Co-reporter:Jiao Chen;Shuai Jiang;Yi-Rong Liu;Teng Huang;Chun-Yu Wang;Shou-Kui Miao;Zhong-Quan Wang;Yang Zhang
RSC Advances (2011-Present) 2017 vol. 7(Issue 11) pp:6374-6388
Publication Date(Web):2017/01/18
DOI:10.1039/C6RA27945G
Oxalic acid and dimethylamine are the most common organic acid and base in the atmosphere, and are recognized as significant precursor species in atmospheric new particle formation. However, the interaction between oxalic acid and dimethylamine in the presence of hydration is not yet understood. In this study, the most stable geometric structures and thermodynamics of (C2H2O4)m(CH3NHCH3)(H2O)n (m = 1–2, n = 0–4) clusters are investigated using M06-2X coupled with the 6-311+G(2d,p) basis set. A high level explicitly corrected CCSD(T)-F12/VDZ-F12 method is utilized to benchmark the density functional theory (DFT) methods. Hydration promotes proton transfer from oxalic acid to dimethylamine for (C2H2O4)(CH3NHCH3)(H2O)n (n = 0–4) clusters, while proton transfer from oxalic acid to dimethylamine occurs without hydration for (C2H2O4)2(CH3NHCH3)(H2O)n (n = 0–4) clusters. With regards to the isomer distribution at the potential energy surface, temperature seems not to be an important parameter, since almost all of the global minima for the investigated size range dominate within the investigated temperature range, except for in the (C2H2O4)m(CH3NHCH3)(H2O)2 clusters. Under atmospheric conditions, the peak hydration distribution shifts from unhydrated clusters to trihydrates for the (C2H2O4)(CH3NHCH3)(H2O)n (n = 0–4) clusters, while for the (C2H2O4)2(CH3NHCH3)(H2O)n (n = 0–4) clusters, unhydrated clusters clearly dominate the cluster distribution, irrespective of whether the humidity is low or high. Finally, the formation free energies obtained from quantum calculations are used to calculate the evaporation rates. We find that evaporation of dimethylamine is preferred compared to oxalic acid for the (C2H2O4)(CH3NHCH3)(H2O)n clusters, while the results are reversed for the (C2H2O4)2(CH3NHCH3)(H2O)n clusters.
Co-reporter:Hui Wen, Gao-Lei Hou, Yi-Rong Liu, Xue-Bin Wang and Wei Huang
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 26) pp:17470-17482
Publication Date(Web):31 May 2016
DOI:10.1039/C6CP01542E
Bicarbonate plays a crucial biochemical role in the physiological pH buffering system and also has important atmospheric implications. In the current study, HCO3−(H2O)n (n = 0–13) clusters were successfully produced via electrospray ionization of the corresponding bulk salt solution, and were characterized by negative ion photoelectron spectroscopy and theoretical calculations. Photoelectron spectra reveal that the electron binding energy monotonically increases with the cluster size up to n = 10 and remains largely the same after n > 10. The photo-detaching feature of the solute HCO3− itself, which dominates in the small clusters, diminishes with the increase of water coverage. Based on the charge distribution and molecular orbital analyses, the universal high electron binding energy tail that dominates in the larger clusters can be attributed to the ionization of water. Thus, the transition of ionization from the solute to the solvent at a size larger than n = 10 has been observed. Extensive theoretical structural search based on the basin-hopping unbiased method was carried out, and a plethora of low energy isomers have been obtained for each medium and large-sized cluster. By comparing the simulated photoelectron spectra and calculated electron binding energies with the experiments, as well as by comparing the simulated infrared spectra with previously reported IR spectra, the best fit structures and the structural evolutionary routes are presented. The nature of bicarbonate–water interactions is mainly electrostatic as implied by electron localization function (ELF) analysis.
Co-reporter:Yan Ma, Jiao Chen, Shuai Jiang, Yi-Rong Liu, Teng Huang, Shou-Kui Miao, Chun-Yu Wang and Wei Huang
RSC Advances 2016 vol. 6(Issue 7) pp:5824-5836
Publication Date(Web):05 Jan 2016
DOI:10.1039/C5RA22887E
Amines have been proposed to participate in the nucleation process, but the electron density analysis and the determination of a temperature dependence of the clusters are still lacking. In this study, the clusters of (H2SO4)m(CH3NHCH3)n (m = 1–2, n = 1–3) are studied using the basin-hopping method coupled with density functional theory (DFT). Considering the high flexibility and complexity of a hydrogen bonding environment, the temperature dependence of the conformational population and the relative population fraction of the clusters are investigated. Moreover, the electron density is analyzed to identify the different types of intra-cluster interactions. The results indicate that the ratio between acid and base is very important for the cluster formation. The main interaction type changes from hydrogen bonding to a weak attraction as the number of bases increase. When the number of dimethylamine molecules is less than or equal to that of the sulfuric acid molecules as the most abundant clusters in the atmosphere, we tentatively suggest that the cluster contains less than two dimethylamine molecules because the critical clusters contain two or fewer sulfuric acid molecules. This means that the sulfuric acid–dimethylamine system can only form three main small clusters in the real atmosphere. Thus, other substances, such as water or organic acids, may be involved to promote the growth of clusters, and they may also affect the nucleation. This work predicts the possible forms of dimethylamine with sulfuric acid when participating in nucleation in a theoretical approach, and provides a reliable reference for the research on the nucleation mechanism containing dimethylamine in the atmosphere.
