl chloride was discovered that can also generate the key intermediate chlorophosphonium salt (CPS), gives better enantioselectivity and possesses significant advantages over other phosphine activators.
Co-reporter:Niall P. Kenny;Dr. Kamalraj V. Rajendran;Elizabeth V. Jennings ; Declan G. Gilheany
Chemistry - A European Journal 2013 Volume 19( Issue 42) pp:14210-14214
Publication Date(Web):
DOI:10.1002/chem.201302907
Abstract
In contrast to tertiary phosphine oxides, the deoxygenation of aminophosphine oxides is effectively impossible due to the need to break the immensely strong and inert PO bond in the presence of a relatively weak and more reactive PN bond. This long-standing problem in organophosphorus synthesis is solved by use of oxalyl chloride, which chemoselectively cleaves the PO bond forming a chlorophosphonium salt, leaving the PN bond(s) intact. Subsequent reduction of the chlorophosphonium salt with sodium borohydride forms the PIII aminophosphine borane adduct. This simple one-pot procedure was applied with good yields for a wide range of PN-containing phosphoryl compounds. The borane product can be easily deprotected to produce the free PIII aminophosphine. Along with no observed PN bond cleavage, the use of sodium borohydride also permits the presence of ester functional groups in the substrate. The availability of this methodology opens up previously unavailable synthetic options in organophosphorus chemistry, two of which are exemplified.
Co-reporter:Damien J. Carr, Jaya Satyanarayana Kudavalli, Katherine S. Dunne, Helge Müller-Bunz, and Declan G. Gilheany
The Journal of Organic Chemistry 2013 Volume 78(Issue 20) pp:10500-10505
Publication Date(Web):September 30, 2013
DOI:10.1021/jo401318g
Racemic 2,3-dihydro-1-phenylbenzo[b]phosphole was obtained by reduction of 1-phenylbenzo[b]phosphole-1-oxide, itself derived by ring-closing metathesis of phenylstyrylvinylphosphine oxide. The title compound was then reoxidized under asymmetric Appel conditions. Comparison of the sense and degree of the stereoselectivity to those obtained with an open-chain analogue indicated that the ring system does not affect the selectivity of the process. This in turn strongly suggests that the stereoselection is not related to pseudorotamer preferences in putative phosphorane intermediates.
Co-reporter:Peter A. Byrne
Journal of the American Chemical Society 2012 Volume 134(Issue 22) pp:9225-9239
Publication Date(Web):May 7, 2012
DOI:10.1021/ja300943z
The true course of the lithium salt-free Wittig reaction has long been a contentious issue in organic chemistry. Herein we report an experimental effect that is common to the Wittig reactions of all of the three major phosphonium ylide classes (non-stabilized, semi-stabilized, and stabilized): there is consistently increased selectivity for cis-oxaphosphetane and its derived products (Z-alkene and erythro-β-hydroxyphosphonium salt) in reactions involving aldehydes bearing heteroatom substituents in the β-position. The effect operates with both benzaldehydes and aliphatic aldehydes and is shown not to operate in the absence of the heteroatom substituent on the aldehyde. The discovery of an effect that is common to reactions of all ylide types strongly argues for the operation of a common mechanism in all Li salt-free Wittig reactions. In addition, the results are shown to be most easily explained by the [2+2] cycloaddition mechanism proposed by Vedejs and co-workers as supplemented by Aggarwal, Harvey, and co-workers, thus providing strong confirmatory evidence in support of that mechanism. Notably, a cooperative effect of ortho-substituents in the case of semi-stabilized ylides is confirmed and is accommodated by the cycloaddition mechanism. The effect is also shown to operate in reactions of triphenylphosphine-derived ylides and has previously been observed for reactions under aqueous conditions, thus for the first time providing evidence that kinetic control is in operation in both of these cases.
Co-reporter:Kamalraj V. Rajendran and Declan G. Gilheany
Chemical Communications 2012 vol. 48(Issue 80) pp:10040-10042
Publication Date(Web):16 Aug 2012
DOI:10.1039/C2CC34136K
Sequential treatment of racemic phosphine oxides with oxalyl chloride and chiral non-racemic alcohol generates the same ratios of diastereomeric alkoxyphosphonium salts obtained in the corresponding asymmetric Appel process, strongly implicating the intermediate chlorophosphonium salt in the stereoselecting step. Subsequent reduction allows a novel synthesis of enantioenriched P-stereogenic phosphines–phosphine boranes.
Co-reporter:Kamalraj V. Rajendran and Declan G. Gilheany
Chemical Communications 2012 vol. 48(Issue 6) pp:817-819
Publication Date(Web):21 Oct 2011
DOI:10.1039/C1CC14856G
A variety of phosphine oxides and sulfides can be efficiently converted directly to the corresponding phosphine boranes using oxalyl chloride followed by sodium borohydride. Optically active P-stereogenic phosphine oxides can be converted stereospecifically to phosphine boranes with inversion of configuration by treatment with Meerwein's salt followed by sodium borohydride.
Co-reporter:Peter A. Byrne, Kamalraj V. Rajendran, Jimmy Muldoon and Declan G. Gilheany
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 17) pp:3531-3537
Publication Date(Web):05 Mar 2012
DOI:10.1039/C2OB07074J
A mild method for the facile removal of phosphine oxide from the crude products of Wittig and Appel reactions is described. Work-up with oxalyl chloride to generate insoluble chlorophosphonium salt (CPS) yields phosphorus-free products for a wide variety of these reactions. The CPS product can be further converted into phosphine.
Co-reporter:Kamalraj V. Rajendran;Jaya S. Kudavalli;Katherine S. Dunne
European Journal of Organic Chemistry 2012 Volume 2012( Issue 14) pp:2720-2723
Publication Date(Web):
DOI:10.1002/ejoc.201200285
Abstract
