Co-reporter:Christopher G. McPherson, Alasdair K. Cooper, Andrius Bubliauskas, Paul Mulrainey, Craig Jamieson, and Allan J. B. Watson
Organic Letters December 15, 2017 Volume 19(Issue 24) pp:6736-6736
Publication Date(Web):December 1, 2017
DOI:10.1021/acs.orglett.7b03470
An organobase-mediated multicomponent reaction of unactivated esters, epoxides, and amines is reported, furnishing functionalized amide derivatives. A wide range of substrates are tolerated under the reaction conditions, including chiral epoxides, which react with no erosion of enantiopurity. Facile modification of the method through replacing the ester derivative with dimethyl carbonate enables access to the corresponding oxazolidinone derivatives.
Co-reporter:Lisa M. Miller, John M. Pritchard, Simon J. F. Macdonald, Craig Jamieson, and Allan J. B. Watson
Journal of Medicinal Chemistry April 27, 2017 Volume 60(Issue 8) pp:3241-3241
Publication Date(Web):January 30, 2017
DOI:10.1021/acs.jmedchem.6b01711
The RGD integrins are recognized therapeutic targets for thrombosis, fibrosis, and cancer, among others. Current inhibitors are designed to mimic the tripeptide sequence (arginine–glycine–aspartic acid) of the natural ligands; however, the RGD-mimetic antagonists for αIIbβ3 have been shown to cause partial agonism, leading to the opposite pharmacological effect. The challenge of obtaining oral activity and synthetic tractability with RGD-mimetic molecules, along with the issues relating to pharmacology, has left integrin therapeutics in need of a new strategy. Recently, a new generation of inhibitor has emerged that lacks the RGD-mimetic. This review will discuss the discovery of these non-RGD-mimetic inhibitors and the progress that has been made in this promising new chemotype.
Co-reporter:Lisa M. Miller, Willem-Jan Keune, Diana Castagna, Louise C. Young, Emma L. Duffy, Frances Potjewyd, Fernando Salgado-Polo, Paloma Engel García, Dima Semaan, John M. Pritchard, Anastassis Perrakis, Simon J. F. Macdonald, Craig JamiesonAllan J. B. Watson
Journal of Medicinal Chemistry 2017 Volume 60(Issue 2) pp:
Publication Date(Web):December 16, 2016
DOI:10.1021/acs.jmedchem.6b01597
Autotaxin (ATX) is a secreted enzyme responsible for the hydrolysis of lysophosphatidylcholine (LPC) to the bioactive lysophosphatidic acid (LPA) and choline. The ATX-LPA signaling pathway is implicated in cell survival, migration, and proliferation; thus, the inhibition of ATX is a recognized therapeutic target for a number of diseases including fibrotic diseases, cancer, and inflammation, among others. Many of the developed synthetic inhibitors for ATX have resembled the lipid chemotype of the native ligand; however, a small number of inhibitors have been described that deviate from this common scaffold. Herein, we report the structure–activity relationships (SAR) of a previously reported small molecule ATX inhibitor. We show through enzyme kinetics studies that analogues of this chemotype are noncompetitive inhibitors, and by using a crystal structure with ATX we confirm the discrete binding mode.
Co-reporter:Christopher G. McPherson;Nicola Caldwell;Iain Simpson;Allan J. B. Watson
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 16) pp:3507-3518
Publication Date(Web):2017/04/18
DOI:10.1039/C7OB00593H
A catalytic amidation protocol mediated by 2,2,2-trifluoroethanol has been developed, facilitating the condensation of unactivated esters and amines, furnishing both secondary and tertiary amides. The complete scope and limitations of the method are described, along with modified conditions for challenging substrates such as acyclic secondary amines and chiral esters with retention of chiral integrity.
Co-reporter:Willem-Jan Keune, Frances Potjewyd, Tatjana Heidebrecht, Fernando Salgado-Polo, Simon J. F. MacdonaldLakshman Chelvarajan, Ahmed Abdel Latif, Sony Soman, Andrew J. Morris, Allan J. B. Watson, Craig JamiesonAnastassis Perrakis
Journal of Medicinal Chemistry 2017 Volume 60(Issue 5) pp:
Publication Date(Web):February 6, 2017
DOI:10.1021/acs.jmedchem.6b01743
Autotaxin produces the bioactive lipid lysophosphatidic acid (LPA) and is a drug target of considerable interest for numerous pathologies. We report the expedient, structure-guided evolution of weak physiological allosteric inhibitors (bile salts) into potent competitive Autotaxin inhibitors that do not interact with the catalytic site. Functional data confirms that our lead compound attenuates LPA mediated signaling in cells and reduces LPA synthesis in vivo, providing a promising natural product derived scaffold for drug discovery.