Co-reporter:Xiu-Qiu Peng, Teng Huang, Shou-Kui Miao, Jiao Chen, Hui Wen, Ya-Juan Feng, Yu Hong, Chun-Yu Wang and Wei Huang
RSC Advances 2016 vol. 6(Issue 52) pp:46582-46593
Publication Date(Web):26 Apr 2016
DOI:10.1039/C6RA03164A
A previous study of the binary system (H2C2O4)(NH3)n (n = 1–6) suggested that an oxalic acid–ammonia complex may participate in atmospheric aerosol formations. However, the mechanism of the hydration of these cores is poorly understood. In this study, the hydration of (H2C2O4)(NH3) and (H2C2O4)(NH3)2 cores with up to three water molecules is investigated with respect to different routes of formation. The results may improve understanding of the nucleation of clusters containing oxalic acid in the atmosphere. Acid dissociation is found to occur during the hydration process, leading to a HC2O4−/NH4+ ion pair. In contrast with the (H2C2O4)(NH3)2 core, water molecules appear to be unfavorable with regard to the formation of hydrates with a (H2C2O4)(NH3) core; additionally, temperature is found to affect the formation of clusters and the distributions of different isomers with the same size, but the impact of relative humidity on the hydrates seems insignificant, implying that the formation of these clusters may be more favorable under cold ambient conditions. The monohydrates and dihydrates of the (H2C2O4)(NH3)2 core may be relatively extensive in (H2C2O4)(NH3)m(H2O)n (m = 1–2, n = 1–3) clusters and may contribute to the atmospheric nucleation. Furthermore, this study presents a first attempt at determining the Rayleigh scattering properties of oxalic acid–ammonia–water pre-nucleation clusters; the results show that adding a water molecule could effectively increase the Rayleigh scattering intensity, but a single ammonia molecule may be able to generate a larger increase in the Rayleigh light scattering intensity than a water molecule. This may also indicate that clusters containing oxalic acid and ammonia show high Rayleigh light scattering intensities, but the more ammonia molecules there are in clusters, the higher the Rayleigh light scattering intensity and the greater the contribution to the extinction properties.
Co-reporter:Hui Wen, Teng Huang, Yi-Rong Liu, Shuai Jiang, Xiu-Qiu Peng, Shou-Kui Miao, Chun-Yu Wang, Yu Hong, Wei Huang
Chemical Physics 2016 Volume 479() pp:129-142
Publication Date(Web):10 November 2016
DOI:10.1016/j.chemphys.2016.09.039
Abstract
Halide ions have been received intense interest in charactering and understanding their implications in atmospheric chemistry since they are related to the ozone destruction in the stratosphere. In the current study, structures, thermodynamic properties, and spectroscopic signatures of hydrated bromide Br−(H2O)n (n = 1–8) clusters are thoroughly studied and compared with available studies, the new global minima were observed for the larger size Br−(H2O)7,8 clusters. The numbers of isomer increase with the increasing water molecules, considering the growing complexity, the isomer populations of each size clusters are provided under a wide temperature range, it was shown that different type of structures possess different temperature dependences. In addition, the bond order of different bond types of hydrated bromide has been systematically investigated for the first time.
Co-reporter:Xiao-Fei Hou, Li-Li Yan, Teng Huang, Yu Hong, Shou-Kui Miao, Xiu-Qiu Peng, Yi-Rong Liu, Wei Huang
Chemical Physics 2016 Volume 472() pp:50-60
Publication Date(Web):15 June 2016
DOI:10.1016/j.chemphys.2016.03.009
Abstract
The equilibrium geometric structures, relative stabilities, electronic stabilities, and electronic and magnetic properties of the AunC and Aun+1 (n = 1–9) clusters are systematically investigated using density functional theory (DFT) with hyper-generalized gradient approximation (GGA). The optimized geometries show that one Au atom added to the Aun−1C cluster is the dominant growth pattern for the AunC clusters. In contrast to the pure gold clusters, the AunC clusters are most stable in a quasi-planar or three-dimensional (3D) structure because the C dopant induces the local non-planarity, with exceptions of the Au6,8C clusters who have 2D structures. The analysis of the relative and electronic stabilities reveals that the Au4C and Au6 clusters are the most stable in the series of studied clusters, respectively. In addition, a natural bond orbital (NBO) analysis shows that the charges in the AunC clusters transfer from the Aun host to the C atom. Moreover, the Au and C atoms interact with each other mostly via covalent bond rather than ionic bond, which can be confirmed through the average ionic character of the Au–C bond. Meanwhile, the charges mainly transfer between 2s and 2p orbitals within the C atom, and among 5d, 6s, and 6p orbitals within the Au atom for the AunC clusters. As for the magnetic properties of the AunC clusters, the total magnetic moments are 1 μB for n = odd clusters, with the total magnetic moments mainly locating on the C atoms for Au1,3,9C and on the Aun host for Au5,7C clusters. However, the total magnetic moments of the AunC clusters are zero for n = even clusters. Simultaneously, the magnetic moments mainly locate on the 2p orbital within the C atom and on the 5d, 6s orbitals within the Au atom.
Co-reporter:Ya-Juan Feng, Teng Huang, Chao Wang, Yi-Rong Liu, Shuai Jiang, Shou-Kui Miao, Jiao Chen, and Wei Huang
The Journal of Physical Chemistry B 2016 Volume 120(Issue 27) pp:6667-6673
Publication Date(Web):June 9, 2016
DOI:10.1021/acs.jpcb.6b01180
Molecular level insight into the interaction between volatile organic compounds (VOCs) and aerosols is crucial for improvement of atmospheric chemistry models. In this paper, the interaction between adsorbed toluene, one of the most significant VOCs in the urban atmosphere, and the aqueous surface of aerosols was studied by means of combined molecular dynamics simulations and ab initio quantum chemistry calculations. It is revealed that toluene can be stably adsorbed on the surface of aqueous droplets via hydroxyl−π hydrogen bonding between the H atoms of the water molecules and the C atoms in the aromatic ring. Further, significant modifications on the electrostatic potential map and frontier molecular orbital are induced by the solvation effect of surface water molecules, which would affect the reactivity and pathway of the atmospheric photooxidation of toluene. This study demonstrates that the surface interactions should be taken into consideration in the atmospheric chemical models on oxidation of aromatics.