An efficient one-pot synthesis has been developed of enantioenriched P-stereogenic phosphanes and phosphane boranes from the corresponding racemic phosphanes in excellent yield under asymmetric Appel conditions. The chiral auxiliary (menthol) can also be recovered unchanged. The simple and efficient protocol significantly expands the scope of our asymmetric Appel process. The crucial step in the preparation involves stereospecific reduction of intermediate diastereomeric alkoxyphosphonium salts, which are obtained in the reaction of phosphane, hexachloroacetone, and menthol. Thereby, reaction with LiAlH4 or NaBH4 gives the corresponding phosphanes or phosphane boranes, respectively.
Co-reporter:Peter A. Byrne, Lee J. Higham, Pádraic McGovern, Declan G. Gilheany
Tetrahedron Letters 2012 Volume 53(Issue 49) pp:6701-6704
Publication Date(Web):5 December 2012
DOI:10.1016/j.tetlet.2012.09.123
Wittig reaction products of keto-stabilised ylides with ortho-substituted benzaldehydes are found to show significantly higher than expected Z-alkene content (up to 50%) compared to analogous reactions of the same ylides with benzaldehyde itself. A cooperative effect is seen whereby the unusual Z-content is further augmented if the ylide bears greater steric bulk in the α′-position. These results are consistent with our previous observations on reactions of all ylide types with aldehydes bearing a β-heteroatom. Significantly, the cooperative effect, previously seen only with semi-stabilised ylides, has now been extended to stabilised ylides. Both the anomalous increase in Z-content and the cooperative effect can be rationalised within the [2+2] cycloaddition mechanism of the Wittig reaction.
Co-reporter:Kamalraj V. Rajendran, Damien J. Carr, Declan G. Gilheany
Tetrahedron Letters 2011 Volume 52(Issue 52) pp:7113-7115
Publication Date(Web):28 December 2011
DOI:10.1016/j.tetlet.2011.10.101
We report a very convenient laboratory preparation of pentachloroacetone (PCA) by selective dechlorination of hexachloroacetone (HCA) via reaction with triphenylphosphine in the presence of methanol or aromatic alcohols.
Co-reporter:Eoin Rafter, Jimmy Muldoon, Helge Müller Bunz, Declan G. Gilheany
Tetrahedron: Asymmetry 2011 Volume 22(16–17) pp:1680-1686
Publication Date(Web):15 September 2011
DOI:10.1016/j.tetasy.2011.09.007
A synthetic route to BINAP disulfide analogues with chirality on one of the phosphorus atoms was developed. The synthesis of these ligands was achieved by step-wise palladium and nickel-catalysed coupling reactions of the precursor binaphthyl triflate with secondary phosphines and phosphine oxides, respectively. C2-Unsymmeric BINAP bissulfide analogues with the same aryl substituents were also synthesized for the sake of comparison. Preliminary studies of the reduction of these sulfides to the free phosphines are also described.(R,R)-2-(Methylphenylthiophosphinyl)-2′-(diphenylthiophosphinyl)-1,1′-binaphthylC39H31P2S2[α]D = +3 (c 1, CH2Cl2)Source of chirality: enantiopure backbone and flash chromatography separation of diastereomersAbsolute configuration: (R,R)(S,R)-2-(Methylphenylthiophosphinyl)-2′-(diphenylthiophosphinyl)-1,1′-binaphthylC39H31P2S2[α]D = −86 (c 0.25, CH2Cl2)Source of chirality: enantiopure backbone and flash chromatography separation of diastereomersAbsolute configuration: (S,R)(R)-2-(Bisanisylthiophosphinyl)-2′-(diphenylthiophosphinyl)-1,1′-binaphthylC46H37O2P2S2[α]D = −61 (c 1, CH2Cl2)Source of chirality: enantiopure backboneAbsolute configuration: (R)(R)-2-(Bis-o-tolylthiophosphinyl)-2′-(diphenylthiophosphinyl)-1,1′-binaphthylC46H37P2S2[α]D = +26.5 (c 1, CH2Cl2)Source of chirality: enantiopure backboneAbsolute configuration: (R)
Co-reporter:Eoin F. Clarke, Eoin Rafter, Helge Müller-Bunz, Lee J. Higham, Declan G. Gilheany
Journal of Organometallic Chemistry 2011 696(23) pp: 3608-3615
Publication Date(Web):
DOI:10.1016/j.jorganchem.2011.08.010
Co-reporter:Kamalraj V. Rajendran;Lorna Kennedy
European Journal of Organic Chemistry 2010 Volume 2010( Issue 29) pp:5642-5649
Publication Date(Web):
DOI:10.1002/ejoc.201000733
Abstract
The effects of aryl ring substitution on the dynamic resolution of aryl(methyl)phenylphosphanes under asymmetric Appel reaction conditions have been studied. As expected, substitution at the ortho position strongly affects the degree ofstereoselection that can be achieved. Unexpectedly, however, there was no variation of stereoselectivity with the electronic nature of the para substituents, which suggests that there are two selection processes in operation. An unusual halogen-exchange process was observed in the arylphenylphosphinous chlorides on route to the required tertiary phosphane substrates.
Co-reporter:Rachel M. Hiney;Lee J. Higham Dr.;Helge Müller-Bunz Dr. Dr.
Angewandte Chemie 2006 Volume 118(Issue 43) pp:
Publication Date(Web):5 OCT 2006
DOI:10.1002/ange.200602143
Gezähmtes Feuer: Enantiomerenreine primäre Phosphane wurden entwickelt, die sowohl im festen Zustand als auch in Lösung bemerkenswert stabil gegen Luftoxidation sind (siehe Beispiel). Die Stabilität dieser neuen Klasse von Liganden scheint auf der Konjugation des ausgedehnten π-Elektronensystems zu beruhen, wobei ein zusätzlicher Ring ausreicht.
Co-reporter:Rachel M. Hiney;Lee J. Higham Dr.;Helge Müller-Bunz Dr. Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 43) pp:
Publication Date(Web):5 OCT 2006
DOI:10.1002/anie.200602143
No rings of fire! Enantiopure primary phosphanes have been developed which are remarkably stable to air oxidation in both solid and solution states (see example). This new class of stable ligand synthon appears to owe its stability to conjugation in their aryl backbones with the extended π ring system. Even one extra ring is good enough.
Co-reporter:Lee J. Higham, Eoin F. Clarke, Helge Müller-Bunz, Declan G. Gilheany
Journal of Organometallic Chemistry 2005 Volume 690(Issue 1) pp:211-219
Publication Date(Web):3 January 2005