Co-reporter:Diana Castagna; David C. Budd; Simon J. F. Macdonald; Craig Jamieson;Allan J. B. Watson
Journal of Medicinal Chemistry 2016 Volume 59(Issue 12) pp:5604-5621
Publication Date(Web):January 8, 2016
DOI:10.1021/acs.jmedchem.5b01599
The autotaxin–lysophophatidic acid (ATX–LPA) signaling pathway is implicated in a variety of human disease states including angiogenesis, autoimmune diseases, cancer, fibrotic diseases, inflammation, neurodegeneration, and neuropathic pain, among others. As a result, ATX–LPA has become of significant interest within both the industrial and the academic communities. This review aims to provide a concise overview of the development of novel ATX inhibitors, including the disclosure of the first ATX clinical trial data.
Co-reporter:Nicola Caldwell, Craig Jamieson, Iain Simpson and Allan J. B. Watson
Chemical Communications 2015 vol. 51(Issue 46) pp:9495-9498
Publication Date(Web):08 May 2015
DOI:10.1039/C5CC02895G
A catalytic amidation method has been developed, employing 2,2,2-trifluoroethanol to facilitate condensation of unactivated esters and amines, enabling the synthesis of a range of amide products in good to excellent yields. Mechanistic studies indicate the reaction proceeds through a trifluoroethanol-derived active ester intermediate.
Co-reporter:Nicola Caldwell, Jonathan E. Harms, Kathryn M. Partin, and Craig Jamieson
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 4) pp:392
Publication Date(Web):February 11, 2015
DOI:10.1021/ml5004553
The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors are a family of glutamate ion channels of considerable interest in excitatory neurotransmission and associated disease processes. Here, we demonstrate how exploitation of the available X-ray crystal structure of the receptor ligand binding domain enabled the development of a new class of AMPA receptor positive allosteric modulators (7) through hybridization of known ligands (5 and 6), leading to a novel chemotype with promising pharmacological properties.Keywords: AMPA receptor; electrophysiology; hybridization; structure-based drug design
Co-reporter:Diana Castagna, Emma L. Duffy, Dima Semaan, Louise C. Young, John M. Pritchard, Simon J. F. Macdonald, David C. Budd, Craig Jamieson and Allan J. B. Watson
MedChemComm 2015 vol. 6(Issue 6) pp:1149-1155
Publication Date(Web):08 May 2015
DOI:10.1039/C5MD00081E
Three novel series were generated in order to mimic the pharmacophoric features displayed by lead compound AM095, a lysophosphatidic acid (LPA1) receptor antagonist. Biological evaluation of this array of putative LPA1antagonists led us to the discovery of three novel series of inhibitors of the ectoenzyme autotaxin (ATX), responsible for LPA production in blood, with potencies in the range of 1–4 μM together with good (>100 μg mL−1) solubility.
Co-reporter:Diana Castagna, Emma L. Duffy, Dima Semaan, Louise C. Young, John M. Pritchard, Simon J. F. Macdonald, David C. Budd, Craig Jamieson and Allan J. B. Watson
MedChemComm 2015 vol. 6(Issue 8) pp:1575-1575
Publication Date(Web):07 Jul 2015
DOI:10.1039/C5MD90032H
Correction for ‘Identification of a novel class of autotaxin inhibitors through cross-screening’ by Diana Castagna et al., Med. Chem. Commun., 2015, 6, 1149–1155.
Co-reporter:Nicola Caldwell, Peter S. Campbell, Craig Jamieson, Frances Potjewyd, Iain Simpson, and Allan J. B. Watson
The Journal of Organic Chemistry 2014 Volume 79(Issue 19) pp:9347-9354
Publication Date(Web):September 7, 2014
DOI:10.1021/jo501929c
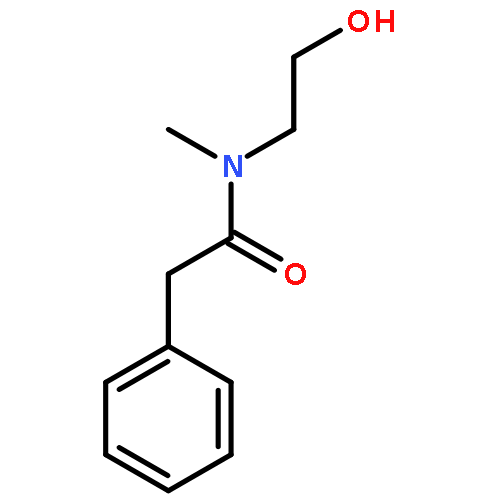
A catalytic protocol for the base-mediated amidation of unactivated esters with amino alcohol derivatives is reported. Investigations into mechanistic aspects of the process indicate that the reaction involves an initial transesterification, followed by an intramolecular rearrangement. The reaction is highly general in nature and can be extended to include the synthesis of oxazolidinone systems through use of dimethyl carbonate.
Co-reporter:Nicola Caldwell, Craig Jamieson, Iain Simpson, and Tell Tuttle
Organic Letters 2013 Volume 15(Issue 10) pp:2506-2509
Publication Date(Web):May 2, 2013
DOI:10.1021/ol400987p
A base-mediated procedure for the amidation of unactivated esters with amino alcohols is reported. Optimization and exemplification of the catalytic process are described, furnishing products in 40–100% isolated yield.
Co-reporter:Nicola Caldwell, Craig Jamieson, Iain Simpson, and Allan J. B. Watson
ACS Sustainable Chemistry & Engineering 2013 Volume 1(Issue 10) pp:1339
Publication Date(Web):July 30, 2013