Co-reporter:Chun-Yu Wang, Yan Ma, Jiao Chen, Shuai Jiang, Yi-Rong Liu, Hui Wen, Ya-Juan Feng, Yu Hong, Teng Huang, and Wei Huang
The Journal of Physical Chemistry A 2016 Volume 120(Issue 15) pp:2357-2371
Publication Date(Web):March 21, 2016
DOI:10.1021/acs.jpca.5b11678
Amino acids are recognized as important components of atmospheric aerosols, which impact on the Earth’s climate directly and indirectly. However, much remains unknown about the initial events of nucleation. In this work, the interaction of alanine [NH2CH(CH3)COOH or Ala], one of the most abundant amino acids in the atmosphere, with sulfuric acid (SA) and water (W) has been investigated at the M06-2X/6-311++G(3df, 3pd) level of theory. We have studied thermodynamics of the hydrated (Ala)(SA) core system with up to four water molecules. We found that Ala, with one amino group and one carboxyl group, can interact with H2SO4 and H2O in two directions and that it has a high cluster stabilizing effect similar to that of ammonia, which is one of the key nucleation precursor. The corresponding Gibbs free energies of the (Ala)(SA)(W)n (n = 0–4) clusters formation at 298.15 K predicted that Ala can contribute to the stabilization of small binary clusters. Our results showed that the hydrate distribution is temperature-dependent and that a higher humidity and temperature can contribute to the formation of hydrated clusters.
Co-reporter:Kang-Ming Xu, Shuai Jiang, Yu-Peng Zhu, Teng Huang, Yi-Rong Liu, Yang Zhang, Yu-Zhou Lv and Wei Huang
RSC Advances 2015 vol. 5(Issue 33) pp:26071-26080
Publication Date(Web):02 Mar 2015
DOI:10.1039/C5RA00131E
Au2P3, the only metastable binary phase of gold phosphide, has been discovered to exhibit remarkable semiconductor properties among metal phosphides. A systematic study on the geometry, the transformation of Au2P3 into different valence states and the different interactions among the atoms of the species is performed by using the density functional theory (DFT) method. The global minimum of Au2P3− is a 3D structure with Cs symmetry. This structure could be distorted from a planar configuration of Au2P3 which decreases the steric effect on it and leads to a new stable configuration. An analogous planar configuration, a local minimum rather than a global minimum, is also found in Au2P3+, due to the electron effect acting on the structure. Natural bond orbital (NBO) analysis reveals the re-distribution progression of the charge within the species. The central located Au atom and another no. 5 positioned P atom play significant roles on the structures. P5, as an electron adjuster, balances the electron distribution at different valence states of the structures. Deformation density analysis supplies information about charge transfer and the bonding type between two adjacent atoms as well. Looking deep into the bonding types, as electron localization function (ELF) suggests, the interaction between two adjacent P atoms (P3 and P4) of Au2P3 belongs to a strong covalent bond. The Au–P interactions among the configurations could be classified as weak classical covalent bonds through the atoms in molecules (AIM) dual parameter analysis. And for the first time, the weak interaction between the two adjacent Au atoms (Au1 and Au2) of the charged states of Au2P3 (Au2P3− and Au2P3+), are verified and different from the neutral Au2P3 through the reduced density gradient (RDG) analysis.
Co-reporter:Shou-Kui Miao, Shuai Jiang, Jiao Chen, Yan Ma, Yu-Peng Zhu, Yang Wen, Miao-Miao Zhang and Wei Huang
RSC Advances 2015 vol. 5(Issue 60) pp:48638-48646
Publication Date(Web):26 May 2015
DOI:10.1039/C5RA06116D
Oxalic acid (OA), one of the most common organic acids in the Earth's atmosphere, is expected to enhance the nucleation and growth of nanoparticles containing sulfuric acid (SA) and water (W); however, the details about the hydration of OA–SA are poorly understood, especially for the larger clusters with more water molecules. We have investigated the structural characteristics and thermodynamics of these clusters using density functional theory at the PW91PW91/6-311++G(3df,3pd) level. The favorable free energies of formation and obvious concentrations of the OA–SA–Wn (n = 0–6) clusters at 298.15 K predict that oxalic acid can contribute to the aerosol nucleation process by binding to sulfuric acid and water until n = 6. There is strong temperature dependence for the complexes formation, and the energy order of these complexes is altered from 100 to 400 K, regardless of different cluster sizes or different isomers within the same cluster size. The lower temperature and higher relative humidity promote the formation of hydrates. Additionally, the investigation of acid dissociation predicts that several acid-dissociated models could coexist in the atmosphere, specifically when more water molecules are present. Fewer waters may be needed to cause the acid dissociation, as the relative acidity of the cluster increases, which plays a key role in forming relatively stable hydrated clusters of OA–SA. Finally, the Rayleigh scattering properties of OA–SA–Wn (n = 0–6) have been systematically investigated for the first time to further discuss its atmospheric implication.
