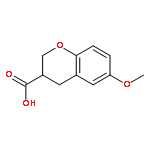
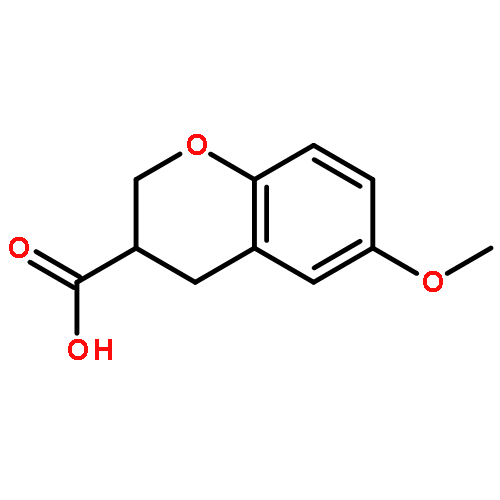
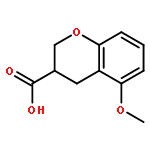
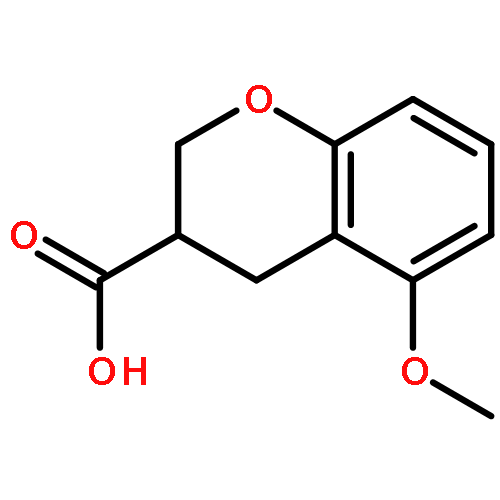
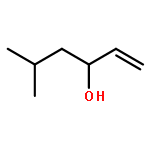
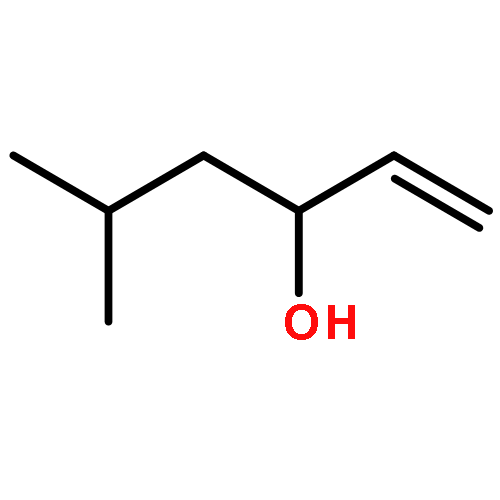
Co-reporter:Reji N. Nair and Thomas D. Bannister
Organic Process Research & Development 2016 Volume 20(Issue 7) pp:1370-1376
Publication Date(Web):June 9, 2016
DOI:10.1021/acs.oprd.6b00128
The importance of the “SEM” group, or the 2-(trimethylsilyl)ethoxymethyl group, as an N-protecting group for special class of pyrrolopyridazinones is reported. Our studies indicate that standard Boc protection on the nitrogen of a pyrrolopyridazinone results in significant decomposition during the incorporation of biaryl groups via Pd-catalyzed cross coupling reactions. However, on using the SEM group, the resulting cross coupling reaction gives excellent yields with no decomposition products, enabling the synthesis of a variety of compounds that were targeted for their antitumor activity.