DOI:10.1021/sc400204g
We describe the development of a sustainable ester amidation process. Base and solvent screening, combined with the application of Design of Experiments methodology was employed to identify an optimized set of reaction conditions using a sustainable protocol. Utilizing these optimized conditions, treatment of a range of ester derivatives with amino alcohols in the presence of a catalytic quantity of potassium phosphate deploying iso-propanol as solvent results in the highly efficient generation of a range of amido-alcohol derivatives in good to excellent yield, accompanied with excellent reaction mass efficiency (RME).Keywords: Amidation; Design of Experiments; Green solvents; Reaction screening; Replacement bases
Co-reporter:Craig Jamieson, John K.F. Maclean, Christopher I. Brown, Robert A. Campbell, Kevin J. Gillen, Jonathan Gillespie, Bert Kazemier, Michael Kiczun, Yvonne Lamont, Amanda J. Lyons, Elizabeth M. Moir, John A. Morrow, John Pantling, Zoran Rankovic, Lynn Smith
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 2) pp:805-811
Publication Date(Web):15 January 2011
DOI:10.1016/j.bmcl.2010.11.098
Starting from compound 1, we utilized biostructural data to successfully evolve an existing series into a new chemotype with a promising overall profile, exemplified by 19.The evolution of an advanced lead compound 19 through a scaffold hopping approach guided by structure-based drug design is described.
Co-reporter:Simon J. A. Grove ; Craig Jamieson ; John K. F. Maclean ; John A. Morrow ;Zoran Rankovic
Journal of Medicinal Chemistry 2010 Volume 53(Issue 20) pp:7271-7279
Publication Date(Web):September 14, 2010
DOI:10.1021/jm1000419
Co-reporter:Craig Jamieson, Robert A. Campbell, Iain A. Cumming, Kevin J. Gillen, Jonathan Gillespie, Bert Kazemier, Michael Kiczun, Yvonne Lamont, Amanda J. Lyons, John K.F. Maclean, Frederic Martin, Elizabeth M. Moir, John A. Morrow, John Pantling, Zoran Rankovic, Lynn Smith
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 20) pp:6072-6075
Publication Date(Web):15 October 2010
DOI:10.1016/j.bmcl.2010.08.063
Starting from lead compound 1, we demonstrate how X-ray structural data can be used to understand SAR and expediently optimize bioavailability in a novel series of AMPA receptor modulators, furnishing 5 with improved bioavailability and robust in vivo activity.The identification of an advanced lead compound 5 guided by structure based drug design is described.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Craig Jamieson, Stephanie Basten, Robert A. Campbell, Iain A. Cumming, Kevin J. Gillen, Jonathan Gillespie, Bert Kazemier, Michael Kiczun, Yvonne Lamont, Amanda J. Lyons, John K.F. Maclean, Elizabeth M. Moir, John A. Morrow, Marianthi Papakosta, Zoran Rankovic, Lynn Smith
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 19) pp:5753-5756
Publication Date(Web):1 October 2010
DOI:10.1016/j.bmcl.2010.07.138
Starting from an HTS derived hit 1, application of biostructural data facilitated rapid optimization to lead 22, a novel AMPA receptor modulator. This is the first demonstration of how structure based drug design can be exploited in an optimization program for a glutamate receptor.Starting from an HTS hit, the evolution of lead compound 22, a positive allosteric modulator of the AMPA receptor is described using structure based drug design.
Co-reporter:Nicola Caldwell, Craig Jamieson, Iain Simpson and Allan J. B. Watson
Chemical Communications 2015 - vol. 51(Issue 46) pp:NaN9498-9498
Publication Date(Web):2015/05/08
DOI:10.1039/C5CC02895G
A catalytic amidation method has been developed, employing 2,2,2-trifluoroethanol to facilitate condensation of unactivated esters and amines, enabling the synthesis of a range of amide products in good to excellent yields. Mechanistic studies indicate the reaction proceeds through a trifluoroethanol-derived active ester intermediate.
Co-reporter:Christopher G. McPherson, Nicola Caldwell, Craig Jamieson, Iain Simpson and Allan J. B. Watson
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 16) pp:NaN3518-3518
Publication Date(Web):2017/04/04
DOI:10.1039/C7OB00593H
A catalytic amidation protocol mediated by 2,2,2-trifluoroethanol has been developed, facilitating the condensation of unactivated esters and amines, furnishing both secondary and tertiary amides. The complete scope and limitations of the method are described, along with modified conditions for challenging substrates such as acyclic secondary amines and chiral esters with retention of chiral integrity.






