Co-reporter:Jiao Chen, Shuai Jiang, Shou-Kui Miao, Xiu-Qiu Peng, Yan Ma, Chun-Yu Wang, Miao-Miao Zhang, Yi-Rong Liu and Wei Huang
RSC Advances 2015 vol. 5(Issue 111) pp:91500-91515
Publication Date(Web):08 Oct 2015
DOI:10.1039/C5RA11462D
Amines have been recognized as important precursor species in the formation of new atmospheric particles. Although dimethylamine–water clusters have been the focus of a large number of theoretical studies during the last few years, some information regarding these clusters, such as the influence of temperature, the analysis of their weak interactions, and their Rayleigh scattering properties, is still lacking. In this study, the equilibrium geometric structures and thermodynamics of (CH3)2NH(H2O)n (n = 1–6) clusters were systematically investigated using density functional theory (PW91PW91) coupled with the 6-311++G(3df,3pd) basis set. To determine the most stable isomer and the order of the different isomers, single-point calculations were executed using a two-point extrapolation method in conjunction with the complete basis set for all isomers. The optimized structures show that the addition of a fifth water molecule changes the most stable configuration from a quasi-planar ring structure to a cage-like configuration. Electron density analysis shows that the interactions of these complexes are mainly medium hydrogen bonds. The dependence on temperature of the conformational population and the Gibbs free energies of the (CH3)2NH(H2O)n (n = 1–6) clusters were determined with respect to temperature (200–300 K). A weak dependence on temperature was found for the formation of (CH3)2NH(H2O)n (n = 1–6) clusters. Dimethylamine–water clusters are favorable at low temperatures, but these clusters may be difficult to form because of the combined effect of Gibbs free energies with small negative values and the low relative concentration of dimethylamine in various atmospheric conditions, and this implies that dimethylamine–water clusters are difficult to form spontaneously in the atmosphere. Finally, the Rayleigh scattering properties of (CH3)2NH(H2O)n (n = 1–6) have been investigated systematically for the first time.
Co-reporter:Kang-Ming Xu, Teng Huang, Yi-Rong Liu, Shuai Jiang, Yang Zhang, Yu-Zhou Lv, Yan-Bo Gai, Wei Huang
Chemical Physics 2015 Volume 456() pp:13-21
Publication Date(Web):29 July 2015
DOI:10.1016/j.chemphys.2015.04.019
•Various 2D/3D structures of the doped clusters were examined.•The higher stability of AunP2- clusters relative to Aun+2- (n = 1–8) clusters was verified.•The possible evolutionary path of AunP2- (n = 1–8) clusters were explored.•The electronic properties of AunP2- (n = 1–8) were then investigated.The geometries of gold clusters doped with two phosphorus atoms, AunP2- (n = 1–8), were investigated using density functional theory (DFT) methods. Various two-dimensional (2D) and three-dimensional (3D) structures of the doped clusters were studied. The results indicate that the structures of dual-phosphorus-doped gold clusters exhibit large differences from those of pure gold clusters with small cluster sizes. In our study, as for Au6P2-, two cis–trans isomers were found. The global minimum of Au8P2- presents a similar configuration to that of Au20-, a pyramid-shaped unit, and the potential novel optical and catalytic properties of this structure warrant further attention. The higher stability of AunP2- clusters relative to Aun+2- (n = 1–8) clusters was verified based on various energy parameters, and the results indicate that the phosphorus atom can improve the stabilities of the gold clusters. We then explored the evolutionary path of AunP2- (n = 1–8) clusters. We found that AunP2- clusters exhibit the 2D–3D structural transition at n = 6, which is much clearer and faster than that of pure gold clusters and single-phosphorus-doped clusters. The electronic properties of AunP2- (n = 1–8) were then investigated. The photoelectron spectra provide additional fundamental information on the structures and molecular orbitals shed light on the evolution of AunP2- (n = 1–8). Natural bond orbital (NBO) described the charge distribution in stabilizing structures and revealed the strong relativistic effects of the gold atoms.
Co-reporter:Xiao-Xiao Lin, Yi-Rong Liu, Teng Huang, Jiao Chen, Shuai Jiang, Wei Huang
Chemical Physics 2015 s 450–451() pp: 21-31
Publication Date(Web):1–15 April 2015
DOI:10.1016/j.chemphys.2015.02.002
•We developed an efficient method called MCTSSM for transition states searching.•We demonstrated MCTSSM’s efficiency and its potential application in atmospheric reactions.•The MCTSSM simplified the time-consuming and tedious manual search procedure.The precise and rapid exploration of transition states (TSs) is a major challenge when studying atmospheric reactions due to their complexity. In this work, a Monte Carlo Transition State Search Method (MCTSSM), which integrates Monte Carlo sampling technique with transition state optimization methods using an efficient computer script, has been developed for transition state searches. The efficiency and the potential application in atmospheric reactions of this method have been demonstrated by three types of test suits related to the reactions of atmospheric volatile organic compounds (VOCs): (1) OH addition, (2) OH hydrogen-abstraction, and (3) the other reactive group (e.g. Cl, O3, NO3), especially for the reaction of β-pinene-sCI (stabilized Criegee Intermediates) with water. It was shown that the application of this method with effective restricted parameters has greatly simplified the time-consuming and tedious manual search procedure for transition state (TS) of the bimolecular reaction systems.
Co-reporter:Shi-Tu Pei, Shuai Jiang, Yi-Rong Liu, Teng Huang, Kang-Ming Xu, Hui Wen, Yu-Peng Zhu, and Wei Huang
The Journal of Physical Chemistry A 2015 Volume 119(Issue 12) pp:3035-3047
Publication Date(Web):March 3, 2015
DOI:10.1021/jp512323k
Although ammonium ion–water clusters are abundant in the biosphere, some information regarding these clusters, such as their growth route, the influence of temperature and humidity, and the concentrations of various hydrated clusters, is lacking. In this study, theoretical calculations are performed on ammonium ion–water clusters. These theoretical calculations are focused on determining the following characteristics: (1) the pattern of cluster growth; (2) the percentages of clusters of the same size at different temperatures and humidities; (3) the distributions of different isomers for the same size clusters at different temperatures; (4) the relative strengths of the noncovalent interactions for clusters of different sizes. The results suggest that the dipole moment may be very significant for the ammonium ion–water system, and some new stable isomers were found. The nucleation of ammonium ions and water molecules is favorable at low temperatures; thus, the clusters observed at high altitudes might not be present at low altitudes. High humidity can contribute to the formation of large ammonium ion–water clusters, whereas the formation of small clusters may be favorable under low-humidity conditions. The potential energy surfaces (PES) of these different sized clusters are complicated and differ according to the distribution of isomers at different temperatures. Some similar structures are observed between NH4+(H2O)n and M(H2O)n (where M represents an alkali metal ion or water molecule); when n = 8, the clusters begin to form the closed-cage geometry. As the cluster size increases, these interactions become progressively weaker. The successive binding energy at the DF-MP2-F12/VDZ-F12 level is better than that at the PW91PW91/6-311++G(3df, 3pd) level and is consistent with the experimentally determined values.