DOI:10.1016/j.jorganchem.2004.09.015
The novel P-chirogenic anisylphenylMOP derivatives (R,R) and (R,S)-2-(anisylphenylphosphino)-2′-methoxy-1,1′-binaphthyl (10a and b) have been synthesized and their corresponding oxides characterised by X-ray crystallography. The results of a parallel screening regimen with various reducing agents highlight the sensitivity of the tertiary phosphine oxides to epimerisation and, interestingly, reveal that the PO, O–CH3 and P–C6H5 bonds can all be cleaved selectively depending on the reducing agents employed. An alternative synthesis was provided by direct coupling of the secondary phosphine with (R)-methoxytriflate 4, which led to the isolation of the optically pure P-chirogenic phosphines via their borane adducts. A brief study of the coordination chemistry of 10a with different rhodium precursors, relevant to the catalytic asymmetric addition of boronic acids to aldehydes is also reported.The synthesis and resolution of the diastereomeric P-chirogenic (R,R) and (R,S) anisylphenylMOP phosphines is presented. In addition, the crystal structures of both corresponding oxides and their unusual behaviour towards a variety of reducing agents is described.
Co-reporter:Lee J. Higham, P. Gabriel Kelly, David M. Corr, Helge Müller-Bunz, Brian J. Walker and Declan G. Gilheany
Chemical Communications 2004 (Issue 6) pp:684-685
Publication Date(Web):11 Feb 2004
DOI:10.1039/B316759C
The Ramirez ylide undergoes electrophilic substitution with acetylenedicarboxylates to form Z and E adducts. The latter can react by cycloaddition with another equivalent of the alkyne to provide a new route to novel tetra-substituted azulenes, which show interesting bond localisation and crystal packing effects.
Co-reporter:E.M. McGarrigle, D.M. Murphy, D.G. Gilheany
Tetrahedron: Asymmetry 2004 Volume 15(Issue 8) pp:1343-1354
Publication Date(Web):19 April 2004
DOI:10.1016/j.tetasy.2004.03.010
A series of Cr(salen) complexes have been synthesised from 5-substituted-3-bromosalicylaldehydes and trans-1,2-cyclohexanediamine. These have been used to probe the Cr(salen)-mediated asymmetric epoxidation of alkenes. No simple correlation was found between the electronic character of the salen-substituents and the enantioselectivity––multiple oxidation pathways are proposed as a possible explanation. Enantioselectivities of up to 90% have been achieved using a novel, synthetically accessible Cr(salen) complex.Graphic(R,R)-(−)-N,N′-Bis(3-bromosalicylidene)-trans-cyclohexane-1,2-diamineC20H20Br2N2O2Ee = 100%[α]D25 = −578 (c 0.601, CH2Cl2)Source of chirality: enantiopure starting materialAbsolute configuration: (R,R)(R,R)-(−)-N,N′-Bis(3-bromo-5-methylsalicylidene)-trans-cyclohexane-1,2-diamineC22H24Br2N2O2Ee = 100%[α]D31 = −498 (c 1.03, CH2Cl2)Source of chirality: enantiopure starting materialAbsolute configuration: (R,R)(R,R)-(−)-N,N′-Bis(3-bromo-5-ethylsalicylidene)-trans-cyclohexane-1,2-diamineC24H28Br2N2O2Ee = 100%[α]D34 = −450 (c 1.06, CH2Cl2)Source of chirality: enantiopure starting materialAbsolute configuration: (R,R)(R,R)-(−)-N,N′-Bis(3-bromo-5-tert-butyl-salicylidene)-trans-cyclohexane-1,2-diamineC28H36Br2N2O2Ee = 100%[α]D32 = −396 (c 1.19, CH2Cl2)Source of chirality: enantiopure starting materialAbsolute configuration: (R,R)(R,R)-(−)-N,N′-Bis(3-bromo-5-methoxysalicylidene)-trans-cyclohexane-1,2-diamineC22H24Br2N2O4Ee = 100%[α]D23 = −432 (c 0.41, CH2Cl2)Source of chirality: enantiopure starting materialAbsolute configuration: (R,R)
Co-reporter:Hans-Jörg Schanz, Michael A Linseis, Declan G Gilheany
Tetrahedron: Asymmetry 2003 Volume 14(Issue 18) pp:2763-2769
Publication Date(Web):19 September 2003
DOI:10.1016/S0957-4166(03)00586-X
Starting from inexpensive l-(+)-tartaric acid, it was possible to resolve and obtain pure both enantiomers of trans-cyclohexane-1,2-diamine 1 and thence both enantiomers of BINOL 2, two of the most powerful, chiral inducing backbones in asymmetric catalysis. The modified method is very economic, not only due to an almost doubling of the overall yields of enantiomerically pure compounds (86% 1, 83% 2) but also due to the easy recovery of resolving agent 1 [66% (R,R)-1, 79% (S,S)-1] in the BINOL resolution. An improvement in the yield of the preparation of racemic BINOL is also recorded.Graphic
Co-reporter:Adrian M. Daly, Declan G. Gilheany
Tetrahedron: Asymmetry 2003 Volume 14(Issue 1) pp:127-137
Publication Date(Web):6 January 2003
DOI:10.1016/S0957-4166(02)00757-7
A complete synthesis of enantiopure trans-cyclopentane-1,2-diamine and trans-cyclobutane-1,2-diamine is described. These diamines have been used as components of novel chiral salen ligands whose chromium and manganese complexes were then evaluated as oxygen transfer agents in the asymmetric epoxidation of alkenes.Graphic(R,R)-(−)-trans-Cyclopentane-1,2-diamine di-(+)-tartrateC13H24N2O12Ee >99%[α]D20=+10.0 (c 2, H2O, lit. +10.1°)Source of chirality: resolution(+)-trans-Cyclobutane-1,2-diamine di-(+)-tartrateC12H22N2O12Ee >99%[α]D20=+28.0 (c 1, H2O)Source of chirality: resolution(R,R)-(−)-N,N′-Bis(3-trifluoromethylsalicylidene)-trans-cyclopentane-1,2-diamineC20H16F6N2O2Ee >99%[α]D20=−437 (c 1, CH2Cl2)Source of chirality: resolution(+)-N,N′-Bis(3-trifluoromethylsalicylidene)-trans-cyclobutane-1,2-diamineC20H16F6N2O2Ee >99%[α]D20=+485 (c 1, CH2Cl2)Source of chirality: resolution(R,R)-(−)-N,N′-Bis(3,5-di-tert-butylsalicylidene)-trans-cyclopentane-1,2-diamineC35H52N2O2Ee >99%[α]D20=−365 (c 1, CH2Cl2)Source of chirality: resolution(+)-N,N′-Bis(3,5-di-tert-butylsalicylidene)-trans-cyclobutane-1,2-diamineC34H50N2O2Ee >99%[α]D20=+400 (c 1, CH2Cl2)Source of chirality: resolution
Co-reporter:James G. Walsh
Journal of Heterocyclic Chemistry 2002 Volume 39(Issue 6) pp:1273-1278
Publication Date(Web):11 MAR 2009
DOI:10.1002/jhet.5570390624