![BENZENEACETAMIDE, N-[2-HYDROXY-1-(HYDROXYMETHYL)ETHYL]-](http://img.cochemist.com/ccimg/223600/223560-31-4.png)
![BENZENEACETAMIDE, N-[2-HYDROXY-1-(HYDROXYMETHYL)ETHYL]-](http://img.cochemist.com/ccimg/223600/223560-31-4_b.png)

















![(7aS)-tetrahydro-1H,3H-Pyrrolo[1,2-c]oxazol-3-one](http://img.cochemist.com/ccimg/158700/158690-57-4.png)
![(7aS)-tetrahydro-1H,3H-Pyrrolo[1,2-c]oxazol-3-one](http://img.cochemist.com/ccimg/158700/158690-57-4_b.png)














![(1S,2S)-2-ethyl-1-({[(3aS,6R,7aS)-6-ethyl-1-oxo-2,3,3a,6,7,7a-hexahydro-1H-inden-4-yl]carbonyl}amino)cyclopropanecarboxylic acid](http://img.cochemist.com/ccimg/62300/62251-96-1.png)
![(1S,2S)-2-ethyl-1-({[(3aS,6R,7aS)-6-ethyl-1-oxo-2,3,3a,6,7,7a-hexahydro-1H-inden-4-yl]carbonyl}amino)cyclopropanecarboxylic acid](http://img.cochemist.com/ccimg/62300/62251-96-1_b.png)



![Benzamide, N-[2-(benzoyloxy)ethyl]-](http://img.cochemist.com/ccimg/16200/16180-99-7.png)
![Benzamide, N-[2-(benzoyloxy)ethyl]-](http://img.cochemist.com/ccimg/16200/16180-99-7_b.png)