Co-reporter:Sha-Sha Lv, Yi-Rong Liu, Teng Huang, Ya-Juan Feng, Shuai Jiang, and Wei Huang
The Journal of Physical Chemistry A 2015 Volume 119(Issue 16) pp:3770-3779
Publication Date(Web):April 6, 2015
DOI:10.1021/acs.jpca.5b00616
Methylamine is the simplest aliphatic amine found in human urine, blood, and tissues. It is thought to play a significant part in central nervous system disturbances observed during renal and hepatic disease. In this work we have investigated the methylamine hydration clusters using a basin hopping (BH) algorithm with the density functional theory (DFT). The results presented herein yield a detailed understanding of the structure and stability for a system consisting of one methylamine molecule and up to seven waters: the most stable geometries arise from a fusion of tetramer or pentamer rings; by the geometrical parameters and topological parameters analysis, the strengths of the H2N···H–O hydrogen bonds of the global minima increase as the sizes of clusters increase, except for n = 5 where there is a slight fluctuation. This work may shed light on the form mechanism of methylamine existing in organisms and the hydration structures of larger molecules containing amino functional groups and their interaction with the water molecules nearby.
Co-reporter:Sha-Sha Lv, Shou-Kui Miao, Yan Ma, Miao-Miao Zhang, Yang Wen, Chun-Yu Wang, Yu-Peng Zhu, and Wei Huang
The Journal of Physical Chemistry A 2015 Volume 119(Issue 32) pp:8657-8666
Publication Date(Web):July 17, 2015
DOI:10.1021/acs.jpca.5b03325
The presence of amines can increase aerosol formation rates. Most studies have been devoted to dimethylamine as the representative of amine; however, there have been a few works devoted to methylamine. In this study, theoretical calculations are performed on CH3NH2(H2SO4)m(H2O)n (m = 0–3, n = 0–3) clusters. In addition to the structures and energetics, we focused on determining the following characteristics: (1) the growth mechanism, (2) the hydrate distributions and the influences of humidity and temperature, (3) Rayleigh scattering properties. We explored the cluster growth mechanism from a thermodynamics aspect by calculating the Gibbs free energy of adding a water or sulfuric acid molecule step by step at three atmospherically relevant temperatures. The relative ease of the reaction at each step is discussed. From the analysis of hydrate distributions, we find that CH3NH2(H2SO4)(H2O)2, CH3NH2(H2SO4)2, and CH3NH2(H2SO4)3 are most likely to exist in the atmosphere. The general trend of hydration in all cases is more extensive with the growing relative humidity (RH), whereas the distributions do not significantly change with the temperature. Analysis of the Rayleigh scattering properties showed that both H2SO4 and H2O molecules could increase the Rayleigh scattering intensities and isotropic mean polarizabilities, with greater influence by the sulfuric acid molecules. This work sheds light on the mechanism for further research on new particle formation (NPF) containing methylamine in the atmosphere.
Co-reporter:Shuai Jiang, Teng Huang, Yi-Rong Liu, Kang-Ming Xu, Yang Zhang, Yu-Zhou Lv and Wei Huang
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 36) pp:19241-19249
Publication Date(Web):29 Jul 2014
DOI:10.1039/C4CP02618G
Cl−(H2O)n (n = 5–6) clusters were investigated using a basin hopping (BH) method coupled with density functional theory (DFT). Structures, energetics, thermodynamics, and vibrational frequencies were obtained using high level ab initio calculations. DF-LMP2 (second-order Møller–Plesset perturbation theory using local and density fitting approximations) with an appropriate basis set were employed for final optimization and frequency calculation, which has been benchmarked in a recent study. The global minimum of Cl−(H2O)5 was verified and the new competitive local minimum of Cl−(H2O)6 was offered. Considering the increasing complexity of the large system and the high flexibility of the hydrogen bonding environment, Boltzmann averaged Gibbs free energy was provided taking into account the contributions of local minima on the potential energy surface. Finally, the temperature dependence of the conformational population for isomers of Cl−(H2O)n (n = 5–6) and Rayleigh scattering properties of Cl−(H2O)n (n = 1–6) have been investigated systematically for the first time.
Co-reporter:Hui Wen, Yi-Rong Liu, Kang-Ming Xu, Teng Huang, Chang-Jin Hu, Wei-Jun Zhang and Wei Huang
RSC Advances 2014 vol. 4(Issue 29) pp:15066-15076
Publication Date(Web):11 Mar 2014
DOI:10.1039/C3RA47873D
Gold sulfur clusters have received much attention because of the dramatic effect that the gold–sulfide interaction produces in thiol-passivated gold nanoparticles. We present a systematic theoretical study of the electronic properties and geometric structures of AuxS0,±1 (x = 1–10) clusters using the basin-hopping global optimization technique coupled with density functional theory (DFT-BH) methods. Higher-level ab initio calculations are performed to aid in structural assignment. The same species with different electric charges possess different configurations. The 2D-to-3D structural transitions of the global minimum structures of cationic, neutral, and anionic AuxS clusters are found at the sizes of x = 3, 6, and 9, respectively. It is found that the Au5S cluster can be regarded as the building-block unit for the evolution of larger Au–S clusters. The tendency toward planarity of each Au–S cluster species, which is similar to that of bare Au clusters, may be attributed to the strong relativistic effects of Au and the similar electronegativity between Au and S. The trends of the binding energies, electron affinities, and bond parameters with increasing cluster size are studied in detail for each species. The results demonstrate that the binding energies and second-order differences exhibit interesting oscillatory behaviors; it is believed that anionic clusters may be the most suitable for catalysis.