Co-reporter:Reji N. Nair, Jitendra K. Mishra, Fangzheng Li, Mariola Tortosa, Chunying Yang, Joanne R. Doherty, Michael Cameron, John L. Cleveland, William R. Roush and Thomas D. Bannister
MedChemComm 2016 vol. 7(Issue 5) pp:900-905
Publication Date(Web):23 Feb 2016
DOI:10.1039/C5MD00579E
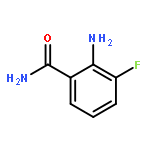
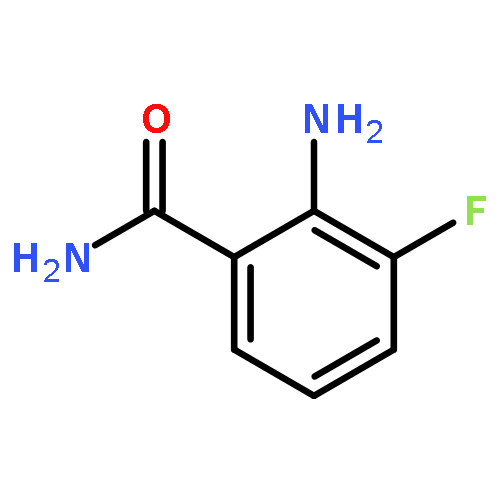
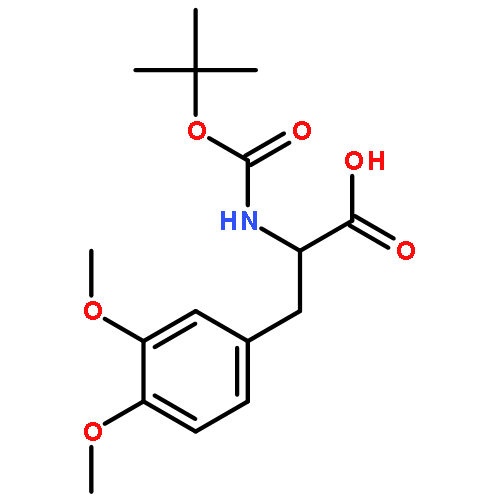
Glutamine and tyrosine-based amino acid conjugates of monocarboxylate transporter types 1 and 2 inhibitors (MCT1/2) were designed, synthesized and evaluated for their potency in blocking the proliferation of a human B lymphoma cell line that expresses the transporters Asct2, LAT1 and MCT1. Appropriate placement of an amino acid transporter recognition element was shown to augment anti-tumour efficacy vs. Raji cells. Amino acid conjugation also improves the pharmacodynamic properties of experimental MCT1/2 inhibitors.
Co-reporter:Xiaohong Pan, Chunying Yang, John L. Cleveland, and Thomas D. Bannister
The Journal of Organic Chemistry 2016 Volume 81(Issue 5) pp:2194-2200
Publication Date(Web):February 1, 2016
DOI:10.1021/acs.joc.6b00022
Sempervirine and analogues were synthesized using a route featuring Sonogashira and Larock Pd-catalyzed reactions. Structure–activity relationships were investigated using three human cancer cell lines. 10-Fluorosempervirine is the most potently cytotoxic member of the family yet described.
Co-reporter:Reji N. Nair
European Journal of Organic Chemistry 2015 Volume 2015( Issue 8) pp:1764-1770
Publication Date(Web):
DOI:10.1002/ejoc.201403491
Abstract
In the course of a structure–activity relationship study of pyrrole[3,4-d]pyridazinones, we optimized conditions for a one-pot directed lithiation/alkylation reaction that also promoted in situ cleavage of a tert-butoxycarbonyl (Boc) protecting group on the pyrrole ring. The efficiency of the process gave access to a number of substituted analogs of interest as possible antitumor agents.
Co-reporter:Hui Wang, Chao Wang, Thomas D. Bannister
Tetrahedron Letters 2015 Volume 56(Issue 15) pp:1949-1952
Publication Date(Web):8 April 2015
DOI:10.1016/j.tetlet.2015.02.051
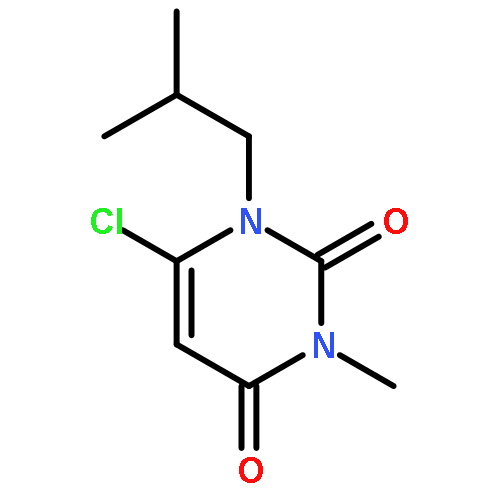
A novel synthetic route to 1,3,5,7-tetrasubstituted pyrimido[4,5-d]pyrimidine-2,4-diones, of interest for potential antitumor activity, is reported. The route uses 1,3-disubstituted 6-amino uracils as starting materials. The key step is a hydrazine-induced cyclization reaction to form the fused pyrimidine ring. By choosing different uracils, acylation reagents, and alkylation reagents, substituents at N-1, N-3, C-5, and C-7 may be selectively varied to provide a structurally diverse set of compounds for biological evaluation.