The reaction of Z,Z-2,3,4,5-tetrahalo-2,4-dien-1,6-dibromides (3, R1 - R4 = Cl, Br) with primary amines in the presence of potassium carbonate leads to both the dihydroazepines 4 and secondary enamines 5. The formation of enamine is suppressed with toluene sulfonamide as nitrogen source. (2Z,4Z)-2,3,4,5-Tetrabromo-hexa-2,4-diene-1,6-diol (2, R1 - R4 = Br) is atropisomeric in solution.
Co-reporter:Peter Brt Dr.;Per-Ola Norrby ;Adrian M. Daly Dr. Dr.
Chemistry - A European Journal 2002 Volume 8(Issue 18) pp:
Publication Date(Web):16 SEP 2002
DOI:10.1002/1521-3765(20020916)8:18<4299::AID-CHEM4299>3.0.CO;2-B
The mechanism of alkene epoxidation by chromium(v) oxo salen complexes has been studied by DFT and experimental methods. The reaction is compared to the closely related Mn-catalyzed process in an attempt to understand the dramatic difference in selectivity between the two systems. Overall, the studies show that the reactions have many similarities, but also a few critical differences. In agreement with experiment, the chromium system requires a change from low- to high-spin in the catalytic cycle, whereas the manganese system can proceed either with spin inversion or entirely on the high-spin surface. The low-spin addition of metal oxo species to an alkene leads to an intermediate which forms epoxide either with a barrier on the low-spin surface or without a barrier after spin inversion. Supporting evidence for this intermediate was obtained by using vinylcyclopropane traps. The chromium(v) oxo complexes can adopt a stepped shape or form a more concave surface, analogous to previous results on manganese salen complexes.
Co-reporter:Niall P. Kenny, Kamalraj V. Rajendran and Declan G. Gilheany
Chemical Communications 2015 - vol. 51(Issue 92) pp:NaN16564-16564
Publication Date(Web):2015/09/28
DOI:10.1039/C5CC06389B
A method is reported for the phosphoryl bond cleavage of O-alkyl phosphinates, phosphinothioates and certain phosphonamidates to furnish the corresponding P(III) borane adducts. The two-step procedure relies upon initial activation of the phosphoryl bond with an alkyl triflate, followed by reduction of the resulting intermediate using lithium borohydride.
Co-reporter:Kamalraj V. Rajendran and Declan G. Gilheany
Chemical Communications 2012 - vol. 48(Issue 80) pp:NaN10042-10042
Publication Date(Web):2012/08/16
DOI:10.1039/C2CC34136K
Sequential treatment of racemic phosphine oxides with oxalyl chloride and chiral non-racemic alcohol generates the same ratios of diastereomeric alkoxyphosphonium salts obtained in the corresponding asymmetric Appel process, strongly implicating the intermediate chlorophosphonium salt in the stereoselecting step. Subsequent reduction allows a novel synthesis of enantioenriched P-stereogenic phosphines–phosphine boranes.
Co-reporter:Peter A. Byrne and Declan G. Gilheany
Chemical Society Reviews 2013 - vol. 42(Issue 16) pp:NaN6696-6696
Publication Date(Web):2013/05/14
DOI:10.1039/C3CS60105F
The mechanism of the Wittig reaction has long been a contentious issue in organic chemistry. Even now, more than 50 years after its announcement, its presentation in many modern undergraduate textbooks is either overly simplified or entirely inaccurate. In this review, we gather together the huge body of evidence that has been amassed to show that the Li salt-free Wittig reactions of non-stabilised, semi-stabilised and stabilised ylides all occur under kinetic control by a common mechanism in which oxaphosphetane (OPA) is the first-formed and only intermediate. The numerous recent significant additions to the subject – including computational studies and experimental material pertinent to both steps of the reaction (OPA formation and its decomposition) are discussed in detail, and the currently accepted explanations for the source of the stereoselectivity in Wittig reactions are given. We also present the other mechanistic proposals that have been made during the history of the Wittig reaction, and show how they are unable to account for all of the experimental evidence that is now available. Details of certain experimental facts to do with Wittig reactions in the presence of Li cation are also included, although the precise mechanistic details of such reactions are yet to be established conclusively. We make the case that a clear distinction should henceforth be made between the unknown “Li-present” and the now well-established “Li salt-free” Wittig mechanisms.
Co-reporter:Peter A. Byrne, Kamalraj V. Rajendran, Jimmy Muldoon and Declan G. Gilheany
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 17) pp:NaN3537-3537
Publication Date(Web):2012/03/05
DOI:10.1039/C2OB07074J
A mild method for the facile removal of phosphine oxide from the crude products of Wittig and Appel reactions is described. Work-up with oxalyl chloride to generate insoluble chlorophosphonium salt (CPS) yields phosphorus-free products for a wide variety of these reactions. The CPS product can be further converted into phosphine.
Co-reporter:Kamalraj V. Rajendran and Declan G. Gilheany
Chemical Communications 2012 - vol. 48(Issue 6) pp:NaN819-819
Publication Date(Web):2011/10/21
DOI:10.1039/C1CC14856G
A variety of phosphine oxides and sulfides can be efficiently converted directly to the corresponding phosphine boranes using oxalyl chloride followed by sodium borohydride. Optically active P-stereogenic phosphine oxides can be converted stereospecifically to phosphine boranes with inversion of configuration by treatment with Meerwein's salt followed by sodium borohydride.
Co-reporter:Kirill Nikitin, Helge Müller-Bunz and Declan Gilheany
Chemical Communications 2013 - vol. 49(Issue 14) pp:NaN1436-1436
Publication Date(Web):2012/12/21
DOI:10.1039/C2CC38363B
Triphenylhalophosphonium halides, Ph3PX2, form crystals comprising bridged linear cations [Ph3P–X–X–X–PPh3]+ where the X3 bridge is shortened from 6.56 Å in Cl–Cl–Cl to 6.37 Å in the Br–Br–Br system. It is proposed that this structure is stabilised by five-centre/six-electron (5c–6e) hypervalent interactions.


