Co-reporter:Xiao-Xiao Lin, Yi-Rong Liu, Teng Huang, Kang-Ming Xu, Yang Zhang, Shuai Jiang, Yan-Bo Gai, Wei-Jun Zhang and Wei Huang
RSC Advances 2014 vol. 4(Issue 54) pp:28490-28498
Publication Date(Web):18 Jun 2014
DOI:10.1039/C4RA04172K
A theoretical study was performed of the reactions of the stabilized Criegee intermediates (sCIs) of β-pinene with H2O and its dimer. Due to the large size of the biogenic sCIs, the transition states of the hydration reactions were explored with the Monte Carlo Transition State Search Program (MCTSSP), which integrated the Monte Carlo sampling technique with a transition state optimization method. The computations were performed with the M06-2X/6-311+G(2d,p) and B3LYP/6-311+G(2d,p) levels of theory. The relative energies showed that the results of the M06-2X functional are in good agreement with the results of the DF-MP2 and CCSD(T) methods. Both the reactions of the β-pinene-sCI with H2O and the β-pinene-sCI with (H2O)2 were found to be strongly exothermic. Activation barrier calculations indicate that the sink reaction with the water dimer may proceed significantly faster than the reaction with the water monomer despite the low concentration of water dimers in the atmosphere. Therefore, the reaction of sCIs with water vapor that includes large water clusters rather than single water molecules should be studied.
Co-reporter:Yi-Rong Liu, Hui Wen, Teng Huang, Xiao-Xiao Lin, Yan-Bo Gai, Chang-Jin Hu, Wei-Jun Zhang, and Wei Huang
The Journal of Physical Chemistry A 2014 Volume 118(Issue 2) pp:508-516
Publication Date(Web):December 30, 2013
DOI:10.1021/jp4109128
Exploration of the low-lying structures of atomic or molecular clusters remains a fundamental problem in nanocluster science. Basin hopping is typically employed in conjunction with random motion, which is a perturbation of a local minimum structure. We have combined two different sampling technologies, “random sampling” and “compressed sampling”, to explore the potential energy surface of molecular clusters. We used the method to study water, nitrate/water, and oxalate/water cluster systems at the MP2/aug-cc-pVDZ level of theory. An isomer of the NO3–(H2O)3 cluster molecule with a 3D structure was lower in energy than the planar structure, which had previously been reported by experimental study as the lowest-energy structure. The lowest-energy structures of the NO3–(H2O)5 and NO3–(H2O)7 clusters were found to have structures similar to pure (H2O)8 and (H2O)10 clusters, which contradicts previous experimental result by Wang et al.(J. Chem. Phys. 2002, 116, 561–570). The new minimum energy structures for C2O42–(H2O)5 and C2O42–(H2O)6 are found by our calculations .
Co-reporter:Yu-Peng Zhu, Yi-Rong Liu, Teng Huang, Shuai Jiang, Kang-Ming Xu, Hui Wen, Wei-Jun Zhang, and Wei Huang
The Journal of Physical Chemistry A 2014 Volume 118(Issue 36) pp:7959-7974
Publication Date(Web):August 21, 2014
DOI:10.1021/jp506226z
While atmosphere is known to contain a significant fraction of organic substance and the effect of acetic acid to stabilize hydrated sulfuric acids is found to be close that of ammonia, the details about the hydration of (CH3COOH)(H2SO4)2 are poorly understood, especially for the larger clusters with more water molecules. We have investigated structural characteristics and thermodynamics of the hydrates using density functional theory (DFT) at PW91PW91/6-311++G(3df,3pd) level. The phenomena of the structural evolution may exist during the early stage of the clusters formation, and we tentatively proposed a calculation path for the Gibbs free energies of the clusters formation via the structural evolution. The results in this study supply a picture of the first deprotonation of sulfuric acids for a system consisting of two sulfuric acid molecules, an acetic acid molecule, and up to three waters at 0 and 298.15 K, respectively. We also replace one of the sulfuric acids with a bisulfate anion in (CH3COOH)(H2SO4)2 to explore the difference of acid dissociation between two series of clusters and interaction of performance in clusters growth between ion-mediated nucleation and organics-enhanced nucleation.
Co-reporter:Kang-Ming Xu, Teng Huang, Hui Wen, Yi-Rong Liu, Yan-Bo Gai, Wei-Jun Zhang and Wei Huang
RSC Advances 2013 vol. 3(Issue 46) pp:24492-24502
Publication Date(Web):14 Oct 2013
DOI:10.1039/C3RA43938K
The geometries of phosphorus-doped gold clusters, AunP− (n = 1–8), have been investigated using different density functionals and basis sets. B3LYP and PBE functionals with 4 basis sets (aug-cc-pVDZ, 6-311++G**, CRENBL ECP and LANL2DZ ECP) are chosen for geometry optimisation. Many low-lying structures are obtained for anionic AunP− clusters. For AunP− (n = 1–7) clusters, each level gives the same global minimum structure. It is found that the evolutionary path of phosphorus-doped gold clusters differs from that of pure gold clusters. Phosphorus atoms induce changes in the structure of pure gold clusters in small cluster sizes. Various 2D–3D structures of doped clusters are also investigated. Clusters with an odd number of gold atoms tend to yield planar 2D structures, while those with an even number of gold atoms tend to yield 3D structures.