Co-reporter:Xiaohong Pan and Thomas D. Bannister
Organic Letters 2014 Volume 16(Issue 23) pp:6124-6127
Publication Date(Web):November 13, 2014
DOI:10.1021/ol5029783
A general synthetic approach to β-carboline-containing alkaloids was developed. Two consecutive palladium-mediated processes, a Sonagashira coupling and a Larock indole annulation reaction, are central to the method. The scope of the approach was investigated and found to be amenable for constructing a variety of biologically significant natural products and also for preparing substituted analogues for optimization and analysis of their biological properties.
Co-reporter:Hui Wang ; Chunying Yang ; Joanne R. Doherty ; William R. Roush ; John L. Cleveland
Journal of Medicinal Chemistry 2014 Volume 57(Issue 17) pp:7317-7324
Publication Date(Web):July 28, 2014
DOI:10.1021/jm500640x
Novel substituted pteridine-derived inhibitors of monocarboxylate transporter 1 (MCT1), an emerging target for cancer therapy, are reported. The activity of these compounds as inhibitors of lactate transport was confirmed using a 14C-lactate transport assay, and their potency against MCT1-expressing human tumor cells was established using MTT assays. The four most potent compounds showed substantial anticancer activity (EC50 37–150 nM) vs MCT1-expressing human Raji lymphoma cells.
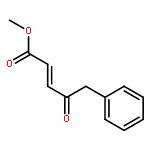
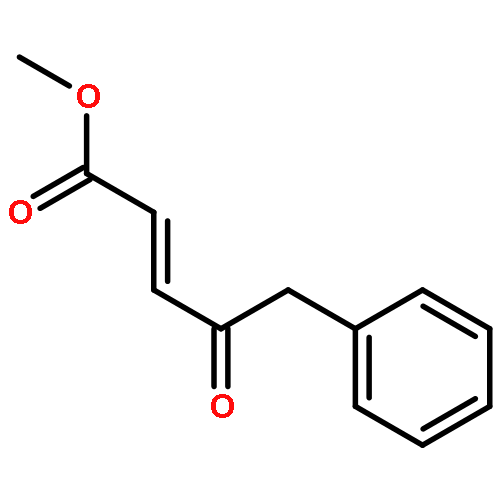
Co-reporter:Reji N. Nair and Thomas D. Bannister
The Journal of Organic Chemistry 2014 Volume 79(Issue 3) pp:1467-1472
Publication Date(Web):January 6, 2014
DOI:10.1021/jo4023606
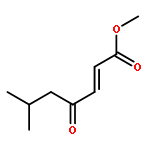
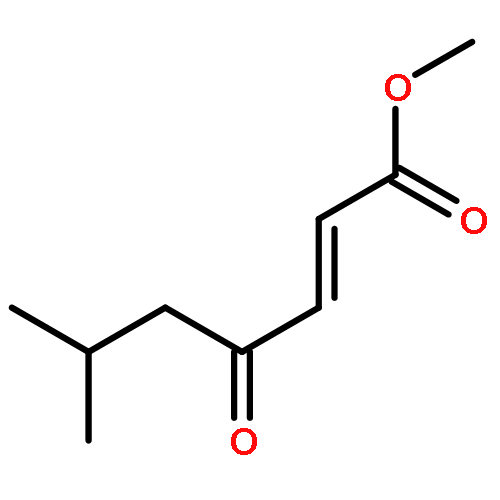
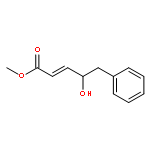
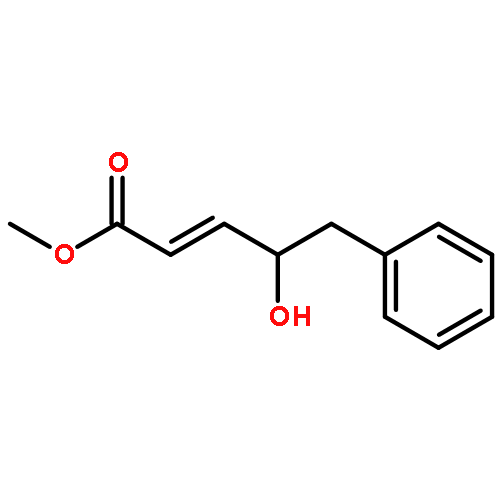
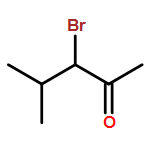
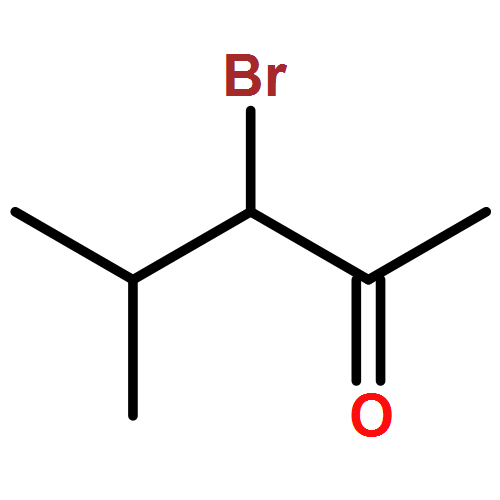
A direct and scalable route to γ-keto-α,β-unsaturated esters, useful intermediates in medicinal chemistry and natural products synthesis, is reported. The key step involves the use of Grubbs’ second-generation olefin metathesis catalyst for cross-metathesis of alkyl acrylates and 2° allylic alcohols. The metathesis step is followed by oxidation to give the desired products in high yield on scales of up to 25 g.