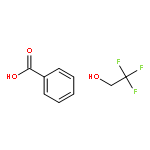
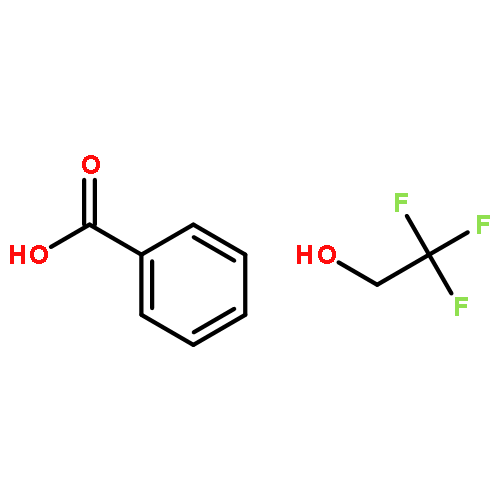


![N-[4-nitro-3-(trifluoromethyl)phenyl]benzamide](http://img.cochemist.com/ccimg/117400/117367-10-9.png)
![N-[4-nitro-3-(trifluoromethyl)phenyl]benzamide](http://img.cochemist.com/ccimg/117400/117367-10-9_b.png)




![2-{[(4-bromophenyl)acetyl]amino}benzamide](http://img.cochemist.com/ccimg/349500/349430-83-7.png)
![2-{[(4-bromophenyl)acetyl]amino}benzamide](http://img.cochemist.com/ccimg/349500/349430-83-7_b.png)


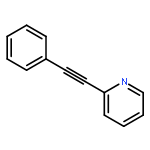
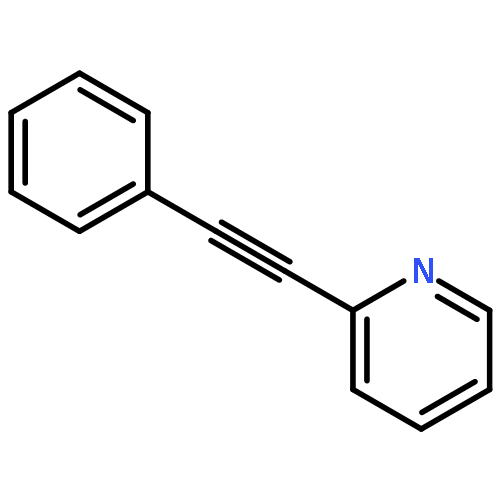
![N-[4-(trifluoromethyl)benzyl]propan-1-amine](http://img.cochemist.com/ccimg/90400/90390-13-9.png)
![N-[4-(trifluoromethyl)benzyl]propan-1-amine](http://img.cochemist.com/ccimg/90400/90390-13-9_b.png)
![1H-Indole, 1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/107800/107734-06-5.png)
![1H-Indole, 1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/107800/107734-06-5_b.png)


![2-[4-[4-[4-[[(1R)-1-(2-CHLOROPHENYL)ETHOXY]CARBONYLAMINO]-3-METHYL-1,2-OXAZOL-5-YL]PHENYL]PHENYL]ACETIC ACID](http://img.cochemist.com/ccimg/1228700/1228690-19-4.png)
![2-[4-[4-[4-[[(1R)-1-(2-CHLOROPHENYL)ETHOXY]CARBONYLAMINO]-3-METHYL-1,2-OXAZOL-5-YL]PHENYL]PHENYL]ACETIC ACID](http://img.cochemist.com/ccimg/1228700/1228690-19-4_b.png)


![Benzamide, N-[(4-methoxyphenyl)methyl]-4-methyl-](http://img.cochemist.com/ccimg/288200/288154-96-1.png)
![Benzamide, N-[(4-methoxyphenyl)methyl]-4-methyl-](http://img.cochemist.com/ccimg/288200/288154-96-1_b.png)










![Benzeneacetamide, N-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/305900/305849-49-4.png)
![Benzeneacetamide, N-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/305900/305849-49-4_b.png)