Co-reporter:Hui Wen, Gao-Lei Hou, Yi-Rong Liu, Xue-Bin Wang and Wei Huang
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 26) pp:NaN17482-17482
Publication Date(Web):2016/05/31
DOI:10.1039/C6CP01542E
Bicarbonate plays a crucial biochemical role in the physiological pH buffering system and also has important atmospheric implications. In the current study, HCO3−(H2O)n (n = 0–13) clusters were successfully produced via electrospray ionization of the corresponding bulk salt solution, and were characterized by negative ion photoelectron spectroscopy and theoretical calculations. Photoelectron spectra reveal that the electron binding energy monotonically increases with the cluster size up to n = 10 and remains largely the same after n > 10. The photo-detaching feature of the solute HCO3− itself, which dominates in the small clusters, diminishes with the increase of water coverage. Based on the charge distribution and molecular orbital analyses, the universal high electron binding energy tail that dominates in the larger clusters can be attributed to the ionization of water. Thus, the transition of ionization from the solute to the solvent at a size larger than n = 10 has been observed. Extensive theoretical structural search based on the basin-hopping unbiased method was carried out, and a plethora of low energy isomers have been obtained for each medium and large-sized cluster. By comparing the simulated photoelectron spectra and calculated electron binding energies with the experiments, as well as by comparing the simulated infrared spectra with previously reported IR spectra, the best fit structures and the structural evolutionary routes are presented. The nature of bicarbonate–water interactions is mainly electrostatic as implied by electron localization function (ELF) analysis.
Co-reporter:Shuai Jiang, Teng Huang, Yi-Rong Liu, Kang-Ming Xu, Yang Zhang, Yu-Zhou Lv and Wei Huang
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 36) pp:NaN19249-19249
Publication Date(Web):2014/07/29
DOI:10.1039/C4CP02618G
Cl−(H2O)n (n = 5–6) clusters were investigated using a basin hopping (BH) method coupled with density functional theory (DFT). Structures, energetics, thermodynamics, and vibrational frequencies were obtained using high level ab initio calculations. DF-LMP2 (second-order Møller–Plesset perturbation theory using local and density fitting approximations) with an appropriate basis set were employed for final optimization and frequency calculation, which has been benchmarked in a recent study. The global minimum of Cl−(H2O)5 was verified and the new competitive local minimum of Cl−(H2O)6 was offered. Considering the increasing complexity of the large system and the high flexibility of the hydrogen bonding environment, Boltzmann averaged Gibbs free energy was provided taking into account the contributions of local minima on the potential energy surface. Finally, the temperature dependence of the conformational population for isomers of Cl−(H2O)n (n = 5–6) and Rayleigh scattering properties of Cl−(H2O)n (n = 1–6) have been investigated systematically for the first time.







![2,5-Bis(2-ethylhexyl)-3,6-di(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione](http://img.cochemist.com/ccimg/1185900/1185885-86-2.png)
![2,5-Bis(2-ethylhexyl)-3,6-di(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione](http://img.cochemist.com/ccimg/1185900/1185885-86-2_b.png)
![4,7-Dibromo-2-octyl-2H-benzo[d][1,2,3]triazole](http://img.cochemist.com/ccimg/960600/960509-83-5.png)
![4,7-Dibromo-2-octyl-2H-benzo[d][1,2,3]triazole](http://img.cochemist.com/ccimg/960600/960509-83-5_b.png)
![3,3'-(5'-(3-(Pyridin-3-yl)phenyl)-[1,1':3',1''-terphenyl]-3,3''-diyl)dipyridine](http://img.cochemist.com/ccimg/921300/921205-03-0.png)
![3,3'-(5'-(3-(Pyridin-3-yl)phenyl)-[1,1':3',1''-terphenyl]-3,3''-diyl)dipyridine](http://img.cochemist.com/ccimg/921300/921205-03-0_b.png)



