Co-reporter:E. Hampton Sessions, Sarwat Chowdhury, Yan Yin, Jennifer R. Pocas, Wayne Grant, Thomas Schröter, Li Lin, Claudia Ruiz, Michael D. Cameron, Philip LoGrasso, Thomas D. Bannister, Yangbo Feng
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 23) pp:7113-7118
Publication Date(Web):1 December 2011
DOI:10.1016/j.bmcl.2011.09.084
Therapeutic interventions with Rho kinase (ROCK) inhibitors may effectively treat several disorders such as hypertension, stroke, cancer, and glaucoma. Herein we disclose the optimization and biological evaluation of potent novel ROCK inhibitors based on substituted indole and 7-azaindole core scaffolds. Substitutions on the indole C3 position and on the indole NH and/or amide NH positions all yielded potent and selective ROCK inhibitors (25, 42, and 50). Improvement of aqueous solubility and tailoring of in vitro and in vivo DMPK properties could be achieved through these substitutions.
Co-reporter:Sarwat Chowdhury, E. Hampton Sessions, Jennifer R. Pocas, Wayne Grant, Thomas Schröter, Li Lin, Claudia Ruiz, Michael D. Cameron, Stephan Schürer, Philip LoGrasso, Thomas D. Bannister, Yangbo Feng
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 23) pp:7107-7112
Publication Date(Web):1 December 2011
DOI:10.1016/j.bmcl.2011.09.083
Rho kinase (ROCK) inhibitors are potential therapeutic agents to treat disorders such as hypertension, multiple sclerosis, cancers, and glaucoma. Here, we disclose the synthesis, optimization, biological evaluation of potent indole and 7-azaindole based ROCK inhibitors that have high potency on ROCK (IC50 = 1 nM) with 740-fold selectivity over PKA (47). Moreover, 47 showed very good DMPK properties making it a good candidate for further development. Finally, docking studies with a homology model of ROCK-II were performed to rationalize the binding mode of these compounds and showed the compounds bound in both orientations to take advantage to H-bonds with Lys-121 of ROCK-II.