![2l5-1,3,2-Diazaphosphorin-2-amine,2-[(1,1-dimethylethyl)imino]-N,N-diethylhexahydro-1,3-dimethyl-](http://img.cochemist.com/ccimg/98100/98015-45-3.png)
![2l5-1,3,2-Diazaphosphorin-2-amine,2-[(1,1-dimethylethyl)imino]-N,N-diethylhexahydro-1,3-dimethyl-](http://img.cochemist.com/ccimg/98100/98015-45-3_b.png)










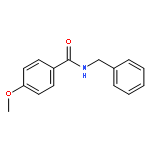
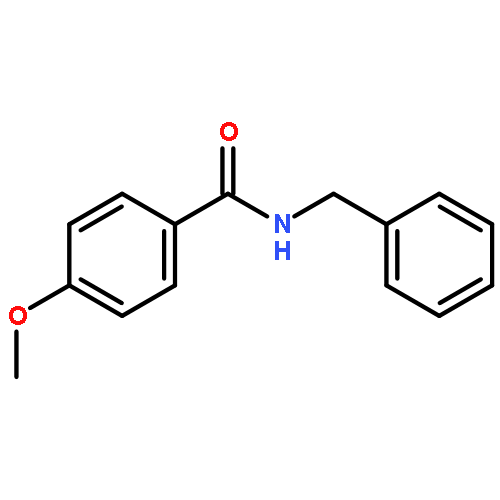
![3H-SPIRO[1-BENZOFURAN-2,4''-PIPERIDINE]](http://img.cochemist.com/ccimg/72000/71916-73-9.png)
![3H-SPIRO[1-BENZOFURAN-2,4''-PIPERIDINE]](http://img.cochemist.com/ccimg/72000/71916-73-9_b.png)
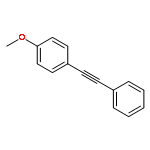
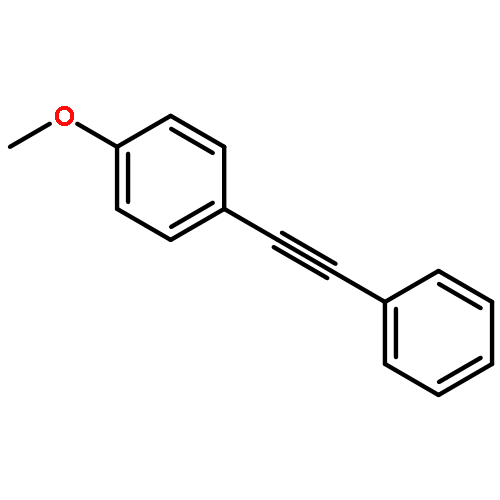



















![SODIUM;2-[4-[4-[3-METHYL-4-[[(1R)-1-PHENYLETHOXY]CARBONYLAMINO]-1,2-OXAZOL-5-YL]PHENYL]PHENYL]ACETATE](http://img.cochemist.com/ccimg/1345700/1345614-59-6.png)
![SODIUM;2-[4-[4-[3-METHYL-4-[[(1R)-1-PHENYLETHOXY]CARBONYLAMINO]-1,2-OXAZOL-5-YL]PHENYL]PHENYL]ACETATE](http://img.cochemist.com/ccimg/1345700/1345614-59-6_b.png)




![[(3-chlorophenyl)hydrazono]malononitrile](http://img.cochemist.com/ccimg/600/555-60-2.png)
![[(3-chlorophenyl)hydrazono]malononitrile](http://img.cochemist.com/ccimg/600/555-60-2_b.png)






![3,5-Dichlorobenzyl 4-(3-oxo-3-(2-oxo-2,3-dihydrobenzo[d]oxazol-6-yl)propyl)piperazine-1-carboxylate](http://img.cochemist.com/ccimg/1144100/1144035-53-9.png)
![3,5-Dichlorobenzyl 4-(3-oxo-3-(2-oxo-2,3-dihydrobenzo[d]oxazol-6-yl)propyl)piperazine-1-carboxylate](http://img.cochemist.com/ccimg/1144100/1144035-53-9_b.png)










![Benzo[g]pteridine-2,4(1H,3H)-dione](http://img.cochemist.com/ccimg/500/490-59-5.png)
![Benzo[g]pteridine-2,4(1H,3H)-dione](http://img.cochemist.com/ccimg/500/490-59-5_b.png)