![5H-Dibenzo[b,f]phosphepin, 10,11-dihydro-5-phenyl-, 5-oxide](/data/chemimg/3726100/30309-75-2.png)
![5H-Dibenzo[b,f]phosphepin, 10,11-dihydro-5-phenyl-, 5-oxide](/data/chemimg/3726100/30309-75-2_b.png)







![Phosphine oxide, methylphenyl[2-(trifluoromethyl)phenyl]-, (S)-](http://img.cochemist.com/ccimg/99000/98933-88-1.png)
![Phosphine oxide, methylphenyl[2-(trifluoromethyl)phenyl]-, (S)-](http://img.cochemist.com/ccimg/99000/98933-88-1_b.png)


![Benzene, 1-methyl-2-[(1Z)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/53500/53423-25-9.png)
![Benzene, 1-methyl-2-[(1Z)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/53500/53423-25-9_b.png)




































![BENZENE, 1-FLUORO-2-[(1E)-2-PHENYLETHENYL]-](http://img.cochemist.com/ccimg/52900/52805-91-1.png)
![BENZENE, 1-FLUORO-2-[(1E)-2-PHENYLETHENYL]-](http://img.cochemist.com/ccimg/52900/52805-91-1_b.png)
![BENZENE, 1-CHLORO-2-[(1Z)-2-(2-METHYLPHENYL)ETHENYL]-](http://img.cochemist.com/ccimg/62700/62640-58-8.png)
![BENZENE, 1-CHLORO-2-[(1Z)-2-(2-METHYLPHENYL)ETHENYL]-](http://img.cochemist.com/ccimg/62700/62640-58-8_b.png)