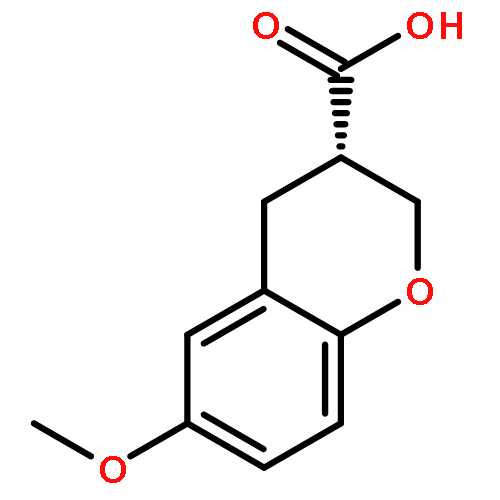
Co-reporter:Yen Ting Chen, Tomas Vojkovsky, Xingang Fang, Jennifer R. Pocas, Wayne Grant, Amiee M. W. Handy, Thomas Schröter, Philip LoGrasso, Thomas D. Bannister and Yangbo Feng
MedChemComm 2011 vol. 2(Issue 1) pp:73-75
Publication Date(Web):06 Dec 2010
DOI:10.1039/C0MD00194E
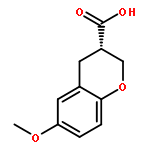
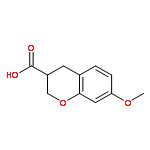
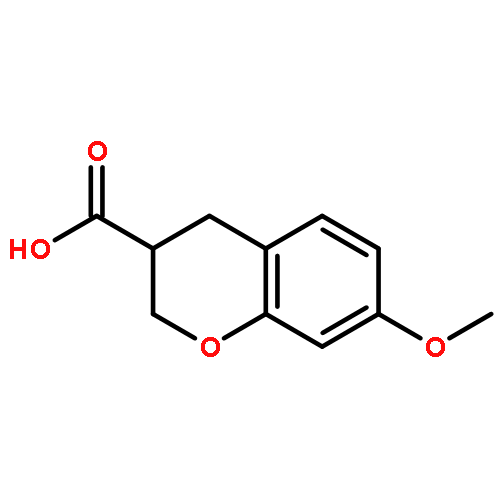
Rho kinase (ROCK) is currently investigated as a target for various diseases such as glaucoma and spinal cord injury. Herein, we report the asymmetric synthesis of chroman 1, a highly potent ROCK inhibitor, and its analogs. The inhibitory properties of these compounds for ROCK-II and a selected set of highly homologous kinases are also discussed.
Co-reporter:E. Hampton Sessions, Michael Smolinski, Bo Wang, Bozena Frackowiak, Sarwat Chowdhury, Yan Yin, Yen Ting Chen, Claudia Ruiz, Li Lin, Jennifer Pocas, Thomas Schröter, Michael D. Cameron, Philip LoGrasso, Yangbo Feng, Thomas D. Bannister
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 6) pp:1939-1943
Publication Date(Web):15 March 2010
DOI:10.1016/j.bmcl.2010.01.124
Rho Kinase (ROCK) is a serine/threonine kinase whose inhibition could prove beneficial in numerous therapeutic areas. We have developed a promising class of ATP-competitive inhibitors based upon a benzimidazole scaffold, which show excellent potency toward ROCK (IC50 <10 nM). This report details the optimization of selectivity for ROCK over other related kinases such as Protein kinase A (PKA).