![2-Octyl-2H-benzo[d][1,2,3]triazole](http://img.cochemist.com/ccimg/112700/112642-69-0.png)
![2-Octyl-2H-benzo[d][1,2,3]triazole](http://img.cochemist.com/ccimg/112700/112642-69-0_b.png)
![Poly[imino-1,5-naphthalenediyliminocarbonyl[(ethoxycarbonyl)phenylen
e]carbonyl[(ethoxycarbonyl)phenylene]carbonyl]](http://img.cochemist.com/ccimg/106500/106493-93-0.png)
![Poly[imino-1,5-naphthalenediyliminocarbonyl[(ethoxycarbonyl)phenylen
e]carbonyl[(ethoxycarbonyl)phenylene]carbonyl]](http://img.cochemist.com/ccimg/106500/106493-93-0_b.png)







![Benzene, 1,1'-[1,8-octanediylbis(oxy)]bis[4-nitro-](http://img.cochemist.com/ccimg/94700/94680-04-3.png)
![Benzene, 1,1'-[1,8-octanediylbis(oxy)]bis[4-nitro-](http://img.cochemist.com/ccimg/94700/94680-04-3_b.png)
![ETHANOL, 2-[ETHYL[4-[(6-NITRO-2-BENZOTHIAZOLYL)AZO]PHENYL]AMINO]-](http://img.cochemist.com/ccimg/91800/91754-59-5.png)
![ETHANOL, 2-[ETHYL[4-[(6-NITRO-2-BENZOTHIAZOLYL)AZO]PHENYL]AMINO]-](http://img.cochemist.com/ccimg/91800/91754-59-5_b.png)
![Benzenamine,4,4'-[1,8-octanediylbis(oxy)]bis- (9CI)](http://img.cochemist.com/ccimg/90100/90076-88-3.png)
![Benzenamine,4,4'-[1,8-octanediylbis(oxy)]bis- (9CI)](http://img.cochemist.com/ccimg/90100/90076-88-3_b.png)


![3,6-bis(4-bromophenyl)-2,5-dihydro-Pyrrolo[3,4-c]pyrrole-1,4-dione](/data/chemimg/1637500/84632-54-2.png)
![3,6-bis(4-bromophenyl)-2,5-dihydro-Pyrrolo[3,4-c]pyrrole-1,4-dione](/data/chemimg/1637500/84632-54-2_b.png)


![cis-()-1-[[2-(2,4-dichlorophenyl)-4-[(prop-2-ynyloxy)methyl]-1,3-dioxolan-2-yl]methyl]-1H-imidazole](http://img.cochemist.com/ccimg/68700/68685-54-1.png)
![cis-()-1-[[2-(2,4-dichlorophenyl)-4-[(prop-2-ynyloxy)methyl]-1,3-dioxolan-2-yl]methyl]-1H-imidazole](http://img.cochemist.com/ccimg/68700/68685-54-1_b.png)










![methyl N-ethyl-N-[4-[(5-nitro-2,1-benzisothiazol-3-yl)azo]phenyl]-β-alaninate](http://img.cochemist.com/ccimg/52300/52239-04-0.png)
![methyl N-ethyl-N-[4-[(5-nitro-2,1-benzisothiazol-3-yl)azo]phenyl]-β-alaninate](http://img.cochemist.com/ccimg/52300/52239-04-0_b.png)
![4-[6-(4-aminophenoxy)hexoxy]aniline](http://img.cochemist.com/ccimg/47300/47244-09-7.png)
![4-[6-(4-aminophenoxy)hexoxy]aniline](http://img.cochemist.com/ccimg/47300/47244-09-7_b.png)
























![1-NITRO-4-[4-(4-NITROPHENOXY)BUTOXY]BENZENE](http://img.cochemist.com/ccimg/14500/14467-68-6.png)
![1-NITRO-4-[4-(4-NITROPHENOXY)BUTOXY]BENZENE](http://img.cochemist.com/ccimg/14500/14467-68-6_b.png)
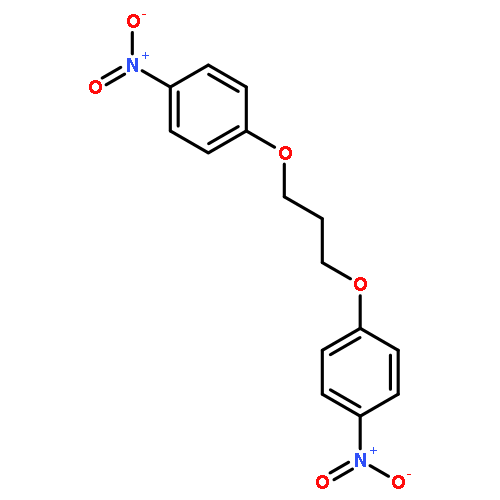
![1-NITRO-4-[5-(4-NITROPHENOXY)PENTOXY]BENZENE](http://img.cochemist.com/data/images/14467-60-8.png)
![1-NITRO-4-[5-(4-NITROPHENOXY)PENTOXY]BENZENE](http://img.cochemist.com/data/images/14467-60-8_b.png)
![Benzoxazolium,3-methyl-2-[3-(3-methyl-2(3H)-benzoxazolylidene)-1-propen-1-yl]-, iodide (1:1)](http://img.cochemist.com/ccimg/14200/14134-79-3.png)
![Benzoxazolium,3-methyl-2-[3-(3-methyl-2(3H)-benzoxazolylidene)-1-propen-1-yl]-, iodide (1:1)](http://img.cochemist.com/ccimg/14200/14134-79-3_b.png)









































![Bicyclo[2.2.1]heptane,2-ethyl-](http://img.cochemist.com/ccimg/2200/2146-41-0.png)
![Bicyclo[2.2.1]heptane,2-ethyl-](http://img.cochemist.com/ccimg/2200/2146-41-0_b.png)








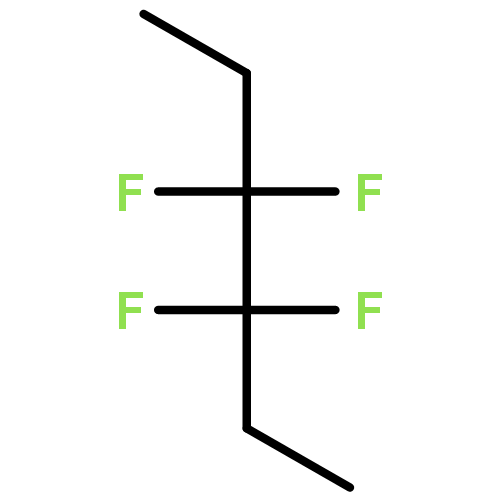








![[2-[(8s,9s,10r,13s,14s,17r)-17-hydroxy-10,13-dimethyl-3,11-dioxo-1,2,6,7,8,9,12,14,15,16-decahydrocyclopenta[a]phenanthren-17-yl]-2-oxoethyl] Acetate](http://img.cochemist.com/ccimg/100/50-04-4.png)
![[2-[(8s,9s,10r,13s,14s,17r)-17-hydroxy-10,13-dimethyl-3,11-dioxo-1,2,6,7,8,9,12,14,15,16-decahydrocyclopenta[a]phenanthren-17-yl]-2-oxoethyl] Acetate](http://img.cochemist.com/ccimg/100/50-04-4_b.png)












![Methanone, bis[4-(diphenylamino)phenyl]-](http://img.cochemist.com/ccimg/2900/2873-76-9.png)
![Methanone, bis[4-(diphenylamino)phenyl]-](http://img.cochemist.com/ccimg/2900/2873-76-9_b.png)