![Butanedioic acid, 2,3-bis(benzoyloxy)-, (2S,3S)-, compd. with2-[[[(1R,2R)-2-aminocyclohexyl]imino]methyl]-4,6-dichlorophenol (1:1)](http://img.cochemist.com/ccimg/870200/870101-87-4.png)
![Butanedioic acid, 2,3-bis(benzoyloxy)-, (2S,3S)-, compd. with2-[[[(1R,2R)-2-aminocyclohexyl]imino]methyl]-4,6-dichlorophenol (1:1)](http://img.cochemist.com/ccimg/870200/870101-87-4_b.png)
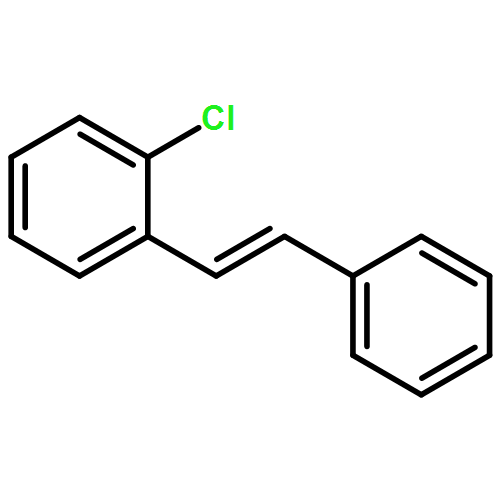
![BENZENE, 1-CHLORO-2-[(1E)-2-(2-METHYLPHENYL)ETHENYL]-](http://img.cochemist.com/ccimg/67500/67456-94-4.png)
![BENZENE, 1-CHLORO-2-[(1E)-2-(2-METHYLPHENYL)ETHENYL]-](http://img.cochemist.com/ccimg/67500/67456-94-4_b.png)






![Phosphine oxide, methyl-1-naphthalenylphenyl-, [P(S)]-](/data/chemimg/1813600/37775-92-1.png)
![Phosphine oxide, methyl-1-naphthalenylphenyl-, [P(S)]-](/data/chemimg/1813600/37775-92-1_b.png)






![BENZENE, [(1E)-2-CYCLOPENTYLETHENYL]-](http://img.cochemist.com/ccimg/40200/40132-68-1.png)
![BENZENE, [(1E)-2-CYCLOPENTYLETHENYL]-](http://img.cochemist.com/ccimg/40200/40132-68-1_b.png)
![BENZENE, 1-BROMO-2-[(1Z)-2-PHENYLETHENYL]-](http://img.cochemist.com/ccimg/4900/4877-77-4.png)
![BENZENE, 1-BROMO-2-[(1Z)-2-PHENYLETHENYL]-](http://img.cochemist.com/ccimg/4900/4877-77-4_b.png)

















![Butanedioic acid, 2,3-bis(benzoyloxy)-, (2S,3S)-, compd. with2-[[[(1R,2R)-2-aminocyclohexyl]imino]methyl]-6-chlorophenol (1:1)](http://img.cochemist.com/ccimg/870200/870101-58-9.png)
![Butanedioic acid, 2,3-bis(benzoyloxy)-, (2S,3S)-, compd. with2-[[[(1R,2R)-2-aminocyclohexyl]imino]methyl]-6-chlorophenol (1:1)](http://img.cochemist.com/ccimg/870200/870101-58-9_b.png)
![Butanedioic acid, 2,3-bis(benzoyloxy)-, (2S,3S)-, compd. with2-[[[(1R,2R)-2-aminocyclohexyl]imino]methyl]-4,6-dibromophenol (1:1)](http://img.cochemist.com/ccimg/870200/870101-63-6.png)
![Butanedioic acid, 2,3-bis(benzoyloxy)-, (2S,3S)-, compd. with2-[[[(1R,2R)-2-aminocyclohexyl]imino]methyl]-4,6-dibromophenol (1:1)](http://img.cochemist.com/ccimg/870200/870101-63-6_b.png)