.jpg)
![(3S)-N-[2-[2-(DIMETHYLAMINO)ETHOXY]-4-(1H-PYRAZOL-4-YL)PHENYL]-6-METHOXY-3,4-DIHYDRO-2H-CHROMENE-3-CARBOXAMIDE](http://img.cochemist.com/ccimg/1273600/1273579-40-0.png)
![(3S)-N-[2-[2-(DIMETHYLAMINO)ETHOXY]-4-(1H-PYRAZOL-4-YL)PHENYL]-6-METHOXY-3,4-DIHYDRO-2H-CHROMENE-3-CARBOXAMIDE](http://img.cochemist.com/ccimg/1273600/1273579-40-0_b.png)


![1H-Benz[g]indolo[2,3-a]quinolizin-6-ium,2,3,4,13-tetrahydro-, inner salt](http://img.cochemist.com/ccimg/600/549-92-8.png)
![1H-Benz[g]indolo[2,3-a]quinolizin-6-ium,2,3,4,13-tetrahydro-, inner salt](http://img.cochemist.com/ccimg/600/549-92-8_b.png)




![6-Chloro-1H-pyrrolo[2,3-b]pyridine-3-carboxylic acid](http://img.cochemist.com/ccimg/915200/915140-96-4.png)
![6-Chloro-1H-pyrrolo[2,3-b]pyridine-3-carboxylic acid](http://img.cochemist.com/ccimg/915200/915140-96-4_b.png)

![6-Chloro-1H-pyrrolo[3,2-c]pyridine-2-carboxylic acid](http://img.cochemist.com/ccimg/800500/800401-54-1.png)
![6-Chloro-1H-pyrrolo[3,2-c]pyridine-2-carboxylic acid](http://img.cochemist.com/ccimg/800500/800401-54-1_b.png)
![4-Isoxazolidinol,
2-[[6-[(3,5-dimethyl-1H-pyrazol-4-yl)methyl]-1,2,3,4-tetrahydro-3-methyl-
1-(2-methylpropyl)-2,4-dioxothieno[2,3-d]pyrimidin-5-yl]carbonyl]-, (4S)-](http://img.cochemist.com/ccimg/496800/496791-37-8.png)
![4-Isoxazolidinol,
2-[[6-[(3,5-dimethyl-1H-pyrazol-4-yl)methyl]-1,2,3,4-tetrahydro-3-methyl-
1-(2-methylpropyl)-2,4-dioxothieno[2,3-d]pyrimidin-5-yl]carbonyl]-, (4S)-](http://img.cochemist.com/ccimg/496800/496791-37-8_b.png)
![1-Propanamine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-N-methyl-](http://img.cochemist.com/ccimg/204600/204580-33-6.png)
![1-Propanamine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-N-methyl-](http://img.cochemist.com/ccimg/204600/204580-33-6_b.png)




![1-Propanamine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/115400/115306-75-7.png)
![1-Propanamine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/115400/115306-75-7_b.png)



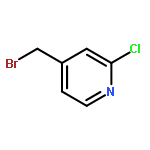
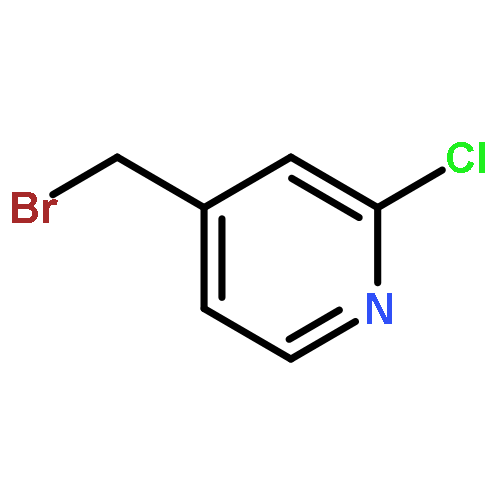


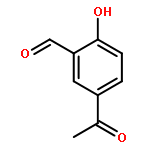
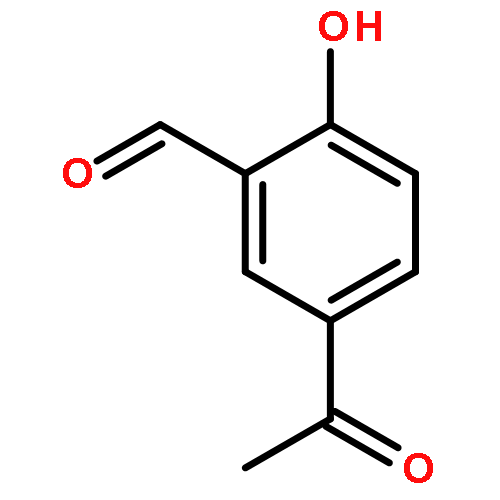
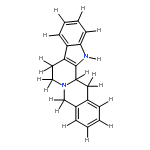
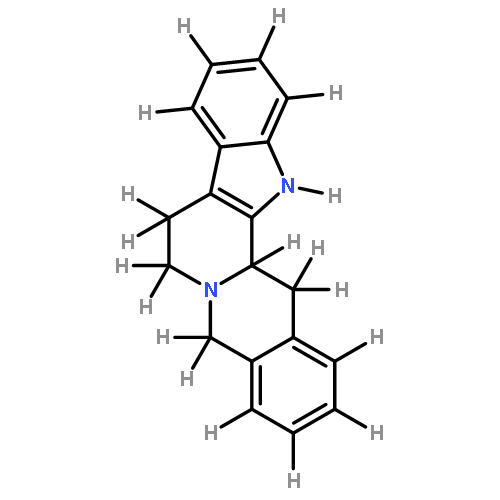




![2-(4-methoxyphenyl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]acetic Acid](http://img.cochemist.com/ccimg/55400/55362-76-0.png)
![2-(4-methoxyphenyl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]acetic Acid](http://img.cochemist.com/ccimg/55400/55362-76-0_b.png)


![BENZ[G]INDOLO[2,3-A]QUINOLIZIN-5(7H)-ONE, 8,13-DIHYDRO-](http://img.cochemist.com/ccimg/51600/51598-75-5.png)
![BENZ[G]INDOLO[2,3-A]QUINOLIZIN-5(7H)-ONE, 8,13-DIHYDRO-](http://img.cochemist.com/ccimg/51600/51598-75-5_b.png)




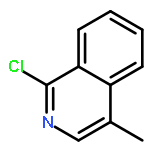
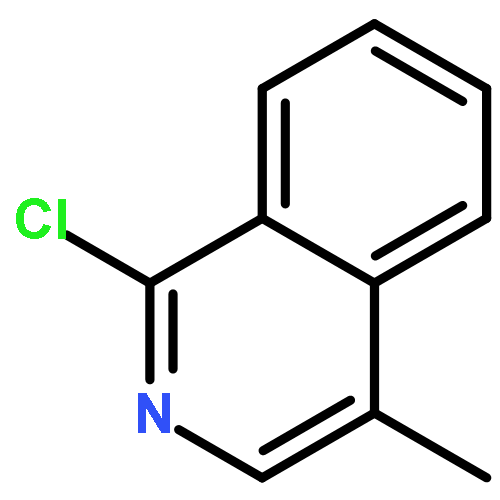
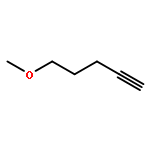
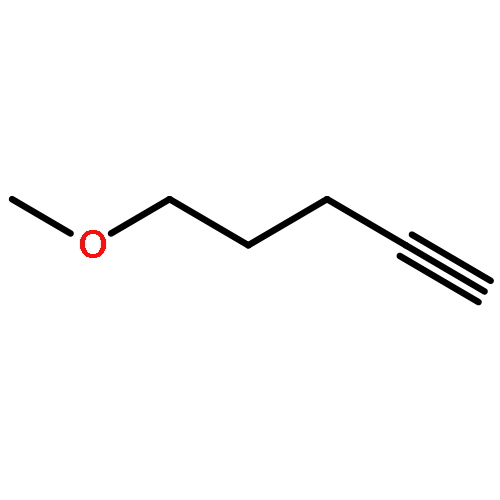


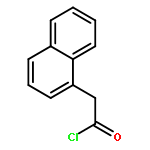

![(E)-Methyl 2-((2S,3S,12bS)-3-ethyl-8-methoxy-1,2,3,4,6,7,12,12b-octahydroindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate](http://img.cochemist.com/ccimg/4100/4098-40-2.png)
![(E)-Methyl 2-((2S,3S,12bS)-3-ethyl-8-methoxy-1,2,3,4,6,7,12,12b-octahydroindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate](http://img.cochemist.com/ccimg/4100/4098-40-2_b.png)




![6-Chloro-1H-pyrrolo[2,3-b]pyridine-2-carboxylic acid](http://img.cochemist.com/ccimg/800500/800402-07-7.png)
![6-Chloro-1H-pyrrolo[2,3-b]pyridine-2-carboxylic acid](http://img.cochemist.com/ccimg/800500/800402-07-7_b.png)