![Butanedioic acid, 2,3-bis(benzoyloxy)-, (2S,3S)-, compd. with2-[[[(1R,2R)-2-aminocyclohexyl]imino]methyl]-3,4,6-trichlorophenol (1:1)](http://img.cochemist.com/ccimg/870200/870101-60-3.png)
![Butanedioic acid, 2,3-bis(benzoyloxy)-, (2S,3S)-, compd. with2-[[[(1R,2R)-2-aminocyclohexyl]imino]methyl]-3,4,6-trichlorophenol (1:1)](http://img.cochemist.com/ccimg/870200/870101-60-3_b.png)




![Benzene, 1-methyl-2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/22300/22257-16-5.png)
![Benzene, 1-methyl-2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/22300/22257-16-5_b.png)
![1-methoxy-2-[(1E)-prop-1-en-1-yl]benzene](http://img.cochemist.com/ccimg/2100/2077-36-3.png)
![1-methoxy-2-[(1E)-prop-1-en-1-yl]benzene](http://img.cochemist.com/ccimg/2100/2077-36-3_b.png)



![Benzene, 1-methoxy-2-[(1Z)-2-phenylethenyl]-](/data/chemimg/1775300/1898-14-2.png)
![Benzene, 1-methoxy-2-[(1Z)-2-phenylethenyl]-](/data/chemimg/1775300/1898-14-2_b.png)
![Benzene, [(1Z)-3-methyl-1-buten-1-yl]-](/data/chemimg/1738300/15325-56-1.png)
![Benzene, [(1Z)-3-methyl-1-buten-1-yl]-](/data/chemimg/1738300/15325-56-1_b.png)















![Phosphonium, [(2,6-dichlorophenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/159800/159783-79-6.png)
![Phosphonium, [(2,6-dichlorophenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/159800/159783-79-6_b.png)


![Phosphorane, [(4-bromophenyl)methylene]triphenyl-](http://img.cochemist.com/ccimg/59700/59625-59-1.png)
![Phosphorane, [(4-bromophenyl)methylene]triphenyl-](http://img.cochemist.com/ccimg/59700/59625-59-1_b.png)






![Phosphonium, [(2,6-difluorophenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/159800/159783-80-9.png)
![Phosphonium, [(2,6-difluorophenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/159800/159783-80-9_b.png)
![Benzene, [(1E)-3-methyl-1-buten-1-yl]-](/data/chemimg/1744400/15325-61-8.png)
![Benzene, [(1E)-3-methyl-1-buten-1-yl]-](/data/chemimg/1744400/15325-61-8_b.png)







![Phosphonium, [(2-iodophenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/62700/62680-65-3.png)
![Phosphonium, [(2-iodophenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/62700/62680-65-3_b.png)




![Phosphonium, [(2-chlorophenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/62700/62640-67-9.png)
![Phosphonium, [(2-chlorophenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/62700/62640-67-9_b.png)






















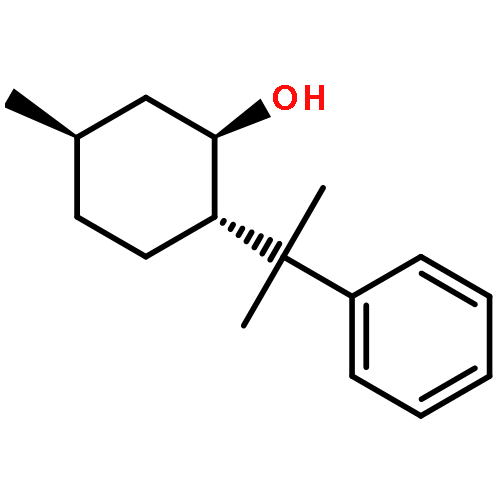
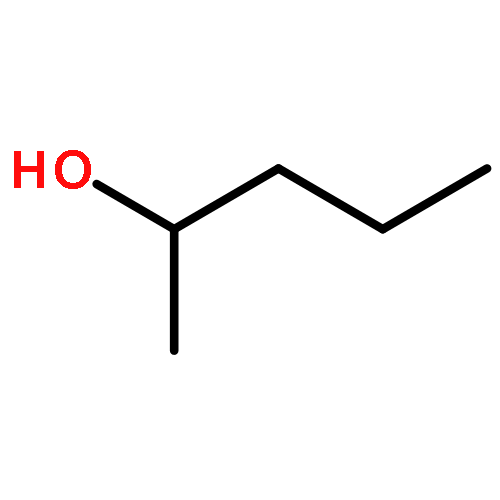
![Bicyclo[3.1.1]heptan-3-ol,2,6,6-trimethyl-, (1R,2R,3R,5S)-](http://img.cochemist.com/ccimg/1200/1196-00-5.png)
![Bicyclo[3.1.1]heptan-3-ol,2,6,6-trimethyl-, (1R,2R,3R,5S)-](http://img.cochemist.com/ccimg/1200/1196-00-5_b.png)


![Phosphonic acid, [2-(2-bromophenyl)ethyl]-, diethyl ester](http://img.cochemist.com/ccimg/500900/500875-62-7.png)
![Phosphonic acid, [2-(2-bromophenyl)ethyl]-, diethyl ester](http://img.cochemist.com/ccimg/500900/500875-62-7_b.png)



















![5-phenyl-5H-benzo[b]phosphindole 5-oxide](http://img.cochemist.com/ccimg/1100/1031-13-6.png)
![5-phenyl-5H-benzo[b]phosphindole 5-oxide](http://img.cochemist.com/ccimg/1100/1031-13-6_b.png)



























![Boron,trihydro[(2-methoxyphenyl)methylphenylphosphine-kP]-, [T-4-(S)]-](http://img.cochemist.com/ccimg/97900/97858-63-4.png)
![Boron,trihydro[(2-methoxyphenyl)methylphenylphosphine-kP]-, [T-4-(S)]-](http://img.cochemist.com/ccimg/97900/97858-63-4_b.png)




![Benzene, 1-methoxy-4-[(4-methylphenyl)sulfinyl]-](http://img.cochemist.com/ccimg/10400/10381-41-6.png)
![Benzene, 1-methoxy-4-[(4-methylphenyl)sulfinyl]-](http://img.cochemist.com/ccimg/10400/10381-41-6_b.png)


![Benzene, 1,3-dichloro-2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/25200/25144-42-7.png)
![Benzene, 1,3-dichloro-2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/25200/25144-42-7_b.png)











![[(E)-3,3,3-TRIFLUORO-1-PHENYLPROP-1-EN-2-YL]BENZENE](http://img.cochemist.com/ccimg/76300/76280-44-9.png)
![[(E)-3,3,3-TRIFLUORO-1-PHENYLPROP-1-EN-2-YL]BENZENE](http://img.cochemist.com/ccimg/76300/76280-44-9_b.png)
















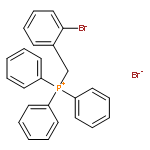
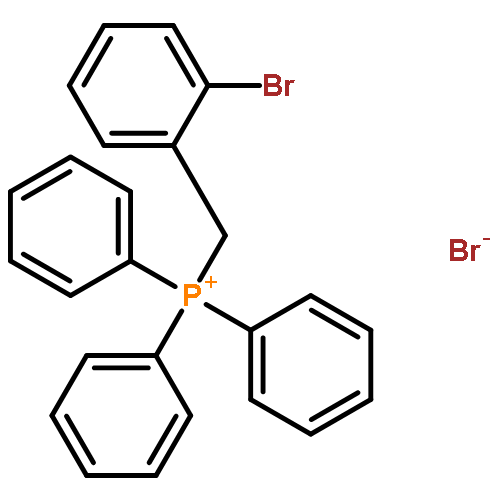


![1-BROMO-2-[2-(2-BROMOPHENYL)ETHENYL]BENZENE](http://img.cochemist.com/ccimg/56700/56667-11-9.png)
![1-BROMO-2-[2-(2-BROMOPHENYL)ETHENYL]BENZENE](http://img.cochemist.com/ccimg/56700/56667-11-9_b.png)







![Phosphonium,[(2-methylphenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/1600/1530-36-5.png)
![Phosphonium,[(2-methylphenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/1600/1530-36-5_b.png)















![1-METHYL-2-[(Z)-2-(2-METHYLPHENYL)ETHENYL]BENZENE](http://img.cochemist.com/ccimg/36900/36888-18-3.png)
![1-METHYL-2-[(Z)-2-(2-METHYLPHENYL)ETHENYL]BENZENE](http://img.cochemist.com/ccimg/36900/36888-18-3_b.png)








![Benzene, [[(1,1-dimethylethyl)sulfinyl]methyl]-](http://img.cochemist.com/ccimg/26800/26756-22-9.png)
![Benzene, [[(1,1-dimethylethyl)sulfinyl]methyl]-](http://img.cochemist.com/ccimg/26800/26756-22-9_b.png)
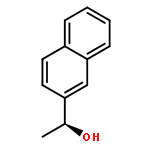



















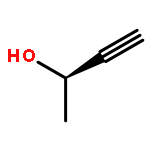
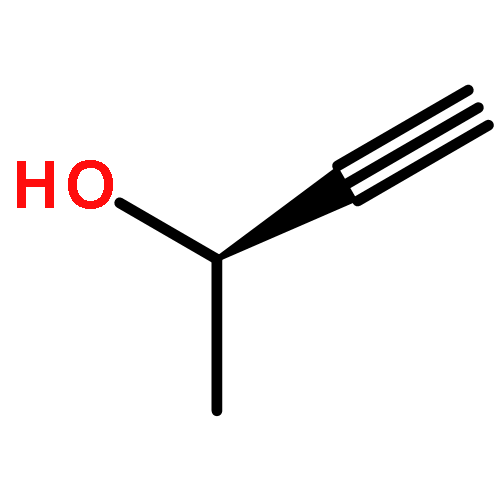


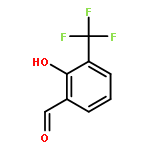




![5-phenylbenzo[b]phosphindole](http://img.cochemist.com/ccimg/1100/1088-00-2.png)
![5-phenylbenzo[b]phosphindole](http://img.cochemist.com/ccimg/1100/1088-00-2_b.png)